
Palladium-catalyzed aromatic C–H alkylation of 1-naphthylamines with dichloroalkanes and its application in fused polycyclic amine synthesis
Yixin Shia,
Ziyi Zhou a,
Yilin Hua,
Siyun Wanga,
Mingzhe Honga,
Kai Cheng*a,
Chun-Xiao Jia*c and
Jie-Ping Wan*bd
a,
Yilin Hua,
Siyun Wanga,
Mingzhe Honga,
Kai Cheng*a,
Chun-Xiao Jia*c and
Jie-Ping Wan*bd
aInstitute of Applied Chemistry, Department of Chemistry, Shaoxing University, Shaoxing 312000, P. R. China. E-mail: chengkai@usx.edu.cn
bCollege of Chemistry and Materials, Jiangxi Normal University, Nanchang 330022, China. E-mail: wanjieping@jxnu.edu.cn
cShandong Provincial Key Laboratory of Monocrystalline Silicon Semiconductor Materials and Technology, College of Chemistry and Chemical Engineering, Dezhou University, Dezhou 253023, P. R. China. E-mail: jiachunxiaodzu@163.com
dInternational Innovation Center for Forest Chemicals and Materials, Nanjing Forestry University, Nanjing 210037, China
First published on 30th July 2025
Abstract
Herein, we report a palladium-catalyzed aromatic C–H bond alkylation reaction of 1-naphthylamines at the C8 site with dichloroalkanes as alkylating agents. DFT calculation demonstrates the distinctive advantage of the separated dichloroalkanes in the reaction by stabilizing the key intermediate via C–H⋯N or C–H⋯π interaction induced by the additional Cl atom. In addition to the specific alkylation site selectivity in the naphthyl ring, the products provided by the current C–H alkylation protocol display important applications in the construction of fused polycyclic amines via a one-step treatment.
The alkylation reaction constitutes the most fundamental tool for forging C–C bonds and increasing the length of the carbon chain in organic molecules.1 However, unlike the well-developed and versatile alkylation reactions in heteroatom sites, the alkylation of C–H bonds, especially aromatic C–H bonds, remains challenging.2 Classical aromatic alkylation reactions, such as Friedel–Crafts alkylation reactions, are hardly applicable because of the challenge in controlling chemo- and regioselectivity.3 In this context, developing alternative methods for the direct alkylation of aromatic C–H bonds is still a central issue in modern organic synthesis. Notably, transition-metal-catalyzed C–H activation has proven to be a useful tool in aromatic C–H bond alkylation reactions.4 However, in most cases, highly functional partners, such as diazo compounds,5 halo-functionalized esters,6 polar alkenes,7 or organometallic reagents,8 are required as alkylating reagents. Reactions with simple and easily accessible alkylating reagents, such as haloalkanes, are still much less available, restricting the flexibility to the synthesis of diverse alkylated arene derivatives.9 According to known work, directed aromatic C–H alkylation with alkyl halides still suffers from the restriction of reaction selectivity. For example, the alkylation of functional naphthalene usually takes place in the C2-site.10 In addition, the chemo-selectivity in reactions using dihaloalkanes, such as 1,2-dichloroalkane, usually involves the transformation of both C–Cl bonds. For example, Yu and co-workers reported the elegant C–H alkylation of benzoic acids with 1,2-dichloroalkane for the synthesis of isochromanones, wherein both C–Cl bonds in the dichloroalkanes were transformed in the resulting products (Scheme 1A).11 Reactions that allow the chemo-selective transformation of one C–X (X = halogen) bond, leaving the other intact for flexible late-stage functionalization is yet a challenge.
 | ||
| Scheme 1 Typical backgrounds. | ||
Notably, haloalkyls such as chloroalkyl-functionalized arenes serve as privileged compounds for drug candidates, as exemplified by the antimalarial agent chloroquine, the anticancer chlorambucil, and a series of other pharmaceutical scaffolds, wherein the chloroalkyl moiety in the aryl contributes directly to bioactivity and metabolic resistance.12 Moreover, chloroalkylated arenes are also fundamental synthons in the construction of numerous other target molecules, with the transformation of the C–X bond.13 Analogously, a fused cyclic amine also constitutes a key structure in many natural alkaloids and pharmaceuticals, such as tolvaptan or isocorydione (Scheme 1B), but the synthesis of fused cyclic amines remains difficult.14,15 Herein, by utilizing quinoline carbonyl as a directing group, we report the first example of selective C8-alkylation of 1-napthylamines with dichloroalkanes as alkylating reagents. Besides the novel site selectivity and the chemo-selectivity in transforming only one of the two C–Cl bonds in dichloroalkanes, the current method enables the titled reactions under solvent-free conditions.16 More critically, the chloroalkylated products resulting from the selective alkylation could be simply transformed into fused polycyclic amines upon treating with NaOH (Scheme 1C), significantly enhancing synthetic efficiency compared to previous multi-step procedures starting from C–H alkylation with halo-functionalized esters.17
To probe the viability of this Pd-catalyzed chloroalkylation, comprehensive efforts were made to optimize the conditions for the reaction of N-(naphthalen-1-yl)quinoline-2-carboxamide 1a and 1,3-dichloropropane 2a (see the SI). After systematic investigation of typical reaction parameters, it was found that the expected product N-(8-(3-chloropropyl)naphthalen-1-yl)quinoline-2-carboxamide 3a could be obtained in 96% yield under the optimal conditions (Table S1, entry 6). A parallel entry using 1,3-dibromopropane to react with 1a led to a complex mixture, probably owing to the high reactivity of the dibromoalkane toward different transformations. Under the optimized conditions, reactions with dichloroalkanes of different chain lengths were executed. The chain length of dichloroalkanes showed an evident influence on reaction efficiency. As illustrated in Scheme 2, geminal dichloromethane and separated 1,2-dichloroalkanes with chains of more than 8 carbons could not work in this alkylation (Scheme 3). Instead, linear dichloroalkanes with 2–7 carbons in the chain could all be practically employed in this protocol (3a–3f, Scheme 3). Notably, 1,3- and 1,4-dichloroalkane (n = 3, 4) achieved excellent yields in this alkylation reaction (3a and 3c, Scheme 3), indicating that the other chlorine atom in a suitable site was crucial for this titled reaction. Notably, gram-scale synthesis of the model reaction provided a satisfactory 75% yield of the product.
 | ||
| Scheme 2 Scope of dichloroalkenes. | ||
 | ||
| Scheme 3 Scope of quinoline component. | ||
Under the optimized reaction conditions, the generality and the limitations of this protocol were examined with a variety of quinoline carbonyl-protected naphthylamines 1 with 1,3-dichloropropane (Scheme 2). According to the results, the reactions proceeded with a broad scope to the corresponding 8-chloropropyl substituted naphthylamines 3 in generally moderate to excellent yields. Both electron-donating (OMe, OAc) and electron-withdrawing (F, Cl, Br and CF3) substituents at the 4-position of the naphthyl moiety were well tolerated, affording the corresponding products (3g–l) with satisfactory results. The substrates with electron-donating groups on the C5, C6 or C7 sites of the naphthyl ring could, as expected, also be smoothly employed for the synthesis (3m–r). Substituents on the C6 or C7 sites of 1 had a negative effect on synthetic efficiency (3p–3r), probably by hampering the formation of the key Pd-complex intermediate (see Fig. 1). Meanwhile, a nitro group or a chlorine atom adjacent to the amino group prevented the titled C–H alkylation, owing to the possible strong electron-withdrawing and/or steric effect against the palladium coordination to the amino site. In addition, attempts were made to perform the reactions using 1-naphthylamine or N-acetyl 1-naphthylamine to react with 2a. No C–H alkylation took place in either entry, confirming the crucial role of the quinoline ring in chelating the metal catalyst.
 | ||
| Fig. 1 The Gibbs free energy profile. | ||
Subsequently, to explore the application of the chloroalkylated products, efforts were made in the synthesis of fused cyclic amines. To our delight, by simply treating 3 with NaOH in heating EtOH, the fused tricyclic amines 4a–4f could be obtained with 74% to 85% yields via sequential N-deprotection and nucleophilic annulation (Scheme 4). The results demonstrated the high value of the alkylated products in the synthesis of useful fused cyclic amines by modifying the multi-step procedures reported in the literature.17 Further attempts using compounds 3c and 3d for the synthesis of 8- and 9-membered cyclic amines were not successful.
 | ||
| Scheme 4 Synthetic application toward fused cyclic amines. | ||
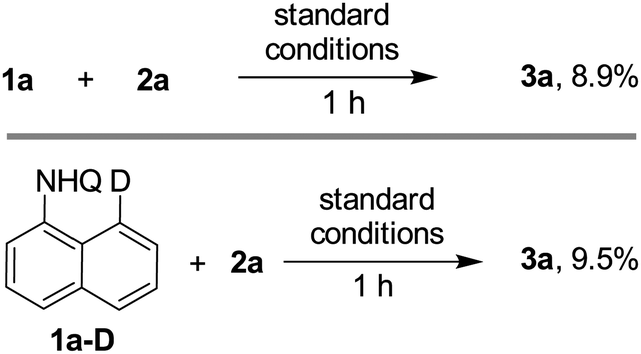
To gain information about the mechanism, a kinetic isotope effect (KIE) experiment was carried out. A primary KIE of 0.94 (kH/kD) (Scheme 2B) between 1a and its deuterated derivative 1a-D was observed in the parallel entries with 1 hour of stirring (see the SI), indicating that activation of the aromatic C–H bond might be the rate-determining step (Scheme 5).
 | ||
| Scheme 5 KIE experiment. | ||
Afterwards, to gain insight into the role of dichloroalkane and the observed chemo-selectivity, DFT calculations were performed by comparing the theoretical reaction modes using chloroethane and 1,2-dichloroethane as substrates. Based on the results (Fig. 1), the calculations revealed that the reaction with 1,2-dichloroethane proceeded with lower activation energy across all proposed transition states and intermediates. For example, the Gibbs free energies of the transition state between metal complex 5 and the oxidative addition intermediate with dichloroethane (6) and chloroethane (6-i) were 27.9 kcal mol−1 (TS1) and 29.1 kcal mol−1 (TSI-i), respectively. Meanwhile, the corresponding transition states between the oxidative addition and reductive elimination intermediates of 7 and 7-i possess a Gibbs free energy of 23.2 and 25.1 kcal mol−1, respectively. Further comparative analyses of the geometric and thermodynamic parameters of substrate-coordinated palladium complexes with chloroethane, 1,2-dichloroethane and 1,3-dichloroethane were performed. As illustrated in Fig. 2, substrate-coordinated complex 5p exhibited a lower free energy (ΔG = 7.7 kcal mol−1 with 1,2-dichloropropane) than 5p-i (ΔG = 8.5 kcal mol−1 with chloromethane). This stabilization could be attributed to stronger C–H⋯N interactions between the geminal hydrogen atom of the substrate and the nitrogen atom in the directing group, as evidenced by shorter hydrogen bond distances (2.58 Å in 5p vs. 2.92 Å in 5p-i). Indeed, substrate-coordinated complex 5p-ii exhibited further increased stability resulting from the C–H⋯π interaction with the hydrogen atom geminal to the chlorine atom (ΔG = 5.5 kcal mol−1 with 1,3-dichloropropane), demonstrating the crucial effect of the additional chlorine atom in the alkane substrate in facilitating C–H activation (Fig. 2).
 | ||
| Fig. 2 Relative Gibbs free energies of Pd-coordinates of different alkyl chlorides. | ||
In conclusion, a solvent-free palladium-catalyzed strategy enabling the site- and chemo-selective C8–H chloroalkylation of 1-naphthylamines with dichloroalkanes has been achieved for the first time. In addition, the chloroalkylated products have been successfully transformed into fused polycyclic amines by a simple one-step operation. The extraordinarily high reactivity of suitable dichloroalkanes in this C–H activation has been illustrated by DFT calculations. As a complementary protocol to those graceful C–H activation reactions reported previously, the novel selectivity and high efficiency of the current reactions, as well as the important application of the synthesized products, make the current work highly attractive.
This work is financially supported by the National Natural Science Foundation of China (21402123), and the Natural Science Foundation of Zhejiang Province (LY18B020006).
K. C. conceived the idea and wrote the manuscript draft, Y. S. and Z. Z. conducted most of the experiments. Y. H., S. W. and M. H. participated in the experimental work and the discussion during the research process. C.-X. J. conducted the calculation part. J.-P. W. reviewed and edited the final manuscript version. J.-P. W. and K. C. supervised the project.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI.Experimental information, synthetic procedures and NMR spectra. See DOI: https://doi.org/10.1039/d5cc03432a
Notes and references
-
(a) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7558–7598 CrossRef CAS PubMed
; (b) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC
-
(a) L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC
- S. Singh and C. K. Hazra, Synthesis, 2024, 368–388 CAS
-
(a) S. B. Ankade, A. B. Shabade, V. Soni and B. Punji, ACS Catal., 2021, 11, 3268–3292 CrossRef CAS
-
(a) B. Zhao, H. Li, F. Jiang, J.-P. Wan, K. Cheng and Y. Liu, J. Org. Chem., 2023, 88, 640–646 CrossRef CAS PubMed
- T. Nishikata, ChemistryOpen, 2024, 13, e202400108 CrossRef CAS PubMed
-
(a) D. Mandal, S. Roychowdhury, J. P. Biswas, S. Maiti and D. Maiti, Chem. Soc. Rev., 2022, 51, 7358–7426 RSC
- K. Takeuchi, Y. Murata, G. Hirata, T. D. Sheppard and T. Nishikata, Chem. Rec., 2019, 20, 403–412 CrossRef PubMed
-
(a) N. Yoshikai and K. Gao, Pure Appl. Chem., 2014, 86, 419–424 CrossRef CAS
-
(a) Y. Mao, X. Song, D. Yuan, J. You, W. Song and Y. Zhang, ACS Catal., 2025, 15, 3247–3255 CrossRef CAS
-
(a) Y. H. Zhang, B. F. Shi and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed
-
(a) A. Sato, L. McNulty, K. Cox, S. Kim, A. Scott, K. Daniell, K. Summerville, C. Price, S. Hudson, K. Kiakos, J. A. Hartley, T. Asao and M. A. Lee, J. Med. Chem., 2005, 48, 3903–3918 CrossRef CAS PubMed
-
(a) Y.-Y. Liang, G.-F. Lv, X.-H. Ouyang, R.-J. Song and J.-H. Li, Adv. Synth. Catal., 2021, 363, 290–304 CrossRef
-
(a) J. K. Ghali, B. Hamad, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2009, 8, 611–612 CrossRef CAS PubMed
-
(a) P. Mátyus, Á. Földi, K. Ludányi and A. Bényei, Synlett, 2010, 2109–2113 CrossRef
-
(a) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed
- L. Huang, K. Cheng, F. Jiang and H. Jin, Chin. J. Org. Chem., 2021, 41, 1691–1702 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |