
Mechanism and origin of stereoselectivity and regioselectivity in cobalt-catalyzed C–H functionalization of arylphosphinamide†
Chuanchuan Luo‡
,
Saibo Cao‡,
Hao-Ran Yang* and
Yang Wang *
*
Department of Material and Chemical Engineering, Zhengzhou University of Light Industry, 136 Science Avenue, Zhengzhou, Henan province, 450001 P. R. China. E-mail: yanghr@zzuli.edu.cn; wangyang@zzuli.edu.cn
First published on 19th June 2025
Abstract
The cobalt-catalyzed C–H functionalization of arylphosphinamides is a promising strategy for constructing P-containing scaffolds but has been mechanistically underexplored. We perform a density functional theory (DFT) study to elucidate the mechanism and origins of selectivities for this transformation. The computational results reveal a stepwise pathway involving sequential N–H and C–H activation followed by alkyne insertion and reductive elimination. The C–H cleavage and alkyne insertion are identified as the stereoselectivity-determining processes, and alkyne insertion is identified as the regioselectivity-determining step. The pronounced S-selectivity arises from a larger number of noncovalent interactions in the low-energy transition state compared with the higher energy transition state. The regioselectivity is determined using a frontier molecular orbital (FMO) analysis. The results of this study provide valuable insights into the underlying chemistry of the Co-catalyzed C–H functionalization of arylphosphinamide.
Introduction
The distinct reactivity of cobalt complexes, particularly for high-valence Co(III) catalysts, renders Co-based systems highly attractive for C–C/C–heteroatom bond formation.1–7 The growing appeal of these systems stems from earth abundance, cost efficiency, and multipurpose catalytic behavior. Two complementary strategies have emerged for Co(III)-catalyzed asymmetric C–H activation. The first approach consists of employing an achiral Cp*Co(III) catalyst in combination with an external chiral carboxylic acid (CCA) ligand to achieve enantiocontrol (Scheme 1A).8–15 The second strategy is pioneered by Cramer and consists of utilizing chiral CpxCo(III) complexes bearing customized cyclopentadienyl (Cpx) ligands (Scheme 1B).16–18 These methodologies have been demonstrated to be remarkably successful, but both fundamentally depend on the use of pseudotetrahedral CpCo(III) catalytic systems, which have inherent limitations. The synthetic challenges associated with CpxCo(III) catalysts, including multistep preparation protocols and limited structural flexibility, have strongly constrained progress in this field. Daugulis and coworkers introduced in situ generated Co(III)-catalyzed C–H functionalization as a more sustainable alternative (Scheme 1C).19 This innovative approach consists of in situ oxidation of commercially available Co(II) salts to form active Co(III) species, bypassing the need for preformed catalysts while offering superior atom economy and cost efficiency.20–26 However, the development of an asymmetric variant of this practical methodology remains an outstanding challenge in the field. | ||
| Scheme 1 (A) Chiral carboxylic acid ligated, (B) chiral cyclopentadienyl ligated, and (C) octahedral Co(III)-catalyzed asymmetric C–H activation strategies. | ||
Chiral phosphorus compounds constitute a privileged class of molecules that have found widespread application as chiral ligands and organocatalysts in asymmetric synthesis.27–30 Consequently, considerable effort has been expended toward developing efficient asymmetric synthetic methods. Recent advances in transition-metal-catalyzed enantioselective C–H functionalization have resulted in a powerful strategy for constructing chiral frameworks, including P-stereogenic phosphorus compounds.31–34 However, existing methodologies predominantly utilize noble metal catalysts, such as Pd, Rh, and Ir systems.35–42 The development of catalytic systems in which earth-abundant 3d transition metals are employed to access these valuable chiral compounds remains largely unexplored to date.
Shi and coworkers recently reported the first successful example of Co-catalyzed enantioselective C–H activation involving arylphosphonamides (Scheme 2).43 Octahedral Co(III) complexes are demonstrated as crucial catalysts for achieving stereocontrol of this transformation. A catalytic paradigm has thus been established for constructing chiral phosphorus compounds through earth-abundant transition-metal catalysis.
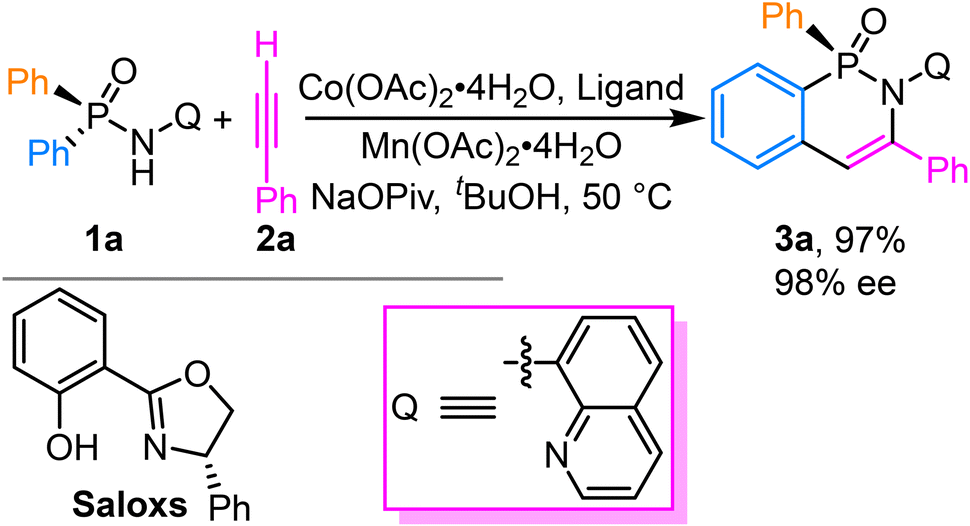 | ||
| Scheme 2 Cobalt-catalyzed asymmetric C–H functionalization of arylphosphinamide. | ||
As illustrated in Scheme 2, selective C–H activation at the phenyl group adjacent to phosphorus enables the formation of P-stereogenic centers with exceptional enantioselectivity (98% ee). Despite the considerable progress made by Shi and coworkers in elucidating the mechanism of this transformation, several critical issues remain to be unresolved: (1) identification of the most energetically favorable reaction pathway remains challenging; (2) the molecular basis for achieving such high stereoselectivity requires further clarification; (3) although [2,1]-insertion of terminal alkynes consistently yields single regioisomeric products, the fundamental origins of this regiocontrol need to be systematically investigated. Building upon our group's extensive expertise in computational mechanistic studies of both organometallic44–47 and organocatalytic48–54 processes, we conduct a comprehensive density functional theory (DFT) investigation into this Co-catalyzed C–H functionalization system. The DFT calculation has been extensively utilized for the mechanistic investigation of reactions catalyzed by transition-metals55–64 and organocatalysts.65–72 The aim of this study is to determine the energetically preferred reaction pathway and thereby uncover the structural and electronic determinants governing stereoselectivity and regioselectivity.
Experimental
Computational details
All quantum chemical calculations are performed using the Gaussian 09 software package.73 Geometry optimizations were conducted using the M06-L functional74,75 at 298 K and 1 atm. The 6-31G(d,p) basis set76,77 is used for the C, H, O, N and P atoms, whereas the SDD basis set78,79 is used for the Co atom. Solvation effects are incorporated into the optimization procedures using the integral equation formalism polarizable continuum model (IEF-PCM)80,81 with 2-methyl-2-propanol (tBuOH) as the solvent. Vibrational frequency analyses are systematically performed at an identical theoretical level to characterize the stationary points in the system: (1) all intermediates were confirmed by the absence of imaginary frequencies and (2) transition states were verified by possessing exactly one imaginary frequency corresponding to the motion of the reaction coordinate. To improve the accuracy of the computed energy, single-point calculations are executed using M06-L with Grimme's D3 dispersion correction combined with the Def2-TZVP basis set82,83 for all atomic centers. An interaction region indicator (IRI) analysis is conducted using the Multiwfn software package,84 and subsequent visualization was performed using the Visual Molecular Dynamics (VMD) molecular graphics system.85To test the accuracy of the results obtained at the M06-L/6-31G(d,p) (sdd for Co atom)//IEF-PCM(tBuOH) level, the structures of the stereoselectivity-determining step are re-optimized using B3LYP,86 B3PW91,87 Cam-B3LYP,88 M06-2X,89 ωB97X-D90 methods. The computational results are summarized in Table S1 of the ESI.† Compared with the results calculated using the M06-L method, the computational outcomes obtained at the different methods have tiny differences and same trends, thus, we think the conclusions based on the results calculated using the M06-L functional are reliable.
Results and discussion
Reaction mechanism
Initial calculations show that the ligand exchange of Saloxs and Co(OAc)2 is exothermic and a feasible process (ΔG = −10.2 kcal mol−1, Fig. S1 of the ESI†), which is then oxidized by Mn(OAc)2 to yield the CoIII–Saloxs species. Thus, the CoIII–Saloxs species should be the catalytically active species and is selected as the starting material in our theoretical study (Scheme 3).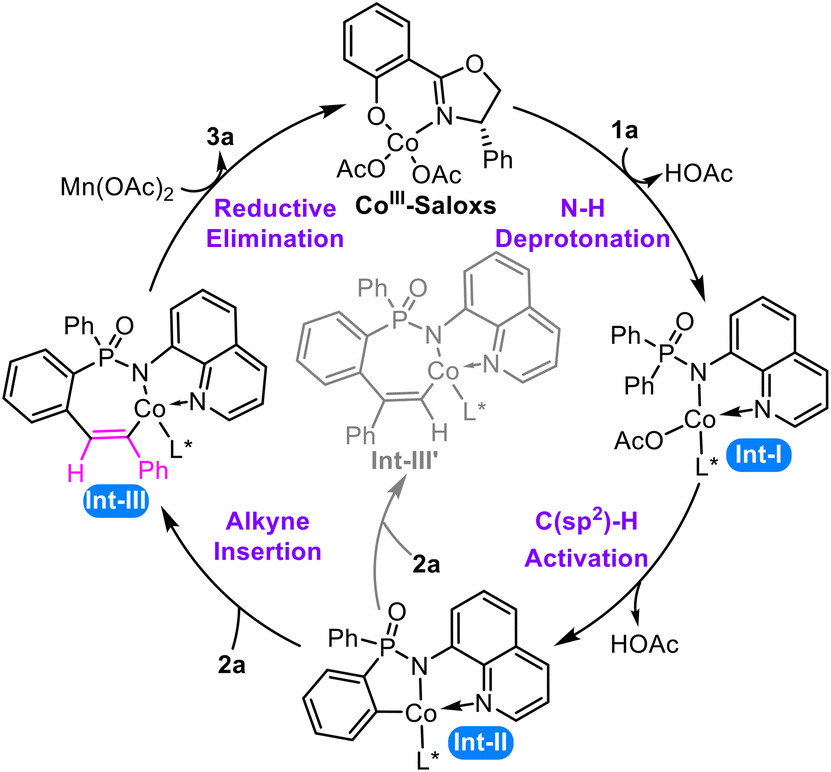 | ||
| Scheme 3 Proposed mechanism of the Co-catalyzed C–H functionalization of arylphosphinamide. | ||
The catalytic process proceeds through four key steps. The cycle commences with ligand exchange, followed by N–H deprotonation to form the N,N-bidentate cobalt intermediate Int-I. This intermediate undergoes concerted metalation–deprotonation (CMD)91,92 to cleave the aromatic C(sp2)–H bond via a six-membered transition state, yielding the cyclometalated intermediate Int-II. This intermediate reacts with the alkyne to induce alkyne insertion,93,94 which can occur through two distinct pathways. The major pathway proceeds through [2,1]-insertion of the terminal alkyne into the Co–C bond to form Int-III, whereas the alternative [1,2]-insertion pathway generates the regioisomeric intermediate Int-III′. Finally, Int-III undergoes reductive elimination followed by reoxidation to release the P-stereogenic product while regenerating the active CoIII–Saloxs species. The reaction mechanism and origins of stereoselectivity and regioselectivity will be discussed in detail in the following sections.
Formation of a five-membered metallacycle
Our computational investigation begins with CoIII–Saloxs as the catalytically active species (Fig. 1). The reaction is initiated by the substrate 1a and CoIII–Saloxs to form the intermediate 4 via an endothermic process (14.7 kcal mol−1).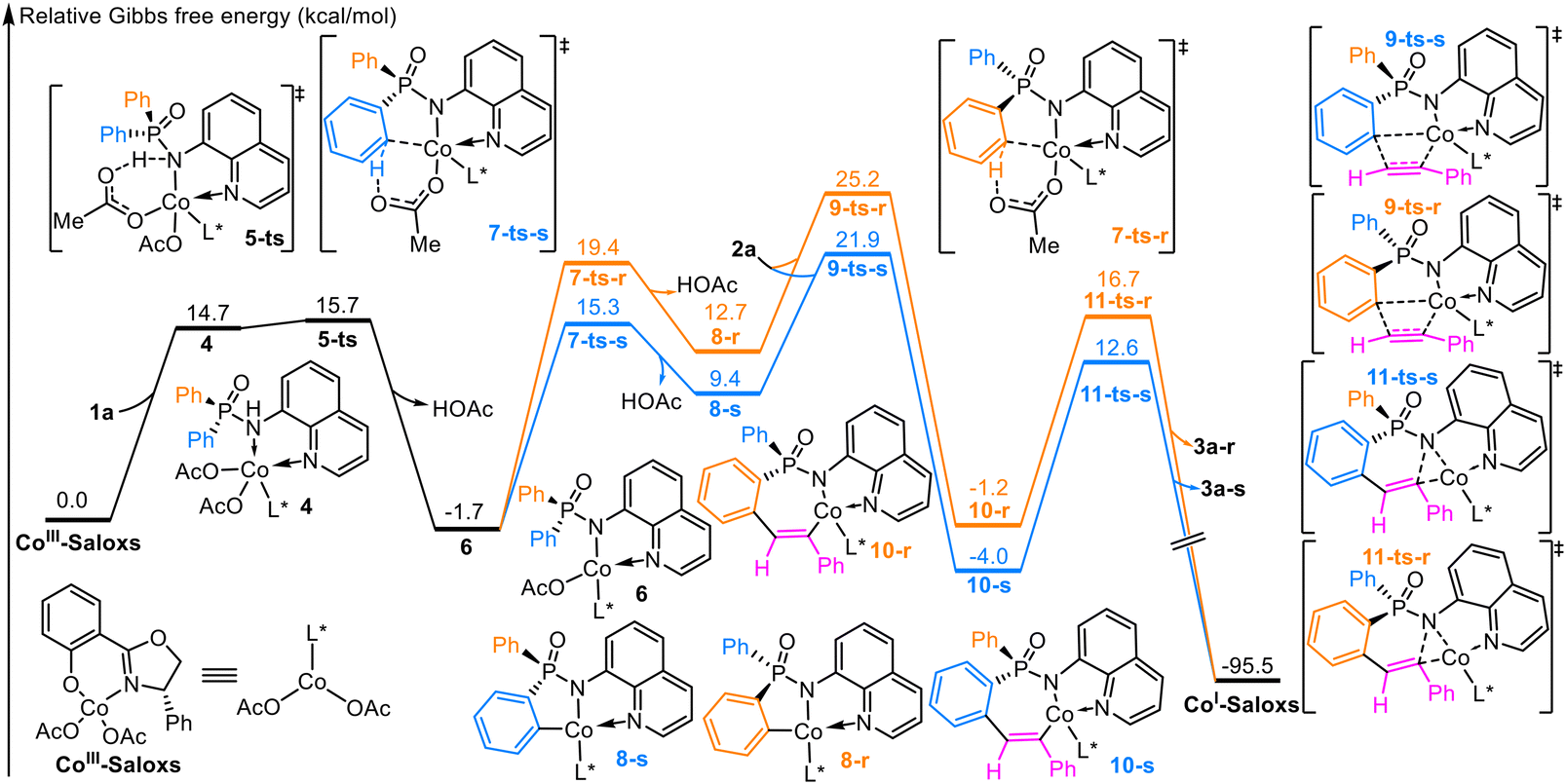 | ||
| Fig. 1 The relative Gibbs free energy profile for the Co-catalyzed C–H functionalization of arylphosphinamide. | ||
As illustrated in Fig. 2, coordination of the N–H bond to the cobalt center induces considerable bond elongation (1.07 Å vs. 1.01 Å in the free substrate), demonstrating catalytic weakening of the N–H bond through Co–N coordination. This activation enables a concerted hydrogen abstraction process to occur, where the acetate ligand (AcO−) facilitates N–H cleavage via a six-membered cyclic transition state (5-ts). This process has an energy barrier of 15.7 kcal mol−1. Subsequent elimination of acetic acid generates the five-membered cobaltacycle intermediate 6, for which the Gibbs free energy is 1.7 kcal mol−1 lower than that of the reactant. In addition, we have also recalculated the energies of N–H deprotonation process at 50 °C to verify the effects of temperature. The computational results show that the Gibbs free energies obtained at 25 °C have a tiny difference from the values obtained at 50 °C. Thus, we think the temperature used in the calculations have the same tendency and little influence on the results.
 | ||
| Fig. 2 The structural parameters for the N–H deprotonation process (distance in Å). | ||
Stereoselective C–H bond activation
Subsequently, the five-membered cobaltacycle intermediate 6 undergoes stereoselective C–H bond activation at either of the prochiral phenyl groups (shown in cyan/orange in Fig. 3), where the absolute configuration is established around the phosphorus atom. As depicted in Fig. 3, the cleavage of the orange-colored phenyl C–H bond forms an R-configured P-stereocenter through the transition state 7-ts-r, whereas the activation of the cyan-colored phenyl C–H bond generates an S-configured P-stereocenter via the transition state 7-ts-s.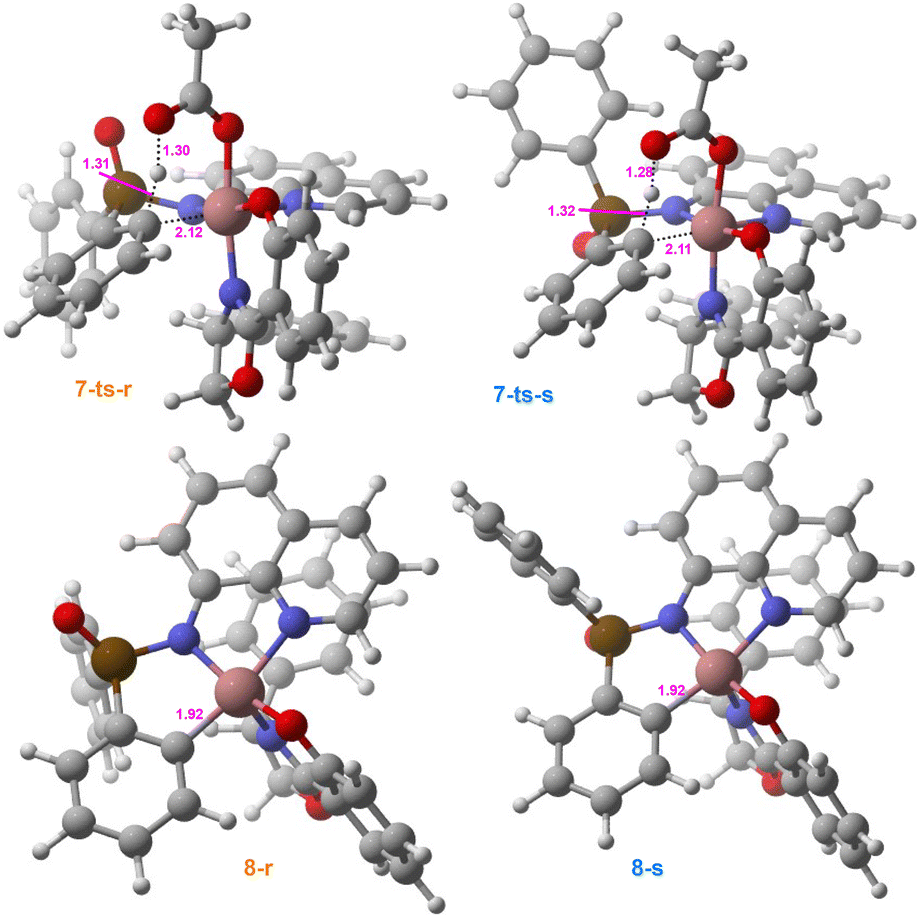 | ||
| Fig. 3 The structural parameters for the stereoselective C–H activation process (distance in Å). | ||
The energy barriers to these two stereoselective pathways associated with transition states 7-ts-s and 7-ts-r are 17.0 and 21.1 kcal mol−1 (Fig. 1), respectively. Thus, the S-configured C–H activation pathway is energetically more favorable than the R-configured C–H activation pathway. The C–H bond is elongated from 1.09/1.08 Å (Fig. 2) in intermediate 6 to 1.31/1.32 Å (Fig. 3) in transition state 7-ts-r/7-ts-s, showing that the C–H bond is broken during the C–H bond activation process.
In the experiment, there is a large excess of NaOPiv in the reaction; the OAc ligands at the metal will be substituted to some degree or other by pivolate. Thus, we have then considered the pivolate-ligated cobalt species promoted C–H functionalization of arylphosphinamide. Three pathways are calculated and compared and the detailed results are provided in Fig. S2 of the ESI.† The computational results show that the energy barriers of N–H deprotonation process are 16.3, 19.5, and 16.6 kcal mol−1, respectively. The energy barriers of pivolate-participated C–H activation process is 22.4 and 18.2 kcal mol−1 associated with R- and S-configured pathways, respectively. These computational results show that the energy barriers involved in pivolate-ligated cobalt species promoted C–H functionalization are disfavored than those in acetate-ligated cobalt species.
Alkyne insertion and reductive elimination
Subsequently, the metallacycle species undergoes the [2,1]-insertion with the terminal alkyne 2a to afford the seven-membered ring intermediates 10-r and 10-s via the transition states 9-ts-r and 9-ts-s, respectively. The energy barriers to the [2,1]-insertion process associated with the transition states 9-ts-r and 9-ts-s are 26.9 and 23.6 kcal mol−1 (Fig. 2), respectively.Alternatively, the metallacycle species can undergo the [1,2]-insertion with the terminal alkyne 2a to give a regioselective isomer. Only S-configured [1,2]-insertion is considered. As depicted in Scheme 4, the energy barrier to the [1,2]-insertion pathway via the transition state 9′-ts-s is 28.1 kcal mol−1, which is 4.5 kcal mol−1 higher than that to the S-configured [2,1]-insertion pathway. This energy difference aligns with the experimental observations that only a single regioisomer is obtained.
 | ||
| Scheme 4 Alternative [1,2]-insertion for the intermediate 8-s with the terminal alkyne 2a (the relative Gibbs free energies are shown in brackets in kcal mol−1). | ||
Next, the reductive elimination of the seven-membered ring intermediate yields the final product and Co(I) species via the transition state 11-ts. The computational results presented in Fig. 1 show that the reductive elimination process can overcome the energy barriers of 16.6 and 17.9 kcal mol−1 associated with 11-ts-s and 11-ts-r, respectively.
The energy barriers to the individual steps for the R-configured pathway are 15.7, 21.1, 26.9, and 17.9 kcal mol−1 and 15.7, 17.0, 23.6, and 16.6 kcal mol−1 for the S-configured pathway. Thus, the S-configured pathway is more energetically favorable than the R-configured pathway and the alternative [1,2]-insertion requires 4.5 kcal mol−1 more energy than [2,1]-insertion. These computational results are in good agreement with the experimental observations.
Origins of selectivities
On the basis of the analyses presented above, the energy barriers to the formation of the P-stereogenic center during the C–H bond activation process for the R- and S-configured isomers are 21.1 and 17.0 kcal mol−1, respectively. The highest energy barrier is associated with the alkyne insertion process, which should therefore be the rate-determining step. Consequently, we identify the sequential C–H activation and alkyne insertion processes as the stereoselectivity-determining step. Other than [2,1]-alkyne insertion, [1,2]-insertion can also occur during the reaction; thus, the alkyne insertion process also determines the regioselectivity, with [2,1]-alkyne insertion proceeding preferentially.Origin of stereoselectivity
The calculated energy barrier to the entire S-configured pathway is calculated to be 3.3 kcal mol−1 lower than that to the entire R-configured pathway (Fig. 1). This energy difference corresponds to an enantiomeric excess (ee) of 99.3%, which aligns well with the experimentally observed 98% ee. To obtain an in-depth understanding of the origin of stereoselectivity, an IRI analysis is performed to visualize the noncovalent interactions in the stereoselective transition states.During the C–H activation process, the C–H⋯N and C–H⋯π noncovalent interactions between the ligand and substrate 1a occur in both 7-ts-r and 7-ts-s. These two types of noncovalent interactions contribute equally to the corresponding transition state. The LP⋯π noncovalent interaction only exists in 7-ts-r, whereas three C–H⋯O noncovalent interactions occur only in 7-ts-s. In conclusion, there are more noncovalent interactions in the transition state 7-ts-s than in 7-ts-r, which is why the S-configured C–H activation process occurs preferentially.
The computational results presented above show that the alkyne insertion process also determines the stereoselectivity; thus, an IRI analysis is also performed on 9-ts. As shown in Fig. 4, the π⋯π and C–H⋯O noncovalent interactions exist in both 9-ts-r and 9-ts-s. For the π⋯π noncovalent interaction, the distances between the phenyl group of the alkyne 2a and the quinoline moiety are 3.04 and 2.98 Å in 9-ts-r and 9-ts-s, respectively, which contribute equally to the corresponding transition state. There is one C–H⋯O noncovalent interaction in 9-ts-r and the distance between the two interacting moieties is 2.52 Å. In 9-ts-s, the distance of the C–H⋯O noncovalent interaction between the two interacting moieties is larger than that in 9-ts-r. Thus, the C–H⋯O noncovalent interactions are stronger in the R-configured transition state than in the S-configured transition state, but this advantage is offset by the unique C–H⋯π and LP⋯π noncovalent interactions in the S-configured transition state 9-ts-s. Therefore, the predominant non-covalent interactions in the S-configured transition state govern the high-level stereoselectivity.
 | ||
| Fig. 4 IRI analysis for the transition states involved in the stereoselectivity-determining processes (the distance between the interacting moieties is shown in brackets in Å). | ||
Origin of regioselectivity
Regioselective alkyne insertion occurs between the intermediates 8 and 2a, where the [1,2]-insertion and [2,1]-insertion modes lead to the formation of the seven-membered cobaltacycles 10′-s and 10-s, respectively. The energy barriers to these two insertion modes are 28.1 and 23.6 kcal mol−1 associated with transition states 9′-ts-s and 9-ts-s, respectively. These results indicate that the [2,1]-insertion mode is more favorable than the [1,2]-insertion mode, which is in accordance with the experimental observations. The regioselectivity of this process is verified by the FMO analysis. As shown in Fig. 5, the FMOs of the regioselective transition states can be divided into two interacting moieties, i.e., 8-s-part and 2a-part. In the [2,1]-insertion mode, the HOMO of 2a directly interacts with the LUMO of 8-s directly to realize the alkyne insertion and the energy gap between the two interacting moieties is 2.23 eV. However, in the [1,2]-insertion mode, the HOMO of 2a interacts with the LUMO+2 of 8-s and the corresponding energy gap (3.69 eV) is higher than that for the [2,1]-insertion mode. This result indicates that a higher energy gap between the two interacting moieties results in a higher energy barrier to [1,2]-insertion, such that [2,1]-insertion occurs preferentially.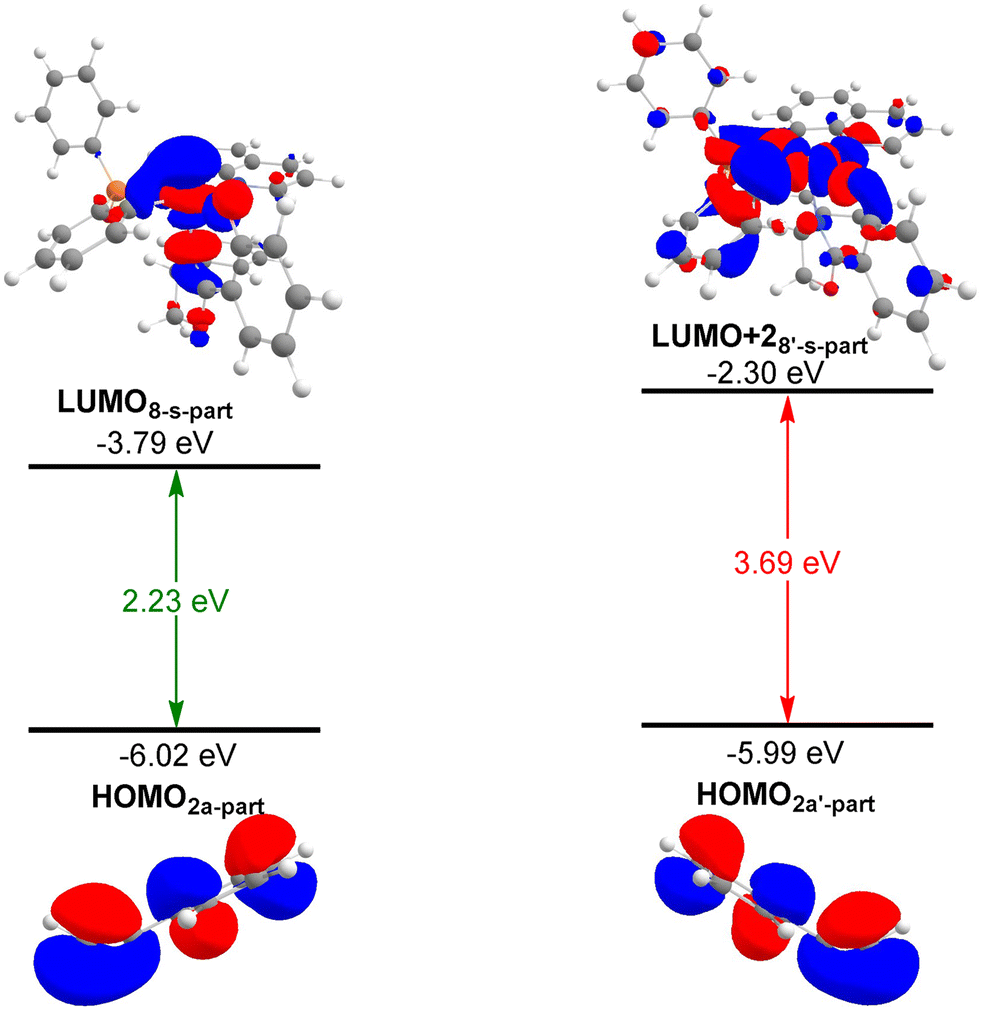 | ||
| Fig. 5 FMO analysis for the regioselective transition states. | ||
Conclusions
In this study, DFT is used to systematically investigate the mechanism and origins of the selectivities (i.e., the stereoselectivity and regioselectivity) of the Co-catalyzed C–H functionalization of arylphosphinamide. The computational results show that the most energetically favorable pathway has four elementary steps: N–H deprotonation, stereoselective C–H bond activation, stereoselective and regioselective alkyne insertion, and reductive elimination. The C–H bond activation and alkyne insertion steps are identified to be the stereoselectivity-determining process, which lead to the formation of the S-configurational isomer forming as the major product. An IRI analysis shows that the predominant non-covalent interactions found in the S-configured transition state considerably lower the relative Gibbs free energy, inducing stereoselectivity. The alkyne insertion process determines the regioselectivity, where the [2,1]-insertion mode is more energetically favorable than the [1,2]-insertion mode. An FMO analysis shows that the regioselectivity is determined by the energy gap between the two interacting moieties in the possible transition states.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Luo C. C. and Cao S. B.: data curation, formal analysis, and investigation; Yang H.-R.: conceptualization, supervision, review & editing; Wang Y.: conceptualization, supervision, funding acquisition, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21903072), the Key R&D and Promotion Projects (Science and Technology Key Projects) of Henan Province (no. 242102310470), and the Key Projects of Colleges and Universities in Henan Province (no. 24B150045). We also thank Edanz (http://www.liwenbianji.cn) for editing a draft of this manuscript.Notes and references
- P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C-H Activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed
.
- Q. Yue, B. Liu, G. Liao and B.-F. Shi, Binaphthyl Scaffold: A Class of Versatile Structure in Asymmetric C-H Functionalization, ACS Catal., 2022, 12, 9359–9396 CrossRef CAS
- M. Moselage, J. Li and L. Ackermann, Cobalt-Catalyzed C-H Activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef PubMed
- S. Sunny and R. Karvembu, Recent Advances in Cobalt-Catalyzed, Directing-Group-Assisted C-H Bond Amidation Reactions, Adv. Synth. Catal., 2021, 363, 4283–4458 CrossRef
- W. W. Xu and M. C. Ye, Enantioselective Cobalt-Catalyzed C-H Functionalization, Synthesis, 2022, 54, 4773–4783 CrossRef CAS
- D. Chandra, Manisha and U. Sharma, Recent Advances in the High-Valent Cobalt-Catalyzed C-H Functionalization of N-Heterocycles, Chem. Rec., 2021, 22, e202100271 CrossRef PubMed
- D. H. Wei, X. J. Zhu, J.-L. Niu and M.-P. Song, High-Valent-Cobalt-Catalyzed C-H Functionalization Based on Concerted Metalation-Deprotonation and Single-Electron-Transfer Mechanisms, ChemCatChem, 2016, 8, 1242–1263 CrossRef CAS
- Q.-J. Yao and B.-F. Shi, Cobalt(III)-Catalyzed Enantioselective C-H Functionalization: Ligand Innovation and Reaction Development, Acc. Chem. Res., 2025, 58, 971–990 CrossRef CAS PubMed
- T. Yoshino and S. Matsunaga, Chiral Carboxylic Acid Assisted Enantioselective C-H Activation with Achiral CpxMIII (M = Co, Rh, Ir) Catalysts, ACS Catal., 2021, 11, 6455–6466 CrossRef CAS
- D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Full Selectivity Control in Cobalt(III)-Catalyzed C-H Alkylations by Switching of the C-H Activation Mechanism, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS PubMed
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Enantioselective C(sp3)-H Amidation of Thioamides Catalyzed by a CobaltIII/Chiral Carboxylic Acid Hybrid System, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed
- F. Pesciaioli, U. Dhawa, J. Oliveira, R. Yin, M. John and L. Ackermann, Enantioselective Cobalt(III)-Catalyzed C-H Activation Enabled by Chiral Carboxylic Acid Cooperation, Angew. Chem., Int. Ed., 2018, 57, 15425–15429 CrossRef CAS PubMed
- Y.-H. Liu, P.-P. Xie, L. Liu, J. Fan, Z.-Z. Zhang, X. Hong and B.-F. Shi, Cp*Co(III)-Catalyzed Enantioselective Hydroarylation of Unactivated Terminal Alkenes via C-H Activation, J. Am. Chem. Soc., 2021, 143, 19112–19120 CrossRef CAS PubMed
- Y.-H. Liu, P.-X. Li, Q.-J. Yao, Z.-Z. Zhang, D.-Y. Huang, M. D. Le, H. Song, L. Liu and B.-F. Shi, Cp*Co(III)/MPAA-Catalyzed Enantioselective Amidation of Ferrocenes Directed by Thioamides under Mild Conditions, Org. Lett., 2019, 21, 1895–1899 CrossRef CAS PubMed
- X. Yu, Z.-Z. Zhang, J.-L. Niu and B.-F. Shi, Coordination-Assisted, Transition-Metal-Catalyzed Enantioselective Desymmetric C-H Functionalization, Org. Chem. Front., 2022, 9, 1458–1484 RSC
- A. G. Herraiz and N. Cramer, Cobalt(III)-Catalyzed Diastereo- and Enantioselective Three-Component C-H Functionalization, ACS Catal., 2021, 11, 11938–11944 CrossRef CAS
- K. Ozols, S. Onodera, L. Woźniak and N. Cramer, Cobalt(III)-Catalyzed Enantioselective Intermolecular Carboamination by C-H Functionalization, Angew. Chem., Int. Ed., 2021, 60, 655–659 CrossRef CAS PubMed
- K. Ozols, Y.-S. Jang and N. Cramer, Chiral Cyclopentadienyl Cobalt(III) Complexes Enable Highly Enantioselective 3d-Metal-Catalyzed C-H Functionalizations, J. Am. Chem. Soc., 2019, 141, 5675–5680 CrossRef CAS PubMed
- L. Grigorjeva and O. Daugulis, Cobalt-Catalyzed, Aminoquinoline-Directed C(sp2)-H Bond Alkenylation by Alkynes, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed
- T. Liu, W. Q. Zhang, C. Xu, Z. H. Xu, D. G. Song, W. Qian, G. Lu, C.-J. Zhang, W. H. Zhong and F. Ling, Synthesis of P-Stereogenic Cyclicphosphinic Amides via Electrochemically Enabled Cobalt-Catalyzed Enantioselective C-H Annulation, Green Chem., 2023, 25, 3606–3614 RSC
- Y.-J. Wu, J.-H. Chen, M.-Y. Teng, X. Li, T.-Y. Jiang, F.-R. Huang, Q.-J. Yao and B.-F. Shi, Cobalt-Catalyzed Enantioselective C-H Annulation of Benzylamines with Alkynes: Application to the Modular and Asymmetric Syntheses of Bioactive Molecules, J. Am. Chem. Soc., 2023, 145, 24499–24505 CrossRef CAS PubMed
- F.-R. Huang, Q.-J. Yao, P. Zhang, M.-Y. Teng, J.-H. Chen, L.-C. Jiang and B.-F. Shi, Cobalt-Catalyzed Domino Transformations via Enantioselective C-H Activation/Nucleophilic [3 + 2] Annulation toward Chiral Bridged Bicycles, J. Am. Chem. Soc., 2024, 146, 15576–15586 CrossRef CAS PubMed
- P.-F. Qian, Y.-X. Wu, J.-H. Hu, J.-H. Chen, T. Zhou, Q.-J. Yao, Z.-H. Zhang, B.-J. Wang and B.-F. Shi, Atroposelective Synthesis of Pyridoindolones Bearing Two Remote Distinct C-N Axes through Cobalt-Catalyzed Enantioselective C-H Activation, J. Am. Chem. Soc., 2025, 147, 10791–10802 CrossRef CAS PubMed
- T. Li, Y. B. Zhang, C. Du, D. D. Yang, M.-P. Song and J.-L. Niu, Simultaneous Construction of Inherent and Axial Chirality by Cobalt-Catalyzed Enantioselective C-H Activation of Calix[4]Arenes, Nat. Commun., 2024, 15, 7673 CrossRef CAS PubMed
- T. Li, L. L. Linlin Shi, X. H. Wang, C. Yang, D. D. Yang, M.-P. Song and J.-L. Niu, Cobalt-Catalyzed Atroposelective C-H Activation/Annulation to Access N-N Axially Chiral Frameworks, Nat. Commun., 2023, 14, 5271 CrossRef CAS PubMed
- X.-J. Si, D. D. Yang, M.-C. Sun, D. H. Wei, M.-P. Song and J.-L. Niu, Atroposelective Isoquinolinone Synthesis Through Cobalt-Catalysed C-H Activation and Annulation, Nat. Synth., 2022, 1, 709–718 CrossRef CAS
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed
- S. Lemouzy, L. Giordano, D. Hérault and G. Buono, Introducing Chirality at Phosphorus Atoms: An Update on the Recent Synthetic Strategies for the Preparation of Optically Pure P-Stereogenic Molecules, Eur. J. Org. Chem., 2020, 2020, 3351–3366 Search PubMed
- G. Q. Xu, C. H. Senanayake and W. J. Tang, P-Chiral Phosphorus Ligands Based on a 2,3-Dihydrobenzo[d][1,3]oxaphosphole Motif for Asymmetric Catalysis, Acc. Chem. Res., 2019, 52, 1101–1112 Search PubMed
- W. J. Tang and X. M. Xumu Zhang, New Chiral Phosphorus Ligands for Enantioselective Hydrogenation, Chem. Rev., 2003, 103, 3029–3070 Search PubMed
- J.-H. Chen, M.-Y. Teng, F.-R. Huang, H. Song, Z.-K. Wang, H.-L. Zhuang, Y.-J. Wu, X. Wu, Q.-J. Yao and B.-F. Shi, Cobalt/Salox-Catalyzed Enantioselective Dehydrogenative C-H Alkoxylation and Amination, Angew. Chem., Int. Ed., 2022, 61, e202210106 CrossRef CAS PubMed
- J. Diesel and N. Cramer, Generation of Heteroatom Stereocenters by Enantioselective C-H Functionalization, ACS Catal., 2019, 9, 9164–9177 Search PubMed
- P.-F. Qian, J.-Y. Li, T. Zhou and B.-F. Shi, Synthesis of P- and S-Stereogenic Compounds via Enantioselective C-H Functionalization, Synthesis, 2022, 54, 4784–4794 CrossRef CAS
- S.-Y. Song, Y. W. Li, Z. F. Ke and S. M. Xu, Iridium-Catalyzed Enantioselective C-H Borylation of Diarylphosphinates, ACS Catal., 2021, 11, 13445–13451 CrossRef CAS
- Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Palladium-Catalyzed Enantioselective C-H Arylation for the Synthesis of P-Stereogenic Compounds, Angew. Chem., Int. Ed., 2015, 54, 6265–6269 CrossRef CAS PubMed
- G. R. Genov, J. L. Douthwaite, A. S. K. Lahdenperä, D. C. Gibson and R. J. Phipps, Enantioselective Remote C-H Activation Directed by A Chiral Cation, Science, 2020, 367, 1246–1251 CrossRef CAS PubMed
- P. J. Hu, L. H. Kong, F. Wang, X. L. Zhu and X. W. Li, Twofold C-H Activation-Based Enantio- and Diastereoselective C-H Arylation Using Diarylacetylenes as Rare Arylating Reagents, Angew. Chem., Int. Ed., 2021, 60, 20424–20429 CrossRef CAS PubMed
- Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, Pd(II)-Catalyzed Enantioselective Synthesis of P-Stereogenic Phosphinamides via Desymmetric C-H Arylation, J. Am. Chem. Soc., 2015, 137, 632–635 CrossRef CAS PubMed
- Y. Sun and N. Cramer, Rhodium(III)-Catalyzed Enantiotopic C-H Activation Enables Access to P-Chiral Cyclic Phosphinamides, Angew. Chem., Int. Ed., 2017, 56, 364–367 CrossRef CAS PubMed
- Y.-S. Jang, M. Dieckmann and N. Cramer, Cooperative Effects between Chiral Cpx-Iridium(III) Catalysts and Chiral Carboxylic Acids in Enantioselective C-H Amidations of Phosphine Oxides, Angew. Chem., Int. Ed., 2017, 56, 15088–15092 CrossRef CAS PubMed
- Y.-S. Jang, L. Woźniak, J. Pedroni and N. Cramer, Access to P- and Axially Chiral Biaryl Phosphine Oxides by Enantioselective CpxIrIII-Catalyzed C-H Arylations, Angew. Chem., Int. Ed., 2018, 57, 12901–12905 CrossRef CAS PubMed
- C.-W. Zhang, X.-Q. Hu, Y.-H. Dai, P. Yin, C. Y. Wang and W.-L. Duan, Asymmetric C-H Activation for the Synthesis of P- and Axially Chiral Biaryl Phosphine Oxides by an Achiral Cp*Ir Catalyst with Chiral Carboxylic Amide, ACS Catal., 2022, 12, 193–199 CrossRef CAS
- Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Cobalt/Salox-Catalyzed Enantioselective C-H Functionalization of Arylphosphinamides, Angew. Chem., Int. Ed., 2022, 61, e202202892 CrossRef CAS PubMed
- X. H. Yu and Y. Wang, Elucidating the Mechanism and Selectivities of Cobalt-Catalyzed Transformation of O-Acyl Oxime, Appl. Organomet. Chem., 2025, 39, e7901 CrossRef CAS
- Y. Wang, K. L. Gong, H. Zhang, Y. Liu and D. H. Wei, Mechanism of a Cobalt-Catalyzed Hydroarylation Reaction and Origin of Stereoselectivity, Catal. Sci. Technol., 2022, 12, 4380–4387 RSC
- Y. Wang, H. Zhang, Y. Liu, K. L. Gong and D. H. Wei, Theoretical Investigation on Cobalt-Catalyzed Hydroacylation Reaction: Mechanism and Origin of Stereoselectivity, Mol. Catal., 2022, 527, 112410 CrossRef CAS
- Y. Wang, C. Du, Y. Y. Wang, X. K. Guo, L. Fang, M.-P. Song, J.-L. Niu and D. H. Wei, High-Valent Cobalt-Catalyzed C-H Activation/Annulation of 2-Benzamidopyridine 1-Oxide with Terminal Alkyne: A Combined Theoretical and Experimental Study, Adv. Synth. Catal., 2018, 360, 2668–2677 CrossRef CAS
- Y. Wang and D. H. Wei, DFT Study in an Asymmetric Organocatalytic Homologation: Mechanism, Origin of Stereoselectivity, Asian J. Org. Chem., 2025, 14, e202400504 CrossRef CAS
- P. X. Liang, H. R. Yang and Y. Wang, Elucidating the Mechanism and Origin of Stereoselectivity in the Activation/Transformation of an Acetic Ester Catalyzed by an N-Heterocyclic Carbene, Phys. Chem. Chem. Phys., 2024, 26, 4320–4328 RSC
- Y. J. Yu, L. J. Zhang and Y. Wang, Theoretical Investigation on the Reaction Mechanism and Origin of Stereoselectivity of a Three-Component Coupling Reaction Under Organocatalysis, Mol. Catal., 2025, 573, 114819 Search PubMed
- Y. R. Zhang and Y. Wang, Elucidating the Mechanism and Origin of Diastereoselectivity in Scandium-Catalyzed β-C(sp3)-H Activation and Transformation of an Aliphatic Aldimine, Org. Chem. Front., 2024, 11, 4391–4401 Search PubMed
- M.-Q. Yang and Y. Wang, Mechanistic Studies on an Isothiourea-Catalyzed Reaction of an Aromatic Ester with an Imine, Mol. Catal., 2024, 567, 114445 CrossRef CAS
- Y. L. Luo, M. Zhao and Y. Wang, Mechanism and Origin of Stereoselectivity of N-Heterocyclic Carbene (NHC)-Catalyzed Transformation Reaction of Benzaldehyde with o-QDM as Key Intermediate: A DFT Study, J. Phys. Chem. A, 2024, 128, 6190–6198 CrossRef CAS PubMed
- S.-L. Liu, Y. Qiao and Y. Wang, Exploring a General Mechanistic Map on NHC-Catalyzed Activation/Transformation Reactions of Saturated Carboxylic Anhydrides, Org. Chem. Front., 2023, 10, 2670–2679 RSC
- G. Lennon, C. O'Boyle, A. I. Carrick and P. Dingwall, Investigating the Mechanism and Origins of Selectivity in Palladium-Catalysed Carbene Insertion Cross-Coupling Reactions, Catal. Sci. Technol., 2023, 13, 372–380 RSC
- Y. X. Liu, J. Zhang, Q. Q. Qu, X. H. Cao, L. J. Liu and G. Cheng, Distinctive C–N Cleavage/C–C Formation Mechanism in Au-Catalyzed Reactions of N-(o-alkynylphenyl)imines and Vinyldiazo Ketones, Catal. Sci. Technol., 2025, 15, 836–844 RSC
- S. Q. Liu, B. P. Ling, S. W. Bi and R. Y. Wang, Mechanism and Origin of Cyclization Selectivity for Ru(ii)-Catalyzed gem-Hydrogenation of 1,3-Enynes: A DFT Study, Catal. Sci. Technol., 2024, 14, 3493–3501 RSC
- Z. Q. Xue, N. Zhang, L. Shi and G. Luo, Origins of Ligand-Controlled Stereoselective Polymerization of ortho-Methoxystyrene by Rare-Earth Catalysts: A Theoretical Perspective, Inorg. Chem., 2024, 63, 9195–9203 CrossRef CAS PubMed
- Y. W. Li, H. Y. Liang, Y. B. Yubang Liu, J. X. Lin and Z. F. Ke, Unraveling the Role of Silyl and Silane in Si-Ni Catalysts for Hydrogenation, ACS Catal., 2023, 13, 13008–13020 CrossRef CAS
- T. T. Liu, K. Lv and X. G. Bao, Mechanistic Differences Between the Ru(ii) and Zn(ii)-Catalyzed Cross-Coupling of Cyclopropenes with Diazo Compounds: A DFT Study, Catal. Sci. Technol., 2024, 14, 6917–6923 Search PubMed
- J. Z. Wang, Y. Y. Yang, C. B. Liu and D. J. Zhang, Theoretical Insight into the Palladium-Catalyzed Prenylation and Geranylation of Oxindoles with Isoprene, Inorg. Chem., 2024, 63, 4855–4866 Search PubMed
- L. Liu, Y. H. Liu, Y. Y. Yang, C. B. Lium and D. J. Zhang, DFT Calculations Reveal the Origin of Controllable Synthesis of β-Boronyl Carbonyl Compounds from Cu/Pd-Cocatalyzed Four-Component Borocarbonylation of Vinylarenes, Catal. Sci. Technol., 2023, 13, 2123–2133 RSC
- J. Ma, S. M. Qi, G. W. Yan, M. A. Kirillov, L. Z. Yang and R. Fang, DFT Study on the Mechanisms and Selectivities in Rh (III)-Catalyzed [5 + 1] Annulation of 2-Alkenylanilides and 2-Alkylphenols with Allenyl Acetates, J. Org. Chem., 2024, 89, 8562–8577 CrossRef CAS PubMed
- J. X. Lin, Y. W. Li and Z. F. Ke, Feature Analysis in High-Dimensional Data: Structure-Activity Relationships of Lewis Acid-Transition-Metal Complex-Catalyzed H2 Activation, J. Phys. Chem. A, 2023, 127, 4375–4387 CrossRef CAS PubMed
- Y.-N. Wang and Y. Wang, Theoretical Investigation of a Chiral Brønsted Acid (CBA)-Catalyzed Isomerization Reaction of BCB: Mechanism and Origin of Stereoselectivity, New J. Chem., 2024, 48, 11360–11365 RSC
- C. H. Liu, P. L. Han, X. X. Hou, S. X. Ge and D. H. Wei, A General Mechanistic Map of Organocatalytic Hydroboration of Alkynes: Polarity Controlled Switchable Selective Pathways, Org. Chem. Front., 2024, 11, 3952–3961 RSC
- P. X. Liang, D. Y. Shi and Y. Wang, Disclosing the Mechanism and Origin of Stereoselectivity of the NHC-Catalyzed Transformation Reaction of Enals with Acyl Azolium as a Key Intermediate, Catal. Sci. Technol., 2024, 14, 4302–4310 RSC
- S. Y. Wang, Z. X. Xia, J. Sheng, J. X. Cui, T. Yao, Y. Liu, C. H. Liu, Z. Y. Liu, J. Tao and Y. Q. Wu, Accurate Control on the Nucleophilic Addition of H2O to Internal Alkynes: An Ag catalyzed Regiospecific Hydration Strategy, ChemCatChem, 2024, 16, e202400735 CrossRef CAS
- S.-L. Liu, X. Q. Liu, Y. Wang and D. H. Wei, Unraveling the Mechanism and Substituent Effects on the N-Heterocyclic Carbene-Catalyzed Transformation Reaction of Enals and Imines, Mol. Catal., 2022, 519, 112122 CrossRef CAS
- C. H. Liu, P. L. Han, X. S. Zhang, Y. Qiao, Z. H. Xu, Y. G. Zhang, D. P. Li, D. H. Wei and Y. Lan, NHC-Catalyzed Transformation Reactions of Imines: Electrophilic versus Nucleophilic Attack, J. Org. Chem., 2022, 87, 7989–7994 CrossRef CAS PubMed
- Y. W. Wang, Y. Liu, K. L. Gong, H. Zhang, Y. Lan and D. H. Wei, Theoretical Study of the NHC-Catalyzed C-S Bond Cleavage and Reconstruction Reaction: Mechanism, Stereoselectivity, and Role of Catalysts, Org. Chem. Front., 2021, 8, 5352–5360 RSC
- C. H. Liu, P. L. Han, D. P. Li, L.-B. Qu, Y. Qiao and Y. Lan, Mechanistic Study of [4 + 3] Cyclization of N,N'-Cyclic Azomethine Imines with Isatoic Anhydrides under Brønsted Acid Catalysis, Mol. Catal., 2022, 525, 112300 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed
- Y. Zhao and D. G. Truhlar, A New Local Density Functional for Main Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical kKnetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-Adjusted ab Initio Pseudopotentials for the First Row Transition Elements, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuss, Energy-Adjusted ab Initio Pseudopotentials for the Second and Third Row Transition Elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS
- B. Mennucci and J. Tomasi, Continuum Solvation Models: A New Approach to the Problem of Solute's Charge Distribution and Cavity Boundaries, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- F. Weigend, Accurate Coulomb-Fitting Basis Sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- T. Lu and Q. X. Chen, Interaction Region Indicator (IRI): A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions, Chem. Methods., 2021, 1, 231–239 CrossRef CAS
- W. Humphrey, A. Dalke and K. V. M. D. Schulten, Visual Molecular Dynamics (version 1.9.3), J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS
- J. P. Perdew, K. Burke and Y. Wang, Generalized gradient approximation for the exchange-correlation hole of a many-electron system, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed
- T. Yanai, D. Tew and N. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. R. Hu and L. Ackermann, Reactivity-controlling factors in carboxylate-assisted C-H Activation under 4d and 3d transition metal catalysis, ACS Catal., 2020, 10, 10551–10558 CrossRef CAS
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, The importance of kinetic and thermodynamic control when assessing mechanisms of carboxylate-assisted C-H activation, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS PubMed
- A. J. Edwards, S. A. Macgregor, A. D. Rae, E. Wenger and A. C. Willis, Regioselectivities of the insertion reactions of unsymmetrical alkynes with the nickelacycles [NiBr(C6H4CH2PPh2-2)(L)] (L = tertiary phosphine), Organometallics, 2001, 20, 2864–2877 CrossRef CAS
- L. A. Hammarback, J. B. Eastwood, T. J. Burden, C. J. Pearce, I. P. Clark, M. Towrie, A. Robinson, I. J. S. Fairlamb and J. M. Lynam, A comprehensive understanding of carbon-carbon bond formation by alkyne migratory insertion into manganacycles, Chem. Sci., 2022, 13, 9902–9913 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00519a |
| ‡ Luo C. C. and Cao S. B. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |