
Photocatalytic Shono-type oxidation of N-alkylamides with hydrogen evolution†
Keitaro Kato,
Shintaro Okumura and
Naoki Ishida*
and
Naoki Ishida*
Department of Synthetic Chemistry and Biological Chemistry, Kyoto University, Katsura, Kyoto 615-8510, Japan. E-mail: ishida.naoki.5m@kyoto-u.ac.jp
First published on 11th July 2025
Abstract
A C–H alkoxylation reaction of N-alkylamides to N-acyl-N,O-acetals provides a straightforward and versatile method for functionalizing the α-position of amides. This reaction has been conventionally carried out by an electrochemical method (Shono oxidation). Although photoinduced variants are also known, they require sacrificial oxidants and/or the pre-functionalization of the amide substrates. We herein report a photoinduced reaction catalysed by an iridium photosensitizer and nickel bromide that eliminates the need for sacrificial oxidants and the pre-installation of reactive handles on the substrates.
A C–H alkoxylation reaction of N-alkylamides to N-acyl-N,O-acetals is a versatile step for the α-functionalization of amides and has been employed in the synthesis of complex targets.1,2 The conventional method, Shono oxidation, is an electrolysis via direct electron transfer from the amide substrates.3 However, this direct electron transfer requires high oxidation potential, significantly limiting the substrate scope. To address this limitation, a variant using mediators has been developed.4,5 The mediator facilitates hydride transfer4 or single electron transfer5 from the amide substrates, enabling functional group tolerance distinct from the original, direct electron transfer method. Alternative approaches are transition-metal catalysed6 and photocatalysed methods.7 While several examples within these approaches have previously emerged,6,7 they require the pre-functionalization of the amide substrates or sacrificial oxidants.
On the other hand, we have been involved in the development of photocatalytic dehydrogenative coupling reactions.8 These coupling reactions proceed without the need for pre-installed reactive handles. In addition, sacrificial oxidants are not required, and gaseous hydrogen evolves as a byproduct. Our ongoing research in this area has recently led us to the discovery of a photocatalytic C–H methoxylation reaction of N-alkylamides with methanol, furnishing N-acyl-N,O-acetals along with the evolution of gaseous hydrogen. Here, we report this photocatalytic variant of the Shono-type oxidation reaction, which dispenses with the pre-installation of reactive handles on the substrates.
A typical example is depicted in entry 1, Table 1. Amide 1a (0.20 mmol) was reacted with methanol (20 equiv.) in the presence of an iridium photoredox catalyst (Ir cat., 2 mol%), NiBr2(dme) (5 mol%), and rac-BINAP (6 mol%) in AcOEt (2.0 mL) under visible light irradiation and a nitrogen atmosphere for 24 h. The N-acyl-N,O-acetal 2a was produced in 76% yield. Purification by column chromatography on silica gel afforded analytically pure 2a in 69% yield. A GC analysis of the headspace of the reaction vessel confirmed quantitative formation of gaseous hydrogen, and the reaction under an oxygen atmosphere yielded <5% of 2a. The use of ethanol in place of methanol resulted in the recovery of 1a (76%), and the yield of the corresponding N-acyl-N,O-acetal was only 13%. Isopropyl and t-butyl alcohol failed to produce the corresponding N-acyl-N,O-acetals. An extensive screening of ligands disclosed that the choice was critical. While triphenylphosphine was totally ineffective (entry 2), bidentate phosphine ligands such as DPPP and DPEphos gave 2a in good yield (entries 3 and 4). An N-heterocyclic carbene ligand afforded the product, albeit in low yield (entry 5). Control experiments showed that visible light irradiation, the iridium photoredox catalyst, and the nickel bromide catalyst were all essential (entries 6–8). It should be noted that a bromide ion was also an indispensable component. Whereas the use of Ni(OAc)2·4H2O as the nickel catalyst resulted in no product at all (entry 10), addition of tetrabutylammonium bromide restored the reactivity to give 2a (entry 11).
|
|||
|---|---|---|---|
| Entry | Ni salt | Ligand | Yield of 2ab (%) |
a Reaction conditions: 1a (0.20 mmol), MeOH (4.0 mmol, 20 equiv.), Ir cat. (0.004 mmol, 2 mol%), Ni salt (0.1 mmol, 5 mol%), ligands (0.012 mmol, 6 mol%), AcOEt (2.0 mL), irradiated with blue LEDs (425 nm) at rt under a nitrogen atmosphere for 24 h.b Determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. Isolated yields are in parentheses.c 12 mol%.d Without light irradiation.e Without Ir cat.f n-Bu4NBr (10 mol%) was added. |
|||
| 1 | NiBr2(dme) | rac-BINAP | 76 (69) |
| 2 | NiBr2(dme) | PPh3c | <1 |
| 3 | NiBr2(dme) | DPPP | 60 |
| 4 | NiBr2(dme) | DPEphos | 44 |
| 5 | NiBr2(dme) | IPr | 14 |
| 6d | NiBr2(dme) | rac-BINAP | 0 |
| 7e | NiBr2(dme) | rac-BINAP | <1 |
| 8 | None | rac-BINAP | 0 |
| 9 | NiBr2(dme) | None | 20 |
| 10 | Ni(OAc)4·H2O | rac-BINAP | 0 |
| 11f | Ni(OAc)4·H2O | rac-BINAP | 17 |

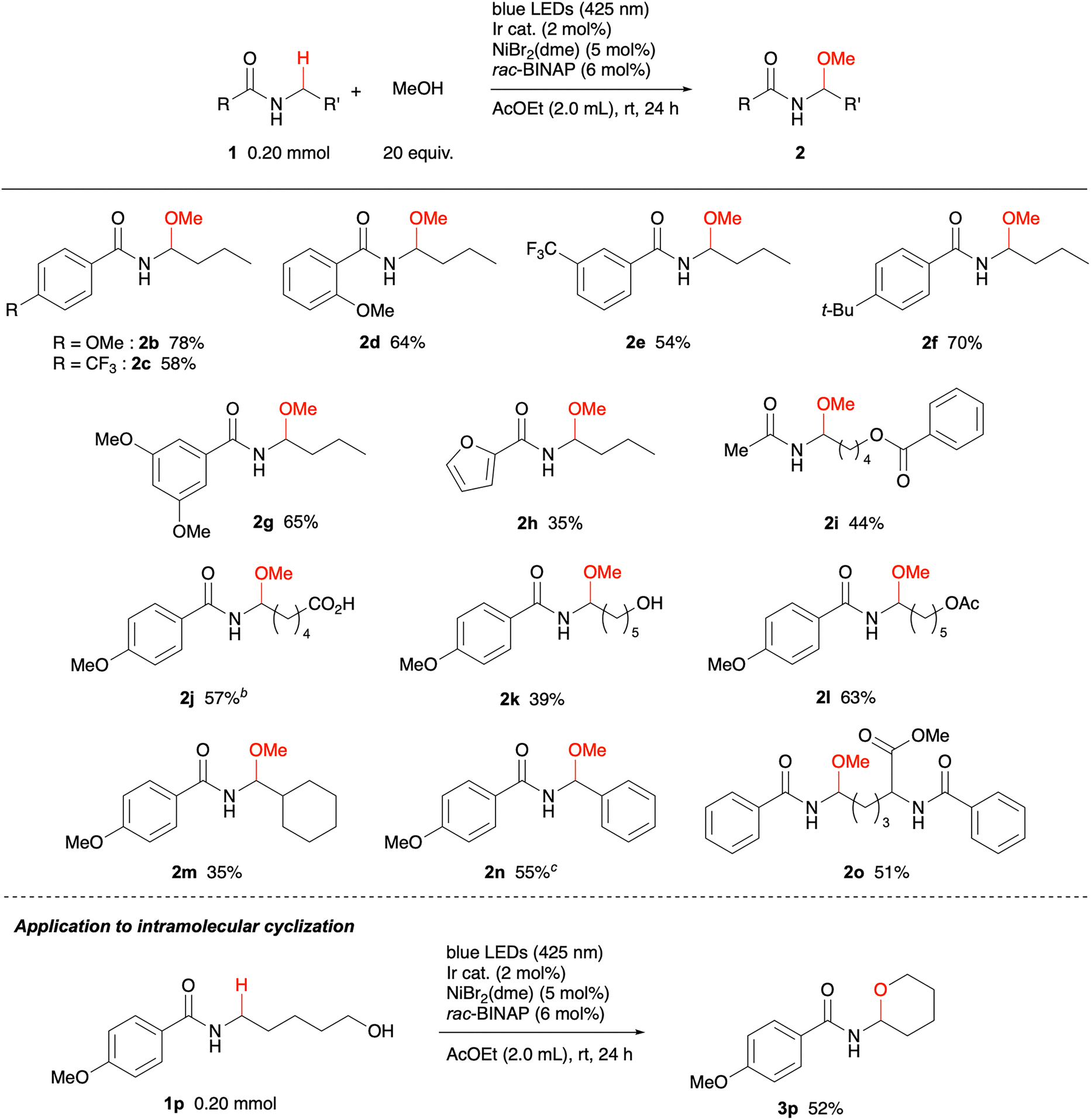
With the optimised conditions in hand, we next investigated the scope of the amide substrate (Table 2). Screening of N-acyl substituents revealed that benzoyl groups bearing methoxy and trifluoromethyl substituents were tolerated, affording the corresponding N-acyl-N,O-acetals 2b–2g in good yields. The reaction of 1b on a 2 mmol scale was also successful; 2b was produced in 71% yield when the reaction time was extended to 40 h using two LED bulbs. A furyl (2h) and an acetyl (2i) group were also compatible. Investigation of N-alkyl substituents demonstrated that primary alkyl groups were successful, while secondary ones were not. Carboxy (2j), hydroxy (2k), and ester (2i, 2l and 2o) groups were retained in the products. Cyclohexylmethylamine 1m and benzylamine 1n were also suitable substrates. However, the yield of the target product 2n from 1n was limited to 19% under standard conditions. Subsequent re-optimisation of the reaction parameters revealed that increasing the methanol to 40 equiv. enhanced the yield to 55%. Lysine derivative 1o possesses two benzamide moieties. Among the two possible reaction sites, the methylene carbon was site-selectively methoxylated presumably for an electronic reason. It should be noted that this methodology could be extended to an intramolecular cyclization reaction of amino alcohol 1p. When 1p was treated with the identical catalyst system, but in the absence of methanol, the cyclic N-acyl-N,O-acetal 3p was produced in 52% yield.
| a Reaction conditions: amide 1 (0.20 mmol), MeOH (4.0 mmol, 20 equiv.), Ir cat. (0.004 mmol, 2 mol%), NiBr2(dme) (0.01 mmol, 5 mol%), rac-BINAP (0.012 mmol, 6 mol%), AcOEt (2.0 mL), irradiated with blue LEDs at rt under a nitrogen atmosphere for 24 h. Isolated yields are shown.b Isolated after esterification with Me3SiCHN2.c MeOH (8.0 mmol, 40 equiv.). |
|---|
 |
To demonstrate the versatility of the obtained N-acyl-N,O-acetal as a synthetic intermediate, we explored derivatization of 2b (Scheme 1). Treatment of 2b with N,N-dimethylaniline in the presence of Ca(NTf2)29 afforded 4b in 84% yield. Similarly, a bismuth-catalysed reaction with 2-methylfuran10 gave 5b in 75% yield. Furthermore, a substitution reaction with thiol proceeded efficiently in the presence of a Brønsted acid,11 yielding N,S-acetal 6b.
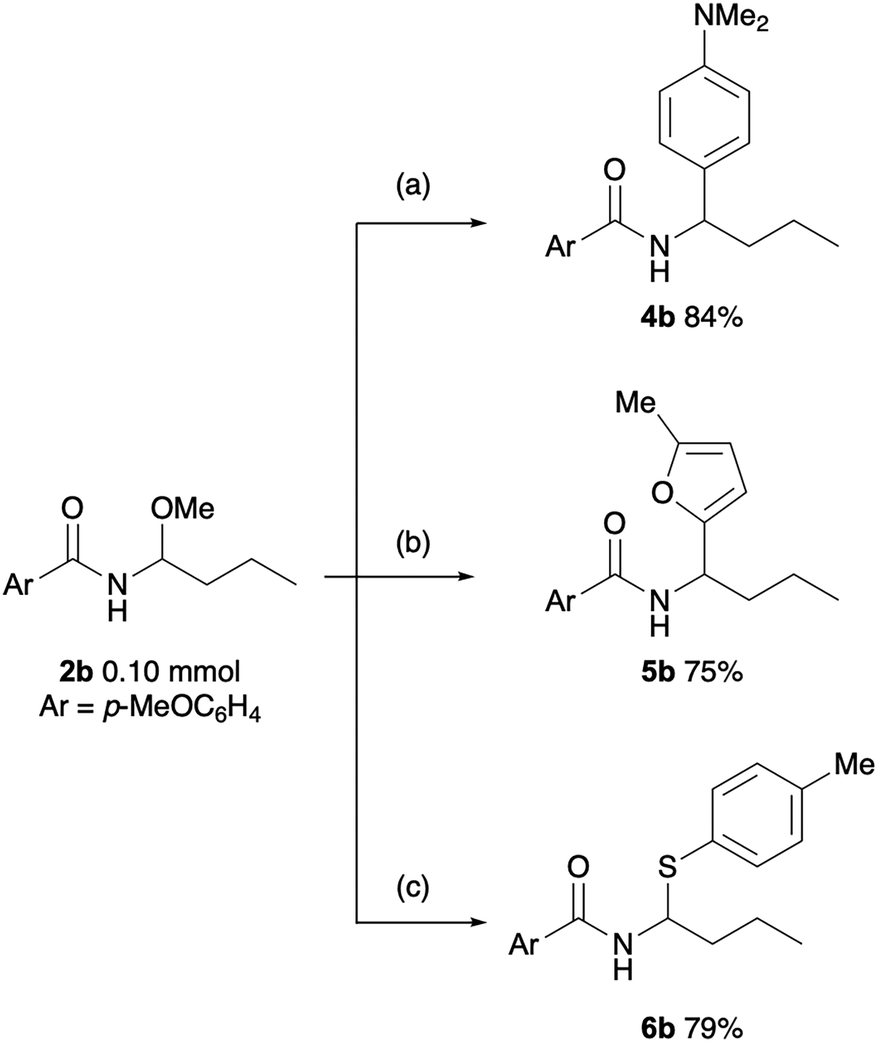 | ||
| Scheme 1 Derivatization. Reaction conditions: (a) N,N-dimethylaniline (1.5 equiv.), Ca(NTf2)2 (5 mol%), (n-Bu)4NPF6 (5 mol%), 1,2-dichloroethane (1.0 mL), 80 °C, 2 h. (b) 2-Methylfuran (4.0 equiv.), Bi(OTf)3 (10 mol%), dichloromethane (1.0 mL), rt, 2 h. (c) 4-Methylbenzenethiol (1.2 equiv.), HCl in 1,4-dioxane (4 M, 1 drop), chloroform (1.5 mL), 1 h. | ||
Next, we turned our attention to the reaction mechanism of the C–H methoxylation reaction. The proposed pathway is depicted in Scheme 2. Initially, the iridium catalyst absorbs light to induce a single electron transfer from the bromide ion to the excited state of Ir[dF(CF3)ppy]2(dtbbpy)PF6.12 The resulting bromine radical then facilitates hydrogen atom transfer (HAT) to cleave the C–H bonds adjacent to the nitrogen, resulting in an alkyl radical and HBr. This alkyl radical subsequently reacts with Ni(II)Br2(binap) to afford an iminium cation intermediate, possibly via a single electron transfer or bromine atom transfer followed by elimination of the bromide ion. Subsequent addition of methanol to the iminium ion forms an N-acyl-N,O-acetal and HBr. Reduction of Ni(I)Br species by the iridium(II) species furnishes Ni(0), which reacts with hydrogen bromide. This reaction leads to the evolution of molecular hydrogen and the regeneration of the Ni(II)Br2(binap) catalyst.13
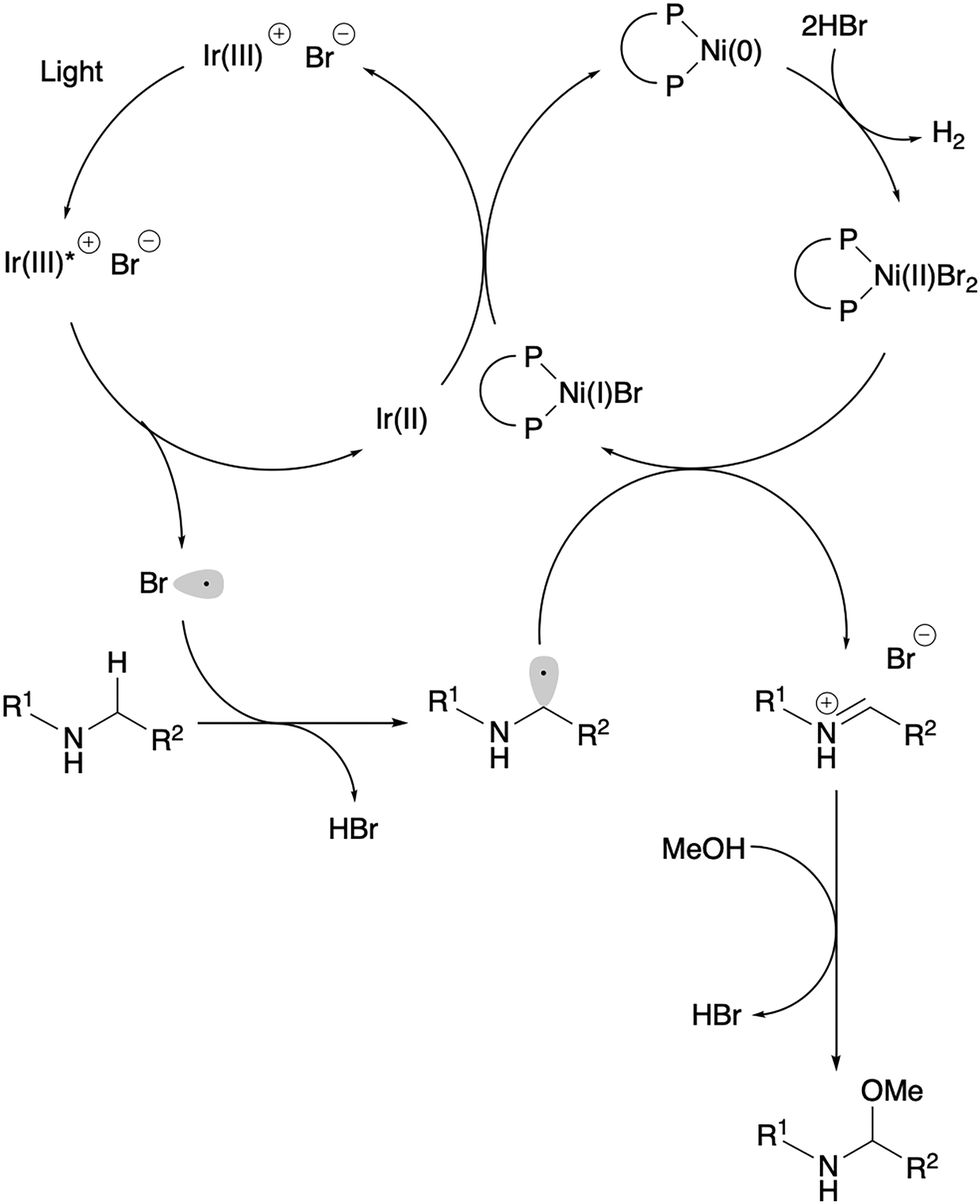 | ||
| Scheme 2 Proposed mechanism. | ||
To test the validity of the proposed mechanism, we conducted several mechanistic studies. While it is well-established that the iridium photoredox catalyst induces photoinduced single electron transfer from a bromide ion,12 it remained unclear whether the iridium complex functions similarly in the presence of a mixture of NiBr2(dme) and BINAP. Thus, we performed a cyclic voltammetry study (Fig. 1). The voltammogram of each reaction component revealed that the oxidation of the bromide ion (Ep/2 = +0.11 V vs. Fc+/Fc) was more facile than those of NiBr2(dme) (Ep/2 = +0.66 V vs. Fc+/Fc) and BINAP (Ep/2 = +0.59 V vs. Fc+/Fc). Furthermore, the voltammogram of the mixture of NiBr2(dme) and BINAP exhibited an oxidation wave with a small shoulder that can be attributed to the oxidation of the bromide ion. These results are consistent with the proposed mechanism.
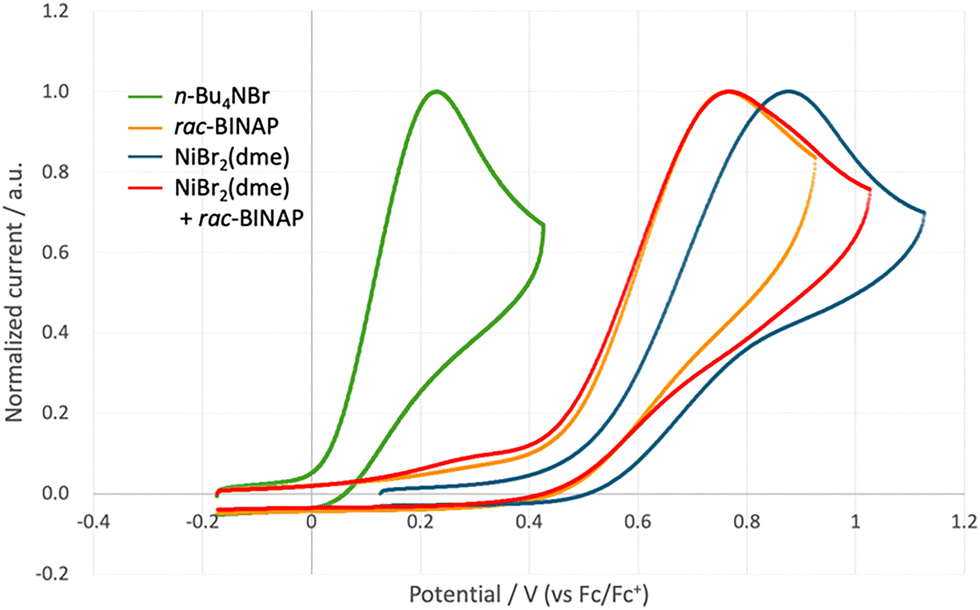 | ||
| Fig. 1 Cyclic voltammogram. All cyclic voltammograms were recorded in AcOEt using n-Bu4NBF4 (0.1 M) as the electrolyte. A glassy carbon anode and Pt cathode were used. Scan rate = 100 mV s−1. Green: n-Bu4NBr, orange: rac-BINAP, blue: NiBr2(dme), red: a mixture of NiBr2(dme) and rac-BINAP. The voltammograms are normalized. | ||
We also conducted a radical trapping experiment. A reaction of N-butylbenzamide with benzalmalononitrile under standard conditions yielded adduct 7a, supporting the generation of the alkyl radical (Scheme 3).
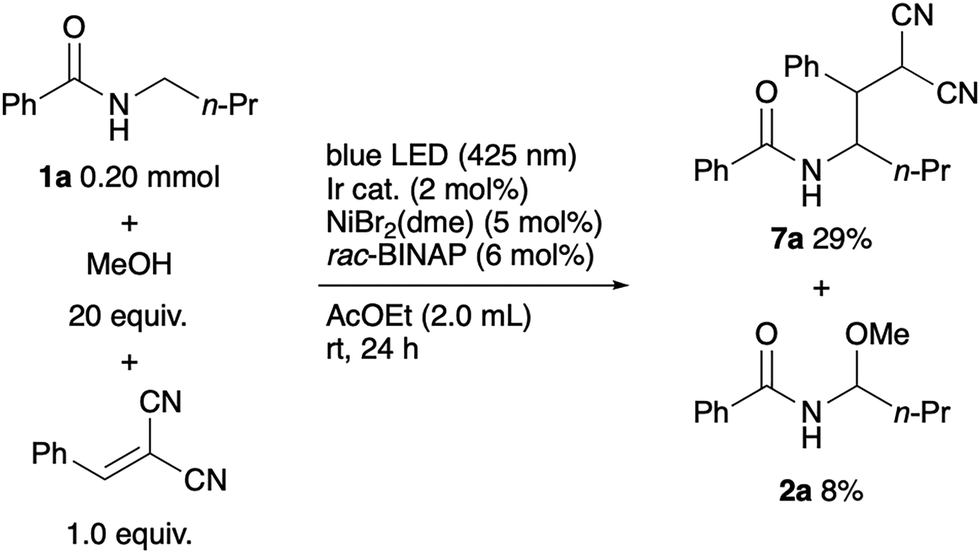 | ||
| Scheme 3 Radical trapping experiment. | ||
In conclusion, we developed a photoinduced C–H methoxylation reaction of N-alkylamides with methanol, catalysed by a combination of an iridium photosensitizer and nickel bromide. This oxidative reaction requires no external oxidant, and hydrogen gas evolves as the byproduct. Mechanistic studies suggest that HAT by a bromine radical is involved as a key elementary step in activating the amide substrates. The present reaction represents a photoinduced variant of Shono-type oxidation, which dispenses with sacrificial oxidants and the pre-installation of reactive handles on the substrates.
This research was supported by Asahi Grass Foundation (to N. I.), Nagase Science and Technology Foundation (to N. I.), JST ACT-X Grant Number JPMJAX23D6 (to S. O.), JSPS KAKENHI Grant Number JP24H01873 (Green Catalysis Science to S. O.) and JP24K17688 (Early-Career Scientists to S. O.). K. K. acknowledges Chihiro Kanagawa Foundation for Future Chemistry for the scholarship.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this communication, including experimental details and characterization data, have been included in the ESI.†Notes and references
- A review: A. M. Jones and C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056–3072 CrossRef PubMed
.
-
(a) T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172–1176 CrossRef CAS
- T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS
-
(a) F. Wang, M. Rafiee and S. S. Stahl, Angew. Chem., Int. Ed., 2018, 57, 6686–6690 CrossRef CAS PubMed
- W. Luo, R. Zhang, Q. Xu, S. Zheng, J. Yang, M. Liu, S. Guo and H. Cai, Synlett, 2023, 2346–2350 CAS
-
(a) Q. Xia and W. Chen, J. Org. Chem., 2012, 77, 9366–9373 CrossRef CAS PubMed
-
(a) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425–4431 CrossRef CAS PubMed
-
(a) T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed
- A. J. Basson and M. G. McLaughlin, Cell. Rep. Phys. Sci., 2023, 4, 101234–101248 CrossRef CAS
- P. Kramer, M. Halaczkiewicz, Y. Sun, H. Kelm and G. Manolikakes, J. Org. Chem., 2020, 85, 3617–3637 CrossRef CAS PubMed
- M. M. Wang, S. Jeon and J. Waser, Org. Lett., 2020, 22, 9123–9127 CrossRef CAS PubMed
-
(a) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed
- D. C. Powers, B. L. Anderson and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 18876–18883 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: https://doi.org/10.1039/d5cc02901e |
| This journal is © The Royal Society of Chemistry 2025 |