
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-supported Ni nanoparticles in CO2 methanation: role of a superficial NiO shell observed by in situ TEM†
Katherine E. MacArthur‡
*a,
Liliana P. L. Gonçalves‡bcd,
Juliana P. S. Sousab,
O. Salomé G. P. Soares cd,
Hans Kungle,
Eva Jodate,
André Karle,
Marc Heggena,
Rafal E. Dunin-Borkowskia,
Shibabrata Basak*e,
Rüdiger-A. Eichelefg,
Yury V. Kolen'kob and
M. Fernando R. Pereira*cd
cd,
Hans Kungle,
Eva Jodate,
André Karle,
Marc Heggena,
Rafal E. Dunin-Borkowskia,
Shibabrata Basak*e,
Rüdiger-A. Eichelefg,
Yury V. Kolen'kob and
M. Fernando R. Pereira*cd
aErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich, 52425 Jülich, Germany. E-mail: k.macarthur@fz-juelich.de
bInternational Iberian Nanotechnology Laboratory (INL), Braga 4715-330, Portugal
cLSRE-LCM – Laboratory of Separation and Reaction Engineering – Laboratory of Catalysis and Materials, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, 4200-465 Porto, Portugal. E-mail: fpereira@fe.up.pt
dALiCE – Associate Laboratory in Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, 4200-465 Porto, Portugal
eInstitute of Energy Technologies – Fundamental Electrochemistry (IET-1), Forschungszentrum Jülich GmbH, Jülich, Germany. E-mail: s.basak@f-juelich.de
fInstitute of Physical Chemistry, RWTH Aachen University, Aachen, Germany
gFaculty of Mechanical Engineering, RWTH Aachen University, Aachen, Germany
First published on 16th July 2025
Abstract
CO2 methanation offers a pathway to produce a carbon-neutral methane fuel. Although a number of research efforts have been conducted on this topic, a greater understanding of the mechanism of the reaction, which is still under debate, is needed. Here, using in situ transmission electron microscopy, we provide direct insights into the dynamics of a metallic nickel catalyst supported on activated carbon during CO2 methanation. The keys to the high performance of the catalyst are the in situ formation and dynamic behavior of a Ni@NiO core@shell nanostructure. Based on the detailed electron microscopy investigation, the mechanism of such nanostructure formation during methanation is proposed. Our studies revealed that the deactivation of the catalyst is not due to the accumulation of carbon coke over nickel nanoparticles, but an increase in the size of the nickel nanoparticles that is responsible for the deactivation of the catalyst over time.
Keywords: CO2 valorization; Hydrogenation; in situ TEM; Carbon catalysts; Core–shell nanoparticles; Microstructure.
1 Introduction
The CO2 methanation reaction has been the subject of intense studies, since it offers an interesting pathway to produce a carbon-neutral methane fuel (e-CH4), thus combating the CO2 emissions to the atmosphere.1–5 Mostly Ni-based catalysts supported on a variety of supporting materials, such as metal oxides,6–8 zeolites,9 metal organic frameworks,10 and carbons,3,11–18 are used for CO2 methanation. Interestingly, the supporting material plays a major role in the catalyst’s performance, and it has been demonstrated that the same active phase, such as Ni nanoparticles, can present different activity, selectivity, and stability, depending upon the supporting material being used.3,5,11,19 Besides the fact that CO2 methanation involves reactant diffusion, adsorption, surface reaction, product desorption, and diffusion, the metal–support interactions can also play a role in the catalytic mode of action, which is consequently reflected in the performance of the catalyst.3,20Notably, the mechanism of CO2 methanation is still under debate. The proposed mechanisms are largely governed by the catalytic system and the reaction conditions, and the methanation could proceed through (i) the associative pathway, in which CO2 is adsorbed associatively with a former adsorbed H atom, forming formate as an intermediate, and (ii) the dissociative pathway, where CO2 is dissociated in CO, which is the main intermediate of the reaction.4 The mechanism behind nickel-catalyzed CO2 methanation involves the dissociation of H2 over the Ni nanoparticles, while CO2 adsorption usually occurs on the supporting material or nickel–support interface with dissociation and subsequent adsorption of the resultant carbonyl on Ni nanoparticles.21
A comprehensive understanding of the behavior of the supported Ni catalyst under reaction conditions, coupled with a thorough elucidation of the associated reaction mechanism, is imperative for facilitating the rational design of high-performance catalyst materials. Hence, in situ techniques are particularly important and have been extensively used to elucidate the mechanism of CO2 methanation over various supported catalytic systems. For instance, in situ infrared (IR) spectroscopy has been used for underpinning the reaction intermediates,20,22–25 while X-ray photoelectron spectroscopy (XPS) and near edge X-ray absorption fine structure (NEXAFS) have been employed to elucidate the chemical nature of the real catalysts under methanation conditions.26,27 At the same time, the fine microstructural modifications, that the supported Ni catalysts undergo during CO2 methanation, have been studied to a less extent. For example, deactivation of the Ni catalysts is often attributed to the sintering of the Ni nanoparticles and/or coke building up on the active phase, which has been investigated by ex situ transmission electron microscopy (TEM).4,28 Importantly, in situ observation of fine microstructural modifications should assist in understanding and further developing active, selective and stable methanation catalysts. Hence, in this study, in situ TEM is applied to elucidate the behavior of the Ni catalyst supported on the reduced activated carbon during CO2 methanation, affording insight into the presently lacking understanding of the catalyst’s real microstructure and its dynamics.
The understanding and atomic level information provided by aberration-corrected TEM is unparalleled, particularly for the study of catalyst nanoparticles. The development of a specialized microelectromechanical system (MEMS)-based sample holder allows us to contain gas near the sample at more than 1 bar pressure with precise control over temperature, as well as gas flow and gas compositions.
In this study, we have utilized a MEMS-based nanoreactor in which two Si/SiNx chips are positioned on top of each other and sealed with an O-ring, allowing for a maximum gas pressure of up to 2 bar. A 5 μm spacer on the bottom chip guides the gas mixture from the gas supply system towards the microheater. The system can be heated up to 1000 °C with small power consumption (in the range of mW), thus offering a low drift, while the 30 nm thin electron transparent SiNx windows, that are present in these chips, allow transmission of electron beams, thus affording structural characterization of the samples under realistic reaction conditions.
2 Results and discussion
Our earlier study3 demonstrated excellent CO2 methanation performance of a Ni catalyst supported on reduced activated carbon (Ni/ACR). Interestingly, after 90 h of CO2 methanation at 450 °C and 0.1 MPa, with 10% H2, 40% CO2 and 50% He, a distinct crystalline shell of Ni oxide was formed on the surface of the Ni nanoparticles supported on ACR observed via ex situ TEM. Fig. 1 shows TEM images of the Ni nanoparticles pre- and post-reaction, with the oxide shell of thickness 2–3 nm highlighted in Fig. 1b. The observation of a Ni oxide shell on the Ni nanoparticles after prolonged CO2 methanation was intriguing, as it contradicted the prevailing expectation that metallic Ni was the active phase of the reaction. However, these samples were transferred in air, after cooling to room temperature, for the post-reaction analysis; therefore, it was unclear whether the oxide shell was formed during the air transfer or as a consequence of the reaction conditions. Understanding this detail, along with the possible deactivation mechanism of the supported Ni catalyst, will not only shed light on the debatable methanation mechanism but also pave the pathway for engineering improved catalysts.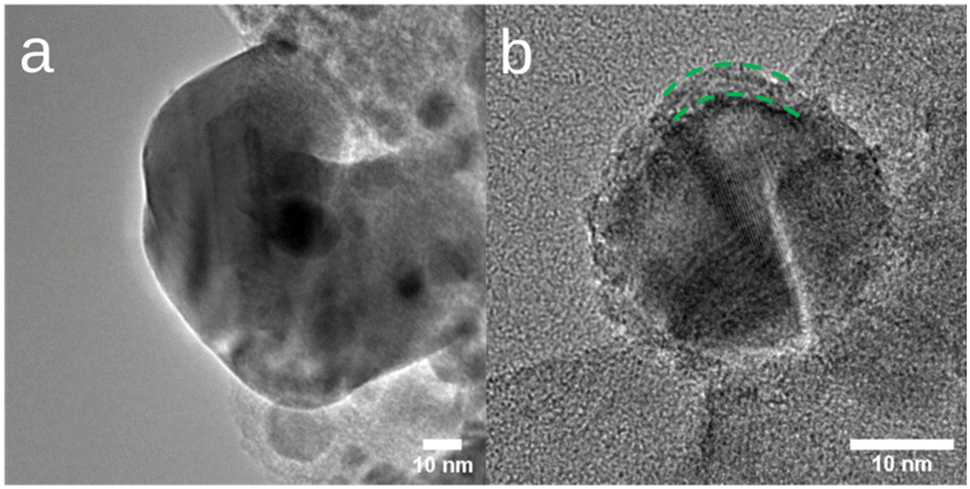 | ||
| Fig. 1 The ex situ TEM analysis results of the Ni/ACR catalyst before (a) and after (b) CO2 methanation for 90 h. The resultant Ni@NiO core@shell nanostructure is highlighted by the green dashed lines in (b). | ||
To exactly replicate the reaction conditions, the drop-cast Ni/ACR catalyst was first reduced under a H2 environment (4% H2 with N2 as the carrier gas at 1 bar pressure) at 400 °C, and then imaged under the reactive H2 and CO2 gases providing methanation conditions. It is found that once a temperature of 450 °C is reached under the reactive gases, the Ni nanoparticles began to dynamically change shape whilst still retaining crystallinity. Fig. 2(a–d) shows the dynamics of such a nanoparticle within the thick ACR supporting material. The arrows highlight the dynamic reduction-growth of the nanoparticle that simulates a ‘breathing’ behaviour. We believe that this ‘breathing’ behavior is the macroscopic manifestation of a dynamic equilibrium at the nanoscale. It likely involves the continuous, competitive oxidation of the nickel surface by CO2 to form NiO, and the simultaneous reduction of this oxide shell by H2. The observed shape changes reflect the local fluctuations in the rates of these opposing reactions. The corresponding breathing process can be better portrayed in Movie S1 in the ESI.† It should be pointed out that the reduction performed at 400 °C during the in situ experiments followed from our previous work on the same Ni on the ACR sample, where during the temperature programmed reduction (TPR) experiment, it was verified that even the Ni species that interact more strongly with the carbon were reduced at ca. 380 °C.3
 | ||
| Fig. 2 In situ time-series of TEM images showing a dynamic active particle away from the edge of the carbon support, from time 2:43 to 2:46 (a–d). The area that has undergone dynamic changes is indicated by the red arrows. | ||
To determine whether the dynamic behavior is impacted by the H2 pre-treatment, Fig. 3 compares in situ results where the sample was initially reduced in an H2 atmosphere to another set of experiments where the H2 pre-treatment was not used. Overall, like the nanoparticle shown in Fig. 2, Fig. 3 shows the dynamic behaviour of Ni nanoparticles towards the edge of the ACR supporting material under the dynamic conditions. The red arrows highlight a few of these changes in two particles, as examples. Interestingly, the presence of an oxide shell is clearly visible in both cases. The green dashed lines in Fig. 3a and j highlight the oxide shell. The high-resolution image in Fig. S1† shows the polycrystalline nature of the oxide layer outside the nanoparticles. The apparent absence of such an oxide layer for the particle shown in Fig. 2 is most likely due to the difficulty to resolve such a layer in the presence of the thick ACR supporting material. Notably, it is clear that the particles towards the edge of the ACR support changed more dynamically and, in some cases, appear almost molten, rolling around within an oxide shell. This is likely due to improved mass transport, allowing for more efficient diffusion of the CO2 and H2 reactant gases to the nanoparticle surface compared to particles embedded deeper within the porous support. The red arrows in Fig. 3a–i indicate one such nanoparticle. This behaviour was seen regardless of whether or not the reducing H2 pre-treatment was applied, as observed in Fig. 3a–i (the sample without H2 reduction) vs. Fig. 3j–n (similar sample, under similar conditions, but with H2 reduction pre-treatment). Primarily, we noticed that the formation of the oxide shell was not a mechanism of the deactivation of the particles. In fact, the most dynamic behaviour was seen for particles exhibiting an oxide shell, thereby suggesting a permeable structure in which the reactive gases can penetrate to reach metallic Ni active sites. Thus, we demonstrated that the observed NiO shell after CO2 methanation for 90 h (Fig. 1) is not a result of the non-reduced catalyst but rather formed under reaction conditions, as observed during the in situ TEM studies.
 | ||
| Fig. 3 Low-magnification in situ time-series of TEM images for active nanoparticles changing dynamically under reaction conditions without H2 pre-treatment (a–i); in situ time-series of TEM images under reaction conditions with H2 pre-treatment showing a non-active particle (pointed by orange arrows) in the top right and an active particle (pointed by red arrows) in the bottom left (j–n). The oxide shell is visible in both cases – marked in green. The timestamp in the top right-hand corner of each image shows the time in minutes and seconds since the methanation reaction conditions were reached. One example of the dynamic behaviour of the nanoparticles is pointed by the red arrows. | ||
Using in situ XPS, Heine and co-workers29 reported the formation of NiO even at room temperature in CO2 gas alone, which requires the transfer of two electrons from metallic Ni to the CO2 molecule, forming Ni2+ due to the oxidation of Ni0 and CO due to the reduction of CO2. However, after the introduction of H2 into the system mimicking methanation conditions, NiO species were found to be reduced again to metallic Ni0. In another interesting study that uses in situ XPS and in situ NEXAFS spectroscopies, Giorgianni and co-workers26 reported a similar behavior, wherein the metallic Ni surface was found to oxidize rapidly upon exposure to CO2. Under methanation conditions (i.e., CO2 + H2), while in situ XPS revealed that Ni is predominantly in a reduced state, in situ NEXAFS indicated that the shell of Ni nanoparticles is prevalently in a Ni2+ state due to incomplete reduction. Interestingly, Mutz and co-workers30 showed that, in the presence of CO2, Ni starts to oxidize after the removal of H2 from the gas flow. When H2 is again introduced into the stream, the reduced fraction of metallic Ni was found to increase to 94%, yet having a 6% fraction of oxidized Ni. In our study, we would not expect the Ni nanoparticles to oxidize, since they are under an atmosphere rich in H2; however, CO2 should be responsible for the oxidation of the surface of the Ni nanoparticles, thus forming the NiO shell according to the following reaction: Ni + CO2 = NiO + CO. The Ni0 species can be oxidized by CO2 to form Ni2+ species, activating the CO2 as well, to form CO.31 Importantly, an experiment where only CO2 gas was used (Fig. S2†) shows that the presence of both H2 and CO2 is required for the oxide-shell formation (i.e., only surface oxidation), since in the presence of CO2 alone, no oxide shell can be seen. Therefore, the synergetic relationship between CO2 and H2 should be responsible for the formation of the Ni@NiO core–shell structure during CO2 methanation, as observed herein.
The formation of this Ni@NiO core@shell structure under reaction conditions might be one of the reasons for the good performance of this catalyst. On the one hand, the NiO formation in close contact with the supporting carbon material, where CO2 is adsorbed, leads to the activation of this molecule,25,31 while the Ni0 in the core is responsible for H2 dissociation, since the dynamic nature of the oxide shell allows for the diffusion of the H2 to the metallic Ni0 core of the nanoparticles. This way, both reactants can be in close contact, which facilitates the reaction. On the other hand, NiO might present oxygen vacancies, which are known to induce the dissociation and activation of CO2 on the surface of the catalyst.32 It has been reported before that the rate-determining step for the CO2 methanation reaction should be related to the availability of adjacent adsorbed H to hydrogenate adsorbed CO;33 thus, this proximity of CO2 activation and CO formation on the NiO shell and H2 dissociation in the Ni core might explain the good performance on the catalyst.3
After examining the dynamic behaviour of the particles under a reactive methanation environment, we then explored the mechanisms of deactivation of the catalyst. The main deactivation mechanism was observed to be related to a particle size increase. This can be observed in Movie S2,† obtained at 450 °C in the presence of both H2 and CO2. The larger particles demonstrated no dynamic changes, while the smaller particles demonstrated to be very active. Upon cooling, the particle structures stabilised to fill the oxide shell again (this can be seen in Fig. S3†), yielding a structure that matches the original ex situ characterisation (Fig. 1b). Finally, post in situ energy dispersive X-ray spectroscopy analysis in scanning TEM mode (STEM-EDX) was performed to confirm the presence of a 2–3 nm thick NiO shell (Fig. 4(a–f)). The heating and gas profile during these in situ experiments can be found in Fig. S4.†
 | ||
| Fig. 4 Post in situ STEM–EDS analysis of the catalyst nanoparticle showing the annular dark-field STEM (a), Ni, O EDS overlay map (b), variation of Ni and O count along the arrow direction (c), and corresponding Ni (d), O (e), and C (f) EDS map. | ||
Particle size distribution was not thoroughly studied in this work; however, since methanation is a known structure-sensitive reaction, the catalyst particle size is an important aspect. Thus, further experiments should be carried out to address this topic.
Our experimental in situ TEM findings have demonstrated that the nickel oxide shell observed on the catalytic Ni nanoparticles supported on reduced activated carbon in our previous study3 is in fact formed under reaction conditions of CO2 methanation, and it is expected to be an integral part of the reaction process. Fig. 5 presents a summary of the findings of this work. We believe that the Ni oxide shell structure, in situ formed during methanation, exhibits a permeable nature, allowing for the penetration of the reaction CO2 and H2 gases to the active metallic Ni0 core, otherwise dynamic changes under reaction conditions would not continue to be observed in the nanoparticles demonstrating a shell. We hypothesize that the Ni oxide shell acts as an enhancer of the CO2 adsorption/activation, while the Ni0 in the core is responsible for H2 dissociation, which leads to both reactants being in close proximity, facilitating the CH4 formation.35,36 Notably, we observed in situ that the main deactivation mechanism was related to a particle size.
 | ||
| Fig. 5 Schematic representation of the hypothesized mechanism of the Ni@NiO nanostructure formation during CO2 methanation over the high-performance Ni/ACR catalyst. Box A – the Ni oxide shell structure is formed in situ (Ni0 + CO2 → NiO + CO), allowing for the penetration of H2 to the active metallic Ni0 core; box B – Ni oxide shell acts as an enhancer of the CO2 adsorption/activation while the Ni0 in the core is responsible for H2 dissociation, leading to both reactants in close proximity, facilitating the CH4 formation. | ||
3 Conclusions
In summary, we have presented an in situ TEM investigation of our previously reported high-performance nickel nanoparticle catalyst supported on reduced activated carbon.3 The unprecedented in situ formation of a NiO shell around the metallic Ni0 core was directly imaged and studied under CO2 methanation conditions. An important proposed feature of real Ni@NiO core@shell catalyst nanoparticles is coupling of the enhanced CO2 adsorption/activation over the NiO shell with H2 dissociation over the Ni0 core, bringing the reactants and intermediates together to accomplish CO2 hydrogenation into CH4. This fundamental understanding of CO2 methanation over the Ni catalyst provides a framework for the judicious design and realization of new high-performance and stable core@shell catalysts for this reaction, in particular, and other interesting CO2 hydrogenation reactions, in general.Data availability
Data for this article are available in the ESI.†Author contributions
Katherine E. MacArthur: conceptualization, data curation, formal analysis, investigation, methodology, and writing – original draft; Liliana P. L. Gonçalves: conceptualization, formal analysis, investigation, methodology, and writing – original draft; Juliana P. S. Sousa: supervision and writing – review & editing; O. Salomé G. P. Soares: conceptualization, supervision, and writing – review & editing; Hans Kungl: writing – review & editing; Eva Jodat: writing – review & editing; André Karl: writing – review & editing; Marc Heggen: writing – review & editing; Rafal E. Dunin-Borkowski: funding acquisition, project administration, and writing – review & editing; Shibabrata Basak: conceptualization, data curation, investigation, methodology, writing – review & editing, and supervision; Rüdiger-A. Eichel: writing – review & editing; Yury V. Kolen'ko: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing; M. Fernando R. Pereira: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of the German Research Foundation (DFG) for supporting this work under grant number HE 7192/1-2. S. B. acknowledges support for the project ‘Electroscopy’ (Grant No. 892916) from the Marie Skłodowska-Curie action. L. P. L. G. thanks the Portuguese Foundation for Science and Technology (FCT) for the PhD grant (SFRH/BD/128986/2017). This work was partially supported by national funds through Fundação para a Ciência e a Tecnologia, I.P./MCTES: LSRE-LCM, UID/50020; ALiCE, LA/P/0045/2020 (DOI: https://doi.org/10.54499/LA/P/0045/2020). O. S. G. P. S. acknowledges FCT funding under the Scientific Employment Stimulus – Institutional Call (CEECINST/00049/2018).References
- M. Bailera, P. Lisbona, L. M. Romeo and S. Espatolero, Power to gas projects review: Lab, pilot and demo plants for storing renewable energy and CO2, Renewable Sustainable Energy Rev., 2017, 69, 292–312 CrossRef CAS
.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, A review of recent catalyst advances in CO2 methanation processes, Catal. Today, 2020, 356, 471–489 CrossRef CAS
- L. P. L. Gonçalves, J. P. S. Sousa, O. S. G. P. Soares, O. Bondarchuk, O. I. Lebedev, Y. V. Kolen'ko and M. F. R. Pereira, The role of surface properties in CO2 methanation over carbon-supported Ni catalysts and their promotion by Fe, Catal. Sci. Technol., 2020, 10, 7217 RSC
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catalysis mechanisms of CO2 and CO methanation, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC
- F. Goodarzi, L. Kang, F. R. Wang, F. Joensen, S. Kegnæs and J. Mielby, Methanation of carbon dioxide over zeolite-encapsulated nickel nanoparticles, ChemCatChem, 2018, 10, 1566–1570 CrossRef CAS
- Z. Zhang, Y. Tian, L. Zhang, S. Hu and J. Xiang, Impacts of nickel loading on properties, catalytic behaviors of Ni/γ–Al2O3 catalysts and the reaction intermediates formed in methanation of CO2, Int. J. Hydrogen Energy, 2019, 44, 9291–9306 CrossRef CAS
- Y. Ma, J. Liu, M. Chu, J. Yue, Y. Cui and G. Xu, Cooperation between active metal and basic support in Ni-based catalyst for low-temperature CO2 methanation, Catal. Lett., 2020, 150, 1418–1426 CrossRef CAS
- X. Wen, L. Xu, M. Chen, Y. Shi, C. Lv, Y. Cui, X. Wu, G. Cheng, C. E. Wu, Z. Miao, F. Wang and X. Hu, Exploring the influence of nickel precursors on constructing efficient Ni-based CO2 methanation catalysts assisted with in-situ technologies, Appl. Catal., B, 2021, 297, 120486 CrossRef CAS
- A. Westermann, B. Azambre, M. C. Bacariza, I. Graça, M. F. Ribeiro, J. M. Lopes and C. Henriques, Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study, Appl. Catal., B, 2015, 174–175, 120–125 CrossRef CAS
- W. Zhen, B. Li, G. Lu and J. Ma, Enhancing catalytic activity and stability for CO2 methanation on Ni@MOF-5 via control of active species dispersion, Chem. Commun., 2015, 51, 1728–1731 RSC
- L. P. L. Gonçalves, A. Serov, G. McCool, M. Dicome, J. P. S. Sousa, O. S. G. P. Soares, O. Bondarchuk, D. Y. Petrovykh, O. I. Lebedev, M. F. R. Pereira and Y. V. Kolen'ko, New opportunity for carbon-supported Ni-based electrocatalysts: Gas-phase CO2 methanation, ChemCatChem, 2021, 13, 4770–4779 CrossRef
- N. N. Ha, N. T. Ha, L. V. Khu and L. M. Cam, Theoretical study of carbon dioxide activation by metals (Co, Cu, Ni) supported on activated carbon, J. Mol. Model., 2015, 21, 322 CrossRef PubMed
- W. Wang, C. Duong-Viet, H. Ba, W. Baaziz, G. Tuci, S. Caporali, L. Nguyen-Dinh, O. Ersen, G. Giambastiani and C. Pham-Huu, Nickel nanoparticles decorated nitrogen-doped carbon nanotubes (Ni/N-CNT); A robust catalyst for the efficient and selective CO2 methanation, ACS Appl. Energy Mater., 2019, 2, 1111–1120 CrossRef CAS
- J. Gödde, M. Merko, W. Xia and M. Muhler, Nickel nanoparticles supported on nitrogen–doped carbon nanotubes are a highly active, selective and stable CO2 methanation catalyst, J. Energy Chem., 2021, 54, 323–331 CrossRef
- P. Kangvansura, L. M. Chew, W. Saengsui, P. Santawaja, Y. Poo-arporn, M. Muhler, H. Schulz and A. Worayingyong, Product distribution of CO2 hydrogenation by K- and Mn-promoted Fe catalysts supported on N-functionalized carbon nanotubes, Catal. Today, 2016, 275, 59–65 CrossRef CAS
- W. Wang, W. Chu, N. Wang, W. Yang and C. Jiang, Mesoporous nickel catalyst supported on multi-walled carbon nanotubes for carbon dioxide methanation, Int. J. Hydrogen Energy, 2015, 41, 967–975 CrossRef
- J. Gao, Q. Jiang, Y. Liu, W. Liu, W. Chu and D. S. Su, Probing the enhanced catalytic activity of carbon nanotube supported Ni-LaOx hybrids for the CO2 reduction reaction, Nanoscale, 2018, 10, 14207–14219 RSC
- M. Romero-Sáez, A. B. Dongil, N. Benito, R. Espinoza-González, N. Escalona and F. Gracia, CO2 methanation over nickel-ZrO2 catalyst supported on carbon nanotubes: A comparison between two impregnation strategies, Appl. Catal., B, 2018, 237, 817–825 CrossRef
- A. I. Tsiotsias, N. D. Charisiou, I. V. Yentekakis and M. A. Goula, Bimetallic Ni-based catalysts for CO2 methanation: A review, Nanomaterials, 2021, 11, 28 CrossRef CAS PubMed
- Y. H. Lee, J. Y. Ahn, D. D. Nguyen, S. W. Chang, S. S. Kim and S. M. Lee, Role of oxide support in Ni based catalysts for CO2 methanation, RSC Adv., 2021, 11, 17648–17657 RSC
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy, Catal. Today, 2013, 215, 201–207 CrossRef CAS
- L. P. L. Gonçalves, J. Mielby, O. S. G. P. Soares, J. P. S. Sousa, D. Y. Petrovykh, O. I. Lebedev, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen'ko, In situ investigation of the CO2 methanation on carbon/ceria-supported Ni catalysts using modulation-excitation DRIFTS, Appl. Catal., B, 2022, 312, 121376 CrossRef
- Z. Hao, J. Shen, S. Lin, X. Han, X. Chang, J. Liu, M. Li and X. Ma, Decoupling the effect of Ni particle size and surface oxygen deficiencies in CO2 methanation over ceria supported Ni, Appl. Catal., B, 2021, 286, 119922 CrossRef CAS
- H. L. Huynh, J. Zhu, G. Zhang, Y. Shen, W. M. Tucho, Y. Ding and Z. Yu, Promoting effect of Fe on supported Ni catalysts in CO2 methanation by in situ DRIFTS and DFT study, J. Catal., 2020, 392, 266–277 CrossRef CAS
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts, Appl. Catal., B, 2020, 265, 118538 CrossRef
- G. Giorgianni, C. Mebrahtu, M. E. Schuster, A. I. Large, G. Held, P. Ferrer, F. Venturini, D. Grinter, R. Palkovits, S. Perathoner, G. Centi, S. Abate and R. Arrigo, Elucidating the mechanism of the CO2 methanation reaction over Ni–Fe hydrotalcite-derived catalysts via surface-sensitive in situ XPS and NEXAFS, Phys. Chem. Chem. Phys., 2020, 22, 18788–18797 RSC
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, Combining in situ NEXAFS spectroscopy and CO2 methanation kinetics to study Pt and Co nanoparticle catalysts reveals key insights into the role of platinum in promoted cobalt catalysis, J. Am. Chem. Soc., 2014, 136, 9898–9901 CrossRef CAS
- P. Strucks, L. Failing and S. Kaluza, A short review on Ni-catalyzed methanation of CO2: Reaction mechanism, catalyst deactivation, dynamic operation, Chem. Ing. Tech., 2021, 93, 1526–1536 CrossRef CAS
- C. Heine, B. A. J. Lechner, H. Bluhm and M. Salmeron, Recycling of CO2: Probing the chemical state of the Ni(111) surface during the methanation reaction with ambient-pressure X-ray photoelectron spectroscopy, J. Am. Chem. Soc., 2016, 138, 13246–13252 CrossRef CAS PubMed
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J. D. Grunwaldt, Methanation of CO2: Structural response of a Ni-based catalyst under fluctuating reaction conditions unraveled by operando spectroscopy, J. Catal., 2015, 327, 48–53 CrossRef CAS
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Role of surface Ni and Ce species of Ni/CeO2 catalyst in CO2 methanation, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS
- Y. Du, C. Qin, Y. Xu, D. Xu, J. Bai, M. Ding, C. Qin, Y. Xu, D. Xu, J. Bai and M. Ding, Ni nanoparticles dispersed on oxygen vacancies-rich CeO2 nanoplates for enhanced low-temperature CO2 methanation performance, Chem. Eng. J., 2021, 417, 129402 CrossRef
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Unravelling structure sensitivity in CO2 hydrogenation over nickel, Nat. Catal., 2018, 1, 127–134 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, together with the additional TEM data and a video. See DOI: https://doi.org/10.1039/d5im00033e |
| ‡ These authors contributed equally. |
| This journal is © Institute of Process Engineering of CAS 2025 |