
Advances in the development of Wnt/β-catenin signaling inhibitors
Minami
Fujita
ab and
Yosuke
Demizu
 *abc
*abc
aDivision of Organic Chemistry, National Institute of Health Sciences, 3-25-26, Tonomachi, Kawasaki, Kanagawa 210-9501, Japan. E-mail: demizu@nihs.go.jp
bGraduate School of Medical Life Science, Yokohama City University, 1-7-29, Yokohama, Kanagawa 230-0045, Japan
cGraduate School of Medicine, Dentistry and Pharmaceutical Sciences, Division of Pharmaceutical Science of Okayama University, 1-1-1 Tsushimanaka, Kita, Okayama 700-8530, Japan
First published on 2nd December 2024
Abstract
The Wnt/β-catenin signaling pathway plays a critical role in various biological processes, including cell proliferation, differentiation, and tissue homeostasis. Aberrant activation of this pathway is strongly associated with the development of various cancers, including colorectal, pancreatic, and gastric cancers, making it a promising therapeutic target. In recent years, inhibitors targeting different components of the Wnt/β-catenin pathway, including small molecules, peptides, and nucleic acid-based therapies, have been developed to suppress cancer cell growth. These inhibitors work by disrupting key interactions within the pathway, thereby preventing tumor progression. Antibody-based therapies have also emerged as potential strategies to block ligand-receptor interactions within this pathway. Despite these advancements, challenges such as the complexity of the pathway and toxicity concerns remain. Innovative approaches, including allosteric inhibitors, proteolysis-targeting chimeras (PROTACs), and peptide-based inhibitors, offer new opportunities to address these challenges. This review provides an overview of the latest progress in the development of Wnt/β-catenin pathway inhibitors and explores future directions in cancer therapy.
Introduction
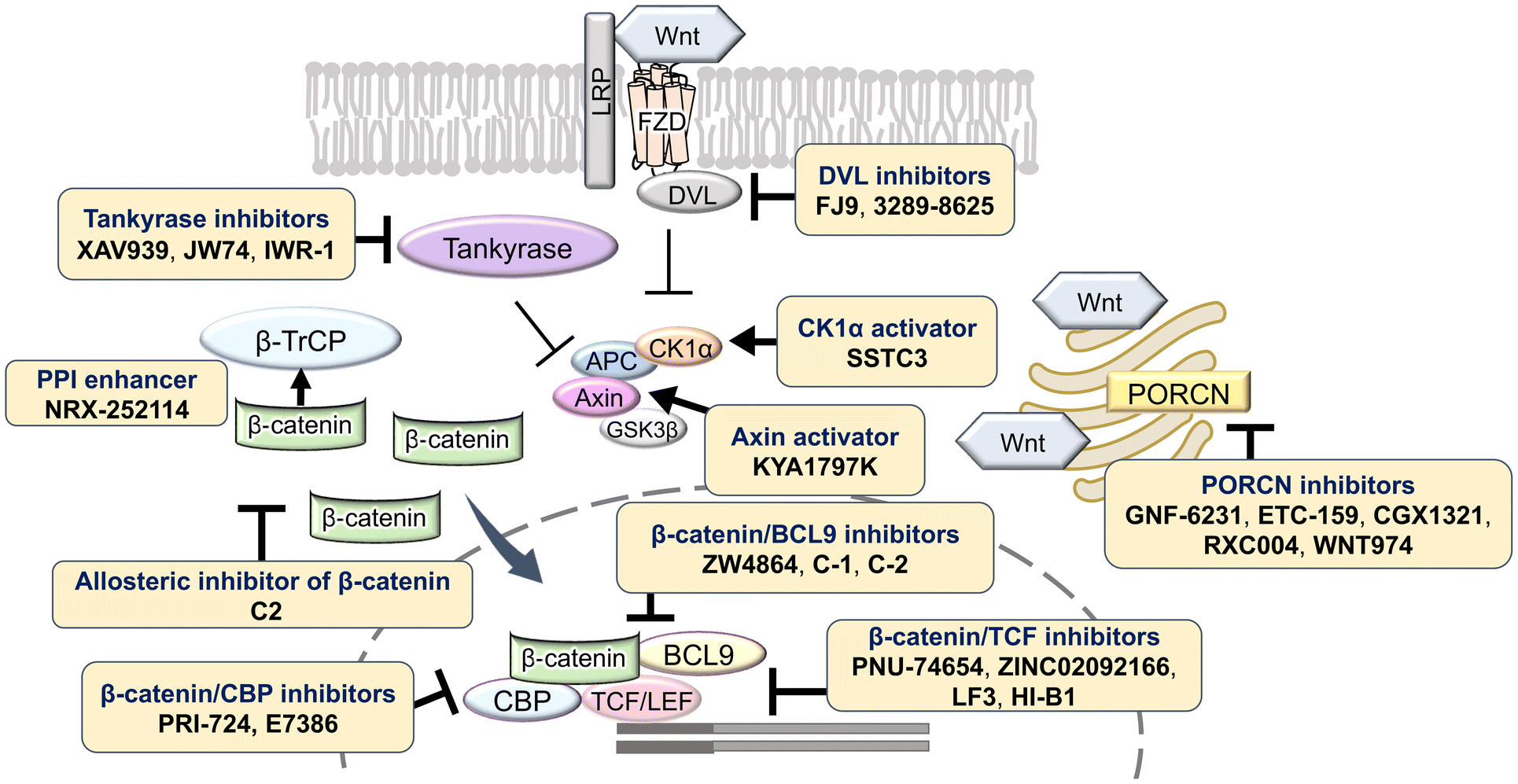
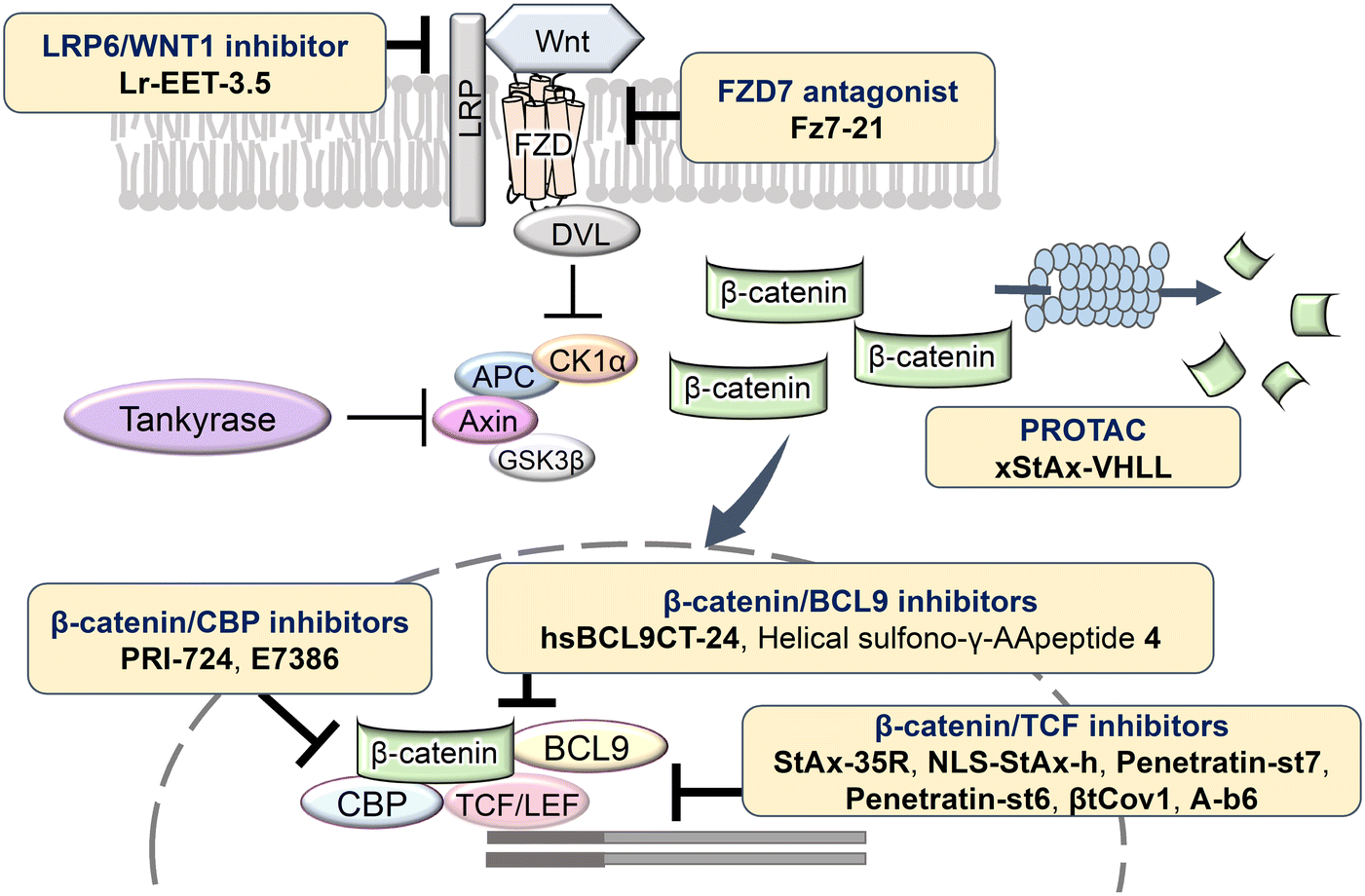
The Wnt/β-catenin signaling pathway plays a critical role in numerous biological processes, including embryogenesis, cell proliferation, differentiation, and tissue homeostasis. Dysregulation of this pathway is linked to a wide range of diseases, such as gastric, colorectal, pancreatic, and prostate cancer, highlighting its importance as a therapeutic target. Under normal conditions, in the absence of Wnt ligands, β-catenin forms a complex with adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), and casein kinase 1α (CK1α), leading to its phosphorylation, ubiquitination, and subsequent proteasomal degradation. This ensures that β-catenin concentrations are kept low in the cytoplasm, preventing its translocation to the nucleus where it can activate gene transcription.1 When Wnt proteins bind to the receptor Frizzled (FZD) and the co-receptor low-density lipoprotein receptor-related protein 5/6 (LRP5/6) on the plasma membrane, Dishevelled (DVL) is recruited to the membrane and inhibits the formation of the β-catenin destruction complex. As a result, β-catenin is stabilized and accumulates in the cytoplasm, eventually translocating into the nucleus. In the nucleus, β-catenin associates with transcription factors such as T-cell factor/lymphoid enhancer factor (TCF/LEF), and coactivators including cAMP-response element-binding protein (CBP) and B-cell lymphoma 9 (BCL9), to drive the expression of target genes involved in cell cycle regulation, survival, and proliferation.2 This aberrant activation of the Wnt/β-catenin pathway is a hallmark of many cancers, where it contributes to tumor initiation, progression, metastasis, and resistance to therapy (Fig. 1). | ||
| Fig. 1 Illustrated Wnt signaling pathway. Abbreviations: APC, adenomatous polyposis coli; CK1α, casein kinase 1α; DVL, Dishevelled; GSK3β, glycogen synthase kinase 3β; LRP, lipoprotein receptor-related protein; TCF/LEF; T-cell factor/lymphoid enhancer factor. | ||
Therapeutic targeting of the Wnt/β-catenin pathway has been an area of intense research, with efforts directed at inhibiting various components of the pathway to suppress cancer cell proliferation and survival. Small-molecule inhibitors have been studied extensively. These include WNT974 (LGK974), which targets porcupine (PORCN), an acyltransferase essential for Wnt ligand palmitoylation and secretion. WNT974 was optimized based on high-throughput screening hits, resulting in a compound with excellent activity, specificity, and pharmacokinetic properties, including high oral bioavailability. WNT974 has shown potent inhibitory effects on Wnt signaling and tumor growth in preclinical models and has progressed into clinical trials.3,4 Similarly, PRI724 disrupts the interaction between β-catenin and CBP, a critical coactivator of β-catenin-mediated transcription, thereby selectively inhibiting Wnt/β-catenin-driven gene expression without affecting the TCF/LEF interaction, which is essential for normal tissue homeostasis.5,6 Another promising small molecule, CWP232291 (CWP291), targets the β-catenin/CBP complex, leading to the inhibition of transcriptional activity and reduced viability of cancer stem cells, which are often resistant to conventional therapies.7,8 Antibody-based therapies have also been explored, with OMP-18R5 (vantictumab) being a prominent example. OMP-18R5 targets multiple Frizzled receptors, blocking Wnt ligand binding and effectively inhibiting pathway activation. Preclinical studies have demonstrated that OMP-18R5 can inhibit the growth of various cancer types, and this drug candidate has now progressed to clinical evaluation for the treatment of solid tumors.9,10 Despite these advancements, the development of Wnt/β-catenin pathway inhibitors has faced significant challenges. The redundancy and complexity of the pathway, which includes multiple Wnt ligands and receptors, make selective targeting difficult. Additionally, Wnt signaling plays critical roles in normal stem cell maintenance and tissue regeneration, raising concerns about potential toxicity and side effects of systemic pathway inhibition.
β-Catenin itself has been a challenging target because of its role as a transcriptional coactivator lacking a defined small-molecule binding pocket, which is typical for enzymes and other druggable proteins. However, recent innovative approaches, such as the use of allosteric inhibitors and proteolysis-targeting chimeras (PROTACs), has opened new avenues for targeting β-catenin. Allosteric inhibitors can modulate protein function by binding to sites distinct from the active site, offering a strategy to inhibit protein–protein interactions (PPIs) involving β-catenin without directly blocking its interaction with TCF/LEF or other coactivators.11 PROTACs, however, are bifunctional molecules that recruit target proteins, such as β-catenin, to E3 ubiquitin ligases, leading to their ubiquitination and proteasomal degradation. This approach has shown promise in preclinical models in degrading β-catenin and suppressing Wnt-driven oncogenesis.12
Furthermore, peptide-based inhibitors and nucleic acid-based therapies, such as small interfering (si)RNAs and antisense oligonucleotides, have also been explored to target the Wnt/β-catenin pathway. Peptide inhibitors can be designed to disrupt specific PPIs, such as those between β-catenin and TCF, thereby inhibiting pathway activation.13 Nucleic acid-based approaches can downregulate the expression of key pathway components at the mRNA level, providing a targeted and potentially less toxic alternative to small molecules and antibodies. For example, siRNAs targeting β-catenin have been shown to reduce its expression and inhibit cancer cell growth in various models.14 Considering the involvement of the Wnt/β-catenin pathway in the maintenance of cancer stem cells, targeting this pathway offers the potential to overcome drug resistance and relapse, which are major challenges in cancer treatment. Cancer stem cells are thought to drive tumor initiation, metastasis, and recurrence, and are often resistant to standard chemotherapy and radiation. Inhibiting the Wnt/β-catenin pathway in these cells could prevent tumor regrowth and improve patient outcomes.15 Despite the lack of approved Wnt/β-catenin pathway inhibitors on the market, ongoing clinical trials and preclinical studies continue to explore and refine these strategies, with the hope of developing effective therapies that can selectively target this pathway in cancer without causing undue harm to normal tissues.
The Wnt/β-catenin signaling pathway remains a highly attractive but challenging target for cancer therapy. Continued research into the pathway's mechanisms, the development of innovative targeting strategies, and a better understanding of the pathway's role in normal versus cancerous cells are essential for advancing Wnt/β-catenin inhibitors toward clinical success. This review aims to provide a comprehensive overview of the progress made over the past decade, highlighting the mechanisms of action, therapeutic potential, and challenges associated with small-molecule inhibitors, peptides, nucleic acids, and macromolecules targeting the Wnt/β-catenin signaling pathway. Expanding therapeutic options by targeting traditionally undruggable proteins within this pathway offers significant potential for new cancer treatments, ultimately contributing to improved patient care and outcomes.
Small-molecule inhibitors
The Wnt/β-catenin signaling pathway plays a crucial role in regulating cell proliferation, differentiation, and survival, making it a key target for therapeutic intervention, particularly in cancer where aberrant activation of this pathway is commonly observed. Within this pathway, DVL functions as a critical mediator that inhibits the β-catenin degradation complex, thereby preventing the degradation of β-catenin and allowing its accumulation in the cytoplasm and subsequent translocation to the nucleus, where it activates Wnt target genes. Targeting the interaction between DVL and FZD receptors has been explored as a strategy to disrupt this signaling cascade. Among the inhibitors developed (Table 1, Fig. 2), FJ9, which features an indole-2-carbinol skeleton, has shown potential in inhibiting DVL function by disrupting the DVL-FZD interaction, leading to reduced Wnt signaling activity.16 Another promising compound, 3289-8625, has demonstrated effective inhibition of DVL, and uniquely, it has been shown to inhibit Wnt signaling not only in vitro but also in vivo.17 This compound operates by binding to the PDZ domain of DVL, thereby blocking the propagation of Wnt signals and resulting in decreased β-catenin stabilization and activity.| Compound | Structure | Target | Phase (biochemical data) |
|---|---|---|---|
| Abbreviations: BCL9, B cell lymphoma 9; CBP, cAMP-response element-binding protein; CK1α, casein kinase 1α; DVL, Dishevelled; PORCN, porcupine; TCF, T cell factor. | |||
| FJ9 |

|
DVL | Preclinical (100 μM, HTC116 cells) |
| 3289-8625 |
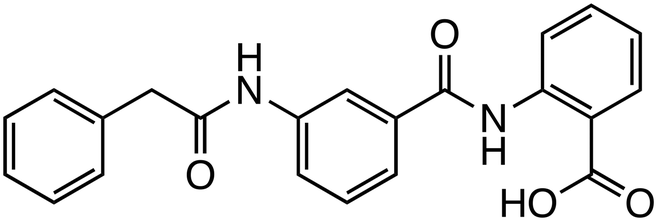
|
DVL | Preclinical (12.5 μM, PC-3 cells) |
| GNF-6231 |

|
PORCN | Preclinical (0.8 nM, TM3 cells) |
| ETC-159 |

|
PORCN | Phase 1 (20 nM, HPAF- II cells) |
| CGX1321 |
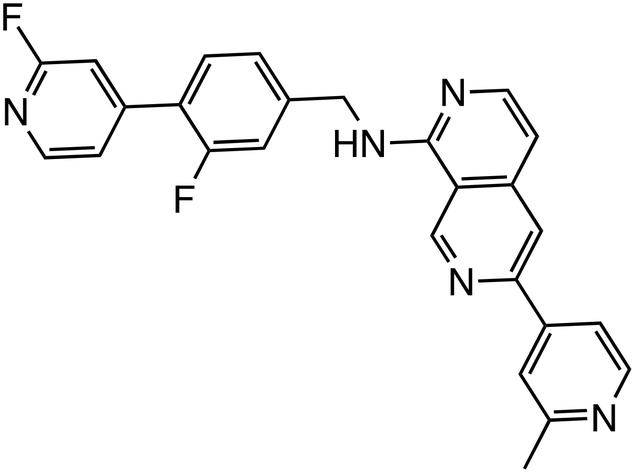
|
PORCN | Phase 1 (18.4 μM, HEK293) |
| RXC004 |

|
PORCN | Phase 2 (64 pM, L-Wnt3a cells) |
| WNT974 |
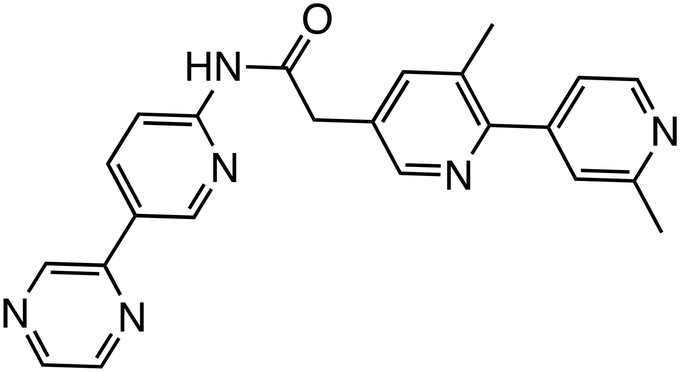
|
PORCN | Phase 2 (0.4 nM, L-Wnt3a cells, TM3 cells) |
| XAV939 |
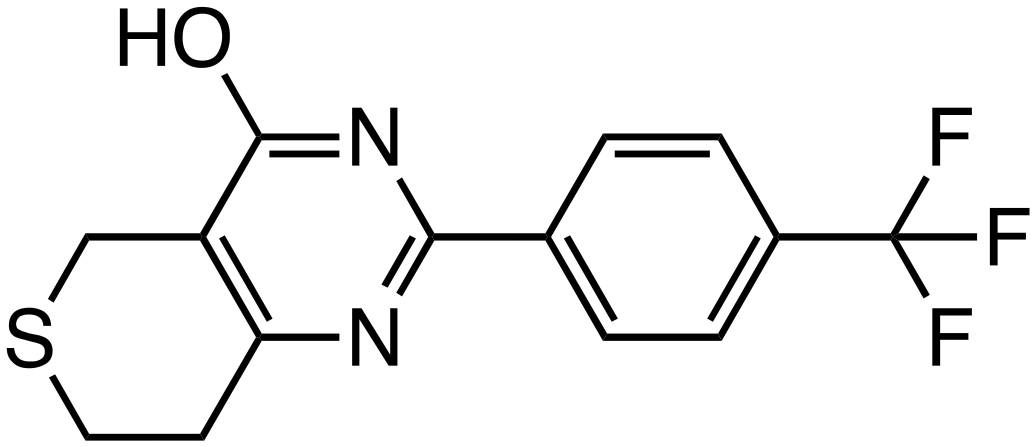
|
Tankyrase | Preclinical (0.3 μM, DLD-1 cells) |
| JW74 |
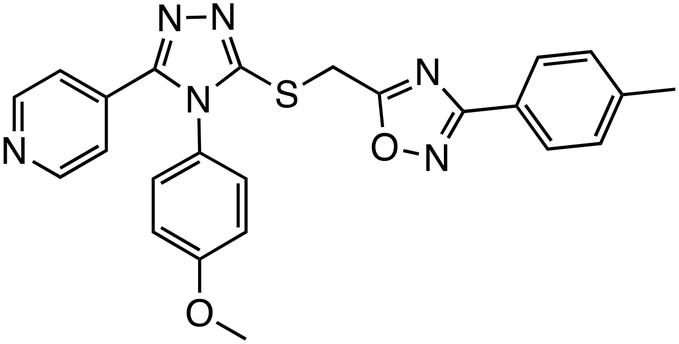
|
Tankyrase | Preclinical (10 μM, U2OS cells) |
| IWR-1 |
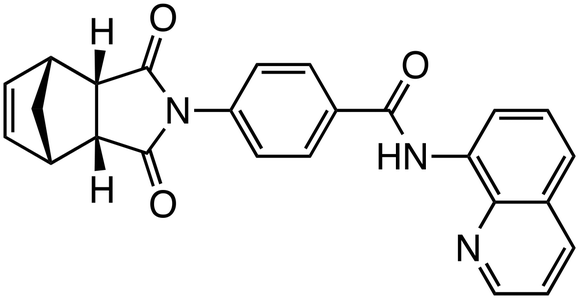
|
Tankyrase | Preclinical (10 μM, MG-63 cells, MNNG-HOS cells) |
| SSTC3 |
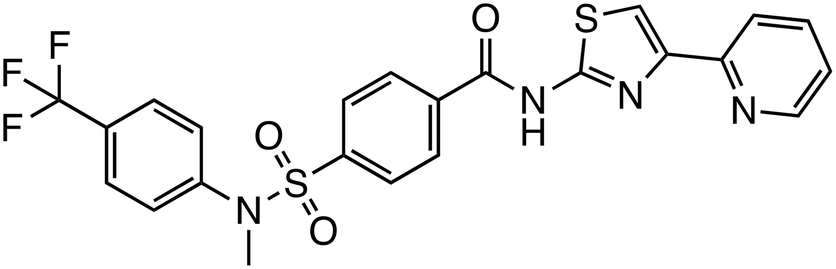
|
CK1α | Preclinical (123 nM, HCT116 cells) |
| SM08502 |

|
CLK | Phase 1 (0.046 μM, SW480 cells) |
| KYA1797K |

|
Axin | Preclinical (0.75 μM, HEK293 cells) |
| ZW4864 |
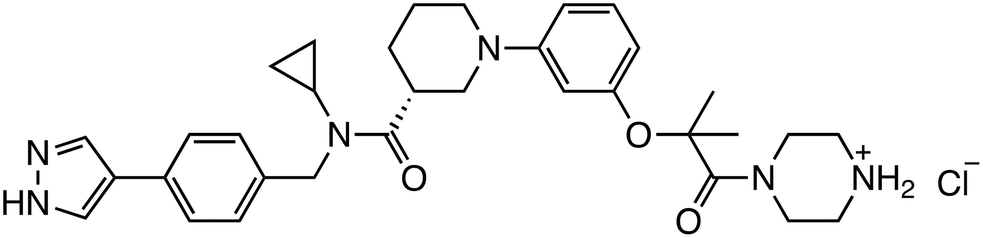
|
β-Catenin/BCL9 | Preclinical (7.0 μM, SW480 cells) |
| C-1 |
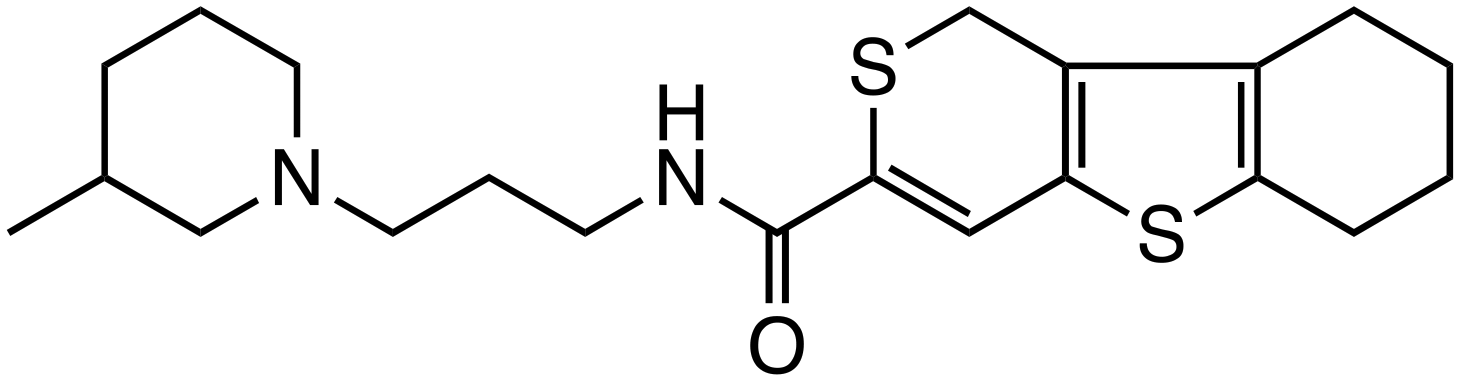
|
β-Catenin/BCL9 | Preclinical (20 μM, Colo320 cells) |
| C-2 |
|
β-Catenin/BCL9 | Preclinical (20 μM, Colo320 cells) |
| PRI-724 |

|
β-Catenin/CBP | Phase 2 (data not shown) |
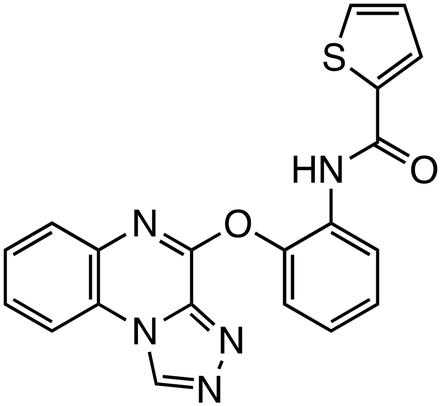
| E7386 |
|
β-Catenin/CBP | Phase 2 (0.0484 μM, HEK293 cells) |
| PNU-74654 |

|
β-Catenin/TCF | Preclinical (129.8 μM, NCI-H295 cells) |
| ZINC02092166 |

|
β-Catenin/TCF | Preclinical (0.71 μM, SW480 cells) |
| LF3 |

|
β-Catenin/TCF | Preclinical (22.2 μM, HEK293 cells) |
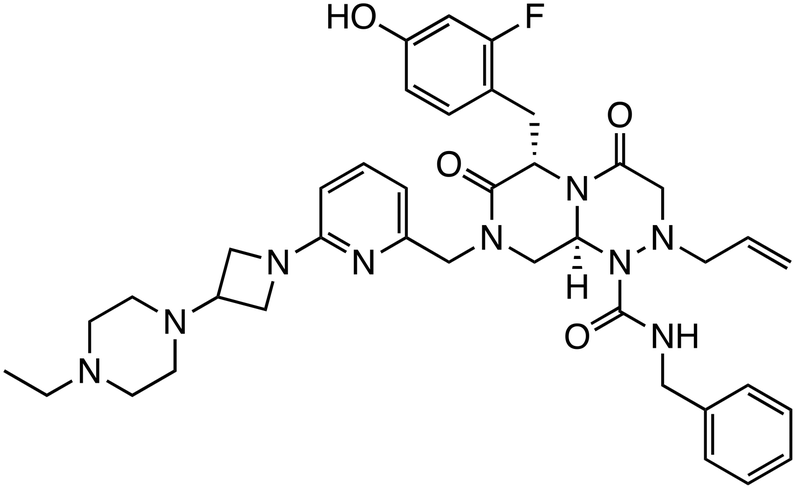
| HI-B1 |
|
β-Catenin/TCF | Preclinical (50 μM, DLD-1 cells) |
| C2 |
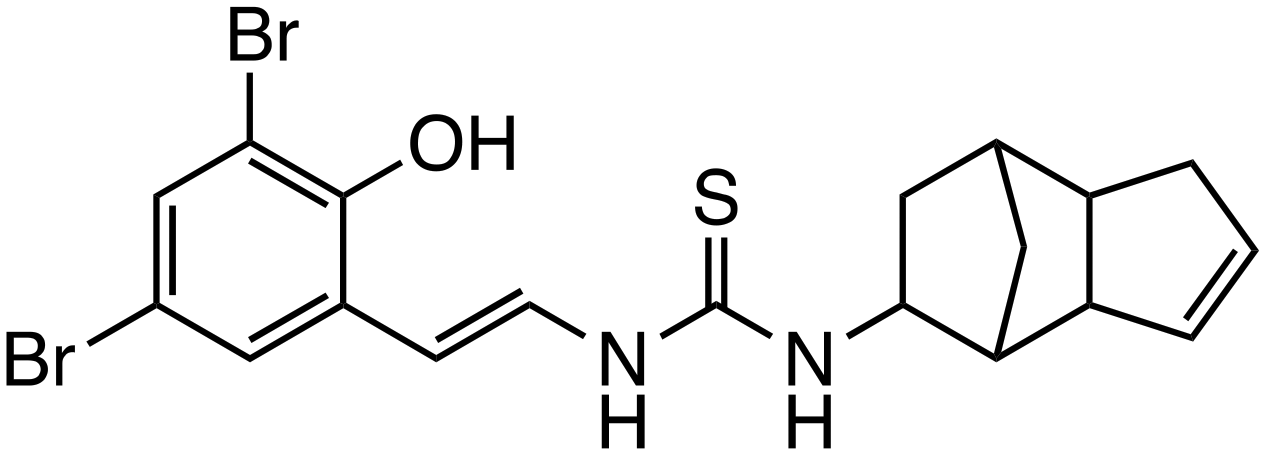
|
β-Catenin | Preclinical (0.8–1.3 μM, DLD-1 cells, SW480) |
| MSAB |
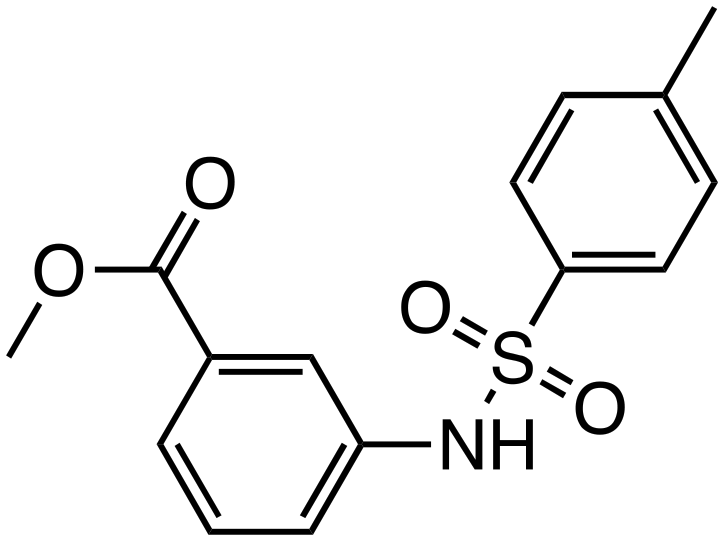
|
β-Catenin | Preclinical (0.583 μM, HCT116 cells) |
| NRX-252114 |

|
β-Catenin | Preclinical (50 μM, HEK293T cells expressing S33E/S37A β-catenin) |
 | ||
| Fig. 2 Small-molecule inhibitors or activators acting on the Wnt/β-catenin signaling pathway. Abbreviations: APC, adenomatous polyposis coli; BCL9, B cell lymphoma 9; CBP, cAMP-response element-binding protein; CK1α, casein kinase 1α; DVL, Dishevelled; FZD, Frizzled; GSK3β, glycogen synthase kinase 3β; LRP, lipoprotein receptor-related protein; PORCN, porcupine; TCF, T cell factor. | ||
PORCN, an O-acyltransferase, regulates Wnt protein secretion and function by palmitoylating Wnt proteins. Inhibitors targeting PORCN, such as GNF-1331 and its derivative GNF-6231,18 were identified from a library comprising 2.4 million compounds, and developed through structure–activity relationship (SAR) studies. SAR analysis revealed that the pyridine nitrogen and thioether moiety are important sites. Replacing the thioether moiety with a phenyl group greatly improved the pharmacokinetic properties and also increased the activity. These have shown robust antitumor effects in the mouse mammary tumor virus (MMTV)-Wnt1 xenograft tumor model. Other PORCN inhibitors currently in clinical trials include ETC-159,19 CGX1321,20 RXC004,21 and WNT974.3,4 RXC004 has exhibited potent antiproliferative effects against Wnt ligand-dependent colon and pancreatic cancer cell lines and inhibited tumor growth in preclinical in vitro and in vivo models. Furthermore, RXC004 enhanced immune system activity when combined with anti-programmed cell death protein (PD)-1 therapy and is currently in clinical trials both as monotherapy and in combination with anti-PD-1 therapy in patients with Wnt ligand-dependent gastrointestinal cancers.
Tankyrase, a member of the poly (ADP-ribose) polymerase (PARP) family, promotes Axin degradation through poly ADP-ribosylation (PARsylation) of Axin. Tankyrase inhibitors, such as XAV939,22 JW74,23 and IWR-1,24 prevent Axin degradation and promote β-catenin degradation. CK1α, a crucial regulator of the Wnt signaling pathway, normally suppresses Wnt signaling by promoting β-catenin degradation. However, many colorectal cancers exhibit aberrant Wnt signaling activation due to mutations in the APC or CTNNB1 genes, leading to reduced CK1α function. SSTC3 (ref. 25) activates CK1α, enhancing β-catenin phosphorylation and degradation. In mouse models, SSTC3 significantly inhibited tumor growth in tumors with APC mutations and in a patient-derived metastatic colorectal cancer model, with minimal effects on normal gastrointestinal tissues, indicating a lack of adverse impact on gastrointestinal stem cells or normal tissue maintenance. SM08502 is an example of kinase targeting, specifically a CDC-like kinase (CLK) inhibitor. SM08502 inhibits the phosphorylation of splicing factors (SRSF), which interferes with the function of the spliceosome. This, in turn, alters the transcriptional activity of the Wnt pathway and the splicing of related genes. This results in the production of abnormal transcripts in cancer cells, thereby effectively inhibiting tumor growth. In a mouse transplant tumor model, oral administration of SM08502 significantly inhibited tumor growth. It was also confirmed to be safe, leading to phase 1 clinical trials on patients with advanced cancer.26
KY1220, which targets Axin, a negative regulator of the Wnt/β-catenin pathway, was identified through screening and further optimized to form KYA1797K.27 KY1220 was able to inhibit the Wnt/β-catenin pathway while simultaneously degrading both β-catenin and Ras; however its poor solubility posed a challenge. To address this, KYA1797K was designed by introducing a potassium salt, which significantly improved solubility. The para-nitro group was identified as critical for maintaining activity, and KYA1797K demonstrated greater efficacy than KY1220.
KYA1797K directly binds to the regulator of G protein signaling (RGS) domain of Axin, promoting the formation of a degradation complex that phosphorylates β-catenin and Ras and thereby reducing their concentrations and inhibiting cancer cell growth. In mice with tumors harboring APC and KRAS mutations, KYA1797K significantly reduced tumor weight and volume, suggesting its efficacy in targeting the Wnt/β-catenin and Ras pathways.
Small molecules that selectively inhibit the interaction between β-catenin and its coactivator BCL9 are also under investigation. ZW4864, designed based on the screening hit compound CP-868388,28 underwent structural modifications to improve its inhibition of the β-catenin/BCL9 interaction. Specifically, the introduction of a piperazine substituent, a cyclopropyl group, and a 1H-pyrazol group improved both activity and selectivity. In patient-derived xenograft models of triple-negative breast cancer, ZW4864 inhibited tumor growth and reduced the expression of β-catenin target genes, while exhibiting high oral bioavailability and tolerability in mice. Additional high-throughput screening identified two lead compounds, C-1 and C-2, which effectively inhibited β-catenin/BCL9 interaction and suppressed colorectal cancer cell proliferation.29 C-1 also significantly inhibited tumor growth in a mouse model with few side effects, underscoring the potential of targeting the β-catenin/BCL9 interaction in cancer therapy.
CBP activates transcription of target genes by binding to β-catenin within the Wnt/β-catenin pathway. β-Catenin/CBP-targeted inhibitors include ICG-001,30,31 its derivative PRI-724,5 and E7386.32 E7386 has demonstrated inhibition of tumor growth in the ECC10 xenograft model and the MMTV-Wnt1 transgenic mouse model, where the Wnt pathway is activated. Additionally, combining E7386 with anti-PD-1 antibodies resulted in potent synergistic effects in MMTV-Wnt1 mouse breast cancer models. E7386 is currently under evaluation in clinical trials for solid tumors, particularly gastrointestinal cancers, and as a combination therapy with other anticancer agents, such as lenvatinib.
The interaction between β-catenin and TCF plays a critical role in the Wnt/β-catenin pathway and is associated with tumorigenesis and progression in various cancers. Inhibitors targeting β-catenin/TCF include PNU-74654,33,34 ZINC02092166,35 LF3,36 and HI-B1.37 PNU-74654, identified through in silico screening, significantly enhanced the efficacy of fluorouracil in colorectal cancer treatment. LF3, discovered via AlphaScreen-based high-throughput screening, underwent further structural analysis, which revealed the importance of the sulfonamide group and benzene tail for its activity. LF3 inhibited endogenous β-catenin/TCF4 interactions in HCT116 cells, suppressing Wnt/β-catenin signaling and tumor growth in mouse models. HI-B1, based on the structure of resveratrol, inhibited β-catenin/TCF4 luciferase activity in a concentration-dependent manner and selectively induced apoptosis in β-catenin-dependent cancer cells. In ApcMin mouse models, HI-B1 suppressed polyp formation, reduced β-catenin-dependent tumorigenesis, and decreased mRNA levels of c-Myc and cyclin D1.
Other inhibitors include allosteric modulators of β-catenin, such as C2,38 which was identified through computational analysis of a newly discovered allosteric site (site C) located in armadillo repeat domains 8 to 10, separate from known binding sites including TCF4, AXIN1, and BCL9. C2 showed selectivity by significantly reducing cell viability at low concentrations (50% inhibitory concentration [IC50]: 0.8–1.3 μM) against high β-catenin-expressing colon cancer cell lines (DLD1 and SW480), while requiring higher concentrations (IC50: 3.45–5.35 μM) for cell lines with low β-catenin expression (HCT116 and SW48). In vivo, C2 markedly inhibited tumor growth and reduced tumor weight in treated mice.
MSAB, discovered through cell-based chemical screening using a luciferase reporter system, promotes β-catenin degradation.39 MSAB strongly inhibited TCF-dependent luciferase activity in a concentration-dependent manner in Wnt-dependent human colon cancer cells (HCT116), with minimal effects on Wnt-independent or normal cells. In mouse xenograft models, MSAB significantly inhibited Wnt-dependent tumor growth and induced apoptosis.
NRX-1532, a small molecule that enhances the interaction between β-catenin and its recognition E3 ligase SCFβ-TrCP, promotes the binding of β-catenin to β-TrCP and facilitates β-catenin degradation in cells.40 Furthermore, derivative molecules such as NRX-103094 and NRX-252114, developed by optimizing the structure from NRX-1532, exhibit improved binding strength and promote the ubiquitination and degradation of β-catenin more efficiently. In particular, NRX-252114 significantly increased the binding affinity of mutated β-catenin to β-TrCP, a normally difficult interaction, and induced the intracellular degradation of β-catenin.
Thus, small-molecule inhibitors targeting various PPIs within the Wnt/β-catenin signaling pathway have been developed to disrupt the aberrant activation of this pathway, which is frequently implicated in cancers and other diseases. These inhibitors aim to interfere with key interactions that drive Wnt signaling, such as those involving DVL, β-catenin, and other critical components of the pathway. The development of small-molecule inhibitors has gained significant attention because of their relatively lower production costs, potential for oral administration, and ease of synthesis compared with larger biologics such as antibodies, which can reduce patient burden and improve accessibility. Despite these advantages, targeting β-catenin directly remains a formidable challenge. The primary obstacle lies in the structure of β-catenin itself. The protein lacks a well-defined binding pocket and its largely flat surface makes it difficult for small molecules to bind effectively and specifically. Traditional small-molecule inhibitors often rely on fitting into grooves or pockets on protein surfaces, a feature that β-catenin does not readily present. Consequently, most small-molecule inhibitors targeting the Wnt/β-catenin pathway act indirectly, by inhibiting upstream components or interactions that facilitate β-catenin stabilization and activity. Additionally, one of the key challenges in the development of small-molecule inhibitors is their potential to cause side effects associated with low target selectivity. This issue arises because small molecules, while adept at entering cells and binding to target proteins, can also interact with off-target proteins, leading to undesirable effects. To mitigate these risks, novel drug discovery approaches are being explored, including the design and use of medium-sized peptides. These peptides have the potential to inhibit PPIs across broader interfaces, offering a middle ground between small molecules and larger biologics. Medium-sized peptides can be engineered to specifically disrupt protein interactions by binding more extensively to the surface of target proteins to increase selectivity and reduce off-target interactions. Overall, while small-molecule inhibitors provide a valuable tool in the modulation of the Wnt/β-catenin pathway, ongoing research and innovation are crucial to address the challenges posed by protein structure and target specificity. The integration of medium-sized peptides into drug development pipelines represents a forward-looking approach that could lead to more effective and safer therapies for conditions driven by Wnt/β-catenin signaling dysregulation. Continued exploration of these strategies will be essential to fully harness the therapeutic potential of targeting this complex signaling network.
Peptide-based inhibitors
While inhibitors of the Wnt/β-catenin pathway have primarily been small-molecule inhibitors, their effectiveness can be limited by the broad interaction surfaces between proteins, which are often not fully inhibited by conventional small molecules. Consequently, medium-sized peptides that can mimic the PPI interface are emerging as a new modality in drug discovery. This section introduces recently developed medium-sized molecular peptides that inhibit the Wnt/β-catenin signaling pathway (Table 2, Fig. 3).| Compound | Structure | Target | Phase (biochemical data) |
|---|---|---|---|
| Abbreviations: BCL9, B cell lymphoma 9; FZD7, Frizzled 7; LRP, lipoprotein receptor-related protein; TCF, T cell factor. | |||
| Fz7-21 | Ac-LPSDDLEFWCHVMY-NH2 | FZD7 | Preclinical (100 nM, HEK293 cells) |
| Lr-EET-3.5 | GCQSNHILKHNRCKQDSDCLAGCVCGPNGFCG | LRP6 | Preclinical (50 nM, HEK293 cells) |
| hsBCL9CT-24 |

|
β-Catenin/BCL9 | Preclinical (191 nM, HCT116 cells) |
| Helical sulfono-γ-AA peptide 4 |
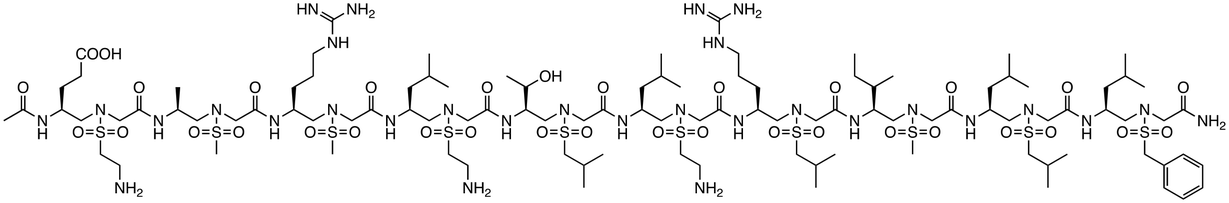
|
β-Catenin/BCL9 | Preclinical (20.6 μM, SW480 cells) |
| StAx-35R | Ac-RRWPRS5ILDS5HVRRVWR-NH2 | β-Catenin/TCF | Preclinical (10 μM, DLD-1 cells, SW480 cells) |
| NLS-StAx-h |

|
β-Catenin/TCF | Preclinical (1.4 μM, HEK293T cells) |
| xStAx-VHLL |

|
β-Catenin | Preclinical (50 μM, HCT116 cells) |
| Penetratin-st7 |
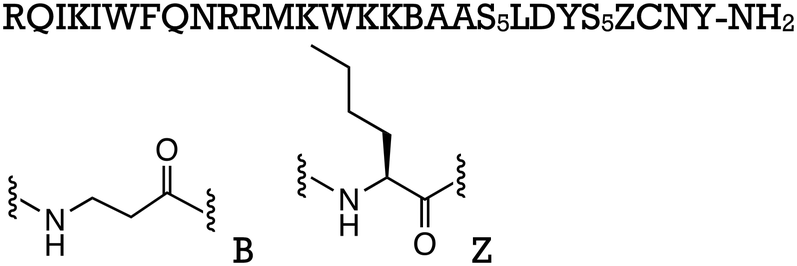
|
β-Catenin/TCF | Preclinical (20 μM, DLD-1 cells) |
| Penetratin-st6 | RQIKIWFQNRRMKWKKBAFS5LLYS5ZRNY-NH2 | β-Catenin/TCF | Preclinical (20 μM, DLD-1 cells) |
| βtCov1 |
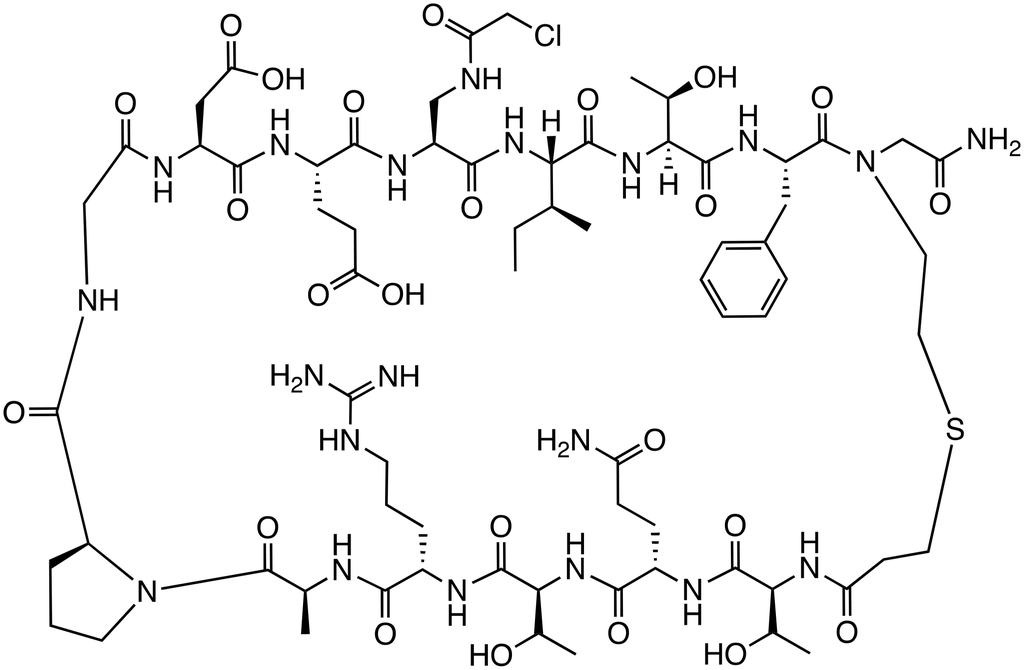
|
β-Catenin/TCF | Preclinical (Ki = 6.0 μM) |
| A-b6 |
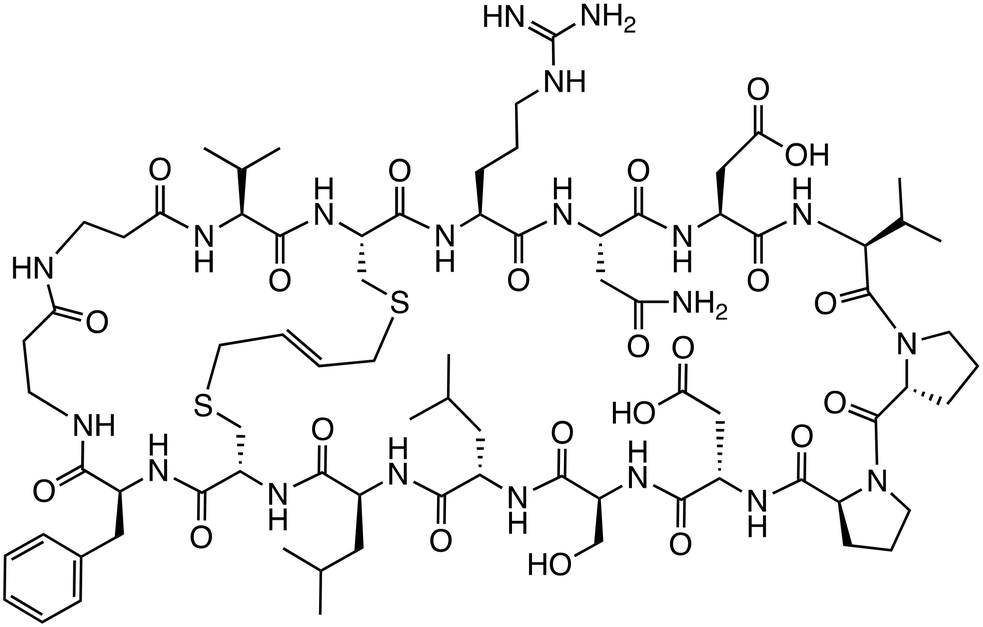
|
β-Catenin/TCF | Preclinical (8 μM, HEK293T) |
 | ||
| Fig. 3 Peptide-based inhibitors acting on the Wnt/β-catenin signaling pathway. Abbreviations: APC, adenomatous polyposis coli; BCL9, B cell lymphoma 9; CBP, cAMP-response element-binding protein; CK1α, casein kinase 1α; DVL, Dishevelled; FZD, Frizzled; GSK3β, glycogen synthase kinase 3β; LRP, lipoprotein receptor-related protein; PROTAC, proteolysis-targeting chimera; TCF/LEF, T cell factor/lymphoid enhancer factor. | ||
The Wnt/β-catenin pathway initiates signaling when Wnt proteins bind to the FZD receptor on the plasma membrane, forming a ternary complex with the co-receptor LRP5/6. Fz7-21, a selective peptide ligand that interferes with the function of the FZD7 receptor, was identified and shown to inhibit the Wnt signaling pathway.41 Additionally, Lr-EET-3.5, a peptide targeting LRP6 identified via phage display from a cystine knot peptide-based library, specifically inhibits Wnt1 binding to the LRP6 receptor.42 Lr-EET-3.5 exhibits high affinity for its target protein (dissociation constant [KD] = 1.01 nM) and specifically inhibits Wnt1-mediated signaling, as demonstrated by luciferase reporter assays.
While inhibitors targeting upstream proteins in the signaling pathway exist, effective inhibition of pathways involved in cancer often requires targeting proteins functioning downstream. Therefore, peptides targeting PPIs that form complexes with β-catenin downstream of the signaling pathway and contribute to transcriptional activation have also been designed. In the Wnt/β-catenin pathway, β-catenin accumulates in the nucleus, interacting with LEF/TCF to participate in the transcription of target genes and binding to the coactivator BCL9 to enhance transcriptional activation. The peptide hsBCL9CT-24 was developed as a PPI inhibitory peptide targeting the β-catenin/BCL9 interaction.43 hsBCL9CT-24 strongly inhibits the transcriptional activity of β-catenin and exhibits marked antitumor effects in animal models. This peptide activates an anti-tumor immune response by decreasing regulatory T cells and increasing dendritic cells in tumors. Furthermore, when combined with PD-1 inhibitors, its therapeutic effect was significantly enhanced, suggesting that hsBCL9CT-24 could be a promising strategy to overcome resistance to immune checkpoint inhibitors through inhibition of the β-catenin/BCL9 interaction. Additionally, helical sulfo-γ-AA peptides that mimic the α-helical HD2 domain of BCL9, which interacts with β-catenin, have been designed to selectively inhibit the β-catenin/BCL9 interaction.44 These peptides are cell-permeable, bind to β-catenin, inhibit the interaction, and suppress cancer cell growth. They are also enzymatically stable, showing resistance to degradation for over 24 h in the presence of pronase. Assays such as MTS and luciferase reporter assays have demonstrated that sulfo-γ-AA peptides exhibit high specificity and potency against Wnt/β-catenin signaling-dependent cancer cells.
Several peptides have also been developed to inhibit the interaction between β-catenin and TCF. Interaction of these two proteins induces the formation of two helices at the C- and N-terminal sides. StAx-35R, derived from Axin, was designed to target the C-terminal helix of TCF.13 Although StAx-35R effectively inhibited the interaction between β-catenin and TCF, its poor intracellular permeability limited its biological activity. To improve cell permeability, Grossmann et al. modified the peptide sequence using arginine derivatives and introduced various hydrophobic and polar groups at the N-terminus. This resulted in the compounds NLS-StAx-h, which exhibited excellent cell membrane permeability and effectively suppressed the expression of target genes.45 Furthermore, a new PROTAC peptide, xStAx-VHLL, was designed to target the Wnt/β-catenin signaling pathway.12 PROTACs are bifunctional molecules that induce the degradation of target proteins, with one end binding to the target protein and the other to an E3 ubiquitin ligase, leading to ubiquitination and proteasomal degradation of the target. xStAx-VHLL binds the Axin-derived peptide xStAx to the von Hippel–Lindau (VHL) ligand, resulting in sustained degradation of β-catenin. Studies have shown that xStAx-VHLL potently inhibits Wnt signaling in cancer cells and effectively suppresses tumorigenesis in mouse models. Furthermore, it significantly inhibited organoid survival in 11 of 12 patient-derived colorectal cancer organoids. xStAx-VHLL represents a promising new therapeutic tool in Wnt signaling-mediated cancer therapy.
The N-terminal region of TCF is also important for its interaction with β-catenin. A PPI inhibitory peptide was developed to target this interaction, as the N-terminal region of TCF overlaps with the binding site of the nuclear receptor liver receptor homolog-1 on β-catenin. To enhance intracellular stability, an amino acid side-chain cross-link was introduced and the peptide was conjugated with the cell-penetrating peptide penetratin. The resulting peptide, Penetratin-st7, exhibited high enzyme tolerance and specifically inhibited the proliferation of DLD-1 cells.46 To further enhance intracellular inhibitory activity, peptide sequences were optimized using computational methods. Penetratin-st6, developed using the Molecular Operating Environment (MOE) software, specifically inhibited the growth of DLD-1 colon cancer cells at approximately 5 μM, showing approximately four times greater inhibitory activity than Penetratin-st7.47 This study demonstrated the feasibility of using rational in silico design for PPI inhibitory peptides targeting β-catenin, paving the way for designing peptide inhibitors targeting other proteins. Additionally, βtCov1, which stabilizes the extended strand region at the TCF N-terminus as a β-hairpin structure, was developed.48 βtCov1 shows specific covalent binding to the cysteine residue of β-catenin, offering a strategy for targeting PPIs via β-strand stabilization. A-b6, a stable β-sheet structure, was designed based on the sequence of E-cadherin binding to β-catenin.49 A-b6 competitively inhibited the interaction of β-catenin with TCF and showed excellent plasma membrane permeability. Furthermore, computational methods have been used to design peptoid PPI inhibitory peptides. Cyclic peptoids developed through these methods effectively inhibited the Wnt signaling pathway and androgen receptor signaling activation.50 In a zebrafish model, these peptoids strongly inhibited Wnt signaling, confirming their therapeutic potential. Thus, cyclic peptoids designed through computational methods effectively inhibited the β-catenin/TCF interaction, showing promise in prostate cancer treatment.
The exploration of peptides as PPI inhibitors has gained momentum in recent years, driven by the recognition that many critical PPIs are challenging to target using traditional small molecules. As highlighted in this review, numerous PPI inhibitory peptides with specialized secondary structures or innovative chemical modifications are being developed to address these challenges. These peptides are designed to fit into the extended, often flat interfaces of PPIs, which are typically considered undruggable by conventional small molecules. Notable among these are α-helical peptides, β-hairpin mimetics, and constrained peptides that have been chemically modified to enhance their stability, binding affinity, and specificity. The design of these specialized PPI inhibitory peptides involves several advanced methodologies. Conventional approaches such as X-ray crystallography provide detailed structural insights into target proteins and their interactions, enabling precise peptide sequence design to disrupt these interfaces effectively. Phage display is another powerful technique widely used for peptide selection as it allows the screening of vast peptide libraries against target proteins, thus facilitating the identification of peptides with high affinity and specificity. This method has been instrumental in discovering peptides that can modulate challenging targets, including those involved in the Wnt/β-catenin pathway.
Computational techniques, including molecular dynamics simulations and in silico docking, have also become mainstream in the peptide design process. These methods allow for the virtual screening of peptide libraries, optimization of peptide sequences, and prediction of binding modes, significantly accelerating the development timeline and reducing the costs associated with experimental screening. Computational tools are particularly valuable for refining peptide structures to improve their drug-like properties, such as cell permeability and resistance to proteolysis, which are critical for their success as therapeutics. One of the most promising advancements in the field is the random nonstandard peptide integrated discovery (RaPID) system,51 which enables the rapid development of cyclic peptides that can bind to a wide range of protein targets with high affinity and specificity. This system utilizes mRNA display combined with nonstandard amino acid incorporation, allowing the generation of cyclic peptides with diverse and complex structures that are otherwise difficult to achieve through conventional synthetic methods. The RaPID system has been increasingly used to develop specialized peptides for pharmaceutical applications. These cyclic peptides can effectively target challenging proteins like β-catenin, which lack well-defined binding pockets, making them otherwise undruggable by small molecules.
The use of these advanced design techniques and systems has the potential to revolutionize cancer therapy by making previously undruggable targets, such as β-catenin, druggable. By disrupting key PPIs within the Wnt/β-catenin signaling pathway, these peptides offer new avenues for therapeutic intervention in cancers where this pathway shows aberrant activation. Furthermore, the versatility and adaptability of peptide-based inhibitors enable the targeting of a broad spectrum of proteins involved in various oncogenic processes, expanding the therapeutic landscape beyond what is achievable with small molecules alone. These innovations underscore a broader shift in drug discovery towards more sophisticated and tailored approaches, leveraging the unique capabilities of peptides to tackle complex biological challenges. As research in this area continues to evolve, it is anticipated that peptide therapeutics will play an increasingly prominent role in the treatment of cancer and other diseases driven by dysregulated PPIs.
Nucleic acid-based inhibitors
The COVID-19 pandemic has profoundly impacted health, society, and the economy worldwide, underscoring the potential of nucleic acid-based therapeutics, particularly as the rapid development and widespread use of mRNA vaccines became a pivotal tool in the fight against the virus. Nucleic acid drugs are chemically synthesized molecules that regulate gene expression using nucleic acid molecules (DNA and RNA). These therapeutics include antisense oligonucleotides (ASOs), which bind to specific mRNAs and inhibit their translation; siRNAs, which suppress gene expression by degrading specific mRNAs; and decoy nucleic acids, which interfere with gene expression by competitively binding to target proteins such as transcription factors. Although the use of this approach remains limited, a few nucleic acid molecules targeting the Wnt/β-catenin pathway have been developed (Table 3, Fig. 4).| Compound | Target | Phase |
|---|---|---|
| Abbreviation: ASO, antisense oligonucleotide. | ||
| DCR-BCAT | CTNNB1 | Preclinical |
| PKMYT1AR ASO#1 | PKMYT1AR | Preclinical |
| PKMYT1AR ASO#2 | ||
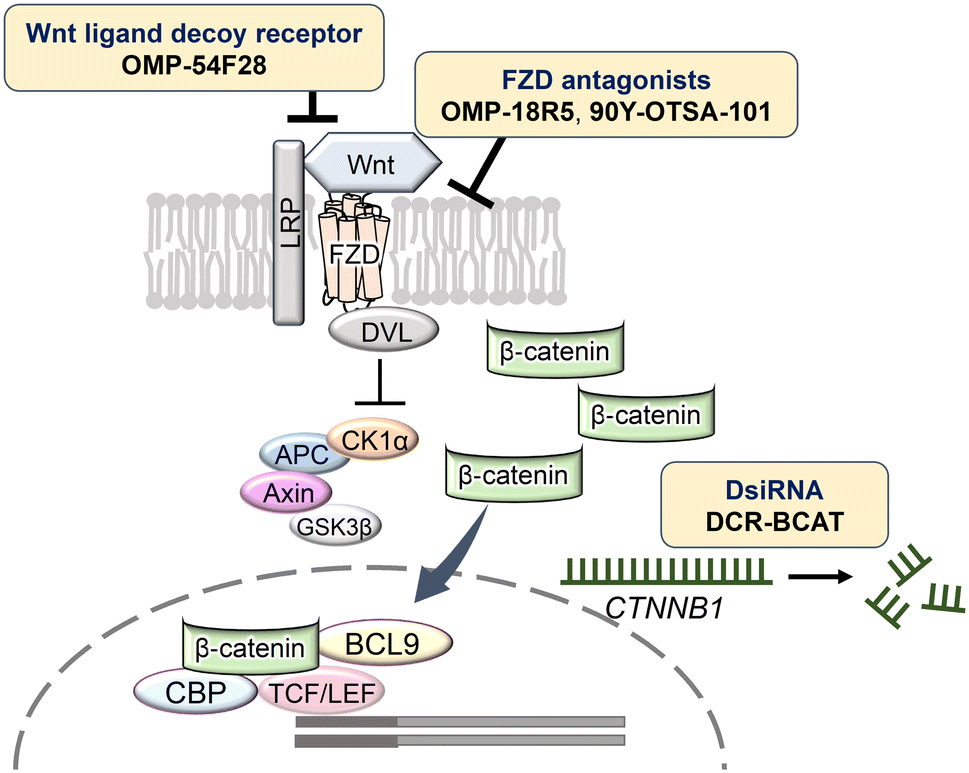 | ||
| Fig. 4 Nucleic acid-based and macromolecular inhibitors acting on the Wnt/β-catenin signaling pathway. Abbreviations: APC, adenomatous polyposis coli; BCL9, B cell lymphoma 9; CBP, cAMP-response element-binding protein; CK1α, casein kinase 1α; DsiRNA, Dicer-substrate RNA; DVL, Dishevelled; FZD, Frizzled; GSK3β, glycogen synthase kinase 3β; LRP, lipoprotein receptor-related protein; TCF/LEF, T cell factor/lymphoid enhancer factor. | ||
Ganesh et al. utilized RNA interference (RNAi) technology to explore strategies for targeting β-catenin, which is challenging to inhibit using conventional small molecules and antibodies. Specifically, the authors developed Dicer-substrate siRNA (DsiRNA) targeting CTNNB1, the gene encoding β-catenin, formulated as DCR-BCAT in lipid nanoparticles.52–54 DsiRNA is a double-stranded RNA molecule designed to target and silence the mRNA of a specific gene, offering greater stability than standard siRNAs. DCR-BCAT, targeting CTNNB1, has been reported to significantly inhibit tumor growth in the MMTV-Wnt1 breast cancer mouse model. Furthermore, DCR-BCAT demonstrated complete tumor regression and increased CD8+ T cell infiltration when combined with PD-1/cytotoxic T-lymphocyte antigen-4 immunotherapy. It also improved survival in a mouse model of human colorectal cancer with liver metastases by reducing CTNNB1 mRNA by more than 50%. Additionally, in combination with the mitogen-activated protein kinase inhibitor trametinib, DCR-BCAT markedly inhibited tumor growth and prolonged survival.
Other researchers have developed ASOs targeting PKMYT1AR, a long non-coding RNA (lncRNA).55 LncRNAs are known to play crucial roles in cancer progression and PKMYT1 is highly expressed in non-small cell lung cancer where it activates the Wnt signaling pathway. Its translation is normally inhibited by miR-485-5p, a tumor suppressor. However, when PKMYT1AR suppresses miR-485-5p, PKMYT1 expression increases, activating the Wnt signaling pathway. To counteract this, two ASOs targeting PKMYT1AR were designed and shown to effectively repress PKMYT1AR transcripts. In a nude mouse model where A549 cells were transplanted subcutaneously, these ASOs inhibited tumor growth and reduced the expression of cancer stem cell markers CD44, Sox2, and β-catenin.
Thus, nucleic acid-based inhibitors targeting the Wnt signaling pathway are being developed and have demonstrated significant tumor suppressive effects. Despite the promising results, the clinical translation of nucleic acid-based inhibitors faces several challenges, including issues related to delivery, stability, and off-target effects. Advances in delivery technologies, such as the use of lipid nanoparticles and conjugation with targeting ligands, are being explored to enhance the stability and specificity of these nucleic acid drugs to improve their therapeutic efficacy and safety profile. Additionally, the use of chemically modified nucleotides, such as locked nucleic acids and peptide nucleic acids, can further enhance the stability and binding affinity of nucleic acid inhibitors, making them more effective in the physiological environment.56 As research in this field continues to evolve, nucleic acid-based drugs hold the potential to become a transformative treatment option for cancers involving the Wnt signaling pathway. By providing a mechanism to precisely target and modulate the expression of key oncogenic drivers within this pathway, nucleic acid inhibitors could complement or even surpass traditional small-molecule and antibody therapies. Ongoing advancements in delivery and design technologies will be crucial in overcoming the current limitations and fully realizing the potential of these innovative therapeutic agents in clinical settings.
Macromolecular inhibitors
While the development of inhibitors targeting β-catenin/TCF in the Wnt/β-catenin pathway has been a primary focus, targeting the undruggable β-catenin remains challenging. Recently, advancements in understanding the activation mechanisms of the Wnt/β-catenin pathway have led to the development of macromolecular inhibitors, including antibodies that target Wnt ligands and receptors involved in Wnt signaling (Table 4, Fig. 4).| Compound | Target | Phase |
|---|---|---|
| a Radiolabeled monoclonal antibody developed for the radioimmunotherapy of synovial sarcoma. Abbreviations: FZD, Frizzled; ROR1, receptor tyrosine kinase-like orphan receptor 1. | ||
| OMP-18R5 | FZD | Phase 1 |
| OMP-54F28 | Wnt ligand | Phase 1 |
| 90Y-OTSA-101a | FZD10 | Phase 1 |
| OMP-131R10 | R-spondin 3 | Phase 1 |
| UC-961 | ROR1 | Phase 2 |
A notable example is OMP-18R5, a monoclonal antibody that targets FZD.9,10 OMP-18R5 has been shown to inhibit Wnt signaling by binding to FZD, thereby suppressing the growth of several tumors, including breast, pancreatic, colon, and lung cancers. OMP-18R5 is currently undergoing clinical trials.
Another significant development is OMP-54F28, a Wnt ligand decoy receptor. This fusion protein comprises the extracellular domain of FZD8 fused to the Fc region of human immunoglobulin G1, which binds to Wnt ligands and inhibits signaling.57,58 In preclinical models, OMP-54F28 has demonstrated tumor growth inhibition when used both as a monotherapy and in combination with chemotherapeutic agents. Phase I clinical trials have been conducted in hepatocellular, ovarian, and pancreatic cancers.
90Y-OTSA-101 is a radiolabeled monoclonal antibody developed for the radioimmunotherapy of synovial sarcoma.59 Its target, FZD10, is part of the seven transmembrane receptor family associated with the Wnt signaling pathway. FZD10 is specifically overexpressed in synovial sarcoma cells and is rarely present in normal adult tissues, allowing the anti-FZD10 antibody (OTSA-101) to bind selectively to tumor cells. The radionuclide yttrium-90 (90Y), a potent beta emitter, is effective in destroying tumor cells. 90Y-OTSA-101 has shown significant tumor shrinkage in mouse models of periosteal osteosarcoma. However, hematological toxicity was observed in clinical trials, indicating the need for molecular redesign, such as switching to a lower-energy radioisotope. Another target, R-spondin 3, an activator of the Wnt signaling pathway, may be inhibited by the monoclonal antibody OMP-131R10.60 In addition, UC-961,61,62 a monoclonal antibody targeting the receptor tyrosine kinase-like orphan receptor 1, has been developed.
Antibody- and fusion protein-based inhibitors have emerged as promising therapeutic agents because of their high specificity and ability to target proteins with complex or extended surfaces that are typically undruggable by small molecules. In the context of Wnt/β-catenin-mediated diseases, these inhibitors can selectively bind to key components of the pathway, such as FZD, Wnt ligands, or β-catenin itself, thereby disrupting the signaling cascade that drives tumorigenesis and other pathological conditions. Because of their precision and high affinity, antibody-based therapies have demonstrated significant therapeutic efficacy in preclinical and clinical settings, offering targeted intervention with minimal off-target effects when compared with conventional therapies. Despite their high specificity and efficacy, antibody- and fusion protein-based inhibitors face several significant challenges. One of the primary limitations is their high manufacturing costs, which can make these therapies expensive and limit access for patients. The production of monoclonal antibodies involves complex processes such as mammalian cell culture, purification, and stringent quality control measures, all of which contribute to the overall cost. Additionally, these biologics are typically administered via intravenous or subcutaneous injection, as they cannot be delivered orally because of degradation in the gastrointestinal tract and poor bioavailability. This mode of administration can be inconvenient for patients, requiring frequent hospital visits and specialized handling. To address these limitations, there is a growing need for the development of small- and medium-molecule inhibitors that can offer similar specificity and efficacy but with lower costs and improved patient convenience. The continued development and refinement of small- and medium-molecule inhibitors are crucial for overcoming the limitations associated with antibody and fusion protein therapies. By combining the specificity and potency of biologics with the practical advantages of small molecules, these new classes of inhibitors have the potential to expand therapeutic options for patients with Wnt/β-catenin-mediated diseases, offering effective and accessible treatment modalities. As research progresses, these inhibitors could play a central role in the next generation of targeted therapies for cancer and other conditions driven by aberrant Wnt signaling.
Conclusions
As highlighted throughout this review, abnormalities in the Wnt/β-catenin pathway are implicated in a wide range of cancers, prompting the design of various inhibitors. However, despite recent progress, no compounds have yet reached the market, underscoring the ongoing need for innovative approaches. This review has summarized the development of inhibitors, including small molecules, peptides, nucleic acids, and polymers.Small-molecule inhibitors have been developed to target multiple points both upstream and downstream of the signaling pathway, with some currently in clinical trials. Nevertheless, challenges such as low target specificity and associated toxicity persist. The efficacy of small-molecule inhibitors is highly dependent on their stability and pharmacokinetic properties, necessitating sufficient stability and effective distribution to the target site. In recent years, high-throughput screening and structure-based drug design have been employed to improve selectivity and physicochemical properties. Specifically, efforts have been focused on enhancing cellular permeability and stability by substituting carboxylic acid groups with bioisosteres and adjusting lipophilicity.63,64 Peptide-based inhibitors have gained increased attention in recent years, with designs leveraging a variety of innovative approaches. These inhibitors show great promise because of their ability to interact with proteins over broader interfaces than small molecules, offering high specificity. They are also amenable to chemical modifications, and some can be administered orally. Recently, advancements have been made in developing next-generation peptide drugs with enhanced degradation resistance and cell membrane permeability through special chemical modifications. The design of cyclic peptides using the RaPID system is also gaining traction. These new technologies and approaches may significantly advance the development of inhibitors targeting the challenging Wnt/β-catenin pathway.
Macromolecular inhibitors, which are expected to provide high target specificity and therapeutic efficacy, are also being developed alongside nucleic acid-based drugs. As discussed, nucleic acid drugs have demonstrated strong inhibitory effects against tumors involving Wnt/β-catenin signaling. Beyond their therapeutic potential, nucleic acid drugs are emerging as a new modality because of their chemical synthesizability, making them potentially more cost-effective than antibody drugs. However, challenges such as cell membrane permeability and in vivo stability remain, necessitating the implementation of drug delivery systems and specific chemical modifications.
As the development of Wnt/β-catenin pathway inhibitors continues, there is a growing recognition of the need for personalized medicine approaches. Identifying biomarkers that can predict responses to Wnt inhibitors will be crucial for tailoring treatments to individual patients and maximizing therapeutic efficacy. For example, tumors with specific mutations in the Wnt pathway, such as APC or CTNNB1 mutations, may be more susceptible to certain inhibitors, allowing for a more targeted treatment approach.65,66 Moreover, combination therapies that include Wnt/β-catenin pathway inhibitors alongside other anticancer agents, such as immune checkpoint inhibitors or chemotherapeutics, hold potential for synergistic effects. By targeting multiple pathways involved in tumor growth and survival concurrently, these combination strategies could overcome resistance mechanisms and improve patient outcomes. The integration of emerging technologies, such as CRISPR-based gene editing and PROTACs, into the development of Wnt pathway inhibitors also presents exciting opportunities. CRISPR technology could be used to selectively knock out key components of the Wnt pathway in cancer cells, while PROTACs offer a novel approach to degrading undruggable proteins like β-catenin. These technologies, when combined with the interdisciplinary approaches discussed, could revolutionize the way Wnt/β-catenin pathway inhibitors are developed and applied in clinical settings. The Wnt/β-catenin pathway remains a complex and challenging target for cancer therapy. However, the integration of computational science, organic chemistry, and biochemistry provides a powerful toolkit for turning previously undruggable targets into viable drug targets. Continued innovation, collaboration, and investment in these interdisciplinary approaches will be essential for advancing the development of effective therapies targeting the Wnt/β-catenin pathway, ultimately leading to improved outcomes for patients with Wnt-driven cancers and other diseases.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
M. F. and Y. D. wrote and edited the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by grants from AMED (grant numbers 24mk0121286, 24ama221127, and 24ak0101185, all to Y. D.). The study also received support from the Japan Society for the Promotion of Science (KAKENHI, grant numbers 21K05320 and 23H04926, both to Y. D.), and JST SPRING (grant number JPMJSP2179 to M. F.). We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.Notes and references
- P. Polakis, Cold Spring Harbor Perspect. Biol., 2012, 4, a008052 CrossRef
.
- B. T. MacDonald, K. Tamai and X. He, Dev. Cell, 2009, 17, 9–26 CrossRef CAS
- J. Liu,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20224–20229 CrossRef CAS PubMed
- J. Rodon,
et al.
, Br. J. Cancer, 2021, 125, 28–37 CrossRef CAS PubMed
- H. J. Lenz and M. Kahn, Cancer Sci., 2014, 105, 1087–1092 CrossRef CAS PubMed
- J. H. Lee,
et al.
, Blood Adv., 2020, 4, 2032–2043 CrossRef CAS PubMed
- S. Pak,
et al.
, J. Exp. Clin. Cancer Res., 2019, 38, 342 CrossRef PubMed
- Y. D. Benoit,
et al.
, Cell Chem. Biol., 2017, 24, 833–844 CrossRef CAS
- A. Gurney,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11717–11722 CrossRef CAS PubMed
- P. N. Le,
et al.
, Mol. Carcinog., 2019, 58, 398–410 CrossRef CAS PubMed
- J. N. Anastas and R. T. Moon, Nat. Rev. Cancer, 2013, 13, 11–26 CrossRef CAS
- H. Liao,
et al.
, Cell Discovery, 2020, 6, 35 CrossRef CAS PubMed
- T. N. Grossmann,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942–17947 CrossRef CAS
- H. Clevers and R. Nusse, Cell, 2012, 149, 1192–1205 CrossRef CAS PubMed
- L. Vermeulen,
et al.
, Nat. Cell Biol., 2010, 12, 468–476 CrossRef CAS
- N. Fujii,
et al.
, Cancer Res., 2007, 67, 573–579 CrossRef CAS PubMed
- D. Grandy,
et al.
, J. Biol. Chem., 2009, 284, 16256–16263 CrossRef CAS
- D. Cheng,
et al.
, ACS Med. Chem. Lett., 2016, 7, 676–680 CrossRef CAS
- Z. Zhong,
et al.
, Oncogene, 2019, 38, 6662–6677 CrossRef CAS
- C. Li,
et al.
, Mol. Ther., 2019, 27, 1558–1567 CrossRef CAS PubMed
- C. Phillips,
et al.
, Cancer Res. Commun., 2022, 2, 914–928 CrossRef CAS
- S. M. Huang,
et al.
, Nature, 2009, 461, 614–620 CrossRef CAS
- E. W. Stratford,
et al.
, Cancer Med., 2014, 3, 36–46 CrossRef PubMed
- S. R. Martins-Neves,
et al.
, Cancer Lett., 2018, 414, 1–15 CrossRef CAS
- B. Li,
et al.
, Sci. Signaling, 2017, 10, eaak9916 CrossRef
- B. Y. Tam,
et al.
, Cancer Lett., 2020, 473, 186–197 CrossRef CAS PubMed
- P. H. Cha,
et al.
, Nat. Chem. Biol., 2016, 12, 593–600 CrossRef CAS PubMed
- Z. Wang,
et al.
, J. Med. Chem., 2021, 64, 12109–12131 CrossRef CAS
- H. Tanton,
et al.
, Sci. Adv., 2022, 8, eabm3108 CrossRef CAS PubMed
- K. H. Emami,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12682–12687 CrossRef CAS
- M. Che,
et al.
, Cancers, 2020, 12, 1476 CrossRef CAS
- K. Yamada,
et al.
, Cancer Res., 2021, 81, 1052–1062 CrossRef CAS PubMed
- F. Amerizadeh,
et al.
, Tissue Cell, 2022, 77, 101853 CrossRef CAS PubMed
- L. F. Leal,
et al.
, Onco Targets Ther, 2015, 6, 43016–43032 Search PubMed
- J. L. Catrow, Y. Zhang, M. Zhang and H. Ji, J. Med. Chem., 2015, 58, 4678–4692 CrossRef CAS PubMed
- L. Fang,
et al.
, Cancer Res., 2016, 76, 891–901 CrossRef CAS
- S. H. Shin,
et al.
, EBioMedicine, 2017, 25, 22–31 CrossRef
- A. Cheltsov,
et al.
, Sci. Rep., 2020, 10, 8096 CrossRef CAS
- S. Y. Hwang,
et al.
, Cell Rep., 2016, 16, 28–36 CrossRef CAS
- K. R. Simonetta,
et al.
, Nat. Commun., 2019, 10, 1402 CrossRef
- A. H. Nile,
et al.
, Nat. Chem. Biol., 2018, 14, 582–590 CrossRef CAS PubMed
- S. Hansen,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207327119 CrossRef CAS
- M. Feng,
et al.
, Sci. Adv., 2019, 5, eaau5240 CrossRef CAS
- P. Sang,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10757–10762 CrossRef CAS PubMed
- L. Dietrich,
et al.
, Cell Chem. Biol., 2017, 24, 958–968 CrossRef CAS
- K. Tsuchiya,
et al.
, Bioorg. Med. Chem., 2022, 73, 117021 CrossRef CAS
- M. Fujita,
et al.
, Bioorg. Med. Chem., 2023, 84, 117264 CrossRef CAS
- S. L. Blosser, N. Sawyer, I. Maksimovic, B. Ghosh and P. S. Arora, ACS Chem. Biol., 2021, 16, 1518–1525 CrossRef CAS
- M. Wendt,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 13937–13944 CrossRef CAS
- J. A. Schneider,
et al.
, Nat. Commun., 2018, 9, 4396 CrossRef PubMed
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 CrossRef CAS
- S. Ganesh,
et al.
, Mol. Cancer Ther., 2016, 15, 2143–2154 CrossRef CAS PubMed
- S. Ganesh,
et al.
, Mol. Cancer Ther., 2018, 17, 544–553 CrossRef CAS PubMed
- S. Ganesh,
et al.
, Mol. Ther., 2018, 26, 2567–2579 CrossRef CAS
- Y. He,
et al.
, Mol. Cancer, 2021, 20, 156 CrossRef CAS
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef CAS
- A. Jimeno,
et al.
, Clin. Cancer Res., 2017, 23, 7490–7497 CrossRef CAS
- P. N. Le, J. D. McDermott and A. Jimeno, Pharmacol. Ther., 2015, 146, 1–11 CrossRef CAS
- A. L. Giraudet,
et al.
, BMC Cancer, 2018, 18, 646 CrossRef PubMed
- M. Zhang,
et al.
, PLoS One, 2020, 15, e0229445 CrossRef CAS PubMed
- M. Y. Choi,
et al.
, Clin. Lymphoma, Myeloma Leuk., 2015, 15, S167–S169 CrossRef
- J. Yu,
et al.
, J. Clin. Invest., 2016, 126, 585–598 CrossRef PubMed
- P. Yang,
et al.
, Eur. J. Med. Chem., 2022, 243, 114789 CrossRef CAS
- X. Zhang,
et al.
, Curr. Top. Med. Chem., 2023, 23, 880–896 CrossRef CAS
- F. C. Geyer,
et al.
, Mod. Pathol., 2011, 24, 209–231 CrossRef CAS
- T. Lau,
et al.
, Cancer Res., 2013, 73, 3132–3144 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |