
Machine learning driven rational design of AuAgPdHgCu HEA catalysts for the two-electron oxygen reduction reaction†
Zhen Chena,
Xi Liua,
Junyi Zhua,
Bihua Hua,
Lin Yang a,
Xin Wang*a,
Shuqin Song*b and
Zhongwei Chen*c
a,
Xin Wang*a,
Shuqin Song*b and
Zhongwei Chen*c
aInstitute of Carbon Neutrality, Zhejiang Wanli University, Ningbo, 315100, China. E-mail: wangx@zwu.edu.cn
bThe Key Lab of Low-carbon Chemistry & Energy Conservation of Guangdong Province, PCFM Laboratory, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, China. E-mail: stsssq@mail.sysu.edu.cn
cPower Battery and System Research Center, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: zwchen@dicp.ac.cn
First published on 25th July 2025
Abstract
This study integrated high-throughput DFT calculations and machine learning to screen AuAgPdHgCu high-entropy alloy catalysts, revealing that negative d-band shifts of Hg/Cu optimize ΔG*OOH for an enhanced 2e− ORR activity. Structure–activity analysis identified an optimal configuration (0.97 ideal active sites), guiding efficient catalyst design.
H2O2, valued as an eco-friendly oxidant in synthesis, textiles, and water treatment,1–5 is mainly produced via the pollutive, energy-intensive anthraquinone process.6 The electrochemical 2e− oxygen reduction reaction (ORR) using renewable energy offers a sustainable alternative,7–10 but requires catalysts suppressing the 4e− ORR pathway by controlling O-intermediate adsorption.11–14 The selectivity of the 2e− ORR is governed by the adsorption strength of *OOH intermediate and the geometric configuration of O2 on active sites.15 Weak *OOH binding prevents O–O bond cleavage, favoring H2O2 formation, while excessively weak adsorption impedes O2 activation. Geometric effects also play a crucial role: O2 adsorption with an end-on configuration preserves O–O bonds better than that with an side-on configuration.16 Despite progress, the complex interplay between electronic/geometric structures and catalytic performance remains unclear, particularly for multicomponent systems.
High-entropy alloys (HEA), containing five or more elements, present unique opportunities for catalyst design, surpassing conventional binary and ternary alloys. This superiority arises from their tunable electronic environments and diverse adsorption sites,17,18 which enable the decoupling of adsorption energies for multiple reaction intermediates—overcoming the limitations of traditional linear scaling relations and allowing for independent optimization.19–21 However, the vast combinatorial space of HEA active sites poses a screening challenge. Traditional density functional theory (DFT) calculations become computationally prohibitive for global exploration.22,23 Machine learning (ML) offers a solution by accelerating catalyst discovery.24 For instance, neural network models have successfully predicted hydrogen evolution reaction activity in Pt–Ru alloys and guided FeCoNiCuMo HEA design for the CO2RR, breaking conventional scaling relationships.25,26
Inspired by binary alloys (Au–Pd, Cu–Hg, etc.) with isolated active sites for the 2e− ORR,27–29 we investigate the AuAgPdHgCu HEA as a potential catalyst. This system combines reactive (Pd and Cu) and inert (Au, Ag, and Hg) elements to balance *OOH adsorption and O–O stabilization; meanwhile, amalgams can form alloys with nearly all metals and exhibit good stability. Through high-throughput DFT (HT-DFT) calculations screening of 1218 initial HEA models and detailed analysis of 80 active sites, we develop neural network-based ML models to predict adsorption energies and identify optimal configurations. The models correlate geometric/electronic descriptors with catalytic activity, revealing multi-element synergy mechanisms. This theoretical screening study establishes a rapid HEA catalyst framework, overcoming computational limits while guiding efficient 2e− ORR design. The methodology extends to other electrocatalysis, advancing the rational design of complex multicomponent catalysts.
In this study, the ORR proceeding via a 2e− pathway was considered in an acidic system, which involves coupled electron–proton transfers (eqn (1) and (2)), governed by the Gibbs free energy of *OOH (ΔG*OOH):
| O2 + * +H+ + e− → *OOH | (1) |
| *OOH + H+ + e− → H2O2 + * | (2) |
 | ||
| Fig. 1 Schematic of the conceptual framework and calculation flow: (a) schematic diagram of high-throughput screening for the most stable AuAgPdHgCu HEA models; (b) the construction of adsorption models and the training process of NN ML models. | ||
Six supervised ML algorithms (LR, RF, GP, SVR, KNN, and NN) were employed to predict the descriptor ΔG*OOH from geometric and electronic configurations. Prediction accuracy, evaluated using MAE and MSE (Fig. 2), is ranked as follows: NN (0.093 eV MAE and 0.011 eV MSE), RF (0.128 eV and 0.029 eV), GP (0.194 eV and 0.065 eV), LR (0.207 eV and 0.065 eV), KNN (0.244 eV and 0.068 eV), and SVR (0.250 eV and 0.101 eV). The NN model achieved the highest accuracy (lowest MAE/MSE), attributed to its ability to self-adjust weights/biases to minimize the loss function and its inherent capacity for effective nonlinear mapping. Similarly, RF captured nonlinear relationships through feature randomness during tree node splitting, yielding superior performance compared to LR, GP, SVR, and KNN. Prediction accuracy for ΔG*OH and ΔG*O follows similar trends (Fig. S9 and S10; parameters in Tables S2–S4, ESI†).
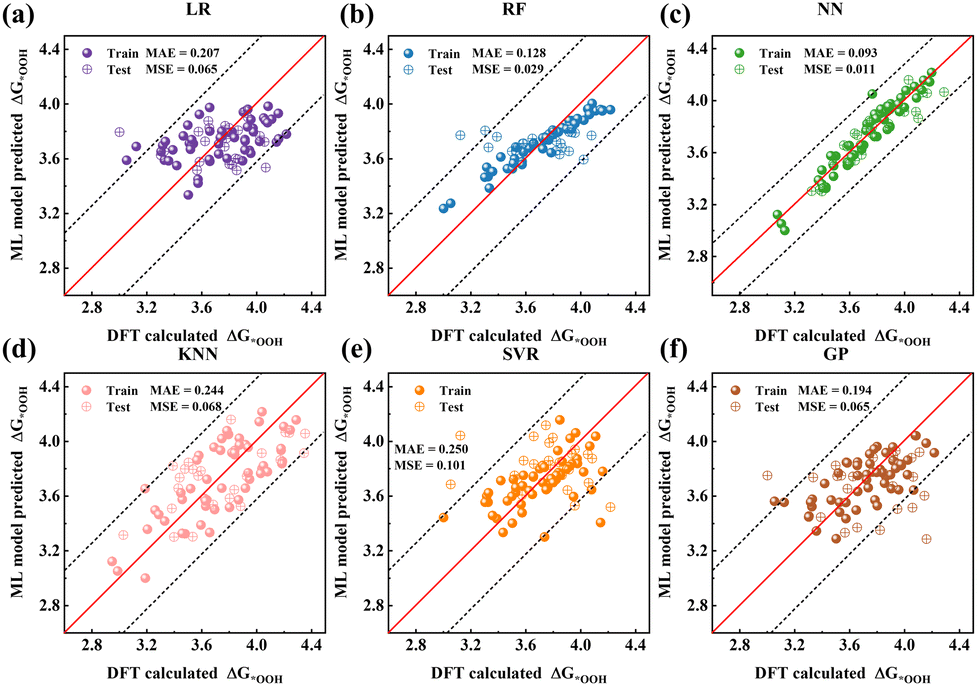 | ||
| Fig. 2 (a) LR, (b) RF, (c) NN, (d) KNN, (e) SVR, and (f) GP model predicted ΔG*OOH values plotted against DFT calculated ΔG*OOH values (70/30% training-testing data splitting). | ||
A key challenge in applying ML to electrocatalyst design is its “black box” nature and the difficulty in deriving concise mathematical equations due to complex structure–performance relationships and instrumental noise. However, explainable ML techniques, like Shapley Additive exPlanations (SHAP),32,33 now allow quantification of individual feature importance. To gain chemical insight, we applied SHAP to interpret the NN model. Fig. S11a (ESI†) ranks the top 20 features by their SHAP value significance in predicting ΔG*OOH. Positive (negative) SHAP values correlate with higher (lower) predicted ΔG*OOH. Notably, NUE features dominate the prediction: 3 of the top 4 features are NUE, and all NUE features for the top and six coordinate atoms rank within the top 20. The influence of NUE depends strongly on the atomic site due to the asymmetric HEA lattice: higher NUE lowers ΔG*OOH for coordinate atoms 2, 3, 5, and 6, increases it for coordinate 4, and shows a variable (high or low) influence with weaker correlation for the top and coordinate 1 atom. In contrast, EN features exhibit relatively weak correlation, appearing only for the top and coordinate 5 atoms in the top 20; the bottom 4 SHAP-ranked features are all EN (Fig. S12, ESI†), contradicting theoretical expectations where EN should significantly influence binding strength (ΔG). Other significant features include GN (5) and PN (6). For non-top atoms, larger GN or PN values correlate with larger ΔG*OOH, potentially due to oxygen proximity. Surface atoms, particularly the top atom (with its NUE, GN, and PN occupying three top-6 positions in Fig. S11a, ESI†), exert the dominant impact, consistent with theoretical understanding.
Fig. S11b (ESI†) shows the SHAP main effect vs. NUE for the top atom. NUE values of 0, 1, and 3 correspond to Au (also Ag and Hg), Cu, and Pd, respectively. NUE = 1 (Cu) provides an overall positive contribution to SHAP values (higher ΔG*OOH), while NUE = 0 or 3 shows unclear patterns. Fig. S11c and d (ESI†) show that a PN of 4 and a GN of 12 increase ΔG*OOH, while a GN of 10 decreases it. Specifically, NUE = 1 and PN = 4 indicate Cu; GN = 10 indicates Ag; GN = 12 indicates Hg. Most calculated ΔG*OOH values are between 3.00 and 4.00 eV, but the ideal value is 4.22 eV. The gain effect from Cu and Hg suggests that the sites containing these elements are more likely to be active sites, despite Au sites having values closer to ideal. This result aligns with the aforementioned intuitive analysis.
To reveal the mechanism for the Cu/Hg gain effect on ΔG*OOH, the electronic structure of surface atoms in 3 sites was analysed (Fig. 3a and c). At Au1 site, Hg atom shows the most negative d-band center, indicating a lower d-electron energy and weaker O interactions. Similarly, Cu1 and Hg2 sites exhibit Cu/Hg atoms with the most negative d-band centers, explaining the reduced ΔG*OOH. This effect stems from Cu/Hg negative d-band shifts in the AuAgPdHgCu alloy. However, the underlying causes differ: Cu acquires electrons from other metals (negative d-band shift), while Hg loses electrons (Fig. 3a–c, inset; Bader charges). In Au2 site, Hg has a +0.248 Bader charge (electron loss), stabilizing its d-band. At Cu1 site, Cu atoms show negative Bader charges (e.g., −0.172, −0.009, and −0.246), lowering d-orbital energy. Under coexisting conditions (Fig. 3c, inset), both Hg and Cu lose electrons, yet Hg's d-band shifts negatively, while Cu's d-band shifts positively. Hg dominates in weakening *OOH binding due to its lower d-band energy. ML analysis reveals that Cu and Hg enhance ΔG*OOH, thereby promoting 2e− ORR activity. To evaluate the overall activity of the HEA (111) facet, we analysed 13 distinct active sites (Au:2, Ag:2, Pd:2, Hg:4, and Cu:3). The free-energy diagram for the O2 to H2O2 pathway (Fig. 3d) shows that at 0.7 V vs. RHE, *OOH formation is exergonic (downhill) across all sites. In contrast, H2O2 formation is endergonic (uphill), establishing it as the rate-determining step (RDS). The calculated RDS energy barriers range significantly from 3 to 199 mV. Furthermore, Fig. 3e correlates the thermodynamic limiting potential (UL, blue) with ΔG*OOH. UL represents the maximum potential, at which both *OOH formation and H2O2 release steps remain exergonic. The thermodynamic overpotential (ηO2/H2O2) is the difference between the H2O2 Nernst potential (UO2/H2O20 = 0.7 V) and UL. Applying a bias equivalent to ηO2/H2O2 minimizes charge transfer barriers. The 13 sites have UL values from 0.501 to 0.697 V vs. NHE and ηO2/H2O2 values from 3 to 199 mV, consistent with the RDS barriers. The selectivity towards H2O2 or H2O is determined by its propensity to break the O–O bond. Although all 13 sites exhibit activity for the 2e− ORR, their selectivity varies significantly. Sites located on the left flank of the 4e− ORR volcano (consequently also on the left flank of the 2e− volcano) predominantly favour the 4e− ORR pathway to H2O. Sites positioned between the 4e− and 2e− ORR volcanoes experience strong competition between the reactions producing H2O2 and H2O. Even at the peak of the 2e− ORR volcano, where the 2e− activity is maximized, the 4e− ORR pathway persists. Other metals like Ti and Mn were also included in Au, Ag, and Hg alloy systems to construct the AuAgTiHgMn HEA by the same HT calculations (Fig. S13, ESI†), so that more active sites are located on the left side of the 2e− volcano. It is found that the Ti3 and Mn2 active sites achieve ΔG*OOH values of 3.751 and 3.866 eV (other Ti and Mn sites have lower ΔG*OOH values) and Au3 and Ag1 sites have higher ΔG*OOH values of 4.436 and 4.267 eV, indicating a relatively higher 2e− ORR selectivity. To describe the entire slab's activity, a total activity metric considering all sites was developed under the following assumptions: (1) no migration, secondary reactions, or interactions; the total rate (rtot) is the sum of individual site rates (ri), with the activation energy (Ea,i) being the RDS barrier; (2) solvent effects are neglected; and (3) constant O2/H2O concentration. According to the Butler–Volmer equation, the current density (j) on the catalyst is proportional to the reaction rate (r) under ideal conditions.34,35 Hence, the relationship between the reaction rate and activation energy of the site could be established as follows:
 | (3) |
 | (4) |
 | (5) |
 | ||
| Fig. 3 (a)–(c) d-orbital LDOS and Bader charges for surface atoms on Au1, Cu1, and Hg2 sites. (d) Free-energy diagram for the 2e− ORR to H2O2 compares ideal and 13 sites. (e) ORR volcano plots show 2e− (blue) and 4e− (red) pathways with limiting potential vs. ΔG*OOH, including equilibrium potentials. | ||
Ne represents the aggregate number of the equivalent ideal site, which can act as a theoretical activity metric for a specific HEA (111) facet. Fig. 4a shows that wi rapidly decreases from 1 to 10−4 as Ea,i increases from 0 to 0.2 eV. Sites with wi < 10−4 are considered inactive, justifying the mapping of only sites with ΔG*OOH < 4.0 eV. Slab 1 exhibits the highest Ne (0.97, Fig. 4b), indicating superior activity. Activity correlates poorly with the number of sites (ΔG*OOH < 4.0 eV) but is exponentially enhanced by lower RDS barriers. The trained NN model predicts Ea,i (via ΔG*OOH) and Ne for any HEA (111) surface atomic arrangement, enabling rapid activity assessment without DFT.
 | ||
| Fig. 4 (a) The change in exp(−Ea,i/kBT) as Ea varies and (b) number of equivalent ideal sites on different slabs. | ||
In summary, HT-DFT calculations on 1218 AuAgPdHgCu HEA models and 320 adsorption structures created a machine learning database for screening 2e− ORR catalysts. Six distinct ML models predicted key intermediate adsorption energies, and the NN model achieved the best accuracy. This process enabled a rapid identification of highly active sites and assessment of overall HEA activity. SHAP and electronic state analysis revealed that Hg and Cu atoms in the surface 7-atom ensemble positively influenced ΔG*OOH, enhancing 2e− ORR activity and selectivity, which correlated with a negative shift in their d-band center. Consequently, an optimal AuAgPdHgCu HEA catalyst was identified, containing 0.97 ideal active sites per surface ensemble. This HT-DFT/ML screening and mechanistic study provide a theoretical guidance for experimental 2e− ORR catalyst development.
We would like to acknowledge the funding support from the National Natural Science Foundation of China (No. 22379047 and 22478450), the General Scientific Research Project of Zhejiang Education Department (Y202353940), and the Yongjiang Talent Program, Talent research start-up project of Zhejiang Wanli University (SC1032380180530). The authors acknowledge the Beijing Super Cloud Computing Center (BSCC) for providing HPC resources that have contributed to the research results reported within this study (https://www.blsc.cn/)..
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- Y. Cui, et al., Chem. Eng. J., 2024, 500, 157070 CrossRef CAS
.
- H. Li, et al., Chem. Eng. J., 2024, 497, 154523 CrossRef CAS
- L. X. Liu, et al., Angew. Chem., Int. Ed., 2023, 62, e202303525 CrossRef CAS
- A. Byeon, et al., Appl. Catal., B, 2023, 329, 122557 CrossRef CAS
- A. Chen, et al., Chem. Commun., 2025, 61, 7101–7104 RSC
- X. Huang, et al., Nano-Micro Lett., 2023, 15, 34 CrossRef PubMed
- Y. H. Tian, et al., Nano-Micro Lett., 2023, 15, 45 CrossRef
- K. Y. Liu, et al., Nano Res., 2023, 16, 10724–10741 CrossRef CAS
- J. Zou, et al., Chem. Commun., 2025, 61, 4228–4231 RSC
- Q. Wang, et al., Adv. Energy Mater., 2023, 13, 27 Search PubMed
- Y. P. Liu, et al., Angew. Chem., Int. Ed., 2024, 63, e202318893 CrossRef
- C. He, et al., Adv. Energy Mater., 2024, 14, 2303233 CrossRef CAS
- S. J. Zhang, et al., Small, 2024, 8, 2310317 Search PubMed
- Y. Wang, et al., Chin. J. Catal., 2019, 40, 523–533 CrossRef CAS
- S. Siahrostami, et al., Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS
- D. Strmcnik, et al., Nat. Chem., 2010, 2, 880–885 CrossRef CAS PubMed
- Y. Wang, et al., Adv. Mater., 2023, 35, 2302499 CrossRef CAS PubMed
- Y. Bu, et al., Adv. Mater., 2024, 36, 2412670 CrossRef CAS
- H. Li, et al., Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS
- Z. Lu, et al., Energy Environ. Sci., 2025, 18, 2918–2930 RSC
- G. Feng, et al., J. Am. Chem. Soc., 2023, 143, 17117–17127 CrossRef PubMed
- L. Chen, et al., J. Mater. Inform., 2022, 2, 20517 Search PubMed
- C. M. Clausen, et al., High-Entropy Alloys Mater., 2023, 1, 120–133 CrossRef
- X. Fan, et al., Adv. Funct. Mater., 2024, 34, 2401887 CrossRef CAS
- Q. Zhou, et al., J. Am. Chem. Soc., 2024, 146, 15167–15175 CrossRef CAS
- Z. W. Chen, et al., ACS Catal., 2022, 12, 14864–14871 CrossRef CAS
- Y. Wang, et al., J. Energy Chem., 2023, 87, 247–255 CrossRef CAS
- S. Siahrostami, et al., ACS Catal., 2020, 10, 7495–7511 CrossRef CAS
- S. Yang, et al., ACS Catal., 2018, 8, 4064–4081 CrossRef CAS
- F. Pedregosa, et al., J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- J. Kiely, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 12588 CrossRef CAS
- P. Raccuglia, et al., Nature, 2016, 533, 73–76 CrossRef CAS
- S. J. Sun, et al., Joule, 2019, 3, 1437–1451 CrossRef CAS
- J. Dumesic, J. Catal., 1999, 185, 496–505 CrossRef CAS
- E. J. Dickinson, et al., J. Electroanal. Chem., 2020, 872, 114145 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Lists of reported stable AuAgPdHgCu structures, distribution of the adsorption free energy, detailed data of DFT calculations and model evolution of ML models. See DOI: https://doi.org/10.1039/d5cc03076e |
| This journal is © The Royal Society of Chemistry 2025 |