
Breathing air into water: dual-pathway H2O2 synthesis via aerating amphiphilic supramolecular films
Bo Yua,
Shuya Liu*b,
Xinyu Lina,
Zhi Zhu a,
Yongsheng Yana,
Yanjun Gongc,
Yanke Chec,
Yan Yan*a and
Weidong Shi*a
a,
Yongsheng Yana,
Yanjun Gongc,
Yanke Chec,
Yan Yan*a and
Weidong Shi*a
aSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang, 212013, China. E-mail: dgy5212004@163.com; swd1978@ujs.edu.cn
bInstitute of Resources and Environment Innovation, Shandong Jianzhu University, Jinan, 250101, China. E-mail: liushuya@sdjzu.edu.cn
cKey Laboratory of Photochemistry CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry Chinese Academy of Sciences, Beijing, 100190, China
First published on 30th July 2025
Abstract
Dual-pathway photosynthesis offers a sustainable method for producing hydrogen peroxide (H2O2) from air and water. The main efficiency-limiting step in existing photocatalytic systems is the simultaneous triphasic enrichment and activation of gaseous air and liquid water at the solid catalyst interface. In this work, utilizing amphiphilic molecular design and hierarchical self-assembly of asymmetric perylene diimide molecules (PDIOH), we developed an O2-permeable 2D organic supramolecular film for dual-pathway H2O2 production. The resulting supramolecular films amplify the amphiphilic characteristics of PDIOH molecules, featuring Janus wettability on both sides, which anisotropically enrich and activate O2 and H2O molecules. Specifically, non-polar O2 is enriched on the hydrophobic surface of the PDIOH films and then photoreduced to the polar O2− species, which subsequently detaches and enters the aqueous phase, mimicking the process of “breathing air into water”. Simply placing PDIOH films at the static air–water interface achieves a high H2O2 yield of 3046.15 μmol g−1 h−1 without the need for stirring, O2 bubbling or small-molecule sacrificial agents. This setup allows for the continuous flow synthesis of H2O2 with a stable efflux concentration of at least ∼800 μM.
Broader contextHydrogen peroxide (H2O2) is a critical chemical commodity that can be used as an environmentally friendly disinfectant and energy storage medium to inactivate pollutants and store energy that can meet the demands of environmental and energy issues. The efficient photocatalytic synthesis of H2O2 requires leveraging the synergistic effects of the oxygen reduction reaction (ORR) and the water oxidation reaction (WOR). However, constructing gas/liquid active sites anisotropically on a single catalyst while preserving the internal charge transfer channels poses a significant challenge. Organic supramolecular systems formed by living self-assembly can be adapted to solve this problem. This research presents a strategy for regulating Janus wettability in PDIOH supramolecular films through the design of asymmetric amphiphilic molecules and multilevel self-assembly across molecules/fibers/films. Amphiphilic PDIOH-assembled supramolecular films anisotropically enrich and photoreduce atmospheric O2 at hydrophobic interfaces, enabling dual-pathway H2O2 synthesis via triphasic “air-to-water” mass transfer at static air–liquid interfaces. |
Introduction
Hydrogen peroxide (H2O2) is a critical chemical commodity, with global demand reaching four million tons annually.1–4 The conventional anthraquinone process used for large-scale production is energy-intensive, generates hazardous emissions, and poses logistical challenges in H2O2 transportation.5 These issues highlight the necessity for more sustainable and decentralized production methods. The photocatalytic synthesis of H2O2, employing ambient air and water and leveraging the synergistic effects of the oxygen reduction reaction (ORR) and the water oxidation reaction (WOR), offers a potential solution for local, emission-free production.6,7 However, most existing photocatalytic systems are prone to significant backreactions (2H2O2 → O2 + 2H2O) and mass transfer challenges at the gas–liquid–solid triphasic interface.8–10 Consequently, these systems fail to fully exploit the dual-pathway potential of ORR and WOR, thus hindering sustainable H2O2 synthesis.Thermodynamically, the WOR at +1.76 V vs. NHE (pH = 0) poses more challenges than the ORR at +0.68 V vs. NHE (pH = 0).11 However, in practical scenarios, the low solubility of oxygen in water at 25 °C and 1 atm, limited to 8 mg L−1, significantly restricts its availability for catalysis in traditional biphasic systems.12,13 Despite efforts like continuous oxygen bubbling, the efficiency of oxygen utilization remains critically low (under 1%), leading to substantial inefficiency under ambient conditions.13 Recent developments in triphasic superhydrophobic floating systems have aimed to improve oxygen accessibility by loading catalyst nanoparticles on floating carriers.10,14–17 Despite these innovations, several common limitations persist: (i) superhydrophobic surfaces limit water contact, restricting WOR and favoring only ORR for H2O2 synthesis; (ii) poor electron transfer between floating carriers and catalysts hampers the synergy of ORR and WOR; (iii) the inert nature of floating carriers blocks light and reactive sites, thereby impairing catalytic efficacy.
We envision that an amphiphilic 2D supramolecular film that inherently facilitates floating and maintains effective interactions with both water and air, circumventing the need for additional carrier supports, could ideally resolve the aforementioned issues and enable efficient and sustainable dual-pathway synthesis of H2O2. Perylene diimide (PDI) derivatives are a class of catalytically active organic supramolecular materials that exhibit excellent self-assembly properties through π–π stacking interactions.18–21 The self-assembly of amphiphilic PDIs typically results in the formation of 1D fibers as building blocks for their aggregates.22–24 By engineering these fibers to self-organize into 2D hierarchical bundles, we can create integrated hierarchical supramolecular films that facilitate floating H2O2 synthesis under ambient conditions. This approach not only leverages the dual-pathway potential of ORR and WOR but also overcomes the limitations typical of triphasic systems.
Results and discussion
In this study, we have designed and synthesized an amphiphilic perylene diimide molecule PDIOH, featuring three key components: a hydrophilic hydroxyl group, a methylene linker providing spatial resistance, and hydrophobic alkyl chains (Fig. 1a). The detailed synthesis procedures and characterizations of PDIOH are provided in the SI (Fig. S1–S4). The asymmetric hydroxyl group and alkyl chain endow PDIOH with its amphiphilic nature, facilitating π–π stacking and self-assembly into 1D fiber units. These units expose perpendicularly oriented hydrophilic base facets and hydrophobic lateral facets (Fig. 1b). This wettability-anisotropy of the PDIOH fibers is a key factor, allowing for the controllable Janus wettability of the ordered 2D hierarchical fiber bundles. | ||
| Fig. 1 Structural characterizations of PDIOH fibers and films. (a) Molecular structure of PDIOH. (b) Molecular packing model of an individual PDIOH fiber. (c) Schematic diagrams illustrating different self-assembly stages, including the primary and secondary structures of PDIOH film; insets show TEM images of M0 and M2 films. (d) HR-TEM image of PDIOH fiber bundles. (e) XRD patterns of PDIOH disordered fibers and hierarchical PDIOH film (M2). (f) AFM profile of primary-structured PDIOH film (M0). SEM images of (g) the hydrophobic side and (h) the hydrophilic side of M2. (i) A comparison of the hydrophobic and hydrophilic sides of M2 and corresponding water contact angles after wetting. (j) UV-vis absorption spectra of PDIOH monomers (in chloroform), disordered PDIOH fibers, and PDIOH film (M2). (k) ATR-FTIR spectra measured on both the hydrophilic and hydrophobic sides of M2 film, along with a disordered fiber sample. (l) CLSM images captured at the edge and center of the M2 film (λex = 405 nm). | ||
A centimeter-scale 2D hierarchical PDIOH film with highly aligned band patterns was self-assembled at the water–chloroform interface (Fig. 1c), while randomly-oriented fibers formed under evaporation in a pure chloroform phase (Fig. S5). An individual PDIOH fiber is highly crystallized, showing clear lattice fringes with a spacing of 0.30 nm (Fig. 1d). The self-assembly behavior of the 2D PDIOH film at the water–chloroform interface is detailed in the SI (Fig. S6 and S7). The primary structure of the PDIOH film, denoted M0, consists of densely packed, laterally aligned fibers with minimal stacking and crossing, as evidenced by laser confocal laser scanning microscopy (CLSM), scanning electron microscopy (SEM), and transmission electron microscopy (TEM) (Fig. S8–S10). The X-ray diffraction (XRD) profiles of both PDIOH film and the disordered fibers exhibit distinct diffraction peaks at 3.9°, which confirms that they share the same single-crystal structure and molecular packing (Fig. 1e). The thickness of M0 measured by atomic force microscopy (AFM) ranged from 6.5 to 6.6 nm (Fig. 1f), further confirming the double-layer arrangement configuration in PDIOH fibers (Fig. 1b). This alignment exposes the hydrophilic base facets of the fiber units on both sides of the film, rendering the M0 surfaces hydrophilic.
When fibers form beyond the constraints of the liquid–liquid interface, they are randomly oriented, exposing lateral alkyl chains and creating a hydrophobic secondary structure (Fig. 1c). Such a design allows for a multilevel hierarchical arrangement (molecules/fibers/film) that realizes a Janus PDIOH film (Fig. 1g and h). The prepared Janus PDIOH film can be easily transferred from the water surface to solid substrates, such as silicon wafers, glass, or copper grids, and can be flipped to display either its hydrophilic or its hydrophobic side (Fig. 1i). By gradually increasing or decreasing the feed of PDIOH into chloroform solution, we can control the growth stages of the secondary structures (denoted M1–M4) with slow evaporation of the chloroform (Fig. S11). UV–vis spectroscopic analysis shows that the PDIOH fibers and the 2D hierarchical film exhibit a redshift in absorption compared to the absorption peak of the PDIOH monomer at 527 nm (Fig. 1j), reflecting the J-type aggregated nature of the fibers. The hydrophobic lateral facet of the PDIOH fibers and the hydrophobic side of the film share a similar exposed surface structure, characterized by a high degree of stacking of long alkyl chains. Observations from attenuated total reflection infrared spectroscopy (ATR-FTIR) confirm this with a triplet band around 2930 cm−1 corresponding to the C–H stretching vibrations of long alkyl chains (Fig. 1k). In contrast, similar signals are nearly absent on the hydrophilic surfaces, further revealing the hierarchical structural details of the PDIOH film. CLSM images show the layered structure of the Janus PDIOH film (M2) at both the center and edges, clearly distinguishing the ordered primary structure from the more disordered secondary growth structures with clear boundaries (Fig. 1l). The increasing freedom of fiber alignment towards the center also reveals that the formation of the secondary structure is inherently linked to the changing interfacial tension as the solvent evaporates.
The anisotropic feature of 2D hierarchical PDIOH films allows for the continuous synthesis of H2O2 at the static air–water interface. The yield of H2O2 was determined by the Ce3+/4+ back-titration method (Fig. S12). The primary-structured PDIOH film (sample M0), with both sides being hydrophilic (WCA < 20°), exhibited an H2O2 production rate of 655.67 μmol g−1 h−1. As the secondary structures gradually developed from M1 to M4 (Fig. 2a, b, d and Fig. S11), the difference in wettability between the hydrophilic and hydrophobic sides increased significantly (Fig. 2e). This evolution enhances the anisotropic functionality of the film surfaces, where the hydrophilic side facilitates water activation, while the hydrophobic side promotes oxygen enrichment and activation (Fig. 2c). Additionally, to demonstrate the scalability of our system, we conducted experiments involving multiple PDIOH films operating concurrently. Specifically, at a total catalyst mass of 0.3 mg (20 PDIOH films), we achieved an H2O2 production rate of 2831.68 μmol g−1 h−1 (Fig. S13). We have also included measurements of the apparent quantum yield (AQY) as a parameter for evaluating photocatalytic performance. The relationship between AQY and incident wavelength was determined using band-pass filters (400–750 nm) to measure H2O2 generation under monochromatic light irradiation. The results show a peak AQY of 0.0516% at 450 nm (Fig. S14 and Table S1). In order to confirm the suitability of this method, we have included SI on H2O2 yield determined using the titanium sulfate colorimetry method. The results indicate that the titanium sulfate method yielded a production rate of 2961.35 μmol g−1 h−1, further confirming the accuracy and reliability of our results (Fig. S15). Note that PDIOH films are not only rich in gaps between fibers but also have a gap of 0.32 nm between PDIOH molecules in π–π stacking (Fig. 1b and Fig. S16), which is sufficient to accommodate O2. This maintains good air permeability between the primary and secondary structures of PDIOH films.
 | ||
| Fig. 2 H2O2 synthesis performance on PDIOH films. A comparison of SEM images of the PDIOH films (M0–M4) on (a) hydrophobic sides and (b) hydrophilic sides (scale bar: 1 μm). (c) Schematic illustration of triphasic dual-pathway H2O2 production via floating Janus PDIOH films; placing the Janus PDIOH film upside-down would disrupt the synergy between WOR and ORR. (d) Photocatalytic H2O2 yields with M0–M4 films under 1000 mW cm−1 xenon lamp irradiation with static air and pure water. (e) The differences in wettability between the hydrophilic and hydrophobic sides of M0–M4 films. (f) Kinetic profiles of the photocatalytic H2O2 yield with M2 in N2, O2, and air atmospheres. (g) Photocatalytic H2O2 yields within the biphasic suspension system using disordered PDIOH fibers and the triphasic floating system with the M2 film. (h) Photocatalytic H2O2 yields using the same M2 film but with the exposed side flipped over. (i) Cyclic tests of photocatalytic H2O2 production using M2 film in 20 h. (j) Time-course for photocatalytic H2O2 accumulation using M2 film. Error bars (in standard deviation) were obtained by statistically repeating identical experimental results three times. | ||
M1–M4 films were assessed by placing them at the static air–water interface with the hydrophilic side submerged and the hydrophobic side exposed to air. As shown in Fig. 2d, under identical xenon lamp irradiation (1000 mW cm−1), films M1–M4 with pronounced Janus wettability (Fig. 2e) all outperformed M0, with the M2 film exhibiting the highest H2O2 production rate at 3046.15 μmol g−1 h−1, ∼5 times that of M0. However, it was observed that an overly thick secondary structure in samples M3 and M4 adversely affected the rate of H2O2 production. In our reaction system, neither small-molecule sacrificial agents nor O2 bubbling were needed. Simply placing the PDIOH film at the static air–water interface enabled efficient H2O2 synthesis. Replacing air with O2 did not significantly alter the H2O2 production rate, as observed in many other reported systems17,21,25 (Fig. 2f).
The floating status of the 2D hierarchical PDIOH film is a decisive factor. Compared to the biphasic suspension system using powdered randomly-oriented PDIOH fibers, the floating triphasic reaction system using M2 film has a 6.18 times higher average H2O2 production rate with the same catalyst mass (Fig. 2g). Moreover, for the same M2 film, by simply reversing its hydrophilic and hydrophobic sides, i.e., placing it upside-down at the same air–water interface, the H2O2 production rate dropped sharply to less than one-fourth, at a mere 744.05 μmol g−1 h−1 (Fig. 2h). This result directly verifies our hypothesis that the hydrophilic and hydrophobic sides of PDIOH film undergo WOR and ORR, respectively, through anisotropic wettability, and reversing the sides disrupts the accumulation and activation of water and O2 on the favorable side, even when using the same catalytic film under identical experimental conditions (Fig. 2c). To elucidate the origin of oxygen produced during the reaction, we conducted isotopic labeling experiments using H218O. When H218O was used as the water source, GC-MS analysis revealed a prominent signal at m/z = 34, corresponding to 16O18O. This result can be explained by the mechanism of hydrogen peroxide formation: one oxygen atom in H2O2 is derived from H218O, while the other typically comes from naturally abundant 16O2 gas. Upon decomposition of H2O2 (2H2O2 → 2H2O + O2), these mixed-isotope H2O2 molecules predominantly generate 16O18O as the main O2 product, along with a small amount of 18O2 (m/z = 36). This isotopic distribution confirms the effective incorporation of 18O from the labeled water and verifies the water oxidation pathway (Fig. S17 and S18). Furthermore, the hierarchical PDIOH film (M2) triphasic system showed no significant decline in H2O2 production after ten repeated experiments (continuous static operation for 20 hours) (Fig. 2i). The unchanged chemical composition and structure before and after the cycles further confirm the stability of our PDIOH film (Fig. S19 and S20).
Fig. 2j depicts the accumulation process for H2O2 in pure water over a longer period. The concentration of H2O2 increased over time, peaking at 140.85 mmol g−1 after 56 hours of illumination. Furthermore, to meet the requirements for large-scale continuous H2O2 production, we developed a scaled-up floating-flow reactor, as shown in Fig. 3a and b. The PDIOH films were statically placed at the air–water interface, with pure water flowing underneath at a consistent rate. Therefore, H2O2 solution with a stable concentration would be continuously produced and collected. Under constant xenon lamp irradiation (1000 mW cm−1), when the flow velocity was set to 0.1 mL min−1, the stable H2O2 eluent concentration was ∼800 μM (Fig. 3c). Note that this value is related to the spread area of the films and the flow velocity, indicating that further optimization of performance is possible, based on these parameters.
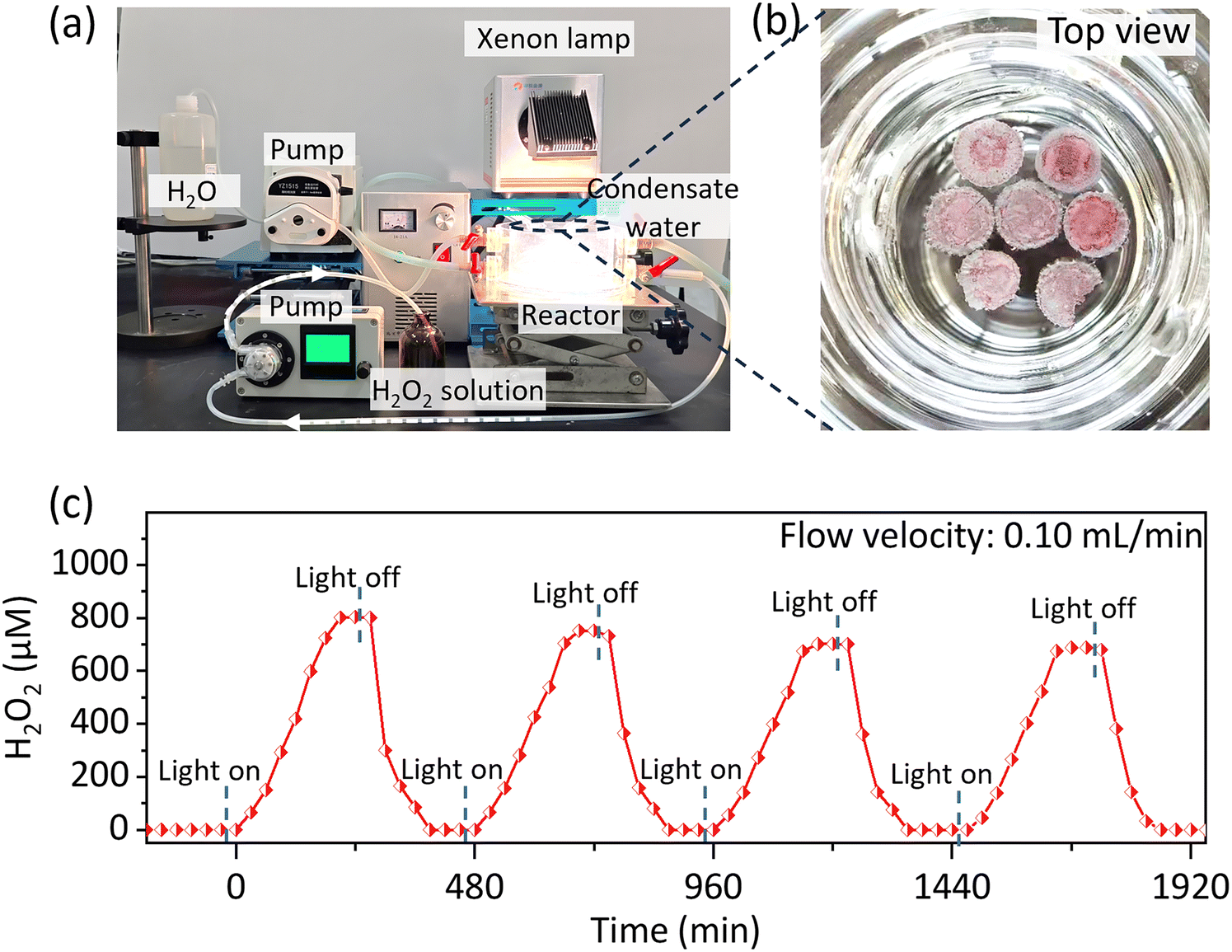 | ||
| Fig. 3 Continuous flow-floating triphasic H2O2 synthesis setup. (a) Photograph of the continuous flow-floating triphasic reaction system for H2O2 photosynthesis. (b) Top view of seven floating PDIOH films in the triphasic reaction chamber before operation. (c) The stable H2O2 output using seven floating M2 films, with data recorded over several cycles of light-on/-off switches. | ||
We reason that the properly oriented PDIOH film favors H2O2 production according to two aspects: (i) the anisotropic distribution of adsorption/reaction sites; and (ii) the unified internal charge transfer within the supramolecular film, promoting synergy between WOR and ORR. For the former, we first assessed the anisotropic adsorption of O2/H2O on the hydrophilic and hydrophobic sides of the PDIOH film (M2) by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). As shown in Fig. 4a, under dark conditions, setting the hydrophilic side of the M2 film in a stable state with dry N2 as the background, when O2 flow carrying water vapor was introduced to the reaction chamber, a broad absorption band increasing at 3380 cm−1 corresponding to the O–H stretching vibration and a weak growth of the H–O–H bending vibration at 1644 cm−1 were detected. The intensity ratio of the 1644 cm−1/3380 cm−1 signal is far lower than that of conventional liquid water (Fig. S21), indicating that H2O is dissociatively adsorbed. O2 adsorption observed on the hydrophilic side of the PDIOH film in the DRIFTS experiments is nearly zero. This is because H2O binds more strongly to the hydrophilic groups than does O2, pushing the O2 signal below the detection limit. The adsorption kinetics and IR spectral absorption profile were nearly identical to those observed under N2/H2O conditions (Fig. S22). Note that O2 itself does not show IR signals, but when adsorbed, O2 can exhibit broad IR absorption bands by changing the dipole moment on adsorption surfaces.26 In contrast, on the hydrophobic side (Fig. 4b), which exposes non-polar long alkyl chains, multiple broad-spectrum infrared absorption bands at 3700, 2845, 2300–1800, and 1500–1000 cm−1 were detected under the same O2/H2O conditions, confirming the ability to adsorb O2 through strong interactions.26,27 Meanwhile, the O–H signal of adsorbed water (3380 cm−1) was completely invisible on the hydrophobic side. Only under N2/H2O conditions, was a very weak O–H signal observed on the hydrophobic side with an intensity ratio of 1640 cm−1/3380 cm−1, far greater than that on the hydrophilic side (Fig. 4e and Fig. S23).
 | ||
| Fig. 4 Anisotropic distribution of O2 and H2O on PDIOH films. In situ DRIFTS collected on (a) the hydrophilic side and (b) the hydrophobic side of M2 films, showing the adsorption and photoreaction processes in the O2/H2O atmosphere. (c) Time profiles showing the rise and decay of the O–H signal intensity at 3447 cm−1 on the hydrophilic and hydrophobic sides of M2. (d) Time profiles of the showing the rise and decay of O2 adsorption signal intensity at 2845 cm−1/2807 cm−1 on the hydrophilic and hydrophobic sides of M2. (e) Peak intensity ratio of 1644 cm−1 to 3447 cm−1 on the hydrophilic and hydrophobic sides of M2. (f) MD time-slides for water molecule diffusion on the hydrophilic and hydrophobic facets of PDIOH fibers. (g) Time profiles of the number of hydrogen bonds formed within 3.5 Å on the hydrophilic and hydrophobic facets. (h) MD time-slides for the primary-structured PDIOH film with water and O2 on both sides. (i) Magnified slide at 500 ps showing the O2/H2O/film triphasic interface. (j) Schematic illustration of the O2 mass transfer of the PDIOH film under light. (k) EPR spectra of PDIOH film in O2 atmosphere under xenon lamp irradiation, using DMPO as the spin-trapping chemical in MeOH solution. | ||
Taking the adsorption equilibrium state under the O2/H2O mixed atmosphere as background, we used a 365 nm monochromatic LED lamp (300 mW cm−1) to illuminate the hydrophilic and hydrophobic sides of the M2 film separately to observe in situ photoreactions. Consistent with our hypothesis, under illumination, only the negative peaks of H2O (at 3300 and 1600 cm−1) were observed on the hydrophilic side, and only the negative peak signals of O2 were observed on the hydrophobic side. Fig. 4c and d demonstrate the distinct O2 and H2O adsorption and reaction kinetics on each side, confirming the anisotropic distribution of adsorption/reaction sites on the PDIOH film. Such an anisotropic distribution facilitates spatial separation of reactive sites for WOR and ORR, minimizing backreactions.
Furthermore, we simulated the interfacial mass transfer process of O2 and H2O on different exposure facets of PDIOH fibers on the hydrophilic side and hydrophobic side through molecular dynamics (MD) simulation. To simplify calculations, a bilayer structure of PDIOH containing 80 molecules was selected as the PDIOH fiber. As shown in Fig. 4f and Video S1, hydroxyl groups are predominantly exposed on the hydrophilic base facet of the DIOH fibers, significantly promoting the dissociative adsorption of water molecules. However, due to the obstruction of internal hydrophobic long alkyl chains, water molecules only attach near hydrophilic hydroxyl groups without penetrating the film, despite the size allowing passage (Fig. S16), ensuring their anisotropic distribution. However, on the hydrophobic facet, the extensive exposure of non-polar long alkyl chains makes it difficult for water clusters to adhere (Fig. 4f, g and Video S2). Unlike water, which easily forms molecular clusters, oxygen molecules exist primarily in a free state, thus easily passing through the molecular gaps in the PDIOH fibers, demonstrating good oxygen permeability of the film (Fig. S24–S26 and Videos S3, S4). However, although O2 can penetrate the hydrophilic surface, the exposed hydroxyl sites are still preferentially occupied by H2O, making it difficult for O2 to adhere to the hydrophilic side of the film (Fig. 4h, i and Video S5). This also explains why O2 adsorption is not observed on the hydrophilic side in in situ DRIFTS (Fig. 4a). Akin to “breathing air into water”, non-polar O2 is initially enriched on the hydrophobic surface of the PDIOH films, where it photoreduces to the polar O2− species. This polar intermediate detaches and enters the liquid phase (Fig. 4j and Fig. S27), resolving the longstanding challenge of triphasic mass transfer. This mechanism is further demonstrated by EPR observations from a triphasic capture experiment (Fig. 4k). Notably, in the absence of PDIOH films or when the O2 atmosphere is replaced with N2, the DMPO–˙O2− signals are not detectable (Fig. S28). Additionally, we conducted additional experiments with benzoquinone (p-BQ, a scavenger of ˙O2−) under identical conditions. The results reveal significant suppression of both the generation of superoxide radicals and the production of H2O2, further substantiating our proposed mechanism (Fig. S29). Collectively, we confirm the anisotropic adsorption of H2O and O2 on the hydrophilic/hydrophobic sides and the rapid internal penetration of O2 through the film into the water phase, forming a synergistic reaction interface for WOR and ORR.
To further reveal the internal carrier transport state between fibers in PDIOH films, femtosecond transient absorption (fs-TA) spectroscopy measurements were performed. Fig. 5(a–c) illustrate the analysis of three parallel samples: disordered PDIOH fibers, the primary-structured film M0, and the hierarchical film M2. All samples share the same molecular structure, differing only in fiber arrangement, as depicted in Fig. 5g. In the disordered PDIOH fiber sample, fibers are isolated, and only a single ground-state bleaching signal at 574 nm and excited-state absorption (ESA) features at 645 nm and 775 nm were observed (Fig. 5a and d), ascribed to intermolecular transitions within the fiber.28–30 However, in the M0 film, where fibers are tightly packed in parallel within 2D bundles, while the ground-state bleaching signal remains nearly the same, the ESA undergoes a substantial shift from 645 nm to 770 nm (Fig. 5b and e), suggesting an enhanced excited-state transition between fibers. Additionally, a new, weaker ground-state bleaching and ESA signal were detected at 532 nm and 515 nm, respectively, indicative of new excitation modes formed through strong fiber–fiber interactions. In contrast, the M2 film exhibits not only fiber–fiber transitions within the primary structure but also between the primary and secondary structure layers (Fig. 5c and f). This arrangement allows for unified molecular-fiber-layer hierarchical transitions within the Janus film (Fig. 5g). Significantly, the 515 nm fiber–fiber transition ESA intensity is notably stronger, and the 644 nm ESA shifts to 665 nm within 1–2 ps, accompanied by altered lifespans (Fig. 5h). This confirms enhanced charge transport between layers after the formation of the secondary structure. These hierarchical transitions in the PDIOH film facilitate uniform internal charge transport, which is crucial for maintaining charge balance at spatially separated anisotropic sites. This arrangement promotes efficient coordination between WOR and ORR, enhancing the overall reaction dynamics. It is worth noting that we compared the decay kinetics of the fs-TAS data for the disordered PDIOH fibers, the primary-structured film M0, and the hierarchical film M2, as shown in Fig. S30. The GSB signal does not fully recover within the 8 ns measurement window (Fig. S30a and b), indicating the presence of a long-lived intermediate state that does not return to the ground state (S0). In contrast, the observed ESA signal decays almost completely within 1 ns (Fig. S30c–f). This clear disparity between the GSB and ESA kinetics strongly suggests the formation of long-lived charge-separation species.31–33 Such a charge-separated state, localized on the PDI moiety, manifests as radical species. EPR measurements of the PDIOH film revealed progressive intensification of the signal with increasing duration of light exposure (Fig. S31), confirming the generation of anionic species within the PDIOH film as active charge-separated intermediates.
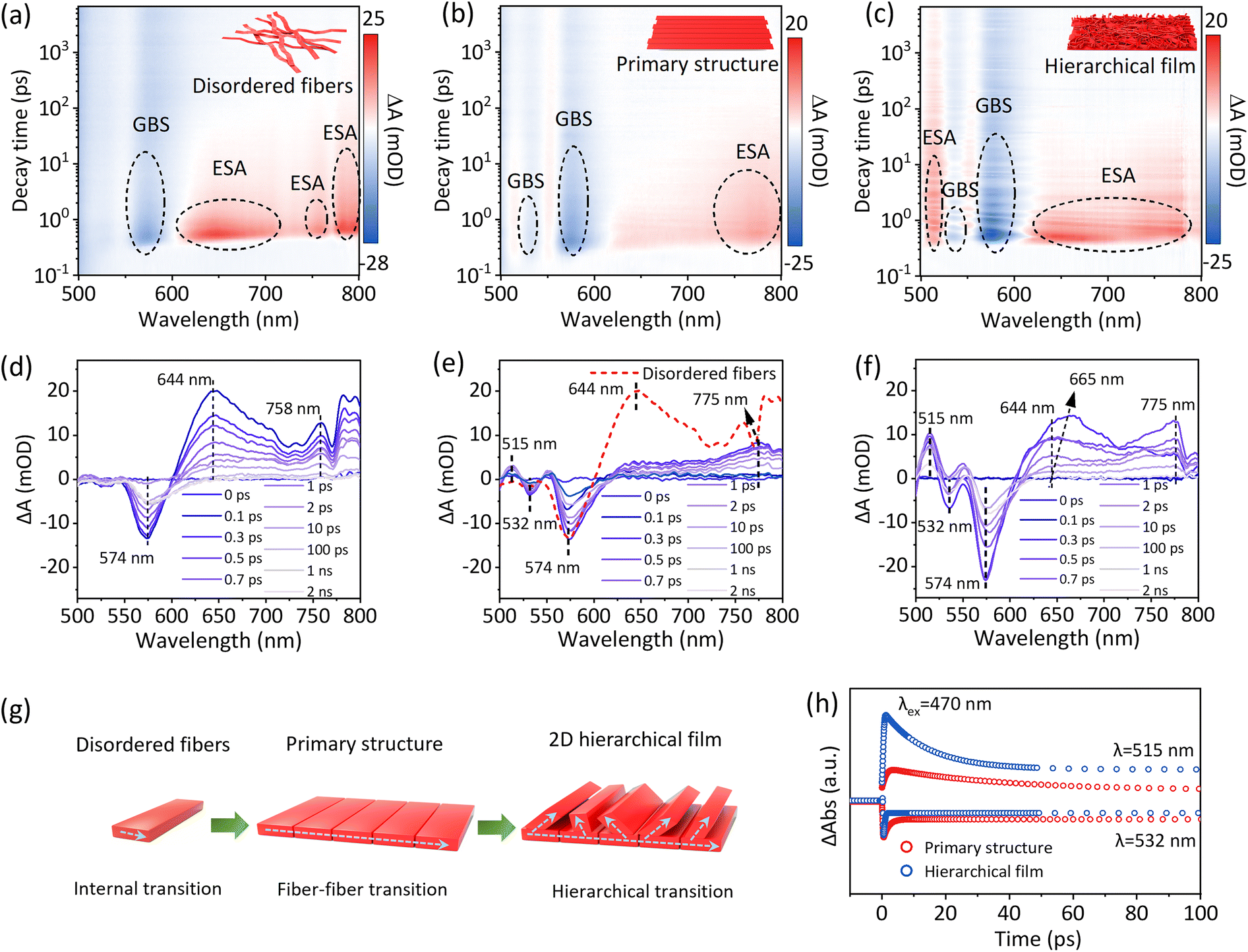 | ||
| Fig. 5 Femtosecond-TA analyses of PDIOH fibers and films. 2D TA maps of (a) PDIOH disordered fibers, (b) the primary-structured film M0, and (c) the hierarchical film M2, and corresponding spectroscopic profiles at selected time points (d)–(f) (λex = 470 nm). (g) Schematic illustration showing the exciton transition mechanism within PDIOH fibers and films; (h) time profiles of fiber–fiber transition bleaching and ESA at 532 nm and 515 nm, respectively, on M0 and M2. | ||
Conclusions
In this proof-of-concept study, we have successfully demonstrated the regulation of Janus wettability in PDIOH supramolecular films through the design of asymmetric amphiphilic molecules and multilevel self-assembly across molecules/fibers/films. This unique supramolecular self-assembly strategy not only ensures the anisotropic enrichment of O2 and H2O but also facilitates the permeation of O2 through intermolecular gaps to the triphasic reactive interface, akin to “breathing air into water”. PDIOH represents a vast family of molecular derivatives, and the effectiveness of our strategy highlights the bright prospects for the use of PDIOH-like family molecules in the well-regulated self-assembly of amphiphilic supramolecular films for multiphase reactions.Author contributions
B. Yu and Y. Yan co-proposed the idea and performed the photocatalytic reactions. S. Liu, X. Lin and Y. Gong designed and synthesized the PDIOH. S. Liu performed molecular dynamics simulation and contributed to perform the experiments. Y. Che, Z. Zhu, W. Shi and Y. Yan contributed to performing and analyzing the experiments. Y. Yan and Y. Gong supervised the whole project. B. Yu, Y. Yan and Y. Gong participated in writing the manuscript with input from all authors.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting the findings of this study are available within the article and its SI.Supplementary methods, figures (Fig. S1–S31), table (Table S1), and references are available. See DOI: https://doi.org/10.1039/d5ee02150b.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (22208127 (Z. Z.), 21806060 (Y.Y.), 22225805 (W. S.), U24A20551 (W. S.), Senior Talent Research Foundation of Jiangsu University (No. 22JD017, (Y. Y.), 23JDG030 (Z. Z.)). Taishan Scholars Program (No. tsqn202306257, (Y. G.)) and the Natural Scientific Foundation of Shandong Province of China (No. ZR2024YQ012, (Y. G.)). We are grateful for the technical support for Nano-X from Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences (SINANO).References
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed
.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Nat. Commun., 2019, 366, 226–231 CAS
- Y. Zhao, P. Zhang, Z. Yang, L. Li, J. Gao, S. Chen, T. Xie, C. Diao, S. Xi and B. Xiao, Science, 2021, 12, 3701 CAS
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS
- X. Chen, Y. Kondo, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Phys. Chem. Chem. Phys., 2020, 22, 14404–14414 RSC
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC
- M. Sun, X. Wang, Y. Li, H. Pan, M. Murugananthan, Y. Han, J. Wu, M. Zhang, Y. Zhang and Z. Kang, ACS Catal., 2022, 12, 2138–2149 CrossRef CAS
- H. Pan, M. Sun, X. Wang, M. Zhang, M. Murugananthan and Y. Zhang, Appl. Catal., B, 2022, 307, 121174 CrossRef CAS
- H. Huang, R. Shi, X. Zhang, J. Zhao, C. Su and T. Zhang, Angew. Chem., Int. Ed., 2021, 133, 23145–23151 CrossRef
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed
- Y. Wang, R. Shi, L. Shang, G. I. Waterhouse, J. Zhao, Q. Zhang, L. Gu and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13057–13062 CrossRef CAS PubMed
- L. Li, L. Xu, Z. Hu and J. Yu, Adv. Funct. Mater., 2021, 31, 2106120 CrossRef CAS
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202200802 CrossRef
- S. Xia, L. Zhen, Z. Ruosha, C. Liping, F. Xinjian and J. Lei, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef PubMed
- Y. Li, Z. Pei, D. Luan and X. Lou, J. Am. Chem. Soc., 2024, 146, 3343–3351 CrossRef CAS
- S. Yan, Y. Li, X. Yang, X. Jia, J. Xu and H. Song, Adv. Mater., 2024, 36, 2307967 CrossRef CAS PubMed
- Y. Li, X. Zhang and D. Liu, J. Photochem. Photobiol., C, 2021, 48, 100436 CrossRef CAS
- Y. Sheng, W. Li, L. Xu and Y. Zhu, Adv. Mater., 2022, 34, 2102354 CrossRef CAS PubMed
- J. Wang, W. Shi, D. Liu, Z. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS
- Z. Luo, F. Wu, T. Zhang, X. Zeng, Y. Xiao, T. Liu, C. Zhong, X. Lu, L. Zhu and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 8520–8525 CrossRef CAS PubMed
- S. Zhao, Y. Jiang, Y. Fu, W. Chen, Q. Zhang, L. He, C. Huang, Y. Liu, X. C. Zeng and Y. Chai, Nat. Commun., 2024, 15, 7423 CrossRef CAS PubMed
- G. Lin, J. Tao, Y. Sun, Y. Cui, I. Manners and H. Qiu, J. Am. Chem. Soc., 2024, 146, 14734–14744 CrossRef CAS
- Y. Zhang, L. Yang, Y. Sun, G. Lin, I. Manners and H. Qiu, Angew. Chem., Int. Ed., 2024, 63, e202315740 CrossRef CAS PubMed
- K. Li, Q. Ge, Y. Liu, L. Wang, K. Gong, J. Liu, L. Xie, W. Wang, X. Ruan and L. Zhang, Energy Environ. Sci., 2023, 16, 1135–1145 RSC
- G. Blyholder and L. Neff, J. Phys. Chem., 1962, 66, 1464–1469 CrossRef CAS
- K. Ueda and K. Yasuda, Mater. Trans., 2008, 49, 2012–2015 CrossRef CAS
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. Wasielewski, Nat. Chem., 2022, 14, 786–793 CrossRef CAS PubMed
- T. Kim, C. Lin, J. D. Schultz, R. M. Young and M. Wasielewski, J. Am. Chem. Soc., 2022, 144, 11386–11396 CrossRef CAS PubMed
- C. Rehhagen, M. Stolte, S. Herbst, M. Hecht, S. Lochbrunner, F. Würthner and F. Fennel, J. Phys. Chem. Lett., 2020, 11, 6612–6617 CrossRef CAS PubMed
- X. Lin, Y. Hao, Y. Gong, P. Zhou, D. Ma, Z. Liu, Y. Sun, H. Sun, Y. Chen, S. Jia, W. Li, C. Guo, Y. Zhou, P. Huo, Y. Yan, W. Ma, S. Yuan and J. Zhao, Nat. Commun., 2024, 15, 5047 CrossRef CAS PubMed
- R. Wang, B. Qian, Y. Xu, D. Zhao, Q. Chen, Y. Wei, C. Zhang, W. Liang, Y. Jiang, H. Zhang and J. Lin, Angew. Chem., Int. Ed., 2024, 64, e202421871 CrossRef PubMed
- H. Zhao and D. Leonori, Angew. Chem., Int. Ed., 2021, 60, 7669–7674 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |