
Mapping the catalytic landscape of triphenylborane (BPh3)-catalyzed CO2-epoxide coupling to carbonates: an in silico approach to solve substrate-dependent selectivity†
Nikunj Kumar a and
Puneet Gupta*ab
a and
Puneet Gupta*ab
aComputational Catalysis Center, Department of Chemistry, Indian Institute of Technology, Roorkee 247667, Uttarakhand, India
bCenter for Sustainable Energy, Indian Institute of Technology Roorkee, Roorkee 247667, Uttarakhand, India. E-mail: puneet.gupta@cy.iitr.ac.in
First published on 9th June 2025
Abstract
The catalytic coupling of CO2 and epoxides is a promising approach for carbon valorization, enabling the synthesis of cyclic-carbonates (CCs) and poly-carbonates (PCs). However, controlling product selectivity remains a challenge. Triphenylborane (BPh3) has emerged as a promising metal-free catalyst, yet the origins of its substrate-dependent selectivity remain unclear. While BPh3 selectively forms CCs with propylene oxide (PO), it exclusively produces PCs with cyclohexene oxide (CHO), highlighting distinct reactivity. To understand this selectivity, we conducted a comprehensive density functional theory (DFT) study, mapping the catalytic landscape of BPh3-mediated CO2-epoxide coupling and comparing it with triethylborane (BEt3) catalysis, which lacks such selectivity. Our calculations reveal that epoxide ring opening is the rate-determining step, consistent with experimental studies. Additionally, high CO2 concentrations can form an inactive species that inhibits epoxide activation, explaining the experimentally observed inverse rate dependence on CO2 concentration. Our distortion/interaction analysis (DIA) and non-covalent interaction (NCI) analysis show that in BPh3-catalyzed CO2 and PO coupling, weaker intermolecular interactions in the epoxide addition step disfavour PC formation, favoring CC formation. Conversely, for CO2 and CHO coupling, the high distortion energy in the ring-closing step makes CC formation unfavourable, leading to PC as the dominant product. In contrast, BEt3 catalysis stabilizes PC formation across both epoxides, eliminating selectivity. This study sketches the catalytic landscape of BPh3-catalyzed CO2-epoxide coupling, revealing how boron substitution governs selectivity and offers insights for designing boron-based catalysts for selective CO2 utilization.
1. Introduction
Carbon dioxide (CO2) is a prominent greenhouse gas naturally regulated through the global carbon cycle, involving terrestrial plants, oceans, and microorganisms.1 The primary drivers of CO2 emissions are the extensive combustion of fossil fuels and various anthropogenic activities.2–4 Since the industrial revolution, CO2 emissions have risen significantly, surpassing the natural absorption capacity due to simultaneous deforestation and ocean acidification, thereby contributing to global warming.5 However, CO2 is not merely an environmental concern; it also represents a promising C1 feedstock that is abundant, non-toxic, non-flammable, and readily available as a sustainable carbon source.6,7 In the context of environmental sustainability and resource valorization, developing innovative technologies for the catalytic conversion of CO2 into high-value products has become a focal point in academia and industry.8 Among the diverse CO2 utilization strategies, its coupling with epoxides to synthesize cyclic-carbonates (CCs) or poly-carbonates (PCs) has garnered significant interest due to its 100% atom economy and high carbon utilization efficiency, aligning with the principles of green chemistry.9,10 CCs serve as green polar aprotic solvents,11 Li battery electrolytes,12 and precursors for pharmaceuticals.13 Meanwhile, PCs are valuable materials for synthesizing polyols used in various applications14 and offer a sustainable alternative to conventional synthetic polymers derived from non-renewable petroleum feedstocks.15In the absence of external catalysts, the coupling reactions between CO2 and epoxides require high temperatures and pressures to overcome the energy barriers associated with CO2 activation and its conversion into CCs or PCs.16,17 To circumvent these obstacles, multifarious catalytic systems have been investigated, including metal porphyrins,18 salen metal complexes,19 metal oxides,20 alkali metals,21 and metal–organic frameworks (MOFs),22 all of which have demonstrated effectiveness in facilitating the CO2 to CC conversions (Fig. 1a). Similarly, for the transformation of CO2 into PCs, the pioneering work of Inoue and coworkers, who first reported the use of diethylzinc for the copolymerization of CO2 and epoxides, laid the foundation for significant advancements in the design of highly efficient catalytic systems.23,24 Over the past decade, a diverse array of metal-based catalysts featuring transition metals and earth-abundant main group metals have been extensively explored for PC production.25–30 Despite their high catalytic activity, metal-based systems often face challenges such as potential metal leaching, recycling difficulties, and complex synthetic procedures, which limit their industrial applicability.31 Consequently, research efforts have increasingly focused on the development of metal-free catalytic systems, driven by their cost-effectiveness, environmental sustainability, and enhanced chemical and thermal stability.32
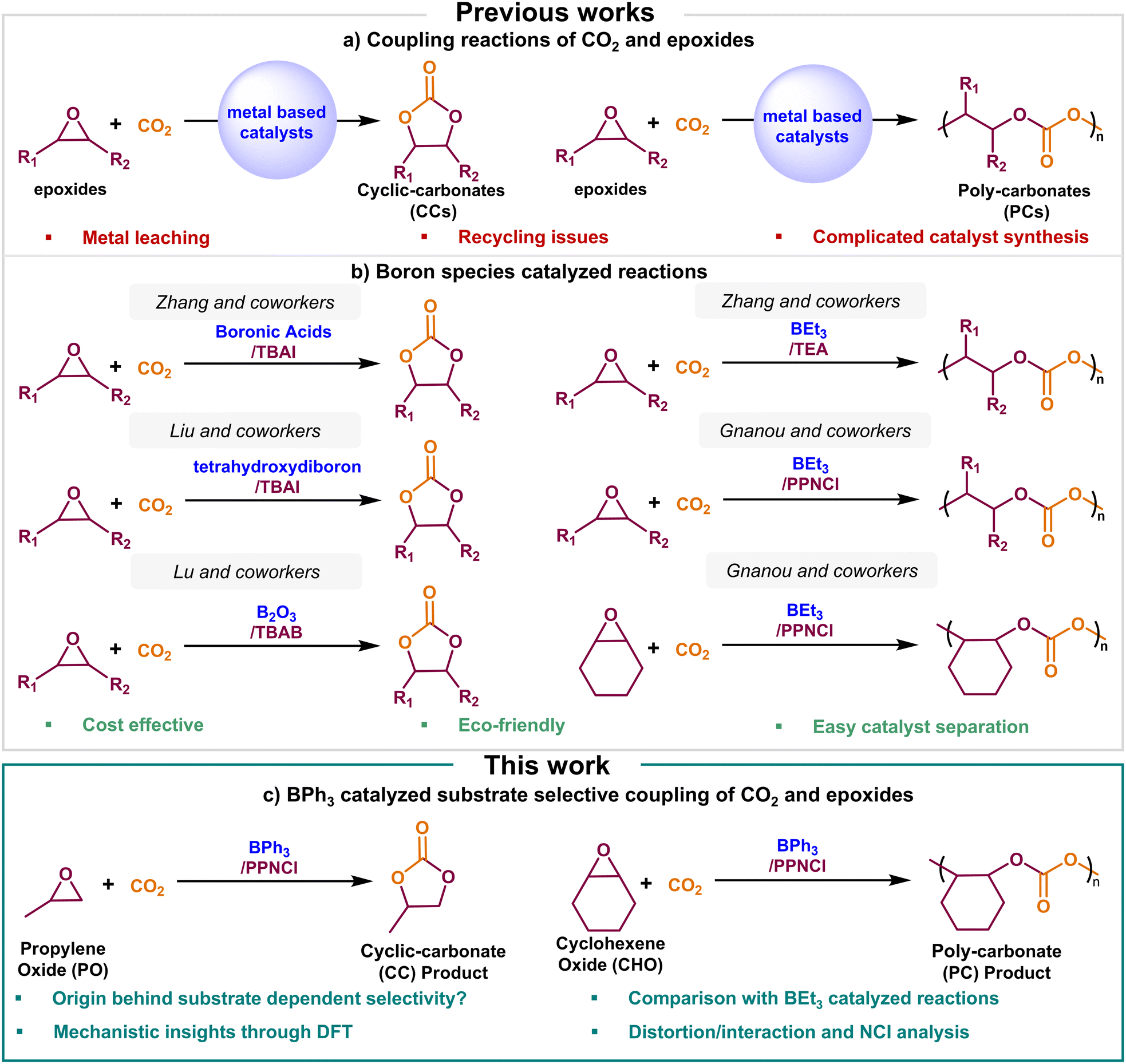 | ||
| Fig. 1 (a) Coupling reactions of CO2 and epoxides; (b) boron species catalyzed reactions; (c) BPh3 catalyzed substrate selective coupling of CO2 and epoxides. | ||
In recent years, boron-based catalysts have emerged as promising candidates in the field of metal-free catalysis of CO2 conversion, primarily due to the intrinsic Lewis acidity of the boron center (Fig. 1b).33–39 Zhang and coworkers demonstrated that boronic acids, in combination with tetrabutylammonium iodide (TBAI), can effectively catalyze the synthesis of CCs from CO2 and various epoxides.34 Liu and coworkers reported a binary catalytic system comprising tetrahydroxydiboron and TBAI, which facilitated the cycloaddition of CO2 with epoxides.35 Independently, Lu and coworkers showed that boron oxide (B2O3), in conjunction with tetrabutylammonium bromide (TBAB), can serve as an efficient catalyst for CO2-epoxide cycloaddition.36 More recently, our research group conducted a computational study that provided valuable mechanistic insights into the B2O3-catalyzed cycloaddition reaction, shedding light on key reaction pathways and activation modes.37 Beyond cyclic-carbonate synthesis, boron-based catalysts have also been explored to copolymerize CO2 with epoxides to produce PCs. Feng, Gnanou, and coworkers reported the first example of a boron-based catalytic system for this transformation, utilizing a binary system comprising triethylborane (BEt3) and onium salts such as bis(triphenylphosphine)iminium chloride (PPNCl) to facilitate the copolymerization of CO2 with both propylene oxide (PO) and cyclohexene oxide (CHO).38 Building on this approach, Zhang and coworkers demonstrated that BEt3, in combination with a tertiary amine (TEA), could also effectively catalyze the copolymerization of CO2 and PO.39
Inspired by the work of Gnanou and coworkers,38 Kerton and colleagues explored the use of triphenylborane (BPh3) as an alternative to BEt3 for catalyzing the coupling of CO2 with epoxides.40 Interestingly, their study revealed substrate-dependent selectivity, leading to the formation of either CC or PC products. Using the same binary catalytic system comprising BPh3 and PPNCl, they observed that PO preferentially formed CC, whereas CHO favoured PC formation (Fig. 1c). Experimental kinetic studies further unveiled a distinctive feature of BPh3 catalysis. Unlike many other metal-free systems, where the reaction order with respect to CO2 is typically one or zero, this system exhibited an inverse dependence, resulting in an overall reaction order of −1 for CO2. However, the fundamental factors governing this unique catalytic behaviour remain unclear, necessitating further investigation. Additionally, the effect of substituting ethyl groups with phenyl groups at the boron center and its role in dictating substrate-dependent selectivity remains an open question. To the best of our knowledge, no comprehensive computational study has sketched the catalytic landscape of BPh3-mediated CO2-epoxide coupling. Herein, we employ density functional theory (DFT) calculations to elucidate the reaction mechanism of BPh3-catalyzed CO2 and epoxide coupling. Through distortion–interaction analysis (DIA) and non-covalent interaction (NCI) analysis, we aim to provide fundamental insights into the origins of substrate-dependent selectivity in this system.
2. Computational details
Computational calculations were performed using the ORCA quantum chemical software package (version 5.0.3).41,42 Geometry optimizations for all minima and transition states were conducted in the gas phase using the hybrid M06-2X43,44 density functional, in combination with the def2-SVP45 basis set, with the defgrid3 tight integration grid. The selection of the M06-2X functional was based on its proven reliability in modelling boron-catalyzed coupling reactions, ensuring accurate predictions of reaction mechanisms.46–48 To enhance computational efficiency, the RIJCOSX49 approximation was employed. Vibrational frequency analysis was performed at the same level of theory to confirm the nature of the stationary points and to obtain zero-point energy (ZPE) and thermal corrections at the corresponding experimental temperatures. Intrinsic reaction coordinate (IRC) calculations50 were carried out to ensure that transition state correctly connects the reactant and product minima on the potential energy surface. Since the experimental studies were conducted under solvent-free conditions,40 no solvation model is applied in our calculations. To further refine the energies, single-point energy calculations were performed on the optimized geometries using the M06-2X functional with the larger def2-TZVP45 basis set. All reported relative Gibbs energies in the mechanistic study are obtained at the M06-2X/def2-TZVP//M06-2X/def2-SVP level of theory. Non-covalent interaction (NCI) analyses were conducted using the Multiwfn program,51 and NCI isosurfaces were visualized with VMD 1.9.3.52 The three-dimensional molecular structures were rendered using the CYLview program.53 The computational model employed in our mechanistic study is illustrated in Fig. 2. | ||
| Fig. 2 Computational model used in the mechanistic study. | ||
3. Results and discussion
The result and discussion section is divided into distinct subsections to systematically analyze the catalytic behaviour of borane systems in CO2-epoxide coupling reactions. We first investigated the BPh3-catalyzed CO2 with propylene oxide (PO) and CO2 with cyclohexene oxide (CHO) coupling reactions, highlighting their mechanistic pathways and product selectivity. To further understand the substrate-dependent selectivity observed in BPh3 catalysis, we performed a detailed computational analysis to elucidate the underlying factors influencing reaction outcomes. Next, we extend our study to BEt3-catalyzed CO2-epoxide coupling reactions, examining the BEt3-catalyzed reactions of CO2 with PO and CHO, respectively. A comparative evaluation of BPh3 and BEt3 systems provides deeper insights into how boron substitution influences product selectivity.3.1 BPh3 catalyzed coupling reaction of CO2 and propylene oxide (PO)
The Gibbs free energy profile for the BPh3-catalyzed coupling reaction of CO2 and PO is depicted in Fig. 3. To account for the role of the bulky PPNCl co-catalyst, its catalytic influence was modelled using a chloride ion (Cl−), as its primary contribution stems from the Cl− active center. This approach aligns with previous computational studies investigating the role of quaternary ammonium halide salts in catalysis.37,54–56 The reaction begins with the formation of adduct Int1_a, in which one BPh3 molecule coordinates to the PO group, activating it, while a second BPh3 forms a complex with a chloride ion (BPh3_Cl−); these two species then associate to form the Int1_a adduct. Subsequently, Int1_a undergoes ring opening via the transition state TS1_a, facilitated by the direct nucleophilic attack of Cl− on the epoxide, leading to concurrent CPO–OPO bond cleavage and CPO–Cl bond formation. This step proceeds with an energy barrier of 27.2 kcal mol−1 (relative to BPh3 + PO). The subsequent introduction of a CO2 molecule and desorption of one BPh3 results in the formation of a stable intermediate Int2_a, characterized by weak van der Waals interactions between CO2 and the alkoxide (O−) species. | ||
| Fig. 3 Gibbs energy profile for BPh3 catalyzed coupling reaction of CO2 and PO using M06-2X/def2-TZVP//M06-2X/def2-SVP level of DFT. Values in the parentheses are the Gibbs energies (relative to BPh3 + PO) at 373.15 K in kcal mol−1. | ||
Introducing an additional BPh3 molecule in the vicinity of CO2 leads to the formation of a new species, Int3_a, which undergoes CO2 insertion via a transition state, TS2_a, with a reduced activation barrier of 20.9 kcal mol−1 (relative to Int2_a). In TS2_a, two BPh3 molecules are involved–one coordinating the epoxide and the other interacting with the CO2 molecule. Following the nucleophilic attack of the alkoxide (O−) on the CO2 carbon center via TS2_a, the reaction proceeds to form the intermediate Int4_a.
From Int4_a, the reaction can proceed via two different pathways: one leading to the formation of cyclic-carbonate (CC) and the other resulting in poly-carbonate (PC) formation. The departure of the weakly coordinated BPh3 molecule from the epoxide oxygen generates the intermediate Int5_a, which undergoes intramolecular cyclization through the transition state TS3_a. This ring-closing transition state involves the formation of a CPO–OCO2 bond, accompanied by the simultaneous cleavage of the CPO–Cl bond. The activation barrier for this step is 23.9 kcal mol−1 (relative to Int5_a), ultimately yielding Int6_a (the CC product coordinated to BPh3 species and regenerated Cl− ion). Alternatively, in the poly-carbonate formation pathway, the dissociation of BPh3 from Int4_a, followed by the introduction of a BPh3-activated PO molecule, leads to the formation of Int7_a. This intermediate subsequently undergoes epoxide addition through the transition state TS4_a, which requires an activation barrier of 28.6 kcal mol−1 (relative to Int2_a). In TS4_a, nucleophilic attack by the alkoxide (O−) on the newly introduced PO molecule results in an epoxide ring opening, yielding Int8_a. This species serves as the key intermediate for poly-carbonate propagation, where further reaction can proceed through alternating CO2 insertion and epoxide addition steps, facilitating polymer chain growth.
A comparison of the activation barriers for the ring-closing step (TS3_a, ΔG‡ = 23.9 kcal mol−1) and the epoxide addition step (TS4_a, ΔG‡ = 28.6 kcal mol−1) indicates that the formation of the CC product is more favorable than PC formation. This aligns well with experimental observations, where the BPh3-catalyzed coupling of CO2 and PO yields the CC product. Among all the mechanistic steps, the epoxide ring-opening transition state TS1_a exhibits the highest activation barrier (ΔG‡ = 27.2 kcal mol−1), identifying it as the rate-determining step. This computational finding is consistent with the experimental proposed rate-determining step and kinetic studies, showing a first-order dependence on PO, PPNCl, and BPh3. Furthermore, experimental investigations by Kerton and coworkers40 revealed that lower CO2 concentrations accelerated reaction rates and enhanced overall substrate conversion. As illustrated in Fig. 4, introducing CO2 to the BPh3 coordinated PO species BPh3_PO leads to the de-coordination of the PO molecule, resulting in the formation of a stable inactive species BPh3_CO2.
 | ||
| Fig. 4 Formation of inactive species in excess of CO2. | ||
This inactive species inhibits the coordination and activation of the epoxide, which is essential for the coupling reaction, thereby suppressing catalytic activity. The formation of this inactive species increases the overall activation barrier to 31.3 kcal mol−1, which is not reasonable for catalysis at 100 °C. This computational insight provides a mechanistic explanation for the experimentally observed inverse rate dependence on CO2 concentration (reaction order with respect to CO2 = −1). It suggests that an excess of CO2 slows down the epoxide ring-opening step by forming an inactive species, thus decelerating the overall reaction.
3.2 BPh3 catalyzed coupling reaction of CO2 and cyclohexene oxide (CHO)
The Gibbs free energy profile for the BPh3-catalyzed coupling reaction of CO2 and CHO is presented in Fig. 5. The reaction begins with formation of adduct Int1_b, followed by ring opening via TS1_b, where nucleophilic attack by the chloride ion induces cleavage of the CCHO–OCHO bond while simultaneously forming the CCHO–Cl bond. This step proceeds with an activation barrier of 22.3 kcal mol−1 (relative to BPh3 + CHO). The subsequent introduction of CO2 and desorption of one BPh3 results in the formation of the oxyanion intermediate Int2_b, characterized by weak van der Waals interactions between CCO2 and the alkoxide O−. In Int2_b, when an additional BPh3 molecule interacts with CO2, leading to the formation of Int3_b. This additional interaction facilitates CO2 activation via the transition state TS2_b, which requires an activation barrier of 20.6 kcal mol−1 (relative to Int2_b). In TS2_b, the alkoxide (O−) attacks the carbon center of CO2, yielding the intermediate Int4_b. | ||
| Fig. 5 Gibbs energy profile for BPh3 catalyzed coupling reaction of CO2 and CHO using M06-2X/def2-TZVP//M06-2X/def2-SVP level of DFT. Values in the parentheses are the Gibbs energies (relative to BPh3 + CHO) at 333.15 K in kcal mol−1. | ||
This boron-carbonate species, Int4_b, serves as a branching point for both CC and PC formation pathways. In the CC formation route, the weakly coordinated BPh3 molecule dissociates from OCHO, generating the intermediate Int5_b, which can subsequently undergo intramolecular cyclization through the five-membered transition state TS3_b. This step features the formation of a CCHO–OCO2 bond, accompanied by the simultaneous breaking of the CCHO–Cl bond, eventually yielding Int6_b (where the CC product remains coordinated to BPh3, and Cl− is regenerated). However, unlike the BPh3-catalyzed reaction of CO2 and PO, the ring-closing step in the CO2–CHO system requires a significantly higher energy barrier (ΔG‡ = 59.1 kcal mol−1, relative to Int2_b), making CC formation kinetically unfavourable. Alternatively, in the PC formation pathway, the dissociation of BPh3 from Int4_b, followed by addition of activated CHO molecule by BPh3, leads to the formation of Int7_b. The polymerization process proceeds via epoxide addition through the transition state TS4_b, with an activation barrier of 18.4 kcal mol−1 (relative to Int2_b). In this step, nucleophilic attack by the alkoxide (O−) on the second CHO molecule induces epoxide ring opening, yielding Int8_b. Once Int8_b is formed, polymer chain growth can continue through alternating CO2 insertion and epoxide addition steps. In summary, the energy barrier required for CC product formation (ΔG‡ = 59.1 kcal mol−1) is significantly higher than the activation barrier of PC formation (ΔG‡ = 18.4 kcal mol−1). This substantial energy difference renders the PC formation pathway kinetically favourable providing a mechanistic rationale for the experimental observation of PC product in BPh3-catalyzed CO2 and CHO coupling reactions.
3.3 Origins of substrate-dependent selectivity in BPh3 catalyzed coupling reactions
To gain deeper insights into the substrate-dependent selectivity observed in BPh3-catalyzed CO2 and epoxide coupling reactions, we performed distortion/interaction analysis (DIA)57–59 on the two key competing transition states: ring-closing (leading to the CC product) and epoxide addition (leading to the PC product) for both CO2 with PO and CO2 with CHO coupling reactions (Fig. 6). In this approach, each transition state was fragmented into two components: the catalyst (Cat) and the substrate (Sub). Single-point energy calculations were then conducted to determine the distortion energy (ΔEdist) and interaction energy (ΔEint) between the fragments (see ESI† section S3 for further details). For the CO2 and PO system, the total distortion energy in the epoxide addition transition state TS4_a (ΔEdist = 30.7 kcal mol−1) is 5.2 kcal mol−1 lower than that in the ring-closing transition state TS3_a (ΔEdist = 35.9 kcal mol−1). However, the interaction energy between the distorted fragments is significantly higher in TS3_a (ΔEint = −12.6 kcal mol−1) compared to TS4_a (ΔEint = −1.8 kcal mol−1). These results suggest that the strong interaction energy between the fragments in TS3_a plays a dominant role in favoring cyclic-carbonate formation in BPh3-catalyzed CO2 and PO coupling. | ||
| Fig. 6 Distortion/interaction analysis (DIA) of BPh3 catalyzed (a) CO2 and PO; (b) CO2 and CHO coupling reaction's selectivity determining transition states. ΔE‡ is the activation energy, ΔECatdist and ΔESubdist are the distortion energies of the Cat and Sub fragment, respectively, ΔEdist is the total distortion energy and ΔEint is the interaction energy between the Cat and Sub fragments. In DIA model, ΔE‡ = ΔEdist + ΔEint. The Sub fragment is highlighted in all the 3D structures and all the numerical values are in kcal mol−1. | ||
In contrast, for the CO2 and CHO system, both the total distortion energy and interaction energy are higher in the ring-closing transition state TS3_b (ΔEdist = 100.1 kcal mol−1, ΔEint = −43.0 kcal mol−1) compared to the epoxide addition transition state TS4_b (ΔEdist = 27.2 kcal mol−1, ΔEint = −11.1 kcal mol−1). The significantly higher distortion energy in TS3_b makes the ring-closing step less favorable than epoxide addition, leading to a preference for poly-carbonate formation. In summary, the differences in distortion and interaction energies between the ring-closing and epoxide addition transition states play a crucial role in determining the product selectivity. In the CO2 and PO system, strong interaction energy in the ring-closing step promotes cyclic-carbonate formation, whereas in the CO2 and CHO system, the high distortion energy in the ring-closing step disfavours cyclic-carbonate formation, making poly-carbonate the preferred product.
In addition, non-covalent interaction (NCI) analysis60 was conducted to further investigate the differences between the ring-closing transition states in the BPh3-catalyzed coupling reactions of CO2 and PO (TS3_a), and CO2 and CHO (TS3_b), respectively. The NCI plots (Fig. 7) for TS3_a and TS3_b reveal a significant increase in steric repulsion around the five-membered ring formation when the substrate changes from PO to the bulkier CHO. The enhanced red isosurfaces in TS3_b indicate pronounced steric hindrance during the ring-closing step, making this transition state energetically less favorable. These findings strongly suggest that the high activation barrier observed for the ring-closing step in the CO2 and CHO system is a direct consequence of increased steric repulsion. Furthermore, this conclusion is consistent with our DIA results, which showed significantly higher distortion energy for TS3_b compared to TS3_a, reinforcing the mechanistic rationale for substrate dependent selectivity in BPh3-catalyzed CO2 and epoxides coupling reaction.
 | ||
| Fig. 7 NCI plots for BPh3 catalyzed coupling reaction of CO2 with PO (TS3_a) and CO2 with CHO (TS3_b) ring closing transition state structures. Blue, green, and red isosurfaces represent strong attractions, weak van der Waals interaction, and steric repulsions, respectively. | ||
3.4 BEt3 catalyzed coupling reaction of CO2 and epoxides
In their experimental study, Gnanou and coworkers utilized BEt3 as a catalyst for the coupling of CO2 and epoxides.38 Unlike the BPh3 system reported by Kerton and coworkers,40 where substrate-dependent selectivity was observed, the BEt3 system exclusively yielded poly-carbonate (PC) products from both propylene oxide (PO) and cyclohexene oxide (CHO). To investigate how substituting ethyl groups with phenyl groups at the boron center influences product selectivity, we extended our mechanistic study to BEt3-catalyzed CO2-epoxide coupling reactions. We first explored the mechanistic pathways of BEt3-catalyzed CO2 and PO coupling, and the corresponding Gibbs free energy profile is provided in Fig. 8. The reaction begins with the formation of the adduct Int1_c, followed by epoxide ring opening via the transition state TS1_c, where chloride ion attacks PO. This step proceeds with an activation barrier of 22.9 kcal mol−1 (relative to BEt3 + PO). The subsequent addition of CO2 and desorption of one BEt3 results in the formation of a stable intermediate Int2_c, where the alkoxide anion interacts with CO2 through weak van der Waals forces. From Int2_c, coordination of an additional BEt3 molecule to CO2 in Int2_c generates Int3_c, which undergoes CO2 insertion via a transition state TS2_c with an activation barrier of 12.4 kcal mol−1 (relative to Int2_c) to yield intermediate Int4_c. The additional BEt3 molecule stabilizes the negative charge on CO2 in TS2_c through interaction with its Lewis acidic boron center, thereby lowering the energy barrier for CO2 insertion step. | ||
| Fig. 8 Gibbs energy profile for BEt3 catalyzed coupling reaction of CO2 and PO using M06-2X/def2-TZVP//M06-2X/def2-SVP level of DFT. Values in the parentheses are the Gibbs energies (relative to BEt3 + PO) at 333.15 K in kcal mol−1. | ||
Following the formation of Int4_c, the reaction can proceed via two competing pathways. In the first pathway, dissociation of the weakly coordinated BEt3 generates Int5_c, which undergoes intramolecular cyclization through a five-membered transition state TS3_c (ΔG‡ = 26.0 kcal mol−1, relative to Int5_c) to yield Int6_c (the CC product coordinated to BEt3 with a regenerated Cl− ion). In the alternative pathway, displacement of BEt3 in Int4_c by an incoming BEt3-activated PO molecule leads to the formation of Int7_c. This intermediate undergoes epoxide addition via transition state TS4_c, requiring an activation barrier of 19.0 kcal mol−1 (relative to Int7_c). Here, nucleophilic attack by the alkoxide ion induces ring opening of the second epoxide, yielding Int8_c, the key intermediate for poly-carbonate propagation. Subsequent alternating CO2 insertion and epoxide addition steps drive poly-carbonate formation. The BEt3-catalyzed coupling of CO2 and CHO follows a similar mechanism, and the corresponding Gibbs free energy profile is presented in Fig. S5 (see ESI† section S5 for more details).
3.5 Comparative evaluation of BPh3 vs. BEt3 catalytic systems
A comparison of the energy profiles for BPh3- and BEt3-catalyzed CO2 and PO coupling reactions (Fig. 4 and 8) reveals a key difference in their reactivity. In the BPh3 system, the pathway leading to CC formation (ΔG‡ = 23.9 kcal mol−1) is more favorable than the PC formation pathway (ΔG‡ = 28.5 kcal mol−1). However, the BEt3 system exhibits the opposite trend, where the transition state TS3_c (ΔG‡ = 26.0 kcal mol−1) associated with CC formation has a 7 kcal mol−1 higher energy barrier than TS4_c (ΔG‡ = 19.0 kcal mol−1), which leads to PC formation. To understand this reactivity difference, we performed DIA on the competing transition states TS3_c and TS4_c (Fig. 9). Although the interaction energy (ΔEint) in TS3_c (−13.6 kcal mol−1) is slightly higher than that in TS4_c (−12.0 kcal mol−1), the total distortion energy (ΔEdist) in TS3_c (38.9 kcal mol−1) is significantly greater than in TS4_c (30.5 kcal mol−1). This indicates that distortion energy plays a dominant role in BEt3-catalyzed CO2 and PO coupling, where the lower distortion energy of the epoxide addition transition state (TS4_c) makes it relatively more stable, favoring PC formation over CC formation. A further comparison of DIA results for the ring-opening and epoxide addition transition states in BPh3 and BEt3 systems (Fig. 6a and 9) reveals additional insights. The overall effect of ΔEdist and ΔEint on the activation barrier for the ring-opening transition states (TS3_a and TS3_c) is similar in both catalytic systems. However, in the epoxide addition transition states (TS4_a and TS4_c), ΔEdist values are nearly identical (30.7 vs. 30.5 kcal mol−1), whereas ΔEint in the BEt3 system (−12.0 kcal mol−1) is significantly higher than in the BPh3 system (−1.8 kcal mol−1). This difference arises from steric factors. In TS4_a, the bulky phenyl groups in the BPh3 catalysts lead to intermolecular steric repulsion, reducing effective attractive interaction between the ‘Cat’ and ‘Sub’ fragments. Such steric hindrance is absent in TS4_c due to small ethyl groups in BEt3, allowing stronger interactions. These results suggest that substituting ethyl groups with bulky phenyl groups at the boron center reduces intermolecular interactions during the epoxide addition step. Consequently, this effect makes PC formation less favorable as compared to CC formation in the BPh3 system, whereas in the BEt3 system, stronger intermolecular interactions facilitate PC formation over CC formation. | ||
| Fig. 9 Distortion/interaction analysis (DIA) of selectivity determining transition states of BEt3 catalyzed coupling reaction of CO2 and PO. | ||
As for the CO2 and CHO coupling reactions, both BEt3 and BPh3 catalytic systems favour PC formation. A comparative DIA analysis of the ring-closing and epoxide addition transition states in both systems (Fig. 6b and S7†) reveals that in both cases, the high distortion energy during the ring-closing step disfavours CC formation, making PC formation the preferred pathway (see ESI† section S5 for more details). In summary, our detailed mechanistic study provides a clear explanation for the substrate-dependent selectivity observed in BPh3-catalyzed CO2-epoxide coupling reactions, while also demonstrating how substituting bulky phenyl groups with ethyl groups at the boron center eliminates this selectivity in BEt3-catalyzed reactions.
Conclusions
In this study, we conducted a detailed mechanistic investigation into the triphenylborane (BPh3)-catalyzed coupling of CO2 with epoxides, aiming to uncover the origins of its substrate-dependent selectivity. While BPh3 catalysis yielded cyclic-carbonate (CC) from propylene oxide (PO), it exclusively favoured poly-carbonate (PC) formation from cyclohexene oxide (CHO), highlighting a distinct selectivity pattern. To further understand this behaviour, we extended our study to triethylborane (BEt3)-catalyzed CO2-epoxide coupling, where both PO and CHO yielded PC products, demonstrating the absence of selectivity. This comparative evaluation provided critical insights into how boron substitution influences catalytic performance and product distribution.Our results indicate that epoxide ring opening is the rate-determining step, aligning with experimental mechanistic studies. Additionally, we found that high CO2 concentrations in the initial stage can lead to the formation of an inactive species that inhibits epoxide coordination and activation, thereby suppressing catalytic activity. This finding provides a mechanistic explanation for the experimentally observed inverse rate dependence on CO2 concentration.
Through distortion/interaction analysis (DIA) and non-covalent interaction (NCI) analysis, we established that in the BPh3-catalyzed CO2 and PO system, the weaker intermolecular interactions in the epoxide addition transition state render PC formation less favorable, leading to a preference for CC formation. However, in the BEt3-catalyzed CO2 and PO system, stronger intermolecular interactions in the corresponding transition state stabilize PC formation, overriding the selectivity observed in BPh3 catalysis. Moreover, for CO2 and CHO coupling reactions, both BPh3 and BEt3 catalytic system exhibit high distortions in the ring-closing transition state, making CC formation kinetically unfavourable, thereby explaining the exclusive formation of PC in both cases.
This study demonstrates that substituting ethyl groups with phenyl groups at the boron center fundamentally alters catalytic behaviour, affecting intermolecular interactions, transition state stability, and overall reaction energetics. Our findings provide a mechanistic rationale for the substrate-dependent selectivity in BPh3 catalysis while also offering valuable insights for the development of tailored boron-based catalysts for selective CO2 utilization in both cyclic-carbonate and poly-carbonate synthesis.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Nikunj Kumar: writing – original draft, visualization, methodology, investigation, formal analysis, data curation, conceptualization. Puneet Gupta: writing – review & editing, supervision, resources, project administration, funding acquisition, formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. K. thanks CSIR for the SRF fellowship (09/143(1026)/2020-EMRI). P. G. acknowledges SERB (CRG/2021/000759) for financial support and National Supercomputing Mission (NSM) for providing computing resources of ‘PARAM Ganga’ at IIT Roorkee, which is implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India.References
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC
.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed
- F. Sher, S. Z. Iqbal, S. Albazzaz, U. Ali, D. A. Mortari and T. Rashid, Fuel, 2020, 282, 118506 CrossRef CAS
- K. H. S. M. Sampath, P. G. Ranjith and M. S. A. Perera, Energy Fuels, 2020, 34, 13369–13383 CrossRef CAS
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 184–187 CrossRef CAS PubMed
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC
- F. D. Meylan, V. Moreau and S. Erkman, J. CO2 Util., 2015, 12, 101–108 CrossRef CAS
- W. D. Jones, J. Am. Chem. Soc., 2020, 142, 4955–4957 CrossRef CAS PubMed
- J. Chen, G. Chiarioni, G. J. W. Euverink and P. P. Pescarmona, Green Chem., 2023, 25, 9744–9759 RSC
- W.-W. Yu, X.-G. Meng, Z.-Y. Gan, W. Li, Y.-L. Zhang and J. Zhou, Catal. Sci. Technol., 2024, 14, 6215–6223 RSC
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed
- V. Besse, F. Camara, C. Voirin, R. Auvergne, S. Caillol and B. Boutevin, Polym. Chem., 2013, 4, 4545–4561 RSC
- J. Wang, H. Zhang, Y. Miao, L. Qiao, X. Wang and F. Wang, Green Chem., 2016, 18, 524–530 RSC
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS
- W. Zhou, Q. W. Deng, G. Q. Ren, L. Sun, L. Yang, Y. M. Li, D. Zhai, Y. H. Zhou and W. Q. Deng, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed
- Z. Li, Z. Su, W. Xu, Q. Shi, J. Yi, C. Bai, N. Wang and J. Li, ChemCatChem, 2020, 12, 4839–4844 CrossRef CAS
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS
- A. I. Adeleye, D. Patel, D. Niyogi and B. Saha, Ind. Eng. Chem. Res., 2014, 53, 18647–18657 CrossRef CAS
- W. Natongchai, S. Posada-Pérez, C. Phungpanya, J. A. Luque-Urrutia, M. Solà, V. D'Elia and A. Poater, J. Org. Chem., 2022, 87, 2873–2886 CrossRef CAS PubMed
- F. N. Al-Rowaili, U. Zahid, S. Onaizi, M. Khaled, A. Jamal and E. M. AL-Mutairi, J. CO2 Util., 2021, 53, 101715 CrossRef CAS
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol.
Chem., 1969, 130, 210–220 CrossRef CAS
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS PubMed
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2011, 50, 4481–4492 CrossRef CAS PubMed
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed
- R. M. Izatt, S. R. Izatt, R. L. Bruening, N. E. Izatt and B. A. Moyer, Chem. Soc. Rev., 2014, 43, 2451–2475 RSC
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed
- Y. Ge, G. Cheng and H. Ke, J. CO2 Util., 2022, 57, 101873 CrossRef CAS
- J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS
- Y. Luo, F. Chen, H. Zhang, J. Liu and N. Liu, J. Org. Chem., 2023, 88, 15717–15725 CrossRef CAS PubMed
- L. Y. Zhao, J. Y. Chen, W. C. Li and A. H. Lu, J. CO2 Util., 2019, 29, 172–178 CrossRef CAS
- N. Kumar and P. Gupta, J. Catal., 2024, 439, 115787 CrossRef CAS
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed
- Y. Wang, J. Y. Zhang, J. L. Yang, H. K. Zhang, J. Kiriratnikom, C. J. Zhang, K. L. Chen, X. H. Cao, L. F. Hu, X. H. Zhang and B. Z. Tang, Macromolecules, 2021, 54, 2178–2186 CrossRef CAS
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, 1–15 Search PubMed
- R. Valero, R. Costa, I. D. P. R. Moreira, D. G. Truhlar and F. Illas, J. Chem. Phys., 2008, 128, 114103 CrossRef PubMed
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- G. W. Yang, Y. Y. Zhang, R. Xie and G. P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed
- Y. Y. Zhang, G. W. Yang, R. Xie, X. F. Zhu and G. P. Wu, J. Am. Chem. Soc., 2022, 144, 19896–19909 CrossRef CAS PubMed
- J. Wang, Y. Zhu, M. Li, Y. Wang, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 61, e202208525 CrossRef CAS PubMed
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- C. Y. Legault, CYLview, ver. 1.0b, Université de Sherbrooke, QC, 2009, http://www.cylview.org Search PubMed
- R. Babu, R. Roshan, Y. Gim, Y. H. Jang, J. F. Kurisingal, D. W. Kim and D. W. Park, J. Mater. Chem. A, 2017, 5, 15961–15969 RSC
- C. J. Zhang, S. Q. Wu, S. Boopathi, X. H. Zhang, X. Hong, Y. Gnanou and X. S. Feng, ACS Sustainable Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS
- J. Tang, M. Li, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 61, 1–8 Search PubMed
- X. Hong, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2017–2025 CrossRef CAS PubMed
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed
- N. Goswami, N. Kumar, S. Bag, P. Gupta and D. Maiti, ACS Catal., 2023, 13, 11091–11103 CrossRef CAS
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00281h |
| This journal is © The Royal Society of Chemistry 2025 |